

Abstract
Human inosine 5′-monophosphate dehydrogenase 2 (hIMPDH2), being an age-old target, has attracted attention recently for anticancer drug development. Mycophenolic acid (MPA), a well-known immunosuppressant drug, was used a lead structure to design and develop modestly potent and selective analogues. The steep structure–activity relationship (SAR) requirements of the lead molecule left little scope to synthesise newer analogues. Here, newer MPA amides were designed, synthesised and evaluated for hIMPDH2 inhibition and cellular efficacy in breast, prostate and glioblastoma cell lines. Few title compounds exhibited cellular activity profile better than MPA itself. The observed differences in the overall biological profile could be attributed to improved structural and physicochemical properties of the analogues over MPA. This is the first report of the activity of MPA derivatives in glioblastoma, the most aggressive brain cancer.
KEYWORDS: Mycophenolic acid, hIMPDH2 inhibitors, anticancer agents, MTT, de novo purine synthesis
Introduction
Cancer is a group of heterogeneous diseases characterised by uncontrolled growth and spread of abnormal cells. Cancer cells adapt to their high metabolic state by increasing energy production, that is increased cellular metabolism. Potential molecular targets for cancer therapeutics are signalling pathways that are preferentially activated in numerous cancers. However, heterogeneity of tumours and frequent oncogenic mutations over a period makes it difficult for a medicinal chemist to design antitumour molecules that target tumour-specific pathways1. Proliferating cells have a high demand for guanine nucleotides (GMP) that generally cannot be sustained by salvage pathways, which explains the importance of inosine 5'-monophosphate dehydrogenase (IMPDH, an enzyme linked with proliferation and malignancy). Non-proliferating cells use an alternative purine nucleotide synthesis pathway, the salvage pathway, to synthesise GMP. These observations pose IMPDH as a potential target to suppress tumour cell growth1.
The IMPDH (E.C.1.1.1.205), the nicotinamide adenine dinucleotide (NAD+)-dependent enzyme that controls de novo synthesis of purine nucleotides, catalyses the oxidation of inosine 5'-monophosphate (IMP) to xanthosine 5'-monophosphate (XMP), which is then converted to guanosine 5'-monophosphate (GMP) by GMP synthase. The IMP also serves as a substrate for the biosynthesis of adenosine 5'-monophosphate (AMP). An adequate pool of purine nucleotides is essential for cell proliferation, cell signalling and as an energy source2. Consequently, inhibition of IMPDH causes a variety of biological responses, such as reduction in guanine nucleotide pools resulting in arrest of cell proliferation (interruption of DNA and RNA synthesis)3, a decline in intracellular signalling (G-protein-mediated signal transduction)4–6, downregulation of c-myc and Ki-ras oncogenes in vitro7. Also, IMPDH inhibition is associated with an upregulation of p53 (commonly mutated protein in human cancers)8, p21, bax and a downregulation of bcl-2, survivin and p27 protein9.
The enzyme human IMPDH exists in two isoforms (type 1 and type 2). These isoforms are of identical size and share 84% sequence identity. However, the type 1 “housekeeping” isoform is constitutively expressed in both normal and neoplastic cells, while type 2 expression is preferentially upregulated in human neoplastic cell lines10. Human IMPDH type 1 (hIMPDH1) has been identified as an anti-angiogenic drug target and mycophenolic acid (MPA) was found to block tumour-induced angiogenesis (in vivo)11 while the disproportionate increase in human IMPDH type 2 (hIMPDH2) activity in neoplastic cells has made this isoform a key target for the development of anticancer drug discovery12,13. Also, hIMPDH2 has become a major drug target for immunosuppression14,15, antiviral16 and parasitic infestations17–20.
Mycophenolic acid (MPA) 1 (Figure 1), a natural product, is a reversible, potent, uncompetitive inhibitor of IMPDH and known to be an anticancer and immunosuppressive agent. Mycophenolate mofetil (MMF, 2, a prodrug of MPA), has been approved for the treatment of acute allograft rejection following kidney transplant. The MPA and its related forms MMF or MPA sodium (3) (MPS) cause dose-limiting gastrointestinal (GI) toxicity. However, adverse effects related to the treatment with MPA-based drugs, such as diarrhoea, leukopenia, sepsis and vomiting, are the barriers to the administration of higher doses and more effective treatment21. The competitive IMPDH inhibitors such as tiazofurin (4), ribavirin (5) and mizoribine (6) (after intracellular activation by phosphorylation) are nucleoside analogues and derivatives thereof22 have unfavourable tolerability profiles too. Thus, there is a urgent need for newer, safer, potent and orally bioavailable IMPDH inhibitors23,24.
Figure 1.
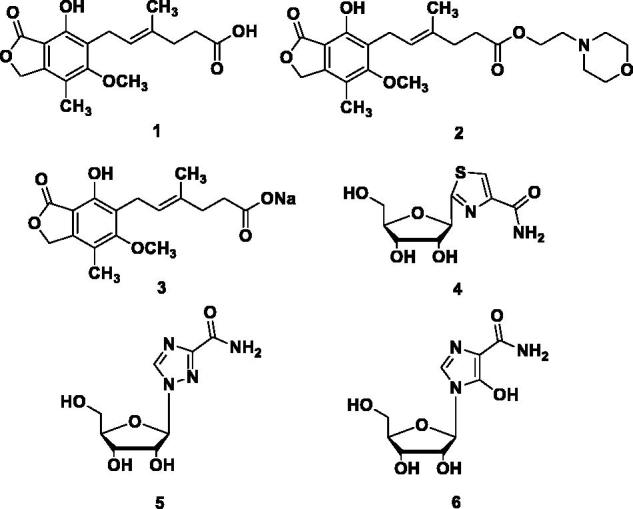
Currently used IMPDH inhibitor drugs.
In an attempt to address this issue, significant efforts were focused on bioisosteric replacements along with various structural modifications of 1 to develop potent IMPDH inhibitors, but with limited success12,25. The MPA has proven to be an effective inducer of differentiation in a number of cancer cell lines (melanoma, leukaemia and prostate cancer). Also, MPA exhibited synergism with imatinib in the treatment of chronic myelogenous leukaemia (CML)26. Furthermore, antitumour activities of several derivatives of 1 are reported27,28. The MPA conjugates possessing amide and ester linkages (7–12, Figure 2) exhibited potent anticancer properties21,29–31. These findings strongly support the role of 1 as a potential anticancer drug. Moreover, MPA, being an acid, is likely to be prevented from entering in the central nervous system (CNS). There are no evidence in the literature citing this fact that 1 could be useful for treating gliomas, cancers of the CNS32.
Figure 2.

Recently reported MPA analogues.
In the present work, we report the design and synthesis of newer analogues of 1 with the aim of improving its cellular potency, selectivity and toxicity profiles. As is previously known, little structural alteration of 1 was detrimental to its IMPDH inhibitory activity29–32. It was crucial to select the design strategy leading to retention of the inhibitory potential against hIMPDH2. The title compounds were further evaluated for their cellular potency in MDA-MB-231 (breast adenocarcinoma), DU145 (prostate carcinoma) and U87 MG (glioblastoma astrocytoma) cell lines. The present study summarises the finding of the medicinal chemistry activities centred on 1.
Methods
Chemistry
The synthetic details of only two representative compounds are described. For the rest of compounds, readers are requested to refer to the Supplementary Material section. We preferentially selected aliphatic amines to balance logP values, particularly benzyl amines and while avoiding the use of anilines which are often mutagenic. Nonetheless, two compounds with substituted anilines were synthesised as a part of structure–activity relationship (SAR) studies.
General procedure for the synthesis of MPA amides(14–28)31,33 (Scheme 1). Method A: Mycophenolic acid 1 (1 eq.), appropriate amines 13a–m (1.14 eq.) and 4(dimethylamino)pyridine (DMAP) (1.14 eq.) were dissolved in anhydrous DMF (3 ml). The reaction mixture was cooled to 0 °C in an ice bath, followed by the addition of EDCI.HCl (1.1 eq.) with stirring. The reaction mixture was stirred at 0 °C for 6 h and left at RT for 48–72 h. After completion of the reaction, the reaction mixture was cooled to 5 °C and poured into cold water (25 ml), extracted with EtOAc (3X20 ml). The combined organic layers were dried over Na2SO4 (anhydrous) and concentrated in vacuo. The crude products were purified with the help of column chromatography using DCM:EtOAc (9:1) as the mobile phase.
Scheme 1.

General scheme for synthesis of mycophenolic acid amides (14–28): Reagents and conditions: Method A: DMAP, EDCI.HCl, DMF, 0 °C 6 h, RT, 48–72 h; Method B: DIPEA, HATU, DMF, 0 °C 6 h, RT, 48–72 h.
Method B: Mycophenolic acid 1 (1 eq.), appropriate amines 13n-o (0.67 eq.) and HATU (1.33 eq.) were dissolved in anhydrous DMF (3 ml). The reaction mixture was cooled to 0 °C in an ice bath and DIPEA (3.4 eq.) was added with stirring. The reaction mixture was stirred at 0 °C for 6 h and left at RT for 48–72 h. After completion of the reaction, the contents of the flask were cooled to 5 °C and poured into cold water (25 ml), extracted with EtOAc (3X20 ml). The combined organic layers were dried over Na2SO4 (anhydrous) and concentrated in vacuo. The crude products were purified with the help of column chromatography using DCM:EtOAc (9:1) as the mobile phase.
(S)(E)-6–(4-hydroxy-6-methoxy-7-methyl-3-oxo-1,3-dihydroisobenzofuran-5-yl)-4-methyl-N-(1-phenylethyl)hex-4-enamide(14). It was synthesised using 1 (0.1 g, 0.31 mmol), (S)-(−)-α-methylbenzylamine 13a (0.045 ml, 0.35 mmol), DMAP (0.044 g, 0.35 mmol) and EDCI.HCl (0.066 g, 0.34 mmol) as per Method A to yield 14 as off-white solid. Yield: 55%; TLC: Rf = 0.71 (DCM: EtOAc, 8:2); purity (HPLC): 95.28%; mp: 114–116 °C; IR (KBr) cm−1 3444 (OH, str), 3284 (NH, str), 1745 (O–C=O, str), 1637 (NH-C=O, str), 1551 (C=C, str), 1134 (C-O, str); 1H-NMR (DMSO-d6, D2O exchange, 400 MHz) δ 7.29–7.12 (m, 5H), 5.21 (s, 2H), 5.09–5.12 (t, J = 7.0 Hz, 1H), 4.79 (q, J = 7.0 Hz, 1H), 3.63 (s, 3H), 3.25 (d, J = 6.8 Hz, 2H), 2.14 (dd, J = 7.1, 11.9 Hz, 4H), 2.04 (s, 3H), 1.69 (s, 3H), 1.23 (d, J = 7.0 Hz, 3H); MS (ESI) m/z: 422 [M–H]−.
(E)-6–(4-hydroxy-6-methoxy-7-methyl-3-oxo-1,3-dihydroisobenzofuran-5-yl)-N-(3-methoxyphenyl)-4-methylhex-4-enamide(27). It was synthesised using 1 (0.1 g, 0.31 mmol), 3-methoxyaniline 13n (0.024 ml, 0.21 mmol), HATU (0.158 g, 0.41 mmol) and DIPEA (0.185 ml, 1.06 mmol) as per Method B to yield 27 as off-white solid. Yield: 45%; TLC: Rf = 0.62 (DCM:EtOAc, 8:2); purity (HPLC): 97.63%; mp: 238–240 °C; IR (KBr) cm−1 3437 (OH, str), 1756 (O–C=O, str), 1689 (NH–C=O, str), 1613 (C=C, str), 1127 (C–O, str); 1H-NMR (DMSO-d6, D2O exchange, 400 MHz) δ 7.2 (s, 1H), 6.8–6.6 (s, 3H), 5.21 (s, 2H), 5.15–5.13 (t, J = 7.0 Hz, 1H), 3.69 (s, 3H), 3.64 (s, 3H), 3.28 (d, J = 6.8 Hz, 2H), 2.30 (t, J = 5.8, 9.5 Hz, 2H), 2.22 (t, J = 7.8 Hz, 2H), 2.04 (s, 3H), 1.76 (s, 3H); MS (ESI) m/z: 424 [M–H]−.
Biological activity
In vitro hIMPDH2 inhibition assay23
The enzyme (hIMPDH2) was purchased from NovoCIB SAS (Lyon, France). A total of 15 molecules were screened at 10 μM concentration for enzyme inhibition and IC50 values were determined for compounds with hIMPDH2 inhibition >70% at 10 μM. The assay was performed in a 200 µl final volume in 96-well UV plates (Tarsons, 980040, Tarsons Products Pvt. Ltd., Kolkata, India) with a reaction buffer composed of 100 mM Tris–HCl (pH 8.6), 100 mM KCl and 5 mM DTT, 4% v/v DMSO plus or minus test compound and 0.15 mU of purified hIMPDH2 enzyme per well (from 1.5 mg/ml stock solution). The final volume of the enzyme stock solution per well was 2 µl which was insignificant to cause any change in the final assay buffer composition. The reaction was initiated by the addition of (substrate buffer) 0.2 mM of IMP and 0.2 mM of NAD+ and the assay was allowed to proceed at 37 °C for 45 min. The generated NADH was measured by reading the absorbance at 340 nm. At this wavelength, a background of <0.1 optical density (OD) was observed with negligible crosstalk between wells. The MPA (10 µM) was used as a positive control and DMSO as a vehicle control. For IC50 determinations, a total of 10 concentrations ranging from 25 µM to 50 nM in triplicates were used. Enzyme inhibition and IC50 values were expressed in % inhibition and μM, respectively.
Cell viability (MTT assay)34
Cancer cell lines such as MDA-MB-231 (breast adenocarcinoma), DU145 (prostate carcinoma) and U87 MG (glioblastoma astrocytoma) were purchased from National Centre for Cell Sciences (NCCS) (Pune, India). Cytotoxic activity of compounds (>80% hIMPDH2 inhibition at 10 μM) was evaluated using colorimetric MTT assay on the above-mentioned cell lines. Briefly, cells were grown in DMEM media supplemented with foetal bovine serum (FBS) 10% and penicillin–streptomycin (50 U/ml, 50 µg/ml) at 37 °C, CO2 (5%) and air (95%). Logarithmically, growing cells were seeded using 96-well plate at different concentrations of the test compounds ranging from 0.01 to 100 μM (seeding density: MDA-MB-231: 10,000 cells/well, DU145: 8000 cells/well, U87 MG: 5000 cells/well). After 24 h of seeding, the cells were observed under microscope and treated with varying drug concentration along with DMSO (vehicle control). Each dilution of test compound was added in triplicate. Following 48-h incubation with the compounds, cells were incubated with MTT reagent (5 mg/ml) for 4 h, and then 100 μl of DMSO was added to dissolve formazan crystals. The absorbance was then measured at 540 nm and 630 nm (background scan) using EPOCH 2 BioTek microplate reader. The IC50 values for the tested compounds were calculated and expressed in μM using mean of triplicate readings. IC50 is defined as the compound concentration required to reduce the viability of the cells by 50% with respect to the control.
Results and discussion
Chemistry
The best operative method for drug discovery is to modify drug substances with known biological activity. Novel analogues are synthesised using molecular modifications to alter the physicochemical properties which may lead to enhancement in efficacy or receptor interactions, administration pathway, toxicity and stability issues. The MPA (1) is a well-known hIMPDH2 inhibitor with several pharmacological activities2. Its antitumour activity was abolished or reduced when any of the modifications, for example reduction or oxidation of the olefin, demethylation of –OCH3, oxidation of methyl group attached to the benzene substructure and altering of the terpene side chain, were made28. We set out with the aim of synthesising MPA amides since the amides were spared from the reduction or loss of biological activity on its structural modification(s). We carefully selected the amines to be coupled with MPA –COOH group based on the predicted structural, pharmacokinetic and toxicity properties of the proposed compounds (Table S1, Supplementary Material section).
The MPA possesses electrophilic centre at the acyl carbon in the lactone ring and free phenol group. Protection of the phenolic –OH using Ac2O (mycophenolic acid acetate), tert-butyldimethylsilyl triflate or tert-butyldimethylsilyl chloride (7-O-TBDMS-mycophenolic acid) and selective hydrolysis of the same has been reported and used for amide formation21. Protection and deprotection approach increases the number of steps leading to lower overall yield. Many reported coupling agents (isobutyl chloroformate, diphenyl phosphorazidate (DPPA)/triethylamine (TEA), 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ)/pyridine, O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU)/N-methylmorpholine (NMM), O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU)/1-hydroxybenzotriazole (HOBt), 1-ethyl-3–(3′-dimethylaminopropyl)carbodiimide (EDCI)/DMAP in the presence of HOBt and so on) were used to form an amide bond between the –COOH of 1 and various amines 13(a-o) (Scheme 1) selectively, without protection of the phenolic –OH30,31. Unfortunately, none of the attempted methods led to the proposed compounds due to low conversion of the substrates, problems with purification and similar reasons. The reaction conditions using EDCI/DMAP without HOBt (Method A) and HATU/DIPEA (Method B) were proved to be suitable for the synthesis of MPA derivatives 14–28 in moderate yields 40–75% (Scheme 1) and higher purity of the products.
Biological activity
In vitro hIMPDH2 inhibition assay
In line with our previous efforts on hIMPDH2 inhibitor discovery and development, all the synthesised molecules were screened for hIMPDH2 inhibition at 10 μM, in triplicate. The results are shown in Table 1. A total of eight derivatives (15, 16, 18–21, 24 and 25) exhibited hIMPDH2 inhibition >70% (subjected to IC50 determination), while remaining compounds showed % inhibition <70. None of the synthesised molecules exhibited hIMPDH2 inhibitory activity superior than MPA (1) (IC50 = 0.25 ± 0.03 μM), although 18 (IC50 = 0.33 ± 0.11 μM) and 24 (IC50 = 0.48 ± 0.02 μM) were close competitors (Table 1, Figure 3). This could be possibly due to the stringent structural requirements and receptor binding space of the enzyme27,28.
Table 1.
hIMPDH2 enzyme inhibition of MPA amides (14–28 at 10 µM).
| Compound | R |
hIMPDH2 enzyme |
|
|---|---|---|---|
| % Inhibitiona | IC50 (µM) ± SDb | ||
| 1 | Mycophenolic acid | 99.90 ± 1.20 | 0.25 ± 0.03 |
| 14 | (S)-(−)-α-Methylbenzyl | 59.15 ± 1.91 | n.d.c |
| 15 | (R)-(+)-α-Methylbenzyl | 73.94 ± 2.84 | 0.82 ± 0.32 |
| 16 | 2-Chlorobenzyl | 72.04 ± 1.11 | 0.73 ± 0.06 |
| 17 | 2-Furfuryl | 67.18 ± 4.16 | n.d.c |
| 18 | 4-Methoxybenzyl | 96.13 ± 2.51 | 0.33 ± 0.11 |
| 19 | 4-Methylbenzyl | 73.42 ± 3.62 | 0.60 ± 0.13 |
| 20 | 4-Chlorobenzyl | 81.36 ± 1.95 | 0.57 ± 0.11 |
| 21 | 4-Pyridylmethyl | 57.38 ± 1.02 | n.d.c |
| 22 | 2-Pyridylmethyl | 59.38 ± 1.81 | n.d.c |
| 23 | 2-Methylbenzyl | 84.00 ± 2.13 | 0.50 ± 0.18 |
| 24 | 4-Phenylbutyl | 93.62 ± 3.17 | 0.48 ± 0.02 |
| 25 | 3,4-(Methylenedioxy)benzyl | 87.09 ± 4.54 | 0.51 ± 0.08 |
| 26 | 2-Morpholinoethyl | 36.05 ± 3.86 | n.d.c |
| 27 | 3-Methoxyphenyl | 30.82 ± 2.82 | n.d.c |
| 28 | 4-Methoxyphenyl | 54.84 ± 1.15 | n.d.c |
aAll the data are expressed as ± SD (results are average of duplicate analysis).
bAll the data are expressed as ± SD (results are average of triplicate analysis). IC50 value was determined when the inhibitory rate of compound is higher than 70% at the concentration of 10 µM.
cn.d.: not determined.
Figure 3.

hIMPDH2 % inhibition of compounds 14–28.
Cell viability (MTT assay)
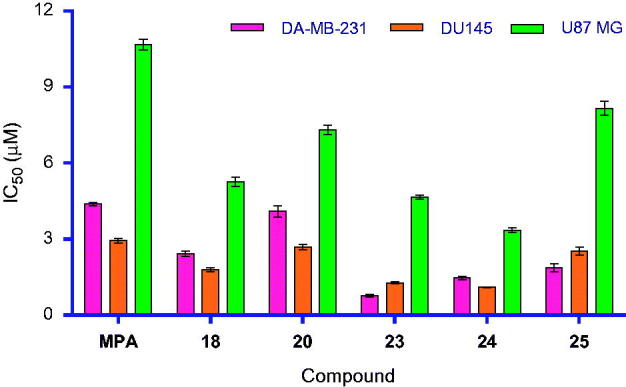
Cytotoxic activity of six compounds (MPA, 18, 20 and 23–25) are reported in Table 2. All MPA amides exhibited better activity than MPA in cell-based assays. This may be due to lower logP value (2.68) of MPA (free acid group), whereas the derivatives tested possessed higher logP (average logP 3.98). Out of five compounds screened (Table 2) against three cell lines, 23 was found to be more potent compared to 18, 20, 24, 25 and five-fold more potent than MPA in MDA-MB-231 (breast adenocarcinoma) cell line (Table 2, Figure 4). Similarly, 24 exhibited greater potency than 18, 20, 23, 25 and three-fold higher potency than MPA in DU145 (prostate carcinoma) cell line (Table 2, Figure 4). For U87 MG, 24 showed promising activity compared to 18, 20, 23, 25 and three-fold more active than MPA in U87 MG (glioblastoma astrocytoma) cell line (Table 2, Figure 4). This is the first time anticancer activity of MPA derivatives on glioblastoma cell lines was demonstrated. Higher lipophilicity and masking of the –COOH in 1 could possibly be responsible for this observation since usually acids are prevented from entering in the CNS.35 Also, compound 24 and 1 were tested for hPBMC proliferation assay (see Supplementary Material section for protocol) and both these compounds exhibited similar % cell viability (63% for 1 and 65% for 24). The possibility of the title compounds behaving as prodrugs could be minute due to the difficulty in hydrolysing the amide linkage inside cells and release of the active form “MPA”. The higher cellular potency of these derivatives over MPA is definitely a progress towards the goal, that is to discover a novel anticancer agent, better than MPA.
Table 2.
Cytotoxicity of MPA amide derivatives
| S. No. | Compound | Cell lines [IC50 (μM) ± SDa] |
||
|---|---|---|---|---|
| MDA-MB-231b | DU145c | U87 MGd | ||
| 1 | Cisplatin | 50.40 ± 1.1 | 34.47 ± 0.78 | 21.32 ± 0.39 |
| 2 | Doxorubicin HCl | 0.54 ± 0.08 | 0.21 ± 0.06 | 0.11 ± 0.02 |
| 3 | MPA (1) | 4.38 ± 0.06 | 2.94 ± 0.09 | 10.69 ± 0.21 |
| 4 | 18 | 2.42 ± 0.11 | 1.8 ± 0.07 | 5.26 ± 0.18 |
| 5 | 20 | 4.10 ± 0.23 | 2.69 ± 0.11 | 7.31 ± 0.17 |
| 6 | 23 | 0.77 ± 0.04 | 1.28 ± 0.04 | 4.66 ± 0.07 |
| 7 | 24 | 1.46 ± 0.07 | 1.09 ± 0.02 | 3.35 ± 0.09 |
| 8 | 25 | 1.86 ± 0.16 | 2.53 ± 0.15 | 8.16 ± 0.27 |
aAll the data are expressed as ± SD (results are average of triplicate analysis).
bMDA-MB-231 (breast adenocarcinoma).
cDU145 (prostate carcinoma).
dU87 MG (glioblastoma astrocytoma).
Bold values indicate the most potent molecule in each cell line.
Figure 4.
Cytotoxicity of MPA amide derivatives.
Conclusions
Our design strategy based on MPA as the lead structure yielded fruitful outcome in terms of slightly better hIMPDH2 inhibitors with improved cellular potency. The potential issues with the steep SAR exhibited by the lead molecule in cells were overcome by virtually modulating the physicochemical properties and then carefully cheery-picking the analogues to be synthesised. Our medicinal chemistry approach yielded modestly more potent molecules in cells. The lead molecules are further being evaluated for their utility as potential anticancer agents.
Supplementary Material
Funding Statement
The authors acknowledge the financial support of their work by Young Scientist’s Grant [No. SERB/LS-639/2013] awarded to PK from Science and Engineering Research Board (SERB), Department of Science and Technology (DST), Government of India, India.
Acknowledgements
CS and PK deeply acknowledge the help received from Dr Kapil Juvale, Assistant Professor with SVKM’s NMIMS, SPP School of Pharmacy and Technology Management, Mumbai, India, and Mr Nitesh Sahu, PhD student in PK’s group during hIMPDH2 inhibition studies and Department of Science and Technology (DST), Science and Engineering Research Board (SERB), Government of India for funding. The authors would also like to thank Dr Vikrant Bhor, Scientist D, National Institute for Research in Reproductive Health (NIRRH), Indian Council of Medical Research (ICMR) for hPBMC assay studies.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Jackson RC, Weber G, Morris HP.. IMP dehydrogenase, an enzyme linked with proliferation and malignancy. Nature 1975;256:331–3. [DOI] [PubMed] [Google Scholar]
- 2.Hedstrom L.IMP dehydrogenase: structure, mechanism, and inhibition. Chem Rev 2009;109:2903–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jayaram HN, Dion RL, Glazer RI, et al. Initial studies on the mechanism of action of a new oncolytic thiazole nucleoside, 2-β-D-ribofuranosylthiazole-4-carboxamide (NSC 286193). Biochem Pharmacol 1982;31:2371–80. [DOI] [PubMed] [Google Scholar]
- 4.Manzoli L, Billi AM, Gilmour RS, et al. Phosphoinositide signaling in nuclei of friend cells: tiazofurin down-regulates phospholipase C beta 1. Cancer Res 1995;55:2978–80. [PubMed] [Google Scholar]
- 5.Mandanas BRA, Leibowitz DS, Gharehbaghi K, et al. Role of p21 RAS in p210 bcr-abl transformation of murine myeloid cells. Blood 1993;82:1838–47. [PubMed] [Google Scholar]
- 6.Kharbanda SM, Sherman ML, Kufe DW.. Effects of tiazofurin on guanine nucleotide binding regulatory proteins in HL-60 cells. Blood 1990;75:583–8. [PubMed] [Google Scholar]
- 7.Vitale M, Zamai L, Falcieri E, et al. IMP dehydrogenase inhibitor, tiazofurin, induces apoptosis in K562 human erythroleukemia cells. Commun Clin Cytom 1997;30:61–6. [PubMed] [Google Scholar]
- 8.Kim HR, Roe JS, Lee JE, et al. A p53-inducible microRNA-34a downregulates Ras signaling by targeting IMPDH. Biochem Biophys Res Commun 2012;418:682–8. [DOI] [PubMed] [Google Scholar]
- 9.Messina E, Gazzaniga P, Micheli V, et al. Guanine nucleotide depletion triggers cell cycle arrest and apoptosis in human neuroblastoma cell lines. Int J Cancer 2004;108:812–7. [DOI] [PubMed] [Google Scholar]
- 10.Konno Y, Natsumedasq Y, Nagai M, et al. Expression of human IMP dehydrogenase types I and I1 in Escherichia coli and distribution in human normal lymphocytes and leukemic cell lines. J Biol Chem 1991;266:506–9. [PubMed] [Google Scholar]
- 11.Chong CR, Qian DZ, Pan F, et al. Identification of type 1 inosine monophosphate dehydrogenase as an antiangiogenic drug target. J Med Chem 2006;49:2677–80. [DOI] [PubMed] [Google Scholar]
- 12.Petrelli R, Vita P, Torquati I, et al. Novel inhibitors of inosine monophosphate dehydrogenase in patent literature of the last decade. Recent Pat Anticancer Drug Discov 2013;8:103–25. [PubMed] [Google Scholar]
- 13.Olah E, Kokeny S, Papp J, et al. Modulation of cancer pathways by inhibitors of guanylate metabolism. Adv Enzyme Regul 2006;46:176–90. [DOI] [PubMed] [Google Scholar]
- 14.Bentley R.Mycophenolic acid: a one hundred year odyssey from antibiotic to immunosuppressant. Chem Rev 2000;100:3801–25. [DOI] [PubMed] [Google Scholar]
- 15.Knight SR, Morris PJ.. Does the evidence support the use of mycophenolate mofetil therapeutic drug monitoring in clinical practice? A systematic review. Transplantation 2008;85:1675–85. [DOI] [PubMed] [Google Scholar]
- 16.Nair V, Shu Q.. Inosine monophosphate dehydrogenase as a probe in antiviral drug discovery. Antivir Chem Chemother 2007;18:245–58. [DOI] [PubMed] [Google Scholar]
- 17.Shah CP, Kharkar PS.. Inosine 5'-monophosphate dehydrogenase inhibitors as antimicrobial agents: recent progress and future perspectives. Future Med Chem 2015;7:1415–29. [DOI] [PubMed] [Google Scholar]
- 18.Shu Q, Nair V.. Inosine monophosphate dehydrogenase (IMPDH) as a target in drug discovery. Med Res Rev 2008;28:219–32. [DOI] [PubMed] [Google Scholar]
- 19.Pankiewicz KW, Petrelli R, Singh R, Felczak K.. Nicotinamide adenine dinucleotide based therapeutics, update. Curr Med Chem 2015;22:3991–4028. [DOI] [PubMed] [Google Scholar]
- 20.Cuny GD, Suebsuwong C, Ray SS.. Inosine-5’-monophosphate dehydrogenase (IMPDH) inhibitors: a patent and scientific literature review (2002–2016). Expert Opin Ther Pat 2017;27:677–90. [DOI] [PubMed] [Google Scholar]
- 21.Cholewinski G, Iwaszkiewicz-Grzes D, Trzonkowski P, Dzierzbicka K.. Synthesis and biological activity of ester derivatives of mycophenolic acid and acridines/acridones as potential immunosuppressive agents. J Enzyme Inhib Med Chem 2016;31:974–82. [DOI] [PubMed] [Google Scholar]
- 22.Chen L, Petrelli R, Olesiak M, et al. Bis (sulfonamide) isosters of mycophenolic adenine dinucleotide analogues: inhibition of inosine monophosphate dehydrogenase. Bioorganic Med Chem 2008;16:7462–9. [DOI] [PubMed] [Google Scholar]
- 23.Dunkern T, Chavan S, Bankar D, et al. Design, synthesis and biological evaluation of novel inosine 5′-monophosphate dehydrogenase (IMPDH) inhibitors. J Enzyme Inhib Med Chem 2014;29:408–19. [DOI] [PubMed] [Google Scholar]
- 24.Dunkern T, Prabhu A, Kharkar PS, et al. Virtual and experimental high-throughput screening (HTS) in search of novel inosine 5′-monophosphate dehydrogenase II (IMPDH II) inhibitors. J Comput Aided Mol Des 2012;26:1277–92. [DOI] [PubMed] [Google Scholar]
- 25.Cholewiński G, Iwaszkiewicz-Grześ D, Prejs M, et al. Synthesis of the inosine 5′-monophosphate dehydrogenase (IMPDH) inhibitors. J Enzyme Inhib Med Chem 2015;30:550–63. [DOI] [PubMed] [Google Scholar]
- 26.Chen L, Wilson DJ, Labello NP, et al. Mycophenolic acid analogs with a modified metabolic profile. Bioorganic Med Chem 2008;16:9340–5. [DOI] [PubMed] [Google Scholar]
- 27.Jones DF, Mills SD.. Preparation and antitumor properties of analogs and derivatives of mycophenolic acid. J Med Chem 1971;14:305–11. [DOI] [PubMed] [Google Scholar]
- 28.Suzui S, Takaku S, Mori T.. Antitumor activity of derivatives of mycophenolic acid. J Antibiot 1976;29:275–85. [DOI] [PubMed] [Google Scholar]
- 29.Dzierzbicka K, Kołodziejczyk AM.. Synthesis and antitumor activity of conjugates of muramyldipeptide or normuramyldipeptide with hydroxyacridine/acridone derivatives. J Med Chem 2003;46:183–9. [DOI] [PubMed] [Google Scholar]
- 30.Malachowska-Ugarte M, Cholewinski G, Dzierzbicka K, Trzonkowski P.. Synthesis and biological activity of novel mycophenolic acid conjugates containing nitro-acridine/acridone derivatives. Eur J Med Chem 2012;54:197–201. [DOI] [PubMed] [Google Scholar]
- 31.Iwaszkiewicz-Grzes D, Cholewinski G, Kot-Wasik A, et al. Synthesis and biological activity of mycophenolic acid-amino acid derivatives. Eur J Med Chem 2013;69:863–71. [DOI] [PubMed] [Google Scholar]
- 32.Domhan S, Muschal S, Schwager C, et al. Molecular mechanisms of the antiangiogenic and antitumor effects of mycophenolic acid. Mol Cancer Ther 2008;7:1656–68. [DOI] [PubMed] [Google Scholar]
- 33.Muraoka T, Ide M, Irie M, et al. Development of a method for converting a TAK1 type I inhibitor into a type II or c-helix-out inhibitor by structure-based drug design (SBDD). Chem Pharm Bull 2016;64:1622–9. [DOI] [PubMed] [Google Scholar]
- 34.Gudipati R, Reddy Anreddy RN, Manda S.. Synthesis, anticancer and antioxidant activities of some novel N-(benzo [d]oxazol-2-yl)-2-(7-or 5-substituted-2-oxoindolin-3-ylidene) hydrazinecarboxamide derivatives. J Enzyme Inhib Med Chem 2011;26:813–8. [DOI] [PubMed] [Google Scholar]
- 35.Kharkar P.Drugs acting on central nervous system (CNS) targets as leads for non-CNS targets. F1000Res 2014;3:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.