

Abstract
Context: The nitrobezoxadiazole derivative NBDHEX is a potent inhibitor of glutathione transferase P1-1 (GSTP1-1) endowed with outstanding anticancer activity in different tumor models.
Objective: To characterize by in vitro biochemical and in silico studies the NBDHEX analogues named MC2752 and MC2753.
Materials and methods: Synthesis of MC2752 and MC2753, biochemical assays and in silico docking and normal-mode analyses.
Results: The presence of a hydrophobic moiety in the side chain of MC2753 confers unique features to this molecule. Unlike its parent drug NBDHEX, MC2753 does not require GSH to trigger the dissociation of the complex between GSTP1-1 and TRAF2, and displays high stability towards the nucleophilic attack of the tripeptide under physiological conditions.
Discussion and conclusion: MC2753 may represent a lead compound for the development of novel GSTP1-1 inhibitors not affected in their anticancer action by fluctuations of cellular GSH levels, and characterized by an increased half-life in vivo.
Keywords: GSH, GSTP1-1, NBDHEX, nitrobenzoxadiazoles, TRAF2
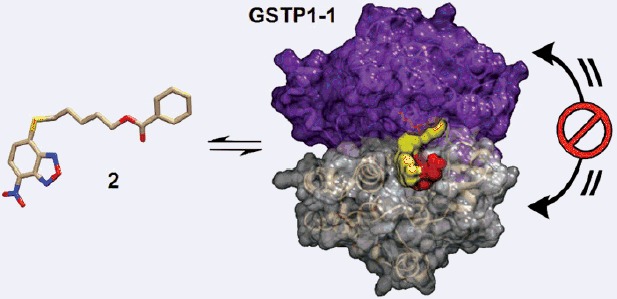
Introduction
Glutathione transferases (formerly glutathione S-transferases; GSTs; EC 2.5.1.18) constitute one of the most important families of detoxifying enzymes and are also involved in the development of multidrug-resistance in tumors1. The ability of GSTs to promote conjugation of the tripeptide glutathione (GSH) with a variety of substrates leads to inactivation of various reactive compounds, including some anticancer drugs. Moreover, tumor cell overexpression of the Pi-class isoform glutathione transferase P1-1 (GSTP1-1) has been associated with inhibition of apoptosis through direct interaction of this enzyme with the mitogen-activated protein kinase (MAPK) named c-Jun-N-terminal Kinase 1 (JNK1), and the scaffold protein TNFα-receptor-associated-factor 2 (TRAF2). Therefore, a new promising strategy for tumor treatment may be the use of agents capable of inducing apoptosis by targeting GSTP1-11.
In a search for selective GST inhibitors, we identified the nitrobenzoxadiazole (NBD) derivative NBDHEX (1), as a potent inhibitor of GSTP1-12. This compound behaves as a mechanism-based inhibitor of human GSTP1-1, by first being recognized as a substrate, followed by conjugation to GSH, to form an intermediate σ-complex which binds very tightly (Kd = 10−9 M) to the protein, with subsequent loss of both enzyme (GSH-conjugating) activity and capability to form complexes with the partner proteins JNK1 and TRAF23–6. Compound 1 was found to exhibit remarkable pro-apoptotic activity towards cultured tumor cells of different origin, as well as therapeutic activity in some human tumor xenograft models1 , 7 , 8. More recently, several analogs of 1 bearing variously substituted alkyl chains or aryl moieties at the C4-sulfur atom have been prepared with the aim of obtaining compounds endowed with more favorable pharmaceutical and/or pharmacological properties9. Here we report the design and synthesis of two novel derivatives of 1, namely compound 2 (MC2753), i.e. the benzoic acid ester of 1, and compound 3 (MC2752), i.e. the acetic acid ester of 1 (see Scheme 1). We focused our attention on 2 since the presence of a hydrophobic moiety in the side chain changes the mode of interaction of the NBD derivative with the target protein (i.e. GSTP1-1), and remarkably decreases its spontaneous reactivity towards GSH.
Scheme 1.

Preparation of compounds 2 and 3. (a) Benzoyl chloride, TEA, dry DCM, 0 °C to rt, 22 h; (b) acetyl chloride, TEA, dry DCM, 0 °C to rt, 6 h.
Materials and methods
Chemicals
Unless specified otherwise, all chemicals used throughout this work were purchased from Sigma-Aldrich (St. Louis, MO). GS-NDB was synthesised as previously described2.
Chemistry
Melting points were determined on a Buchi 530 melting point apparatus (Flawil, Switzerland) and are uncorrected. 1H-NMR spectra were recorded at 400 MHz on a Bruker AC 400 spectrometer (Billerica, MA); reporting chemical shifts in δ (ppm) units relative to the internal reference tetramethylsilane (Me4Si). All compounds were routinely checked by TLC and 1H-NMR. TLC was performed on aluminum-backed silica gel plates (Merck DC, Alufolien Kieselgel 60 F254, Kenilworth, NJ) with spots visualized by UV light. Yields of all reactions refer to the purified products. All chemicals were of the highest purity. Mass spectra were recorded on an API-TOF Mariner by Perspective Biosystem; samples were injected by an Harvard pump using a flow rate of 5–10 μL/min, infused in the Electrospray system. Elemental analyses were performed by a PE 2400 (Perkin-Elmer, Waltham, MA) analyzer and have been used to determine purity of the described compounds, which is >95%. Analytical results are within ±0.40% of the theoretical values.
General procedure for the preparation of 2 and 3: Example: 6-((7 nitrobenzo[c][1,2,5]oxadiazol-4-yl)thio)hexyl benzoate (2)
A solution of benzoyl chloride (94.55 mg, 0.078 mL, 0.672 mmol) in dry DCM (2 mL) was added dropwise at 0 °C to a solution of 1 (100 mg, 0.336 mmol) and TEA (85.08 mg, 0.117 mL, 0.840 mmol) in dry DCM (3 mL), and the resulting mixture was stirred at room temperature for 22 h. The mixture was then diluted with 10 mL of DCM and washed with HCl 2 N (2 × 5 mL), NaHCO3 saturated solution (3 × 5 mL) and finally with brine (1 × 5 mL). The organic phase was dried over Na2SO4, filtered and evaporated under vacuum providing a crude solid, which was purified by a silica gel flash chromatography (SNAP 50, Biotage Isolera One™, Uppsala, Sweden) using a linear gradient of AcOEt (5–30%) in Hexane to give 2 as a pure yellow solid. Yield: 76%. M.p.: 84–85 °C. 1H-NMR (CDCl3) δ 1.56–1.58 (m, 2H CH2CH2CH2CH2OCO), 1.60–1.67 (m, 2H, CH2CH2CH2OCO), 1.80–1.84 (m, 2H, SCH2CH2), 1.86–1.93 (m, 2H, CH2CH2OCO), 3.28–3.32 (t, 2H, SCH2), 4.35–4.38 (t, 2H, CH2OCO), 7.14–7.16 (d, 1H, CH benzoxadiazole ring), 7.44–7.47 (t, 2H, CH benzene ring), 7.56–7.60 (t, 1H, CH benzene ring), 8.04–8.06 (d, 2H, CH benzene ring), 8.40–8.42 (d, 1H, CH benzoxadiazole ring). 13C-NMR (CDCl3) δ 25.3, 28.4, 30.5, 30.8, 31.4, 64.5, 119.1, 124.4, 126.2, 128.4 (2C), 129.7 (2C), 130.1, 133.2, 135.6, 144.1, 143.9, 165.8 ppm. Anal. (C19H19N3O5S) Calcd. (%): C, 56.85; H, 4.77; N, 10.47; S, 7.99. Found (%): C, 56.15; H, 4.62; N, 10.59; S, 8.07. MS (ESI), m/z: 402 [M + H]+. 3: Yield: 73%. M.p.: 57–58 °C. 1H-NMR (CDCl3) δ 1.41–1.48 (m, 2H CH2CH2CH2CH2OCO), 1.58–1.62 (m, 2H, CH2CH2CH2OCO), 1.64–1.71 (m, 2H, SCH2CH2), 1.83–1.91 (m, 2H, CH2CH2OCO), 2.05 (s, 3H, OCOCH3), 3.26–3.30 (t, 2H, SCH2), 4.06–4.09 (t, 2H, CH2OCO), 7.11–7.18 (d, 1H, CH benzoxadiazole ring), 8.40–8.42 (d, 1H, CH benzoxadiazole ring). 13C-NMR (CDCl3) δ 20.7, 25.3, 28.3, 28.9, 30.4, 30.9, 64.8, 119.1, 124.7, 126.4, 135.8, 143.6, 143.9, 170.1 ppm. Anal. (C14H17N3O5S) Calcd. (%): C, 49.55; H, 5.05; N, 12.38; S, 9.45. Found (%): C, 49.01; H, 4.94; N, 12.51; S, 9.59. MS (ESI), m/z: 340 [M + H]+.
Evaluation of solubility and molar extinction coefficient (ɛ) of compounds 2 and 3
Each compound was first dissolved in DMSO, and then diluted in a buffer (0.1 M potassium phosphate, 0.1 mM EDTA, pH 6.5) supplemented with different amounts of Triton X-100. The UV–visible spectra of the obtained solutions were then recorded. Standard solutions of both compounds were prepared in the above buffer supplemented with 0.1% (v/v) Triton X-100, and the molar extinction coefficients were obtained by linear regression analysis of plots of absorbance at 430 nm versus compound concentration.
Kinetic analysis
The enzymatic activity of GSTP1-1 (20 nM subunits) was spectrophotometrically assayed at 340 nm (ɛ = 9600 M−1 cm−1) and at 25 °C, by measuring the rate of CDNB conjugation with GSH as a function of time. The assay mixture contained 1 mM GSH, and 1 mM CDNB in 1 mL of 0.1 M potassium phosphate buffer (pH 6.5) containing 0.1 mM EDTA and 0.1% (v/v) Triton X-100 (buffer A) or in the same buffer lacking the detergent (buffer B). The inhibitory efficacy of compounds 1, 2 and 3 was determined by recording the activity of GSTP1-1 in the presence of various amounts of the selected NBD derivative (0.01–40 μM). The K iapp values for each compound were obtained by fitting the data points to a hyperbolic saturation curve, using GraphPad Prism 5 software (La Jolla, CA). For multiple inhibition analysis, the GST activity was measured in the presence of variable amounts of 2 (0.1–10 μM) at different fixed concentrations of compound 1 (0.2–10 μM).
Spectrophotometric analysis
The spontaneous reactivity of 1 and 2 with GSH was first determined by recording at 25 °C the UV–visible spectrum of each compound (20 μM), dissolved in buffer A (pH 6.5), before and at different times after the addition of GSH (1 mM). The ability of GSTP1-1 to stabilize the σ-complex was evaluated by recording the spectrum of these compounds before and after the addition of GSH (1 mM) and a stoichiometric amount of GSTP1-1 (20 μM). The same experiments were then repeated at 37 °C and pH 7.4 to mimic the physiological environment in terms of pH and temperature. All spectra were recorded against buffer A.
Spectrofluorometric analysis
The emission spectra of both 1 and 2 (4 μM in buffer A, pH 6.5) were recorded at 25 °C, using the following conditions: λ exc = 405 nm, λ em = 450–700 nm; slits, 5–10 nm, with a 455 nm cutoff filter. Spectra were also recorded upon addition of variable amounts of GSTP1-1 (1–20 μM) and 1 mM GSH.
The interaction of 2 with GSTP1-1 was also analyzed at 25 °C and pH 6.5 by following the quenching of the intrinsic fluorescence of the protein (λ exc = 280 nm and λ em = 300–450 nm; slits 5–5 nm). Spectra were measured before and after the addition of 2 (20 μM) to GSTP1-1 (4 μM) dissolved in buffer B, or buffer B containing 1 mM GSH. Fluorescence spectra were recorded in a single photon counting spectrofluorometer (Fluoromax-4, Horiba, Irvine, CA) and data were corrected both for dilution and for inner filter effects.
ELISA for protein–protein interaction analysis
The complex formation between TRAF2 and GSTP1-1 was studied as previously described6. Briefly, 200 μL of His-tagged TRAF2 C-terminal domain (0.005 μM in 20 mM Tris–HCl, pH 7.6 containing 150 mM NaCl and 10% glycerol) was added to each well of a 96-well His-Sorb plate (Qiagen, Hilden, Germany) and incubated overnight at 4 °C on a rocking platform. Afterward, wells were washed with PBS and incubated for 30 min with GSTP1-1 (concentration range from 0.1 to 8 μM) in 10 mM potassium phosphate buffer, pH 7.0, containing 0.1 mM EDTA. Incubation with GSTP1-1 was also performed in the presence of 1 (8 μM) or 2 (20 μM), both in the absence and presence of GSH (1 mM). At the end of incubation, the wells were washed with PBS and then filled with 200 μL of a mouse anti-GSTP1-1 antibody for 2 h at room temperature. Subsequently, wells were washed with PBS and incubated with an anti-mouse IgG antibody for 45 min at room temperature. The immunocomplexes were detected by the addiction of 200 μL per well of the 1-Step-Turbo TMB substrate solution (Pierce, Los Angeles, CA). The reaction was stopped after 10 min by the addition of 50 μL of 2 M H2SO4, and the absorbance was measured at 450 nm. Data were analyzed by fitting to Equation (1) where ν is the percentage of the saturated binding sites; [P]t and [L]t are the total concentrations of monomeric GSTP1-1 and TRAF2, respectively10.
| (1) |
Statistical analysis
Unless specified otherwise, experiments were repeated at least three times, and results are presented as mean ± SD.
Results
Chemistry
The ester derivatives 2 and 3 were obtained by an acylation reaction of 1, prepared as reported previously9, with the appropriate commercial acyl chloride under basic conditions as shown in Scheme 1.
Chemical and physical properties of compounds 2 and 3
The aqueous solubility of compounds 2 and 3 is lower than that of 1. Indeed, the UV–visible spectra of 2 and 3 in saline solution, acquired at a nominal concentration of 0.1 mM, showed a high background, indicative of formation of aggregates causing light scattering; this phenomenon was not observed for 1 under the same conditions. To prevent aggregation, 2 and 3 were dissolved in a buffer (0.1 M potassium phosphate, 0.1 mM EDTA, pH 6.5) containing 0.1% (v/v) Triton X-100, that is the maximum amount of detergent that we found to not affect GST enzyme activity (buffer A). Under these conditions, the solubility limit of both 2 and 3 approached 0.05 mM. Thus, unless stated otherwise, in all of the experiments, compounds 2 and 3 were dissolved in buffer A. The molar extinction coefficients (ɛ) of 2 and 3 were obtained by measuring the absorbance at 430 nm of diluted standard solutions prepared in buffer A. The calculated ɛ were 12 500 M−1 cm−1 and 14 400 M−1 cm−1 for 2 and 3, respectively.
Compound 2 inhibits GSTP1-1 activity by 50%
We performed GSTP1-1 inhibition experiments by recording enzyme activity in the presence of different amounts of 2 dissolved in buffer A (pH 6.5). Surprisingly, no more than 50% of GSTP1-1 activity was inhibited by this compound with an apparent binding affinity constant (K iapp) of 3.9 ± 0.2 μM (Figure 1, panel A). When 1 was dissolved in buffer A, the GSTP1-1 activity was fully inhibited, although the presence of the nonionic surfactant decreased the affinity of 1 (K iapp = 1.9 ± 0.3 μM) compared to that previously reported (0.8 ± 0.1 μM)2 (Figure 1, panel B). To understand whether the partial inhibition by 2 was due to the presence of Triton X-100, the same experiment was performed dissolving compound 2 in a buffer lacking the detergent (buffer B). Even if 2 was not completely soluble in the absence of Triton X-100, the half-site inhibition pattern did not change (Figure 1, panel C). Instead, the K iapp value was six-fold lower (0.6 ± 0.3 μM). Under these conditions, compound 3 caused a complete inhibition of GSTP1-1 with a K iapp of 0.6 ± 0.1 μM (Figure 1, panel D). Therefore, the partial inhibition of GSTP1-1 by 2 was related neither to its low water solubility, nor to the presence of Triton X-100. HPLC evaluation of compound stability proved that, under the assay conditions (pH 6.5 and 25 °C), compounds 2 and 3 were stable up to 20 min (data not shown). Therefore, we can also rule out the possibility that the different inhibition percentages obtained with the two compounds are ascribable to a different rate of ester hydrolysis.
Figure 1.

Inhibition of GSTP1-1 by NBD derivatives. Inhibition curves of GSTP1-1 by 2 (A) or 1 (B) dissolved in buffer A (pH 6.5), at 25 °C. Inhibition curves of GSTP1-1 by 2 (C) or 3 (D), dissolved in buffer B (pH 6.5), at 25 °C. Data points represent the mean ± SD from three independent experiments. Error bars smaller than the symbols are not visible. Calculated K iapp values for each condition are reported.
Compounds 1 and 2 show different spontaneous reactivity towards GSH
In order to clarify the peculiar kinetic findings, we focused our attention on 2. The spontaneous reactivity of 2 (20 μM) towards the nucleophile GSH (1 mM) was analyzed by recording changes in the UV–visible spectrum of the molecule at different time points after addition of the thiol. The results were compared to those obtained with 20 μM compound 1. In both cases, no significant changes in the UV–visible spectrum were detected in a 1-h incubation period at 25 °C and pH 6.5 (Figure 2, panel A), as previously reported for 1 2. However, a remarkable reactivity was observed when 1 was incubated with GSH at 37 °C and pH 7.4 (Figure 2, panel B). Under these conditions, the spectral band centered at 433 nm rapidly decreased (t 1/2 ∼ 1 h), with a simultaneous increase of absorbance at 350 nm. This new optical species represents the intermediate σ-complex between 1 and GSH. On the contrary, compound 2 was stable at 37 °C and pH 7.4, demonstrating that the insertion of a benzoyl moiety in the side chain of 1 strongly affects the reactivity of C4 towards nucleophilic substitution reactions. A confirmation of these results comes from HPLC analysis of incubations of compound 1 or 2 (10 μM) with 1 mM GSH carried out at 37 °C in phosphate buffer at pH 7.4. As shown in Figure S1 (see Supplemental material), compound 2 was relatively stable, with less than 20% chromatographic peak area loss after 30 min (panels A and B). By contrast, compound 1 reacted rapidly with GSH (∼90% peak area loss after 30 min) giving rise to the NBD S-conjugate of GSH (GS-NBD; panels A and C). No peak corresponding to compound 1 was observed in HPLC analysis of compound 2 incubated in phosphate buffer (pH 7.4) at 37 °C up to 60 min (not shown). These data indicate that compound 2 is stable both to GSH-mediated thiolysis and spontaneous hydrolysis at pH 7.4.
Figure 2.

Evaluation of 1 and 2 reactivity with GSH in the absence or in the presence of GSTP1-1. Spectrophotometric analysis. UV–visible spectra of 20 μM 1 (left) and 2 (right) dissolved in buffer A, pH 6.5, recorded at 25 °C (panels A and C) or dissolved in buffer A, pH 7.4, recorded at 37 °C (panels B and D), before and at different times (0–60 min) after the addition of 1 mM GSH (panels A and B) or 1 mM GSH and 20 μM of GSTP1-1 (panels C and D).
Compounds 1 and 2 form different amounts of σ-complex with GSH in the presence of GSTP1-1
The next step was to analyze the reaction between 2 and GSH in the presence of GSTP1-1, and compare these results with those obtained with 1. The results obtained at 25 °C with 1 dissolved in buffer A (pH 6.5) were in agreement with previously published data obtained in the absence of the detergent2; the addition of GSH and GSTP1-1 caused a decrease of ∼80% in the absorption band centered at 433 nm. Moreover, a net increase of the absorbance was observed at 350 nm, corresponding to a band representing the σ-complex intermediate stabilized by the active site of GSTP1-12 (Figure 2, panel C). It is worth mentioning that, when the reaction was followed at 37 °C and pH 7.4, the absorption band at 350 nm, which appeared immediately upon GSH addition, rapidly decreased and the band at 433 nm shifted to 410–420 nm. Based on previously published data, we can identify this new optical species as the adduct between GSH and NBD, which absorbs at 419 nm2. Therefore, the σ-complex between GSH and 1 rapidly evolves toward GS-NBD under physiological pH and temperature conditions (Figure 2, panel D). GSTP1-1 activates also compound 2 towards nucleophilic attack by GSH; indeed, a decrease of the band at 433 nm was immediately recorded when 2 was incubated with both GSH and GSTP1-1, at 25 °C in buffer A, pH 6.5 (Figure 2, panel C). However, after a 1-h incubation, the extent of reduction was nearly half of that observed with 1, and only a modest increase of the absorbance was observed at 350 nm. Moreover, unlike 1, compound 2 was not affected by the increase of pH and temperature: at 37 °C and pH 7.4, the optical species at 433 nm did not shift to 410–420 nm (Figure 2, panel D), suggesting that the σ-complex between GSH and 2 is not rapidly converted into GS-NBD.
Fluorescence spectroscopy suggests that compounds 1 and 2 have different modes of binding to GSTP1-1
Additional details of the interaction between 2 with GSTP1-1 were obtained by fluorescence spectroscopy. We previously reported a strong quenching of the intrinsic fluorescence of 1 upon formation of the σ-complex with GSH within the GST active site2. Here we show that binding of GSTP1-1 to 1 quenches the emission spectrum of the molecule, even in the absence of GSH (Figure 3, panel A left). However, this spectral perturbation is strongly increased after the addition of the thiol and the subsequent formation of the σ-complex (Figure 3, panel A right), in accordance with previous findings2.
Figure 3.

Spectrofluorometric analysis of the interaction between 1 or 2 and GSTP1-1. The emission spectra of (A) 1 (4 μM) and (B) 2 (4 μM), dissolved in buffer A (pH 6.5), were recorded at 25 °C before and after the addition of increasing amounts of GSTP1-1 (1–20 μM), in the absence (left) or in the presence (right) of 1 mM GSH. (C) The emission spectrum of GSTP1-1 (4 μM in buffer B, pH 6.5) was recorded at 25 °C before and after the addition of 2 (20 μM), in the absence (left) or in the presence (right) of 1 mM GSH.
In the case of 2, a decrease in the fluorescence intensity and a red shift (12 nm) were observed in the presence of GSH (Figure 3, panel B right), while spectral changes were negligible in the absence of the substrate (Figure 3, panel B left). In order to analyze protein–ligand association also by following changes in the emission spectrum of GSTP1-1, the binding was evaluated in the absence of Triton X-100 (buffer B), since the detergent absorbs in the same spectral region of the aromatic side chains (Figure 3, panel C). We previously reported that binding of 1 to GSTP1-1 perturbs the intrinsic fluorescence of the enzyme even in the absence of GSH2; a similar behavior was observed for 2, although the extent of fluorescence quenching was smaller in the absence of GSH (Figure 3, panel C).
These data confirm the different mode of binding of 1 and 2 to GSTP1-1.
Co-inhibition experiments reveal that 2 does not prevent a complete GSTP1-1 inhibition by 1
We have previously demonstrated that GSTP1-1 displays allosteric properties11. Therefore, we investigated whether the partial inhibition of GSTP1-1 was the result of the binding of compound 2 to one subunit, an event preventing the binding of a second inhibitory species (i.e. 1) to the vacant subunit. We thus performed co-inhibition experiments of GSTP1-1 activity by using a mixture of 1 and 2 dissolved in buffer B, pH 6.5 (Figure 4). When the amount of 2 was increased and the concentration of 1 was kept low, a partial additive inhibitory effect was observed up to an enzyme inhibition of approximately 50%. However, when 1 concentration was sufficiently high, the compound caused a complete inhibition of the enzyme, irrespective of the presence of 2 in the sample. Therefore, the presence of 2 does not prevent enzyme inhibition by 1.
Figure 4.

Co-inhibition experiments involving 1 and 2. Inhibition of GSTP1-1 activity was evaluated at 25 °C and pH 6.5, in the presence of both 1 and 2 in a concentration range between 0.1 and 10 μM. The substrates GSH and CDNB were kept constant at 1 mM. Data points represent the mean ± SD from three independent experiments. Error bars smaller than the symbols are not visible.
GSH is not required by 2 to decrease the interaction between GSTP1-1 and TRAF2
We have previously shown that, in the presence of GSH, both 1 and its analogue MC3181 strongly reduce the affinity of GSTP1-1 for TRAF26, 12. Similar results were obtained with compound 2 (Figure 5, panel A). We have also shown that the ability of 1 to decrease the affinity between the GSTP1-1 and TRAF2 is more evident at high levels of GSH6. Surprisingly, further experiments (Figure 5, panel B) showed that compound 2 was able to affect the formation of the TRAF2–GSTP1-1 complex even in the absence of GSH, whilst 1 was completely ineffective. As a control, we evaluated the occurrence of a direct interaction between 2 and TRAF2. Addition of 2 to a solution of TRAF2 (5 μM) did not perturb the intrinsic fluorescence of the protein, ruling out this possibility (data not shown).
Figure 5.

Effect of 1 and 2 on TRAF2–GSTP1-1 interaction. (A) His-tagged TRAF2 (0.005 μM) was immobilized on Ni-NTA-coated plates and incubated with increasing amounts of GSTP1-1 (from 0.1 to 8 μM) in the absence (-•-) or in the presence of 8 μM 1 (-□-) or 20 μM 2 (-Δ-), and 1 mM GSH. (B) The same experiment as in (A), performed in the absence of GSH. Data points represent the mean ± SD from three independent experiments. Error bars smaller than the symbols are not visible.
Molecular docking-obtained orientations for 2 in the GSTP1-1 active site
A docking analysis was performed to determine possible structures for the 2/GSTP1-1/GSH complex (see Supplemental Material for protein–ligand docking conditions). Figure 6 shows two representative binding poses for 2 among the clusters with the most favorable binding energies. Along with an expected binding pose similar to the crystal structure obtained for the complex of GSTP1-1 with 1, thus lining the H-binding site of GST, the other pose shows the NBD moiety of 2 shifted slightly deeper in the cavity and, most importantly, with its benzoic ring placed at the interface between the protein monomers, in close proximity with the side chain of Tyr49. This residue is located in a loop between the α2-helix and the β3-strand in a flexible region of the protein, and is involved in a lock-key motif that plays a critical role in catalysis and in the inter-subunit communication13. Furthermore, Lys102, located in the α4-helix of the other monomer, is at less than 3 Å from the oxygen atom of the ester function of 2, thus allowing for a hydrogen bond formation. This interaction can strongly perturb the relative motions of the two monomers as also supported by normal mode analysis. Direct visualization of non-trivial modes shows large amplitude fluctuations of the two monomers with sliding movements of the α4-helix pivoting around Tyr49 (Figure S2), as already established in the literature13.
Figure 6.

Protein–ligand docking analysis. Two docking-obtained orientations for 2 (in medium grey); GSH is in white, residues Tyr49 from monomer A and Lys102 from monomer B are shown in sticks inside the protein surface and labeled for clarity. For comparison, the position of 1 in the crystal structure is also shown in dark grey. Ribbon and surface representation of the remaining part of the protein is shown with subunit A (bottom, darker surface) and subunit B (top, ligther surface).
Discussion
Our results show that the insertion of an aromatic ring into the side chain of 1 strongly affects the interaction between the NBD derivative 2 and GSTP1-1. The first difference between 1 and 2 is that the second compound inhibits only ∼50% of the enzyme activity. One possible explanation for this finding is the occurrence of a strong negative cooperative interaction between the two active sites of GSTP1-1; upon the binding of 2 to the first site, this interaction would prevent the binding of a second molecule of 2 to the vacant active site while still allowing the binding of the substrate. This phenomenon has been already described for GSTP1-1 interaction with other ligands, and has been interpreted as a self-preservation mechanism that avoids the full inactivation of the enzyme11.
Co-inhibition experiments show that 1 fully inactivates GSTP1-1 even in the presence of high 2 concentrations. This may occur either because binding of 2 to one subunit does not hinder association of 1 to the other, or because 1 is able to displace 2 from the active site by removing the strong negative cooperativity, and allowing a complete inhibition of the enzyme by 1.
Another difference between 1 and 2 is the ability of 2 to decrease the affinity of the TRAF2-GSTP1-1 complex even in the absence of GSH, while 1 requires GSH to affect the TRAF2-GSTP1-1 complex. Therefore, 2 does not require GSH to affect the flexibility of the α2-helix (residues 35–46) of GSTP1-1, which contains a TRAF2 binding motif (TWQE)6 , 14, and is likely involved in the TRAF2–GSTP1-1 interaction.
These data confirm that compounds 1 and 2 bind to the active site of GSTP1-1 in a different fashion, in agreement with the different spectral behavior of their complexes with GSTP1-1 (see Figures 2 and 3).
The results of the docking analysis sustain this hypothesis, showing that one of the two representative binding poses has the benzoyl moiety of 2 placed near the dimer interface pointing towards Tyr49 and Lys102 of the other monomer (Figure 6). The interaction of 2 with these residues can strongly perturb the relative motions of the two monomers of GSTP1-1, as also supported by normal mode analysis (Figure S2). Restraint of inter-subunit motions caused by 2 could account for long range effects in protein regions involved in the GSTP1-1 cooperativity and in the protein–protein interaction with TRAF2.
A critical aspect of most of the NBD-based GST inhibitors so far reported is their propensity to react spontaneously with GSH9. This may limit their clinical efficacy as the resulting GSH adduct may be extruded from the cells by efflux pumps recognizing GSH conjugates. It is noteworthy that the presence of a bulky benzoyl moiety in the side chain of compound 2 confers high stability towards the nucleophilic attack by GSH in solution, and also inhibits σ-complex conversion into the final GS-NBD adduct in the GSTP1-1 active site (Figure 2). The most important consequence of the present finding is that compound 2 may serve as a lead compound for the development of novel NBD derivatives not affected in their anticancer action by fluctuations of GSH levels, and likely characterized by increased in vivo half-life.
Disclosure statement
The authors report no conflicts of interest.
Supplementary Material
Funding Statement
Associazione Italiana per la Ricerca sul Cancro, 10.13039/501100005010 [AIRC-TRIDEO 2015 (Id.17515)] Ministero dell'Istruzione, dell'Università e della Ricerca [PRIN 2012 (prot.2012CTAYSY) and PRIN 2015 (prot. 20157WW5EH_007)]
References
- 1. Sau A, Pellizzari Tregno F, Valentino F, et al. Glutathione transferases and development of new principles to overcome drug resistance. Arch Biochem Biophys 2010;500:116–22. [DOI] [PubMed] [Google Scholar]
- 2. Ricci G, De Maria F, Antonini G, et al. 7-Nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione S-transferases. Mechanism of action of potential anticancer drugs. J Biol Chem 2005;280:26397–405. [DOI] [PubMed] [Google Scholar]
- 3. Turella P, Cerella C, Filomeni G, et al. Proapoptotic activity of new glutathione S-transferase inhibitors. Cancer Res 2005;65:3751–61. [DOI] [PubMed] [Google Scholar]
- 4. De Luca A, Federici L, De Canio M, et al. New insights into the mechanism of JNK1 inhibition by glutathione transferase P1-1. Biochemistry 2012;51:7304–12. [DOI] [PubMed] [Google Scholar]
- 5. Sau A, Filomeni G, Pezzola S, et al. MTargeting GSTP1-1 induces JNK activation and leads to apoptosis in cisplatin-sensitive and -resistant human osteosarcoma cell lines. Mol Biosyst 2012;8:994–1006. [DOI] [PubMed] [Google Scholar]
- 6. De Luca A, Mei G, Rosato N, et al. The fine-tuning of TRAF2–GSTP1-1 interaction: effect of ligand binding and in situ detection of the complex. Cell Death Dis 2014;5:e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pellizzari Tregno F, Sau A, Pezzola S, et al. In vitro and in vivo efficacy of 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) on human melanoma. Eur J Cancer 2009;45:2606–17. [DOI] [PubMed] [Google Scholar]
- 8. Tentori L, Dorio AS, Mazzon E, et al. The glutathione transferase inhibitor 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX) increases temozolomide efficacy against malignant melanoma. Eur J Cancer 2011;47:1219–30. [DOI] [PubMed] [Google Scholar]
- 9. Rotili D, De Luca A, Tarantino D, et al. Synthesis and structure–activity relationship of new cytotoxic agents targeting human glutathione-S-transferases. Eur J Med Chem 2015;89:156–71. [DOI] [PubMed] [Google Scholar]
- 10. Stella L, van de Weert M.. Fluorescence quenching and ligand binding: a critical discussion of a popular methodology. J Mol Struct 2011;998:144–50. [Google Scholar]
- 11. Ricci G, Caccuri AM, Lo Bello M, et al. Glutathione transferase P1-1: self-preservation of an anti-cancer enzyme. Biochem J 2003;376:71–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. De Luca A, Rotili D, Carpanese D, et al. A novel orally active water-soluble inhibitor of human glutathione transferase exerts a potent and selective antitumor activity against human melanoma xenografts. Oncotarget 2015;6:4126–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hegazy UM, Mannervik B, Stenberg G.. Functional role of the lock and key motif at the subunit interface of glutathione transferase p1-1. J Biol Chem 2004;279:9586–96. [DOI] [PubMed] [Google Scholar]
- 14. Wu Y, Fan Y, Xue B, et al. Human glutathione S-transferase P1-1 interacts with TRAF2 and regulates TRAF2-ASK1 signals. Oncogene 2006;25:5787–800. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.