

Abstract
Three series of 2-arylpyridothieno[3,2-d]pyrimidin-4-ones 3a–j, pyridothienotriazolopyrimidines 6–8 and 4-imino-pyridothieno[3,2-d]pyrimidines 9a,b were prepared to improve the pim-1 inhibitory activity of the previously reported 2-arylpyridothieno[3,2-d]pyrimidin-4-ones. All the test compounds showed highly potent pim-1 inhibition with IC50 in the range of 0.06–1.76 µM. No significant difference was detected between the pim-1 inhibitory activity of the 4-pyrimidinone and the 4-imino (=NH) or the cyclised triazolopyrimidine derivatives. The most active compounds were tested for their cytotoxic activity on MCF7 and HCT116 and showed potent activity on both the cell lines.
Keywords: Pyridothienopyrimidine, pyridothienotriazolopyrimidine, cytotoxic activity, pim-1 inhibitors
Introduction
The proviral integration site for Moloney murine leukaemia virus-1 (known as pim-1) is a serine/threonine kinase that controls many cellular functions including cell cycle, cell differentiation, cell survival, apoptosis and drug resistance1–3. High levels of pim-1 kinase are associated with many types of cancer such as myeloid leukaemia, breast cancer and prostatic cancer1–4. The identification of the role of pim-1 in controlling the growth of cancer stem cells and promotion of multiple drug resistance added more to the importance of developing potent pim-1 inhibitors as anticancer agents that can overcome the drug resistance developed by cancer stem cells1,5.
Within the past 20 years, many pim-1 inhibitors have been identified, developed and are currently under preclinical studies or clinical trials as anticancer agents. Examples include the thiazolidin-2,4-dione derivative AZD1208 (I)1,2,6 and the benzonaphthyridine derivative CX-4595 (II)2,7,8 (Figure 1).
Figure 1.

Pim-1 inhibitors under clinical studies.
Recently, we had reported the identification of pyridothienopyrimidin-4-one derivatives IV as potent pim-1 inhibitors9. These derivatives were developed through structure rigidification strategy via ring closure of their precursors thieno[2,3-b]pyridines III10 to ensure the presence of the carbonyl group at proper orientation for binding with the enzyme. Indeed, our efforts led to significant improvement in the enzyme inhibitory activity as well as the cytotoxic activity. The most potent inhibitors were the 2-aryl-2,3-dihydro derivatives Va–c that exhibited pim-1 inhibition in the range of 1.18–1.97 µM9. However, the aryl groups used in that study were all bearing ortho substitution to mimic the structure of the previously published benzofuropyrimidinone derivative VI11 (Figure 2).
Figure 2.

Previous work published on pim-1 inhibitors.
In continuation to these efforts, we reported herein the SAR study of the effect of substitution on different positions of the aryl group on the pim-1 inhibitory activity. Thus, para substitution, disubstitution and trisubstitution on the phenyl ring were all investigated (compounds 3a–j). Besides, the effects of isosteric replacement of the C=O with = NH to give 4-imino derivatives (9a,b) or cyclisation into pyridothienotriazolopyrimidines (6–8) on pim-1 inhibition were investigated. It is noteworthy that this is the first published work describing the pim-1 inhibitory activity of pyridothienotriazolopyrimidine derivatives (Figure 3).
Figure 3.

Designing pyridothienopyrimidinones as pim-1 inhibitors.
All the new compounds were tested for their pim-1 enzyme inhibitory activity and the most active compounds were further tested for their anti-proliferative activity using two different cell lines MCF7 and HCT116.
Experimental part
General notes
Stuart SMP20 apparatus was used to determine the melting points and they were uncorrected. The IR spectra were recorded on Shimadzu IR 435 spectrophotometer (Kyoto, Japan) and the values were represented in cm−1. The 1H NMR and 13C NMR spectra were recorded on Bruker 400 and 100 MHz spectrophotometer, respectively. TMS was used as an internal standard and the chemical shifts were recorded in ppm on δ scale. Both IR and NMR spectra were carried out at Faculty of Pharmacy, Cairo University, Cairo, Egypt. The electron impact mass spectra were recorded on Thermo Scientific ISQLT single quadrapole mass spectrometer. Both mass spectra and elemental analyses were carried out at the regional centre for mycology and biotechnology, Al-Azhar University, Cairo, Egypt. All reagents and solvents were purified and dried by standard techniques. 3-Amino-5-bromo-4,6-dimethylthieno[2,3-b]pyridine-2-carbonitrile (1) and 3-amino-5-bromo-4,6-dimethylthieno[2,3-b]pyridine-2-carboxamide (2) were prepared according to the published methods9,12.
General procedure for the synthesis of 2-aryl-8-bromo-7,9-dimethyl-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-ones 3a–j
A mixture of 3-amino-5-bromo-4,6-dimethylthieno[2,3-b]pyridine-2-carboxamide (2) (0.6 g, 0.002 mol) and the appropriate aldehyde (0.002 mol) in glacial acetic acid (10 ml) was heated under reflux for 12 h. The reaction mixture was cooled and the product was filtered, dried and crystallised from acetic acid except 3j which was crystallised from DMF.
2-(4-(Benzyloxy)phenyl)-8-bromo-7,9-dimethyl-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-one (3a)
Yield: 61%; mp: 255–256 °C; IR (cm−1): 3421, 3394 (NH), 2924, 2854 (CH-aliphatic), 1654 (C=O); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.70 (s, 3H, CH3), 2.85 (s, 3H, CH3), 5.08 (s, 2H, OCH2), 5.80–5.83 (dd, 1H, CH-2, J = 2.96 Hz, J = 2.92 Hz), 7.00–7.50 (m, 10H, Ar-H + NH), 8.46 (s, 1H, NH); MS m/z: 495 [(M + 2)+, 1.77%], 493 [M+, 1.89%], 312 [(M + 2-C6H4OCH2C6H5)+, 100%], 310 [(M-C6H4OCH2C6H5)+, 93.00%]; Anal. calcd for C24H20BrN3O2S: C, 58.30; H, 4.08; N, 8.50. Found: C, 58.53; H, 4.17; N, 8.79.
8-Bromo-2-(4-bromophenyl)-7,9-dimethyl-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-one (3 b)
Yield: 63%; mp: >300 °C; IR (cm−1): 3421, 3394 (NH), 2924, 2854 (CH-aliphatic), 1654 (C=O); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.69 (s, 3H, CH3), 2.87 (s, 3H, CH3), 5.88 (s, 1H, CH-2), 7.18 (d, 1H, NH), 7.46–7.57 (m, 4H, Ar-H), 8.59 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ ppm 19.9, 26.7 (CH3), 65.4 (CH-2), 110.7, 121.4, 121.6, 124.5, 129.1, 131.5, 141.3, 144.2, 144.8, 157.4, 159.5 (Aromatic C), 161.4 (C=O); MS m/z: 469 [(M + 4)+, 41.60%], 467 [(M + 2)+, 100%], 465 [M+, 96.83%], 312 [(M + 2-C6H4Br)+, 17.25%], 310 [(M-C6H4Br)+, 14.59%]; Anal. calcd for C17H13Br2N3OS: C, 43.71; H, 2.80; N, 8.99. Found: C, 44.05; H, 2.93; N, 9.18.
8-Bromo-2-(4-chlorophenyl)-7,9-dimethyl-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-one (3c)
Yield: 68%; mp: >300 °C; IR (cm−1): 3394, 3363 (NH), 2924, 2854 (CH-aliphatic), 1666 (C=O); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.69 (s, 3H, CH3), 2.87 (s, 3H, CH3), 5.90 (s, 1H, CH-2), 7.20–7.53 (m, 4H, Ar-H), 8.30 (s, 1H, NH), 8.60 (s, 1H, NH); Anal. calcd for C17H13BrClN3OS: C, 48.30; H, 3.10; N, 9.94. Found: C, 48.61; H, 3.28; N, 10.11.
8-Bromo-2-(2,4-dihydroxyphenyl)-7,9-dimethyl-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-one (3d)
Yield: 68%; mp: 290–291 °C; IR (cm−1): 3502–3394 (NH/OH), 2954, 2900 (CH-aliphatic), 1654 (C=O); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.62 (s, 3H, CH3), 2.80 (s, 3H, CH3), 6.33–6.38 (dd, 1H, CH-2, J= 2.12 Hz, J = 2.12 Hz), 7.91–8.26 (m, 3H, Ar-H), 8.26 (s, 1H, NH), 10.23 (s, 1H, NH), 12.26 (br s, 2H, OH); Anal. calcd for C17H14BrN3O3S: C, 48.58; H, 3.36; N, 10.00. Found: C, 48.75; H, 3.27; N, 10.32.
8-Bromo-2-(3,4-dihydroxyphenyl)-7,9-dimethyl-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-one (3e)
Yield: 71%; mp: >300 °C; IR (cm−1): 3525 (OH), 3344, 3275 (NH), 2958, 2935 (CH-aliphatic), 1650 (C=O); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.68 (s, 3H, CH3), 2.84 (s, 3H, CH3), 5.69–5.72 (dd, 1H, CH-2, J= 2.88 Hz, J= 2.88 Hz), 6.69-6.95 (m, 4H, Ar-H + NH), 8.35 (d, 1H, NH, J= 2.92 Hz), 8.90 (s, 1H, OH), 8.95 (s, 1H, OH); 13C NMR (100 MHz, DMSO-d6) δ ppm 19.9, 26.7 (CH3), 66.2 (CH-2), 110.5, 114.5, 115.5, 118.1, 121.3, 124.5, 132.3, 144.5, 144.6, 145.3, 145.6, 157.1, 159.4 (Aromatic C), 161.8 (C=O); MS m/z: 421 [(M + 2)+, 9.58%], 419 [M+, 7.50%], 378 [100%]; Anal. calcd for C17H14BrN3O3S: C, 48.58; H, 3.36; N, 10.00. Found: C, 48.90; H, 3.49; N, 10.21.
8-Bromo-2-(4-fluorophenyl)-7,9-dimethyl-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-one (3f)
Yield: 48%; mp: >300 °C; IR (cm−1): 3410, 3170 (NH), 2993, 2954 (CH-aliphatic), 1662 (C=O); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.70 (s, 3H, CH3), 2.87 (s, 3H, CH3), 5.85 (s, 1H, CH-2), 7.12–7.57 (m, 4H, Ar-H), 8.32 (s, 1H, NH), 8.55 (s, 1H, NH); MS m/z: 312 [(M + 2-C6H4F)+, 10.05%], 310 [(M-C6H4F)+, 11.58%]; Anal. calcd for C17H13BrFN3OS: C, 50.26; H, 3.23; N, 10.34. Found: C, 50.41; H, 3.35; N, 10.57.
8-Bromo-2-(4-hydroxy-3-methoxyphenyl)-7,9-dimethyl-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-one (3 g)
Yield: 66%; mp: >300 °C; IR (cm−1): 3502 (OH), 3394, 3325 (NH), 2997, 2935 (CH-aliphatic), 1647 (C=O); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.69 (s, 3H, CH3), 2.84 (s, 3H, CH3), 3.76 (s, 3H, OCH3), 5.75–5.76 (d, 1H, CH-2), 6.73–7.13 (m, 3H, Ar-H), 7.13 (s, 1H, NH), 8.37 (s, 1H, NH), 9.04 (s, 1H, OH); 13C NMR (100 MHz, DMSO-d6) δ ppm 19.9, 26.7 (CH3), 56.0 (OCH3), 66.7 (CH-2), 111.1, 111.4, 115.3, 119.6, 121.3, 124.6, 131.8, 144.6, 144.7, 147.0, 147.8, 157.1, 159.5 (Aromatic C), 161.8 (C=O); Anal. calcd for C18H16BrN3O3S: C, 49.78; H, 3.71; N, 9.68. Found: C, 50.02; H, 3.89; N, 9.82.
8-Bromo-2-(4-methoxyphenyl)-7,9-dimethyl-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-one (3 h)
Yield: 57%; mp: >300 °C; IR (cm−1): 3390, 3367 (NH), 2931, 2908 (CH-aliphatic), 1658 (C=O); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.74 (s, 3H, CH3), 2.85 (s, 3H, CH3), 3.73 (s, 3H, OCH3), 5.82 (s, 1H, CH-2), 6.91–7.45 (m, 4H, Ar-H), 8.22 (d, 1H, NH), 8.46 (s, 1H, NH); MS m/z: 419 [(M + 2)+, 10.28%], 417 [M+, 14.49%], 301 [100%]; Anal. calcd for C18H16BrN3O2S: C, 51.68; H, 3.86; N, 10.05. Found: C, 51.84; H, 3.98; N, 10.31.
8-Bromo-7,9-dimethyl-2-(3,4,5-trimethoxyphenyl)-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-one (3i)
Yield: 59%; mp: >300 °C; IR (cm−1): 3236, 3167 (NH), 2989, 2962 (CH-aliphatic), 1651 (C=O); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.71 (s, 3H, CH3), 2.87 (s, 3H, CH3), 3.70 (br s, 6H, OCH3), 3.77 (s, 3H, OCH3), 5.81 (s, 1H, CH-2), 6.90–7.00 (m, 3H, Ar-H + NH), 8.48 (s, 1H, NH); Anal. calcd for C20H20BrN3O4S: C, 50.22; H, 4.21; N, 8.78. Found: C, 50.49; H, 4.37; N, 8.90.
8-Bromo-7,9-dimethyl-2-(thiophen-3-yl)-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-one (3j)
Yield: 54%; mp: 280–281 °C; IR (cm−1): 3275, 3190 (NH), 2924, 2854 (CH-aliphatic), 1639 (C=O); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.69 (s, 3H, CH3), 2.86 (s, 3H, CH3), 5.89–5.91 (dd, 1H, CH-2), 7.06–7.51 (m, 3H, Ar-H), 8.37 (d, 1H, NH), 8.52 (d, 1H, NH); 13C NMR (100 MHz, DMSO-d6) δ ppm 19.9, 26.8 (CH3), 63.5 (CH-2), 111.1, 121.3, 123.2, 123.4, 124.8, 127.1, 143.5, 144.4, 144.8, 157.2, 159.4 (Aromatic C), 161.5 (C=O); MS m/z: 395 [(M + 2)+, 94.00%], 393 [M+, 100%]; Anal. calcd for C15H12BrN3OS2: C, 45.69; H, 3.07; N, 10.66. Found: C, 45.82; H, 3.22; N, 10.92.
Synthesis of ethyl N-(5-bromo-2-cyano-4,6-dimethylthieno[2,3-b]pyridin-3-yl)formimidate (4)
A mixture of 3-amino-5-bromo-4,6-dimethylthieno[2,3-b]pyridine-2-carbonitrile (1) (0.56 g, 0.002 mol), triethyl orthoformate (6 ml) and acetic anhydride (4 ml) was heated under reflux for 12 h. The reaction mixture was cooled and the product was filtered, dried and crystallised from acetic acid. Yield: 83%; mp: 158–159 °C; IR (cm−1): 2989, 2850 (CH aliphatic), 2206 (CN); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.37–1.40 (t, 3H, CH3CH2O, J = 6.64 Hz), 2.73 (s, 3H, CH3), 2.76 (s, 3H, CH3), 4.42–4.44 (q, 2H, CH3CH2O, J = 6.76 Hz), 8.35 (s, 1H, =CH); 13C NMR (100 MHz, DMSO-d6) δ ppm 14.5 (CH3CH2), 19.83, 26.9 (ring CH3), 64.0 (CH3CH2), 90.9, 105.0, 114.5, 122.9, 145.9, 152.1, 158.1, 159.0, 159.9 (Aromatic C and CN); Anal. calcd for C13H12BrN3OS: C, 46.16; H, 3.58; N, 12.42. Found: C, 45.93; H, 3.72; N, 12.69.
Synthesis of 8-bromo-4-imino-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-3(4H)-amine (5)
Compound 4 (0.67 g, 0.002 mol) was mixed with hydrazine hydrate (99%, 6 ml) in absolute ethanol (10 ml). The mixture was heated under reflux for 10 h, allowed to cool and the product was filtered, dried and crystallised from acetic acid. Yield: 90%; mp: >300 °C; IR (cm−1): 3367, 3329, 3294 (NH/NH2), 2951, 2920 (CH aliphatic), 1658 (C = N); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.76 (s, 3H, CH3), 3.15 (s, 3H, CH3), 4.95 (s, 2H, NH2), 8.57 (s, 1H, =CH), 9.18 (s, 1H, =NH); Anal. calcd for C11H10BrN5S: C, 40.75; H, 3.11; N, 21.60. Found: C, 40.91; H, 3.24; N, 21.93.
Synthesis of 8-bromo-7,9-dimethylpyrido[3′,2′:4,5]thieno[2,3-e][1,2,4]triazolo[1,5-c]pyrimidine (6)
A mixture of compound 5 (0.64 g, 0.002 mol) and formic acid (3 ml) was heated under reflux for 5 h. The reaction mixture was allowed to cool and poured onto ice-cold water (20 ml), then the product was filtered, dried and crystallised from acetic acid. Yield: 73%; mp: 277–278 °C; IR (cm−1): 2985, 2962 (CH aliphatic), 1616 (C = N); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.76 (s, 3H, CH3), 3.12 (s, 3H, CH3), 8.82 (s, 1H, =CH), 9.94 (s, 1H, =CH); Anal. calcd for C12H8BrN5S: C, 43.13; H, 2.41; N, 20.96. Found: C, 43.50; H, 2.53; N, 21.08.
Synthesis of 8-bromo-2,7,9-trimethylpyrido[3′,2′:4,5]thieno[2,3-e][1,2,4]triazolo[1,5-c]pyrimidine (7a) and 8-bromo-7,9-dimethyl-2-(trifluoromethyl)pyrido[3′,2′:4,5]thieno[2,3-e][1,2,4]triazolo[1,5-c]pyrimidine (7 b)
A mixture of compound 5 (0.64 g, 0.002 mol) and acetic anhydride or 2,2,2-trifluoroacetic anhydride (4 ml) was heated under reflux for 5 h and then allowed to cool. The reaction mixture was poured onto ice-cold water (100 ml) and the product was filtered, dried and crystallised from acetic acid.
8-Bromo-2,7,9-trimethylpyrido[3′,2′:4,5]thieno[2,3-e][1,2,4]triazolo[1,5-c]pyrimidine (7a)
Yield: 93%; mp: 218–219 °C; IR (cm−1): 2939, 2850 (CH aliphatic), 1612 (C = N); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.91 (s, 3H, CH3), 2.80 (s, 3H, CH3), 3.19 (s, 3H, CH3), 9.18 (s 1H, =CH); Anal. calcd for C13H10BrN5S: C, 44.84; H, 2.89; N, 20.11. Found: C, 45.12; H, 3.01; N, 20.43.
8-Bromo-7,9-dimethyl-2-(trifluoromethyl)pyrido[3′,2′:4,5]thieno[2,3-e][1,2,4]triazolo[1,5-c]pyrimidine (7 b)
Yield: 68%; mp: 199–200 °C; IR (cm−1): 2958, 2924 (CH aliphatic), 1612 (C = N); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.67 (s, 3H, CH3), 3.01 (s, 3H, CH3), 8.69 (s, 1H, =CH); 13C NMR (100 MHz, DMSO-d6) δ ppm 19.1, 26.5 (CH3), 117.2, 119.0, 123.2, 123.9, 135.7, 139.7, 141.7, 145.4, 146.5, 148.8, 158.5 (Aromatic C and CF3); Anal. calcd for C13H7BrF3N5S: C, 38.82; H, 1.75; N, 17.41. Found: C, 39.07; H, 1.89; N, 17.68.
Synthesis of ethyl 2-(8-bromo-7,9-dimethylpyrido[3′,2′:4,5]thieno[2,3-e][1,2,4]triazolo[1,5-c]pyrimidin-2-yl)acetate (8)
A mixture of compound 5 (0.64 g, 0.002 mol) and diethyl malonate (5 ml) was heated under reflux for 2 h and then allowed to cool. The product was filtered, dried and crystallised from acetic acid. Yield: 27%; mp: 198–199 °C; IR (cm−1): 2978, 2924 (CH aliphatic), 1735 (ester C=O); 1H NMR (400 MHz, DMSO-d6) δ ppm 1.22–1.25 (t, 3H, CH3CH2, J = 7.04 Hz), 2.51 (s, 3H, CH3), 2.84 (s, 3H, CH3), 4.11-4.19 (q, 2H, CH2CH3, J = 7.04 Hz), 4.59 (s, 2H, CH2COO), 9.40 (s, 1H, =CH); Anal. calcd for C16H14BrN5O2S: C, 45.72; H, 3.36; N, 16.66. Found: C, 45.98; H, 3.54; N, 16.93.
Synthesis of 8-bromo-N-(substituted benzylidene)-4-imino-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-3(4H)-amines 9a,b
A mixture of compound 5 (0.64 g, 0.002 mol) and 4-bromobenzaldehyde or 2-(trifluoromethyl)benzaldehyde (0.002 mol) in glacial acetic acid (10 ml) was heated under reflux for 7 h and then allowed to cool. The product was filtered, dried and crystallised from acetic acid.
8-Bromo-N-(4-bromobenzylidene)-4-imino-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-3(4H)-amine (9a)
Yield: 90%; mp: >300 °C; IR (cm−1): 3197 (NH), 2920, 2850 (CH aliphatic); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.81 (s, 3H, CH3), 3.32 (s, 3H, CH3), 7.78–8.22 (m, 5H, Ar-H + NH), 8.32 (s, 1H, CH-2), 8.80 (s, 1H, N=CH); Anal. calcd for C18H13Br2N5S: C, 44.01; H, 2.67; N, 14.26. Found: C, 43.89; H, 2.80; N, 14.38.
8-Bromo-4-imino-7,9-dimethyl-N-(2-(trifluoromethyl)benzylidene)pyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-3(4H)-amine (9 b)
Yield: 78%; mp: >300 °C; IR (cm−1): 3178 (NH), 2970, 2819 (CH aliphatic); 1H NMR (400 MHz, DMSO-d6) δ ppm 2.67 (s, 3H, CH3), 3.05 (s, 3H, CH3), 7.61–7.89 (m, 5H, Ar-H + NH), 8.51 (s, 1H, CH-2), 8.68 (s, 1H, N=CH); Anal. calcd for C19H13BrF3N5S: C, 47.51; H, 2.73; N, 14.58. Found: C, 47.82; H, 2.86; N, 14.74.
Pim-1 kinase inhibitory activity
The kinase inhibitory activity of the synthesised compounds was determined using Human proto-oncogene serine/threonine-protein kinase pim-1 (PIM1) ELISA Kit (catalog #MBS7573). All the compounds were tested for their inhibitory activity against pim-1 kinase at 0.2, 1, 5 and 25 µM using Staurosporine as a reference standard. The results were displayed in terms of percent inhibition and IC50. Table 1 and Figure 4 show the obtained results.
Table 1.
Results of pim-1 kinase inhibition achieved by the test compounds.
 | |||
|---|---|---|---|
| Compound number | R or Ar | % Inhibition at 25µM | IC50 (µM) |
| 3a | 4-C6H5CH2OC6H4 | 83 | 0.96 |
| 3b | 4-BrC6H4 | 96 | 0.06 |
| 3c | 4-ClC6H4 | 92 | 0.14 |
| 3d | 2,4-(OH)2C6H3 | 81 | 1.34 |
| 3e | 3,4-(OH)2C6H3 | 93 | 0.06 |
| 3f | 4-FC6H4 | 89 | 1.00 |
| 3g | 3-OCH3-4-OHC6H3 | 92 | 0.08 |
| 3h | 4-OCH3C6H4 | 84 | 1.17 |
| 3i | 3,4,5-(OCH3)3C6H2 | 81 | 0.93 |
| 3j | 3-Thienyl | 95 | 0.10 |
| 6 | H | 81 | 1.08 |
| 7a | CH3 | 76 | 1.76 |
| 7b | CF3 | 93 | 0.08 |
| 8 | CH2COOC2H5 | 95 | 0.10 |
| 9a | 4-BrC6H4 | 88 | 0.52 |
| 9b | 2-CF3C6H4 | 89 | 0.22 |
| Staurosporine | – | 94 | Not determined |
Figure 4.

IC50 of the synthesised compounds in µM on pim-1 kinase.
In vitro cytotoxic activity
Cell culture protocol
Cell line cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). The cell lines used in this study were human breast adenocarcinoma (MCF7) and human colon adenocarcinoma (HCT116). The cells were cultured using DMEM (Invitrogen/Life Technologies) supplemented with 10% FBS (Hyclone), 10 µg/mL insulin (Sigma) and 1% penicillin-streptomycin. All of the other chemicals and reagents were from Sigma or Invitrogen.
Plate cells (cells density 1.2–1.8 × 10,000 cells/well) in a volume of 100 µL complete growth medium and 100 µL of the test compound per well were prepared in a 96-well plate for 24 h before the MTT assay. The culture medium was removed to a centrifuge tube. The cell layer was rinsed with 0.25% (w/v) Trypsin 0.53 mM EDTA solution to remove all traces of serum which contains Trypsin inhibitor. Then, 2–3 ml of Trypsin EDTA solution was added and the cells were observed under an inverted microscope until the cell layer is dispersed (usually within 5–15 min.). A volume of 6 to 8 ml of complete growth medium was added and the cells were aspirated by gently pipetting. The cell suspension was transferred to the centrifuge tube with the culture medium and centrifuged at approximately 125 ×g for 5 to 10 min. The supernatant was discarded and the cells were suspended in fresh growth medium then incubated at 37 °C for 24 h. After treatment of the cells with the serial concentrations of the test compounds or the control Staurosporine in DMSO (at concentrations of 200, 100, 10, 1 and 0.1 µM), incubation was carried out for 48 h at 37 °C, then the plates were examined under the inverted microscope and proceeded for the MTT assay.
Cytotoxicity assay by 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT)
The MTT method of monitoring in vitro cytotoxicity is well suited for use with multiwell plates. For best results, cells in the log phase of growth should be employed and final cell number should not exceed 106 cells/cm2. Each test should include a blank containing complete medium without cells. Cell cultures were removed from the incubator into laminar flow hood. Each vial of MTT [M-5655] was reconstituted to be used with 3 ml of medium or balanced salt solution without phenol red and serum and the reconstituted MTT was added in an amount equal to 10% of the culture medium volume. Then the cultures were returned to the incubator for 2–4 h depending on the cell type and the maximum cell density. After the incubation period, the cultures were removed from incubator and the resulting formazan crystals were dissolved by adding an amount of MTT solvent (solubilisation solution) [M-8910] equal to the original culture medium volume. Gentle mixing in a gyratory shaker enhanced dissolution. Occasionally pipetting up and down [trituration] may be required to completely dissolve the MTT formazan crystals. The absorbance was measured at a wavelength of 450 nm using Bioline ELIZA reader. The background absorbance of the multiwell plates was measured at 690 nm and subtracted from the 450 nm measurement. The results are shown in Table 2 and graphically represented in Figure 5.
Table 2.
Results of log P and in vitro cytotoxic screening of compounds 3b, 3c, 3e, 3g, 3j, 7b and 8 on two cell lines.
| IC50 μMa |
|||
|---|---|---|---|
| Compound number | MCF-7 | HCT116 | log P |
| 3b | 1.84 ± 0.12 | 2.74 ± 0.15 | 5.02 |
| 3c | 0.39 ± 0.07 | 0.47 ± 0.03 | 4.88 |
| 3e | 0.22 ± 0.04 | 0.37 ± 0.02 | 3.24 |
| 3g | 0.46 ± 0.09 | 0.38 ± 0.05 | 3.55 |
| 3j | 0.31 ± 0.02 | 4.82 ± 0.21 | 3.79 |
| 7b | 4.62 ± 0.14 | 0.65 ± 0.04 | 3.89 |
| 8 | 0.41 ± 0.03 | 0.42 ± 0.01 | 3.38 |
The values given are means of three experiments.
Figure 5.
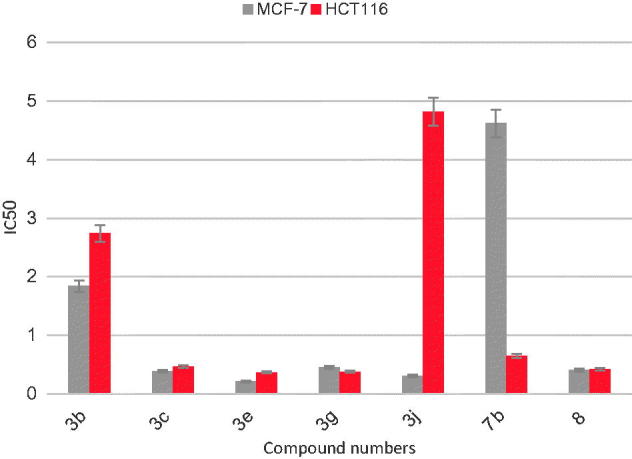
IC50 in µM of compounds 3b, 3c, 3e, 3g, 3j, 7b and 8 on MCF7 and HCT116 cell lines.
Results and discussion
Chemistry
Schemes 1 and 2 outline the synthesis of the target compounds. In the current work, 3-amino-5-bromo-4,6-dimethylthieno[2,3-b]pyridine-2-carboxamide (2) was prepared by alkaline hydrolysis of the 3-aminothieno[2,3-b]pyridine-2-carbonitrile 1 as reported by Naguib et al9. Compound 2 was then reacted with different aldehydes in glacial acetic acid to afford 2-aryl-8-bromo-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-ones 3a–j. The IR spectra of compounds 3a–j showed two NH bands at 3502–3167 cm−1 together with C=O band at 1666–1639 cm−1. Their 1H NMR spectra revealed the presence of a signal at δ 5.69–6.38 ppm corresponding to CH-2 proton. Besides, two signals appeared at δ 6.90–8.37 ppm and δ 8.35–10.23 ppm corresponding to two NH protons together with the aromatic protons signals at δ 6.69–8.26 ppm. The 13C NMR spectra of compounds 3b, 3e, 3g and3j showed a signal at δ 63.5–66.7 ppm corresponding to CH-2 carbon and a signal at δ 161.4–161.8 ppm corresponding to C=O carbon. Finally, the mass spectra of compounds 3a, 3b, 3e, 3h and 3j showed their corresponding molecular ion peaks.
Scheme 1.

Synthesis of the target compounds 3a–j.
Scheme 2.

Synthesis of the target compounds 6–9.
On the other hand, reacting 3-aminothieno[2,3-b]pyridine-2-carbonitrile 1 with triethyl orthoformate and acetic anhydride afforded the ethyl formimidate derivative 4. The formation of the latter compound was confirmed by IR spectroscopy that revealed the disappearance of the characteristic NH2 forked bands. Its 1H NMR spectrum showed triplet and quartet signals at δ 1.37–1.40 ppm and δ 4.42–4.44 ppm corresponding to CH3CH2 protons, respectively. Moreover, a singlet signal appeared at δ 8.35 ppm corresponding to N=CH proton. The 13C NMR spectrum of compound 4 revealed the presence of CH3CH2 carbons at δ 14.5 and 64.0 ppm, respectively.
Compound 4 was allowed to react with hydrazine hydrate to give 8-bromo-4-imino-7,9-dimethylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-3(4H)-amine (5). The IR spectrum of compound 5 showed three bands at 3367, 3329, 3294 cm−1 corresponding to NH/NH2 groups. The 1H NMR spectrum of compound 5 showed two singlet signals at δ 4.95 ppm and δ 9.18 ppm corresponding to NH2 and = NH protons, respectively, in addition to a singlet signal at δ 8.57 ppm corresponding to CH-2 proton.
Reacting compound 5 with formic acid or different anhydrides afforded pyrido[3′,2′:4,5]thieno[2,3-e][1,2,4]triazolo[1,5-c]pyrimidine derivatives 6 and 7a,b, respectively. Their IR and 1H NMR spectra revealed the absence of NH/NH2 groups.
The acetate derivative 8 was obtained via reacting compound 5 with diethyl malonate. The IR spectrum of compound 8 indicated the presence of the ester carbonyl band at 1735 cm−1. The 1H NMR spectrum revealed the appearance of a singlet signal at δ 4.59 ppm assigned to the methylene protons in addition to triplet and quartet signals at δ 1.22–1.25 ppm and δ 4.11–4.19 ppm corresponding to the ethyl protons.
Finally, the reaction of compound 5 with different aldehydes in glacial acetic acid furnished N-(substituted benzylidene)-4-imino-pyridothienopyrimidin-3-amines 9a,b. Their IR spectra indicated the disappearance of NH2 band, while their 1H NMR spectra revealed the presence of singlet signal of N=CH proton as well as the introduced aromatic protons.
Pim-1 kinase inhibitory activity
All the compounds were tested for their ability to inhibit pim-1 kinase at 0.2, 1, 5 and 25 µM using Staurosporine as a reference compound and the results in terms of percentage inhibition and IC50 were displayed in Table 1 and represented graphically in Figure 4.
The results indicated that all the test compounds exhibited highly potent pim-1 inhibitory activity in the range of 76–96% at 25 µM and IC50 in the range of 0.06–1.76 µM. The most potent compounds were 3b (96% inhibition, IC50= 0.06 µM), 3c (92% inhibition, IC50= 0.14 µM), 3e (93% inhibition, IC50= 0.06 µM), 3g (92% inhibition, IC50= 0.08 µM), 3j (95% inhibition, IC50= 0.10 µM), 7b (93% inhibition, IC50= 0.08 µM) and 8 (95% inhibition, IC50= 0.10 µM).
SAR study of the 2-aryl-8-bromo-7,9-dimethyl-2,3-dihydropyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4(1H)-ones 3a–j as pim-1 inhibitors indicated the following points:
Regarding the para-substituted derivatives 3a–c, f and h, the best results were obtained with halogens especially 3b (4-bromophenyl) with IC50 = 0.06 µM and 3c (4-chlorophenyl) with IC50 = 0.14 µM. Increasing the size of the substituents from 3h (4-methoxyphenyl) to 3a (4-benzyloxyphenyl) improved the inhibitory activity slightly.
Regarding the disubstituted derivatives 3d, e, g, better results were obtained with 3,4-disubstituted than with 2,4-disubstituted analogues. Both 3g (3-OCH3-4-OH derivative) and 3e (3,4-dihydroxy derivative) gave highly potent pim-1 inhibitory activity with IC50 of 0.08 and 0.06 µM, respectively.
Increasing the number of substitution in 3i (3,4,5-trimethoxyphenyl) was accompanied by reduction in the inhibitory activity (IC50=0.93 µM).
The presence of the heterocyclic ring (3-thienyl) in 3j gave highly potent pim-1 inhibitor as well (IC50=0.10 µM).
Upon designing the target pyridothienopyrimidinones, it was assumed that the substituent at position 2 of the ring occupied a solvent exposed area in pim-1 kinase active site and thus can accommodate various groups. Introduction of H-bonding and hydrophilic groups (like OH and methoxy groups) on the aryl ring especially at meta and para positions seemed to make extra H-bonding to the enzyme active site which might account for the higher kinase inhibitory activity exerted by compounds 3e and 3g.
On the other hand, SAR study of the pyridothienotriazolopyrimidines 6–8 indicated the following remarks:
The unsubstituted triazole derivative 6 showed potent pim-1 inhibition with IC50= 1.08 µM.
Substitution with methyl group in compound 7a reduced the inhibitory activity. However, substitution with CF3 group in compound 7b or C2H5OCOCH2 moiety in compound 8 improved the inhibitory activity significantly.
Finally, the 4-imino derivative 9a, b showed also potent pim-1 inhibitory activity with IC50= 0.52 and 0.22 µM, respectively. It is noteworthy that no significant difference was detected between the pim-1 inhibitory activity of the 4-pyrimidinone and the 4-imino (=NH) or the cyclised triazolopyrimidine derivatives.
In vitro cytotoxic activity
The most active pim-1 inhibitors in this work (compounds 3b, 3c, 3e, 3g, 3j, 7b and 8) were tested for their cytotoxic activity against MCF7 and HCT116 cell lines using MTT method13,14. The results in terms of IC50 in µM are given in Table 2 and represented graphically in Figure 5.
All the compounds showed potent cytotoxic activity on both cell lines. The cytotoxic activity on MCF7 cell line ranged between 0.22 and 1.84 µM. Five compounds (3c, 3e, 3g, 3j and 8) exerted potent cytotoxic activity with IC50 values below 0.5 µM. Whilst, the cytotoxic activity on HCT116 ranged between 0.37 and 4.82 µM. Here five compounds (3c, 3e, 3g, 7b and 8) showed potent cytotoxic activity with IC50 values below 1 µM.
Compound 3e [the 2-(3,4-dihydroxyphenyl) derivative] displayed the most potent cytotoxic effect on both cell lines with IC50 values of 0.22 and 0.37 µM, respectively. These results were consistent with its high kinase inhibitory activity (IC50= 0.06 µM). While compound 3b showed the least potent cytotoxic activity on both cell lines with IC50 values of 1.84 and 2.74 µM, respectively.
Notably, compounds 3b and 3e exhibited the same pim-1 inhibitory activity with IC50 of 0.06 µM. The dramatic variation in their cytotoxic activity might be explained by examining their calculated log p values15 (Table 2). The calculated log P of compound 3b was 5.02, while that of 3e was 3.24.
It is noteworthy that significant improvement was observed in the cytotoxic activity of the tested compounds compared to their precursors Va–c. The latter cytotoxicity range was between 30 and 180 µM on the same cell lines9.
Conclusions
In this work, series of 2-arylpyridothieno[3,2-d]pyrimidin-4-ones 3a–j, pyridothienotriazolopyrimidine 6–8 and 4-imino-pyridothieno[3,2-d]pyrimidines 9a,b were prepared to improve the pim-1 inhibitory activity of the previously reported 2-arylpyridothieno[3,2-d]pyrimidin-4-ones. The lead optimisation strategies used were: isosteric replacement of C=O by =NH and ring closure of the imino derivatives into triazolo ring. Besides, a brief SAR study of the substitution on the 2-aryl moiety was done. All the test compounds showed highly potent pim-1 inhibition with IC50 in the range of 0.06–1.76 µM. Compounds 3g (3-OCH3-4-OH derivative) and 3e (3,4-dihydroxy derivative) were the most potent pim-1 inhibitors in this study with IC50 of 0.08 and 0.06 µM, respectively. No significant difference was detected between the pim-1 inhibitory activity of the 4-pyrimidinone and the 4-imino (=NH) or the cyclised triazolopyrimidine derivatives. The most active compounds were tested for their cytotoxic activity on two cell lines [MCF7 and HCT116] using MTT method. All the compounds showed potent cytotoxic activity on both cell lines but compound 3e [the 2–(3,4-dihydroxy) derivative] displayed the most potent cytotoxic effect on both cell lines with IC50 values of 0.22 and 0.37 µM and these results were consistent with its high kinase inhibitory activity (IC50 = 0.06 µM). A remarkable improvement in the cytotoxic activity was also noticed compared to the previously reported pyridothienopyrimidinones. Further work on pyridothienopyrimidine scaffold is still needed to obtain more potent pim-1 inhibitors and to improve the physicochemical properties of these derivatives.
Supplementary Material
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- 1.Tursynbay Y, Zhang J, Li Z, et al. . Pim-1 kinase as cancer drug target: an update (Review). Biomed Rep 2016;4:140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Le BT, Kumarasiri M, Adams JRJ, et al. . Targeting pim kinases for cancer treatment: opportunities and challenges. Future Med Chem 2015;7:35–53. [DOI] [PubMed] [Google Scholar]
- 3.Narlik-Grassow M, Blanco-Aparicio C, Carnero A.. The PIM family of serine/threonine kinases in cancer. Med Res Rev 2014;34:136–59. [DOI] [PubMed] [Google Scholar]
- 4.Zha W, Qiu R, Li P, Yang J.. PIM1: a promising target in patients with triple-negative breast cancer. Med Oncol 2017;34:142. [DOI] [PubMed] [Google Scholar]
- 5.Xie Y, Bayakhmetov S.. PIM1 kinase as a promise of targeted therapy in prostate cancer stem cells (Review). Mol Clin Oncol 2016;4:13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keeton EK, McEachern K, Dillman KS, et al. . AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood 2014;123:905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siddiqui-Jain A, Drygin D, Streiner N, et al. . CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res 2010;70:10288–98. [DOI] [PubMed] [Google Scholar]
- 8.Pierre F, Stefan E, Nedellec AS, et al. . 7-(4H-1,2,4-Triazol-3-yl)benzo[c][2,6]naphthyridines: a novel class of Pim kinase inhibitors with potent cell antiproliferative activity. Bioorg Med Chem Lett 2011;21:6687–92. [DOI] [PubMed] [Google Scholar]
- 9.Naguib BH, El-Nassan HB, Abdelghany TM.. Synthesis of new pyridothienopyrimidinone derivatives as Pim-1 inhibitors. J Enzyme Inhib Med Chem 2017;32:457–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naguib BH, El-Nassan HB.. Synthesis of new thieno[2,3-b]pyridine derivatives as pim-1 inhibitors. J Enzyme Inhib Med Chem 2016;31:1718–25. [DOI] [PubMed] [Google Scholar]
- 11.Tsuhako AL, Brown DS, Koltun ES, et al. . The design, synthesis, and biological evaluation of PIM kinase inhibitors. Bioorg Med Chem Lett 2012;22:3732–8. [DOI] [PubMed] [Google Scholar]
- 12.Madkour HMF, Afify AAE, Abdalha AA, et al. . Synthetic utility of enaminonitrile moiety in heterocyclic synthesis: synthesis of some new thienopyrimidines. Phosphorus Sulfur Silicon Relat Elem 2009;184:719–32. [Google Scholar]
- 13.Mosmann T.Rapid colorimetric assay for cellular growth andsurvival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63. [DOI] [PubMed] [Google Scholar]
- 14.Scudiere DA, Shoemaker RH, Paul KD, et al. . Evaluation of a soluble Tetrazolium/Formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res 1988;48:4827–33. [PubMed] [Google Scholar]
- 15. Molinspiration Cheminformatics company, Slovak Republic. Available from: http://www.molinspiration.com/cgi-bin/properties [last accessed 17 Oct 2017].
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.