

Abstract
We demonstrate a 15-fold enhancement of solid-state NMR signals via dynamic nuclear polarization (DNP) based on a stable, naturally occurring radical in a protein: the flavin mononucleotide (FMN) semiquinone of flavodoxin. The linewidth of flavodoxin’s EPR signal suggests that the dominant DNP mechanism is the solid effect, consistent with the field-dependent DNP enhancement profile. The magnitude of the enhancement as well as the bulk-polarization build-up time constant (τB) with which it develops are dependent on the isotopic composition of the protein. Deuteration of the protein to 85 % increased the nuclear longitudinal relaxation time T1n and τB by factors of five and seven, respectively. Slowed dissipation of polarization can explain the two-fold higher maximal enhancement than that obtained in proteated protein, based on the endogenous semiquinone. In contrast, the long τB of TOTAPOL-based DNP in non-glassy samples was not accompanied by a similarly important long T1n, and in this case the enhancement was greatly reduced. The low concentrations of radicals occurring naturally in biological systems limit the magnitude of DNP enhancement that is attainable by this means. However, our enhancement factors of up to 15 can nonetheless make an important difference to the feasibility of applying solid-state NMR to biochemical systems. We speculate that DNP based on endogenous radicals may facilitate MAS NMR characterization of biochemical complexes and even organelles, and could also serve as a source of additional structural and physiological information.
Keywords: DNP, solid-state NMR, Deuteration, Flavodoxin, Flavin Semiquinone
TOC image
Introduction
Dynamic nuclear polarization (DNP) is a powerful method for enhancing signal intensities in liquid and solid-state NMR experiments, and enhancement factors of up to 200 for 1H nuclei have been observed for model systems and biomolecules1–5. In a DNP experiment, the large Boltzmann electron polarization of a paramagnetic polarizing agent is transferred to nuclei in close proximity by irradiating the sample with microwave radiation near the electron paramagnetic resonance (EPR) transition frequency. Depending on the sizes of the inhomogeneous (Δ) and homogeneous (δ) breadths of the EPR signal relative to the nuclear Lamor nuclear frequency (ω0n) the DNP process at high magnetic fields (> 5 T) can either occur through the solid effect (SE, ω0n > Δ, δ) or through the cross-effect (CE, Δ> ω0n > δ)6–9. Currently the largest signal enhancements are observed in DNP-enhanced solid-state NMR experiments that exploit the CE using exogeneous biradicals such as TOTAPOL as polarizing agents10,11. In this approach the solvent, usually a glassy matrix such as glycerol/H2O, is polarized and 1H-1H spin diffusion distributes the polarization uniformly throughout the sample. In a second step the enhanced 1H polarizaton is transferred to 13C, 15N or other nuclei in solutes of interest. However for optimum DNP performance a large amount of the exogenous polarizing agent has to be added to the sample; this is generally assumed to be distributed randomly throughout the matrix and to not affect the integrity of the molecules of interest12,13.
In addition to exogenous polarizing agents biology offers several endogenous radicals whose use does not perturb the system and moreover offers control over the location of the radical, and coupling of its occurrence to a particular state of the sample. Biological systems are typically more complex and more dilute than model systems, so the enhancements possible by DNP would be particularly valuable. With a view to enabling DNP-enhanced NMR to address intact biological systems, we propose the use of endogeneous radicals that occur naturally in many proteins as sources of polarization.
Some 10 % of biological macromolecules possess a paramagnetic center such as a paramagnetic metal ion (eg. Cu2+, Fe3+, Mn2+), metal cluster (eg. FexSy) or transient or stable organic radical (quinone, flavin, tyrosine, etc.). Based on a recent (January 2012) search of the Protein Data Bank (www.rcsb.org), the number of protein structures whose annotation included the name of a metal ion that readily adopts a paramagnetic oxidation state accounted for 9 % of all protein structures, and the number of structures whose annotation included either ‘FMN’, ‘FAD’, ‘quinone’ or ‘radical’ accounted for 3 %. When protein structures that posses diamagnetic cations Zn2+, Mg2+ and Ca2+ were also included, the total percentage of structures containing a metal ion came to 40%, in qualitative agreement with more formal determinations14. Diamagnetic cations can often be replaced by paramagnetic metal ions to produce a paramagnetic site, for example for studies by EPR15–18. These cofactors are essential to the activity of many redox-active enzymes and electron transport proteins that participate in the respiratory or photosynthetic electron transport chains. Many of the above cofactors alternate among different oxidation and spin states depending on the physiological state of the system, leading to the possibility that DNP enhancement could be used as a probe of physiological status.
Our proposed use of a naturally-occurring endogenous radical for DNP-enhanced NMR spectroscopy is supported by numerous successes with related techniques. A popular implementation of chemically-induced DNP (CIDNP) has exploited photoexcited flavins that can react with Trp, His and Tyr side chains to produce a radical pair. When the recombination efficiency is greater in the presence of one nuclear spin state than the other, for example based on electron-nuclear hyperfine facilitated intersystem crossing, then the resulting diamagnetic ground state retains net nuclear polarization19,20. In the case of dye-supported photo-CIDNP, flavins free in solution serve as the dye and can produce enhanced NMR resonances from exposed aromatic amino acid side chains19,21. Photogenerated endogenous radicals in bacterial reaction centers have also been used to obtain thousand-fold enhancements of 13C and 15N solid-state NMR signals of the pigments that participate in photosynthetic electron transfer, and several different mechanisms have been proposed22–24. In this case, effects are restricted to nuclei in hyperfine contact with the endogenous radicals so the method yields primarily local information. Moreover its reliance on a pair of highly divergent electron recombination rates for the two participating spin states, and large electron-hyperfine coupling, limits the general applicability of the method. This combination of spin properties has been proposed to be an evolved characteristic of photosynthetic reaction centers23, but the effect has recently been demonstrated in a photoactive flavoprotein as well25. Stable naturally-occurring radicals have also been used to measure important distances in biochemical systems26–28. A recent example of double electron-electron resonance (DEER) exploits the naturally occurring flavin radical of the electron transfer flavoprotein (ETF) that is generated specifically upon turnover of a coupled enzyme29. Thus, the resulting signal is not only a probe of structure, but also a signature of physiological state. In contrast, the diverse opportunities natural radicals offer to DNP-enhanced NMR spectroscopy have barely been explored.
In this first example of using an endogenous stable radical for DNP-enhanced NMR spectroscopy we exploit the flavin mononucleotide (FMN) radical semiquinone (SQ) of flavodoxin (FD). FDs are small well-behaved electron transport proteins containing a single non-covalently bound FMN. FDs accept a single electron at low potential (for example from photosystem I) and then serve as electron donors to a variety of enzymes and processes in microbial metabolism (for example nitrogen fixation). Thus, FD’s bound FMN cycles between a diamagnetic oxidized state and a stable neutral SQ radical formed upon acquisition of an electron and a proton. The FD from Desulfovibrio vulgaris has been extensively characterized electrochemically30 and by various forms of optical spectroscopy31. Many mutants have been characterized32–34, the crystal structures of all three oxidation states have been solved35,36 and NMR resonance assignments have been published for both the diamagnetic states37–39. The structure of flavodoxin is depicted in Figure 1 and is unaffected by the flavin oxidation state, with the exception of residues 59-62 and 95-102 immediately adjacent to the flavin36,38–40. Thus, FD provides a robust and familiar system in which to explore the practicality of using a stable endogenous radical that participates in biological function, as a source of DNP.
Figure 1.
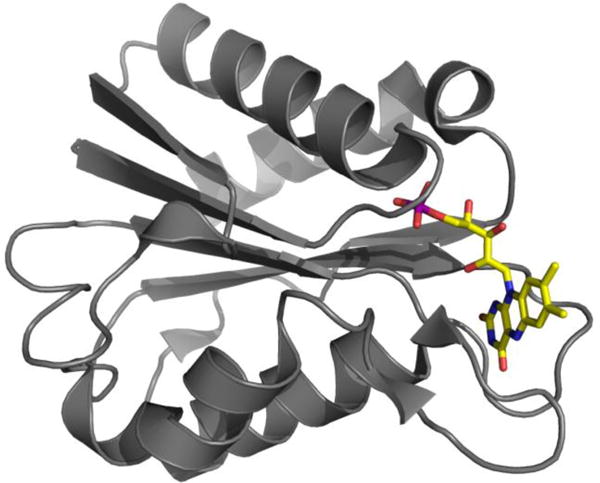
Ribbon structure of FD depicting elements of secondary structure with ribbon segments and intervening stretches of backbone with cord. Amino acid side chains are omitted for simplicity however the flavin is shown in atomic detail with N atoms in blue, O atoms in red and the P atom in purple. H atoms are not shown and C is in yellow consistent with FMN’s intense yellow color. Figure generated using Pymol41.
In this paper we report a 15-fold DNP enhancement of 1H magic-angle spinning solid-state NMR (MAS NMR) spectra despite the ten-fold lower concentration of our FD SQ as compared to typical concentrations used of exogenous polarization agents for DNP-enhanced NMR spectroscopy. In addition we show that the enhancement is responsive to the isotopic composition of the FD protein and that the increased enhancement upon deuteration can be attributed to a longer T1n. The enhancement we observe conforms to other expectations for SE DNP, as anticipated based on the breadth of the SQ EPR signal. Thus we show that an endogenous radical can support DNP just as exogenous radicals do, albeit with smaller enhancements most likely as a result of its lower concentration.
Materials and Methods
Overexpression and Purification of Flavodoxin (FD)
The gene encoding the FD of Desulfovibrio vulgaris was cloned into the pET23d vector, which provides a C-terminal Leu-Glu-His6 tag. The base substitution required to produce an NcoI site also produced a Pro -> Ala amino acid substitution at position 2. These changes had no apparent effect on FD’s optical properties or EPR signal. The resulting FD-pET23d plasmid was transformed into Escherichia coli BL21 (DE3) competent cells.
Unlabelled FD
Unlabelled FD was expressed in Luria-Bertani (LB) medium containing 50 μg/mL ampicillin, upon addition of IPTG to 1 mM when the optical density at 600 nm (OD600) reached approximately 0.6. The temperature was lowered from 37 °C to 32 ˚C and after an additional 4 hours of shaking, cells were harvested by centrifugation. The cell pellet was resuspended in lysis buffer (50 mM potassium phosphate, pH = 7.30, containing 300 mM KCl, 10 mM imidazole) with 0.5 mg/mL 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF) and 0.1 mM FMN, and lysed by passage through a French press twice at 12,000-15,000 psi. The suspension was centrifuged at 4 °C, 15000 × g for 45 min. to remove cell debris. Supernatant from about 10 g of cells, wet weight, was applied to a 20 mL Ni-NTA (nitrilotriacetate) agarose column equilibrated with 200 mL of lysis buffer. The flow-through was collected and reloaded three more times. The column was washed with wash buffer (50 mM potassium phosphate, pH = 7.30, containing 300 mM KCl, 20 mM imidazole) to remove non-specifically bound protein until the OD280 of the eluant was constant and lower than 0.02. His-tagged FD was eluted using high imidazole elution buffer (50 mM potassium phosphate, pH = 7.30, containing 300 mM KCl, 250 mM imidazole). Fractions were collected and their OD280 was monitored to identify those containing protein. The eluted protein’s identity was determined on the basis of molecular weight by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and yellow color. Fractions containing pure His-tagged FD were combined and dialyzed against dialysis buffer (200 mM potassium phosphate, pH = 7.50). The protein was concentrated using an Amicon Ultra-15 centrifugal filter device (10 kDa nominal molecular weight limit). The concentration of FD was determined based on measured values of OD273 and OD458 using a HP8452B diode array spectrophotometer. (ε273 = 46,300 M−1cm−1, ε458 = 10,700 M−1cm−1 30). Resulting FD had an A458/A273 ratio of 0.196 or higher, and was electrophoretically pure.
[13C, 1H] labeled FD
[13C, 1H] labeled FD was expressed in 1 L M9 medium including only 3 g 13C D-glucose (vs. the standard 10 g) prepared as a separate filter-sterilized stock solution. Cells were produced by growing 1 L of LB culture at 37 °C to an OD600 of 0.5, harvesting by centrifugation, washing the pellet with 20 ml of the 13C-glucose-free M9 medium and then suspending the cells in 1 L 13C-glucose-complete M9 supplemented with 10 μM FeCl2, 4 mM MgSO4, 100 μM CaCl2, 50 μg/mL carbenicillin, 0.5 mM IPTG and 1 mL of micronutrient solution. The culture was incubated at 32 °C with shaking for 12 hours before harvesting. FD was purified as above.
[13C, 2H] labeled FD
[13C, 2H] labeled FD was expressed as above but using 1 L of M9 medium made up in 2H2O and filter-sterilized. Stock solutions of Fe, Mg, Ca, micronutrients, carbenicillin and IPTG were made up in 1H2O. FD was purified as above. The protein was estimated to be 85% deuterated in aliphatic positions based on the intensity of aliphatic 1H NMR signals vs. 1H backbone amide signals, which had acquired 1H from 1H2O.
Sample preparation for DNP
Purified FD was concentrated to > 2 mM and exchanged into a solution that was 70 % by volume 2H-glycerol and 30 % by volume perdeuterated-buffer (200 mM potassium phosphate pH 7.5, 0.02 % NaN3 in 2H2O) by repeated dilution and re-concentration. The samples were rendered anaerobic and then reduced to the SQ state using a calibrated anaerobic solution of dithionite also in 70 % 2H-glycerol: 30 % perdeuterated buffer (v:v). Calibration was achieved by titrating a known amount of FMN with the dithionite solution and using optical spectrophotometry to determine when the sample had been fully reduced (oxidized FMN’s ε445 of 12.5 mM−1cm−1 42 and the known volume were used to determine the amount of FMN in use). The dithionite was used within 2 hours of calibration, during which time it was kept anaerobic and on ice. Sample reductions were performed in septum-sealed anaerobic reactivials (Pierce) and each NMR sample’s color was monitored until the transition from yellow (oxidized) to purple (neutral SQ radical) was complete (~15 min.). Working in a glove bag, the material was then loaded into degassed NMR rotors, sealed, frozen and stored in Ar-filled sealed vials. Such samples remained in the SQ state for months at − 80 °C.
For samples prepared with the exogenous biradical polarizing agent TOTAPOL11, the FD was used in its diamagnetic oxidized state and TOTAPOL was added to a final concentration of 10 mM (20 mM unpaired e−).
Data shown here were all obtained using samples made up in fully deuterated buffer and glycerol. This is important because the paramagnetic flavin of FD is exposed at the surface of the protein (Figure 1), so that bleeding of the 1H polarization into the solvent matrix can readily occur.
DNP experiments
DNP experiments were performed using a custom-built DNP MAS NMR spectrometer operating at a nuclear Larmor frequency of 212 MHz for 1H (140 GHz for electrons) corresponding to a magnetic field strength of 5 T. Experiments were performed in a custom-designed 4 mm triple-resonance (e−, 1H, 13C) low-temperature MAS NMR probe, or a similar 2.5 mm triple resonance probe using a commercial stator (Revolution NMR Inc.). Microwave radiation was generated by a gyrotron operating at a frequency of 139.662 GHz43,44. The data presented here were obtained using a microwave power of approximately 2.5 W (estimated at the position of the sample). The sample was contained in a 4.0 mm or a 2.5 mm sapphire rotor (Insaco, Quakertown, PA USA).
DNP-enhanced 1H MAS NMR spectra were obtained at approximately 90 K using direct 1H detection at 212 MHz. While MAS NMR experiments using the naturally-occurring SQ radical where performed using either a 4 mm sapphire rotor typically spinning at ωr/2π ≈ 3 kHz or a 2.5 mm sapphire rotor, all experiments using TOTAPOL as the polarizing agent were performed using a 2.5 mm rotor at a spinning frequency of ωr/2π ≈ 5 kHz. For all experiments a 1H pre-saturation sequence was applied (saturation train of 16 120º pulses interspersed with 2 ms delays) followed by polarization for a time period tmw and then a single 90° detection pulse (83 kHz field strength). During the polarization period the sample was either irradiated with microwave radiation (“on-signal”) or not (“off-signal”). The free induction decay (FID) was typically digitized for 2048 μs before the sequence was repeated. No additional recycle delay was necessary. To record the DNP enhancement profile the applied magnetic field was varied using a superconducting sweep coil (± 75 mT). At each point in the profile, the actual field strength was measured using a field/frequency lock system accessory built into the spectrometer45. DNP enhancement profiles were recorded using a tmw of 5 s.
For DNP buildup curves, the static field (B0) was adjusted to 4987.8 mT so that the microwave irradiation frequency coincided with ω0e + ω0n, and tmw was varied. In each case two series of 1H MAS NMR spectra were obtained, one with application of microwave irradiation to the sample and one without microwave radiation. The first such series yields the polarization build-up time τB as described below and the second yields the corresponding T1n via the same analysis.
1H detection suffers from technical artifacts, such as incompletely resolved rotational sidebands, or receiver ring-down, which can interfere with other measurements. However since such artifacts are not related to the DNP mechanism, they remain constant throughout the enhancement profiles and series of 1H spectra were used to construct build-up curves and field profiles.
For 13C-labeled samples containing TOTAPOL as the polarizing agent the 1H polarization was transferred to 13C with a 0.7 ms cross-polarization (CP) step using a 50 kHz field strength on the 1H and 13C channels46. This enabled us to observe only polarization that diffused into the protein because only the protein was uniformly 13C labelled whereas glycerol in the buffer was 12C-glycerol.
NMR Data Processing and Analysis
Processing of 1H detected FIDs included left-shifting of the data set by two points (to eliminate a large contribution of receiver ring-down) and zero-filling to twice the number of experimental data points followed by magnitude-mode Fourier transformation. The MAS NMR signal intensity was calculated as the peak-to-trough height of the center-band in order to minimize contributions from broad background signals. Build-up of bulk-polarization was characterized by fitting observed 1H MAS NMR signal intensity S to a mono-exponential function of the microwave polarization time tmw
| Eq. 1 |
where M∞ is the (maximal) steady-state signal amplitude, and τB is the bulk-polarization build-up time. All fits were performed in Matlab (Mathworks) using home-written routines. The DNP enhancement of the 1H MAS NMR signal (εH) was calculated based on the tmw → ∞ steady-state asymptotes of the buildup curves, M∞, as the ratio of the value obtained with microwave irradiation (‘on-signal’) divided by the value obtained without (‘off-signal’). We estimate errors on the order of 5-10 % based on reproducibility of the results obtained from different data sets and different samples.
EPR experiments
EPR experiments were performed on a custom-designed high-field EPR spectrometer operating at a microwave frequency of 139.504 GHz47,48. The sample, with a volume of approximately 250 nL, was transferred to a Suprasil quartz tube (o.d. 0.55 mm) and frozen to 40 K, the temperature at which data were collected. EPR spectra were recorded using a two-pulse echo sequence (π/2–τ–π–τ–echo) using a 80 ns π/2 pulse length and a τ of 200 ns. The data were recorded at a 1 kHz repetition rate and 10 points per field position were averaged. To obtain an EPR spectrum the integrated echo intensity was plotted as a function of the magnetic field over a range of 4976 ± 10 mT. The spectrometer was equipped with a field/frequency lock system45 to ensure accurate field measurements at each point. Pseudo-modulated spectra were calculated from the absorption-mode spectra using a modulation amplitude of 0.3 mT as per Hyde et al.49.
X-band EPR data were recorded using a Bruker EMX spectrometer in continuous wave mode using a microwave power of 0.2 mW and a modulation amplitude of 0.5 G with the sample (250 μl, 4 mm quartz tube) immersed in liquid N2 (at 77 K).
Results and Discussion
Optical Titration of Flavodoxin and Production of the Semiquinone
To maximize the efficiency of DNP, FD was quantitatively reduced to the neutral (blue) flavin SQ state via one-electron reduction with dithionite under anaerobic conditions30 (see Scheme 1). Anaerobic formation of the SQ state of FD was monitored optically between small additions of a calibrated solution of dithionite. The isosbestic at 515 nm shared by the spectra ranging from black through red in Figure 2 confirms simple conversion of the yellow oxidized state (λmax = 462 nm) to the violet SQ (λmax = 581 nm), prior to onset of the second reductive phase (violet to grey traces, not detailed) in which the SQ is converted to the faintly colored hydroquinone30. Our absorption maxima are very close to the literature values of 458 nm and 580 nm, respectively. The use of a calibrated solution of dithionite permitted virtually quantitative production of FD SQ at the high, optically opaque concentrations required for MAS NMR spectroscopy (central region of lower inset).
Scheme 1.
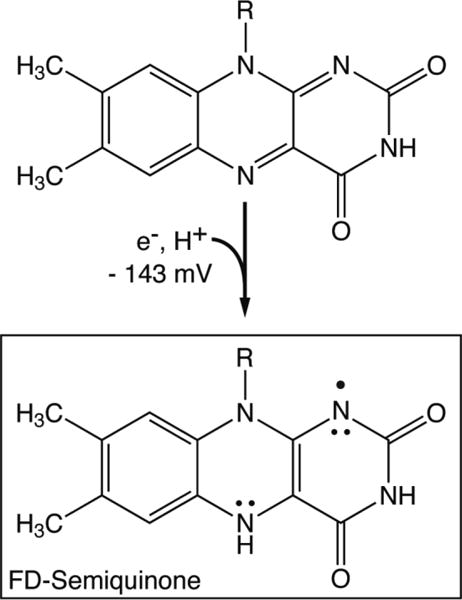
Reduction of FMN cofactor by one electron. The reduction potential indicated is that of D. vulgaris FD (vs. N.H.E)30 and R = ribose phosphate. Only one of several resonance structures is shown for the SQ.
Figure 2.
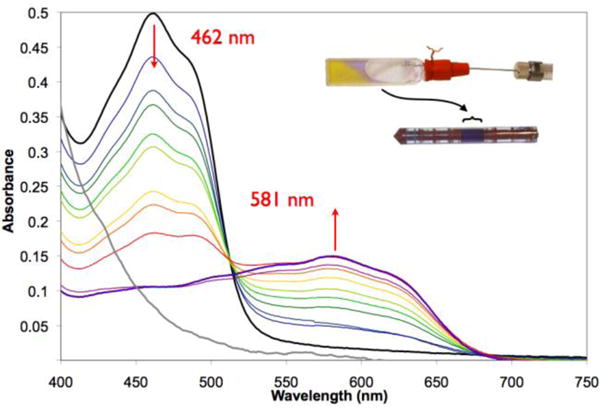
Optical profile of conversion of diamagnetic oxidized FD (black trace and yellow material in the inset cuvette) to the neutral SQ radical form (purple trace and violet material in the inset cuvette and MAS NMR rotor). Experimental conditions are described in the Methods section. The MAS NMR rotor (lower inset) is magnified relative to the optical cuvette (upper inset), to improve visibility of the purple-coloured FD SQ in the region indicated by the brace symbol.
High-field EPR Spectroscopy and DNP Enhancement Profile
To verify the paramagnetic state of the FD SQ sample we performed 9 GHz and 140 GHz EPR spectroscopy prior to DNP experiments. The 140 GHz high-field EPR absorption spectrum of the FD SQ radical together with its pseudo-modulated spectrum49 are shown in Figure 3 (supplemental Figure S1 shows the X-band signal). The spectrum is very similar to high-field EPR spectra of the FMN SQ radical of photolyase40,50. Schnegg et al. have explained the features of the photolyase signal in terms of g-anisotropy and large hyperfine interactions between the electron spin and a hydrogen plus two nitrogen nuclei.40,51. A simulation of the 140 GHz EPR spectrum of the FD SQ radical using parameters obtained by Schnegg et al. is given in Figure S2 of the supplemental material.
Figure 3.
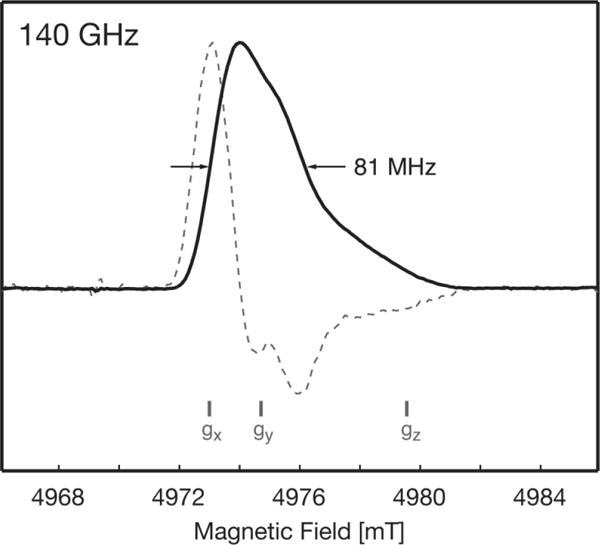
140 GHz pulsed EPR spectrum of FD SQ in 2H8-glycerol/2H2O buffer (60:40 v:v) at 40 K (solid line). The pseudo-modulated spectrum was calculated using a modulation amplitude of 0.3 mT (dashed line).
At a magnetic field strength of 5 T, an inhomogeneous breadth of Δ = 81 MHz was observed (full width at half height), which is smaller than the 1H Larmor frequency ω0n = 212 MHz at the same magnetic field strength. Therefore, 1H-DNP from this SQ is expected to occur through the SE mechanism1.
The SE can be understood via a two-spin system composed of an electron (S = ½) and a nucleus (I = ½). In this system the energy levels can be described by a four-level system including two allowed NMR transitions of resonant frequency ω0n and two allowed EPR transitions of resonant frequency ω0e. In the presence of an anisotropic hyperfine interaction electron-nuclear state-mixing occurs, and the formally-forbidden zero- and double-quantum transitions (ZQ, DQ) become partially allowed. Excitation of these forbidden transitions, which occur at frequencies of ω0e ± ω0n (matching conditions), mediates DNP through the SE7,52,53. Since the SE relies on excitation of forbidden transitions reflecting admixture of Zeemann states, the efficiency of SE decreases at high magnetic fields in proportion to B0−2, and the observed enhancements are typically smaller than those observed via the CE54. Fortunately, FD’s EPR linewidth is sufficiently narrow that no differential SE is expected, which would otherwise result in overlap and cancellation of negative and positive enhancements, effectively decreasing the observed overall DNP enhancement52,55.
For DNP via the SE maximal enhancement of the NMR signal intensity is expected when the microwave frequency satisfies a matching condition, and the sign of the signal is expected to depend on which matching condition is being met. Because our gyrotron oscillator operates at a fixed frequency of 139.662 GHz, we measured the MAS NMR signal intensity as a function of the static field B0 (effectively varying ω0n = γnB0 and ω0e = geβeB0/ħ) to record the field-dependent 1H-DNP enhancement profile shown in Figure 4 (where βe is the Bohr magneton, ge is the electron g-value and γn is the nuclear gyromagnetic ratio).
Figure 4.
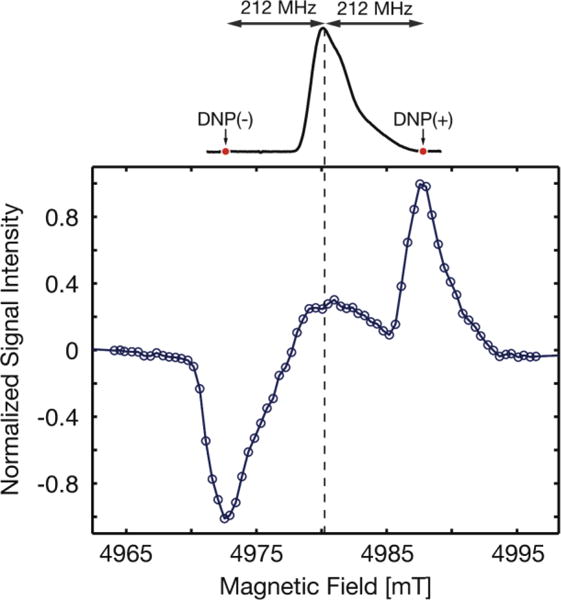
Field dependence of the DNP enhancement and EPR spectrum of the FD SQ radical. Bottom: 1H-MAS NMR detected DNP enhancement as a function of magnetic field for DNP based on the FD SQ radical; ωr/2π = 3.0 kHz. Top: 140 GHz echo-detected EPR spectrum of FD in 2H8-glycerol/2H2O (60:40), T = 40 K.
The 1H-DNP enhancement profile for the FD SQ radical shows a maximum at a magnetic field of 4987.8 mT (DNP(+)) and a minimum at 4972.6 mT (DNP(-)) (Figure 4). These features are separated by exactly twice the 1H Larmor frequency (2*212 MHz ≡ 15.2 mT at 5 T) as expected for well-resolved SE52. The overall shape of the DNP enhancement profile also reflects the strong asymmetry of FD’s EPR signal, as expected11,52,56.
Maximum enhancement
At a temperature of 90 K, we obtained a steady-state 1H enhancement of εH = 7 for 1H-FD. While this number is low, it can be understood in terms of the DNP mechanism in effect, the concentration of the polarization source and the isotopic composition of the system. Comparable enhancements of 10 and 15 were obtained via SE DNP based on added polarizing agents BDPA or trityl radical at concentrations of 50 and 40 mM, respectively48,57. This is in contrast to enhancements obtained via DNP based on the CE. Depending on the CE polarizing agent used (e.g. TEMPO or TOTAPOL) enhancement factors of 120 and up to 200 have been reported for nanocrystals of the polypeptide GNNQQNY or model samples, respectively10,13,58. Thus, the lower enhancement produced by FD’s SQ is attributable in part to the relatively low efficiency of the SE mechanism compared to the CE, especially as optimized by biradicals.
Our enhancements also reflect the relatively low 2 mM concentration of the SQ. Published DNP enhancement factors have also been found to depend on the concentration of the polarization agent. For the example of TEMPO radical the observed enhancements increased with increasing TEMPO concentration up to approximately 40 mM, beyond which accelerated paramagnetic relaxation eroded the MAS NMR signal strength59,60. Qualitatively similar behaviour is observed for biradicals except that the optimal concentration is lower at approximately 10 mM, consistent with the two-fold higher stoichiometry of unpaired electrons per molecule and more efficient polarization11. However in the case of endogenous radicals, the ratio of unpaired electrons to nuclei is fixed by the choice of protein, and 40 mM concentrations of radical are only possible for protein crystals or other highly condensed phases. Thus, two disadvantages of using endogenous radicals are that we is limited by the CE vs. SE nature of the naturally occurring radical as well as by achievable concentrations, which may not be ideal for spectroscopy. The MAS NMR signal intensities will be smaller than those possible with exogenous radicals, although we show here that they are still much larger than MAS NMR in the absence of DNP.
On the other hand the isotopic composition of the biochemical system can easily be manipulated by overexpression in deuterated medium. By using FD that was 85% deuterated (2H-FD) we were able to double the enhancement to εH = 15 (see Figure 5). This is consistent with published reports that deuteration can slow down nuclear spin-lattice relaxation and yield larger enhancements for DNP based on exogenous radicals61,62, as well as our own experience with FD, wherein enhancements were only half as large when the buffer used was made up with 1H2O (see also Rosay60). Similarly, Abkey et al. demonstrated a four-fold larger TOTAPOL-based enhancement upon perdeuteraton of the protein under study, and also reported that the enhancement obtained depends strongly on the 1H content (at exchangeable sites). Thus, although deuteration decreases the concentration 1H available for detection or cross polarization, it also confers important advantages, which moreover can be deployed strategically via selective deutration, as appropriate.
Figure 5.
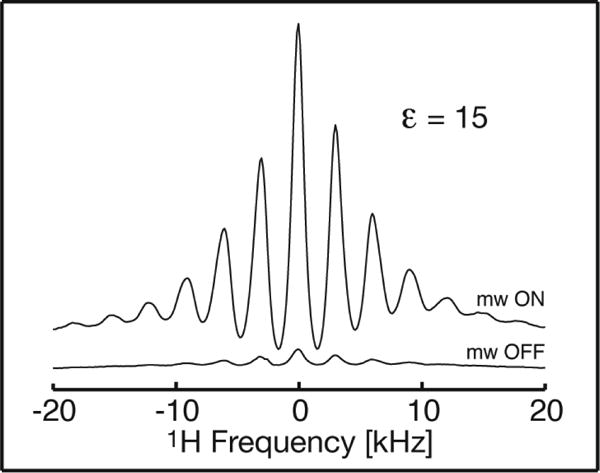
DNP-enhanced 1H MAS NMR spectrum of 2H-FD based on the endogenous SQ radical as the source of polarization. The signal collected with microwave pre-irradition of 500 s (upper trace) is compared to the signal obtained with a simple 500 s delay (lower trace) for the same sample. Spectra were collected at 90 K at a field position corresponding to DNP(+) in Figure 4, spinning at ωr/2π = 3 kHz, as described in the Methods section.
For both 1H-FD and 2H-FD samples, the 1H enhancement was observed to increase with increased microwave power to the maximum power obtainable with our apparatus (supplementary Figure S3). Indeed, since SE DNP occurs via forbidden electron-nuclear transitions with small transition moments at high magnetic fields, we do not expect that the DNP effect can be saturated with our gyrotron. However larger SE enhancements could in principle be obtained if larger microwave field strengths could be achieved at the position of the sample. Upon extrapolation of the microwave power dependence (supplementary Figure 3S) we obtain a DNP enhancement of 25 for 1H-FD at infinite microwave power.
The steady-state 1H enhancement expected for the well-resolved SE can be approximated by
| Eq. 2 |
where Ne is the number of unpaired electron spins per m−3, B1e is the amplitude of the microwave field in mT, τB is the proton bulk-polarization build-up time and Δ is the inhomogeneous breadth of the EPR signal55. Using our values for the enhancement and the bulk-polarization build-up times given in Table 1 we can estimate an average microwave field strength of B1e of 0.35 MHz (0.013 mT) at the position of the sample, which is similar to values reported for analogous instrumentation55. In our experiments, the microwave radiation is delivered to the sample using a rectangular fundamental waveguide. However, since no microwave resonator is employed, the coupling efficiency of the TE011 mode of the rectangular waveguide to the sample is very poor. More recent probe designs employ a circular corrugated waveguide and irradiate the sample using a Gaussian-like mode. This approach yields larger B1e field strengths at the position of the sample and values of 0.84 MHz (0.031 mT) have been reported for such systems63. Using this value for B1e we would estimate that steady-state enhancement factors of 23 and 147 would be achievable for DNP experiments based on the SQ radical in proteated and 85% deuterated FD, respectively, consistent with our extrapolation result for 1H-FD (above). Thus, ongoing advances in DNP MAS NMR probe design, which can yield large microwave fields at the position of the sample, are producing exciting improvements for DNP enhancements, especially for SE DNP (Personnal communication, B. Corzilius, A. Smith). Regardless, even our observed enhancement of 15 can make hitherto-impractical experiments viable for biological systems.
Table 1.
Bulk polarization build-up times τB and T1n relaxation times for 1H-FD and 2H-FD, along with DNP enhancement factors εH.
| 1H-FD | 2H-FD | ||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| [e-] (mM) | τB (s) | T1n (s) | εH | τB (s) | T1n (s) | εH | |
| SQ | 2 | 21 | 18 | 7 | 140 | 100 | 15 |
| TOTAPOL | 20 | 4 | 4 | 25 | 5 | 4 | 30 |
Bulk-Polarization Build-Up Times and their Dependence on Isotopic Composition of the Protein
The propagation of 1H polarization through the sample during the DNP process can be characterized by recording the amplitude of the 1H MAS NMR signal as a function of the duration of microwave irradiation tmw to obtain the bulk-polarization build-up curve. Repetition of the same experiment but with no microwave irradiation (off-signal) yields analogous longitudinal magnetization recovery curves from which T1n can be determined. In Figure 6 the build-up curves of 1H-FD and 2H-FD for SE DNP based on the flavin SQ (‘SQ’) and CE DNP based on 10 mM TOTAPOL (‘T’) are shown.
Figure 6.
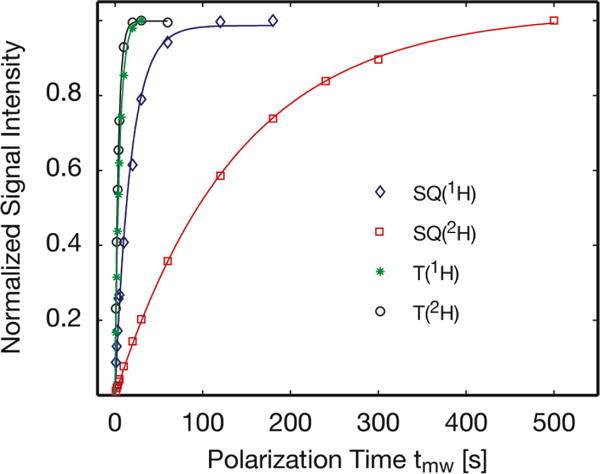
1H bulk-polarization build-up curves for SE DNP based on the endogenous SQ radical (SQ) or CE DNP based on TOTAPOL (T). The kinetics of 1H polarization were compared for 1H-FD and 85% deuterated 2H-FD, for the SE and the CE DNP mechanisms. Measurements were performed at a field position corresponding to DNP(+) in Figure 4. Data points were fit using a mono-exponential recovery curve (Eq. 1, methods section) to obtain build-up time constants, τB. All traces are normalized to a maximum signal intensity of one.
For SE DNP based on the SQ, 85 % deuteration caused the build-up time constant to increase seven-fold from τB = 21 s for 1H-FD to 140 s for 2H-FD (Table 1). Similarly, the T1n increased by a factor of five upon 85 % deuteration. The changes in both τB and T1n can be explained by slowed spin-diffusion when 1H atoms are diluted to greater distances from another.
The buildup curves describe the propagation of nuclear polarization to the more distant nuclei whose resonances are observed at unperturbed chemical shifts (bulk nuclei), by spin diffusion64–66. Based on the 18.2 nm3 volume of FD35 and FD’s 969 H atoms, we calculate a 1H concentration of 88 M for 1H-FD and 13 M for 2H-FD (see supporting information). Both are high compared to the 1H concentration of 0.8 M of the medium that makes up the rest of our samples (see supporting information). Thus we anticipate that for both 1H-FD and 2H-FD, polarization will spread much more efficiently within the protein than in the medium, and that the buildup curve of DNP based on the SQ will reflect primarily polarization of the Hs within the same protein as the SQ polarization source.
A lower concentration of 1Hs will tend to attenuate the spin-diffusion process due to the greater average distance between 1H nuclei and thus the lower strength of homonuclear dipolar coupling. In the case of inhomogenously broadened lines, the spin diffusion constant D is given by:
| Eq. 3 |
where 〈r2〉 is the average squared distance between nuclei participating in the spin-diffusion process and Δv1/2 is the full line width at half intensity67,68. The average 1H-1H distance within 1H-FD is 2.9 Å and that in 2H-FD is 5.5 Å, based on the proton concentration inside each protein (see supporting information). Thus, we estimate that the diffusion constant in the proteated protein is some 4-fold larger than that in the 85% deuterated protein, using equation 3. This is in reasonable agreement with the factor of 5 increase in the 1H T1n relaxation time (see Table 1).
When 10 mM TOTAPOL was used as the polarization source, the τB and T1n were drastically shorter, moreover neither τB nor T1n changed much upon deuteration of the protein. Most likely T1n relaxation of 1H inside the protein is not limited by spin-diffusion69 as in the case of the SQ-containing samples, but is foreshortened, or quenched, by the high concentration of the paramagnetic polarization agent. Indeed, paramagnetic impurities or dopants are known to sharply decrease the relaxation times of surrounding nuclei in an effect called paramagnetic relaxation enhancement (PRE)70,71.
The much shorter T1n values observed in the presence of TOTAPOL can be understood as consequences of the higher concentration of unpaired electrons overall, as well as a different distribution of distances from 1H to the closest unpaired electron in FD. Our 10 mM concentration of the bi-radical TOTAPOL creates an unpaired electron concentration ten times that present in the FD SQ samples. This will increase the importance of PRE by shortening the average distance between an H atom and an unpaired electron. In addition, the spatial distribution of the flavin SQ and the TOTAPOL are very different and create contrasting distance distributions from a FD 1H atom to the nearest unpaired electron. Because PRE depends on the inverse sixth power of the electron-nuclear separation, PRE drops off very fast with increasing separation and is dominated by the contribution of the free radical’s closest approach to a given 1H atom. For the case of PRE due to flavin SQ, 1H atoms in the protein have fixed distances to the flavin, the distribution of which is shown in Figure 7. For the case of TOTAPOL in solution, no information is available as to possible discrete binding sides for TOTAPOL on the FD protein. Therefore, we calculated the closest possible distance of approach of TOTAPOL to each H in FD via the proxy of calculating the distance to the closest solvent water, for each H in FD, using the positions of crystallographic waters in the structure of D. vulgaris FD (4FX2.pdb). Thus we assume that TOTAPOL will not penetrate the FD protein or get closer than a surface water molecule could. While individual distances from the surface to specific 1H atoms are expected to be only approximate, the set of the closest approach distances should to reproduce the shape of the distribution of closest-approach distances of TOTAPOL. This distribution of closest approaches to the surface is compared with the distribution of distances between an H and the centre of the flavin central ring in Figure 7.
Figure 7.
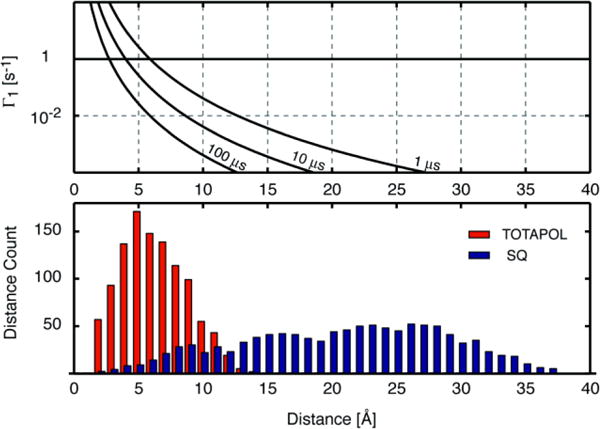
Top: Paramagnetic relaxation enhancement (Γ1 = 1/T1,p) for 1H vs. distance from an electron with T1e = 1, 10 or 100 μs (a logarithmic scale is used for Γ1). Bottom: Numbers of 1H atoms at different distances from the closest high-occupancy water (red) and at different distances from the centre of the flavin (blue). The average distance to the closest high-occupancy water is 6.3 Å. The average distance to the centre of the flavin’s central ring is 21 Å. (Further details are given in the supporting information and Figure S4).
In FD containing SQ radical, the majority of the H atoms are more than 10 Å away from the flavin ring with an average separation of 21 Å. This is in strong contrast to the scenario in the presence of added TOTAPOL, in which case the distance of closest calculated approach between a TOTAPOL molecule and a FD H atom is usually less than 10 Å, with an average of approximately 6 Å. Thus TOTAPOL’s ability to approach from all sides exposes more of FD’s Hs to PRE. The effect was modelled based on the distance dependence of PRE (see Bertini et al. equation 3.1972) for a range of electron spin-lattice relaxation times T1e since precise electron spin relaxation rates are not known for these samples under the conditions used (see Figure 7). Even for a very short T1e of 1 μs, only 1H nuclei at a distance of less than 6 Å would be significantly influenced by the paramagnetic center (Γ1 ≥ 1). Thus only in the samples containing TOTAPOL do we expect that a significant fraction of the 1Hs of FD will be affected by PRE. In addition the SQ is expected to have a much longer T1e than TOTAPOL, further decreasing the importance of PRE effects in case of DNP experiments based on the endogenous SQ radical73. We can therefore explain the shorter T1n relaxation times in the TOTAPOL samples on the basis of PRE from both more and closer unpaired electrons. This will also affect τB since it has been found to correspond closely to the T1n relaxation time on numerous occasions59.
We also note that the DNP enhancements obtained based on TOTAPOL are considerably lower than have been reported previously10,13. This may be because the values in Table 1 were obtained from direct detection of total 1H polarization. Bulk 1H polarization is typically not a good measure of DNP enhancement due to large probe ring-down effects in conjunction with the very broad signals of 1H 74. When 1H polarization was transferred to 13C nuclei for detection using cross-polarization 46 we measured a greater-than 100-fold enhancement of 1H polarization based on TOTAPOL, consistent with other work 10,11 (supplementary Figure S5). In addition, buildup times measured via 13C detection agreed well with values obtained via direct 1H detection (supplementary Figure S6). However we used 1H detection for comparative studies and time courses because the lower SQ concentration and enhancements attainable made 13C detection of FD more difficult than for model samples. Thus the enhancements reported here are likely under-estimates of the 13C enhancement that may be possible based on endogenous SQ in 2mM FD.
Importance of a good glass for DNP experiments
The samples described above were all prepared in mixtures of glycerol and water that form a glass at low temperatures, ensuring homogenous distribution of exogenous polarizing agents while also providing cryoprotection for the protein. Moreover studies have shown that non-glass-forming solvent matrices typically yield smaller DNP enhancements74. We studied this effect by preparing samples with lower glycerol content that formed fragile glasses, or did not form a glass at cryogenic temperature (Supplementary Figure S7). We found that non-glassy samples had bi-exponential bulk-polarization build-up curves with a very slow buildup rate (long τB) characterizing a large fraction of the population (Figure 8 and Table 2). We speculate that this reflects slow polarization transfer across microscopic domain boundaries in the frozen sample, whereas the faster buildup rate would reflect polarization of 1H nuclei in the same domain as a TOTAPOL molecule because the faster buildup occurs on a timescale similar to that observed in homogenous (glassy) samples. However even in samples that did not form a glass, T1n displayed only a small contribution from a slow phase and the T1n value characterizing most of the signal was similar to the values obtained for glassy samples (Table 2).
Figure 8.
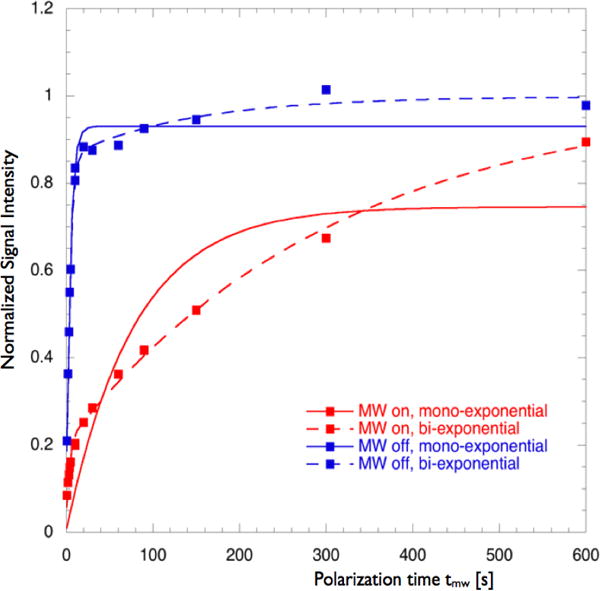
1H-DNP bulk-polarization build-up (shown in red) and saturation recovery traces (shown in blue) for a non-glass sample (12% 2H6-glycerol vol%). Signal intensities are scaled by the steady-state polarization M∞ (tmw → ∞). Original data were fit to eq. 1 (mono-exponential buildup) or (bi-exponential buildup) where f1 is the fraction of the population with the buildup time τB1 and τB2 is the buildup time that describes the balance of the population.
Table 2.
Bulk-Polarization build-up times τB, T1n recovery times and DNP enhancements εH for DNP in glassy and non-glass samples.a
| Glycerol [vol%] | Σtate upon freezing | τB [s] | T1 [s] | εH |
|---|---|---|---|---|
| 70 | glass | 4.9 ± 0.1 | 4.0 ± 0.1 | 30 |
| 50 | fragile glass | 3.0 ± 0.1 | 3.8 ± 0.1 | 24 |
| 12 | no glass | 310 ± 40 (80 %) 3.2 ± .5 (20 %) |
3.9 ± 0.2 (86 %) 140 ± 70 (14 %) |
14 |
DNP was based on 10 mM TOTAPOL. Data were fit with the equation when possible (mono-exponential buildup) or (bi-exponential buildup) when it was necessary to include two time constants.
MAS NMR studies of polymer blends have employed spin-diffusion measurements to determine the the sizes of domains68. Further experiments are planned to assess the applicability of such models for FD in glassy and non-glassy matrices.
Non-glass samples dominated by a large τB but a normal T1n achieved enhancements of only 14 compared to 30 for samples whose τB and T1n were comparable. Thus we confirm that the enhancement is related inversely to τB.61 Since τB was increased in SQ-based DNP of 2H-FD, and this alone should have caused a decrease in enhancement, we can conclude that the net increase in enhancement obtained upon deuterating FD reflects additional direct dependence of εH on T1n.
Kagawa et al. have already shown that the propagation of polarization and the spin lattice relaxation need not have identical mechanisms or kinetics61. They found that the magnitude of steady-state enhancement increases in proportion with the inverse of τB but that it also increases with T1n. Thus, reduced spin lattice relaxation rates (longer T1n) are expected to increase the enhancement. This is consistent with the two-fold larger enhancement produced by our 85% deuterated FD with its 5-times longer T1 than 1H-FD, although the effect is tempered by a larger τB as well.
Potential Applications of DNP based on Endogenous Radicals
For maximum DNP enhancement the best choice of polarization agent is currently a biradical such as TOTAPOL that supports efficient DNP via the CE11,74,75. However, for many of the naturally-occurring radicals the less effective SE will be the active DNP mechanism. Nonetheless there are compensating advantages. Unique features of the use of an endogenous radical are that the built-in radical occupies a defined location in the protein under study, it is genuinely non-perturbative and its oxidation state may be related to the physiological state of the system. The positional invariance of the built-in radical suggests the possibility of using the magnitude and build-up rate of DNP enhancement as sources of insight as to whether a species being observed (based on selective isotopic labeling) is closely associated with the protein containing the radical. Thus a 13C-labelled protein might gain stronger enhancement when complexed with a protein containing a radical than when the two are independent. Similarly, cofactors that adopt a radical state only under certain physiological conditions should produce DNP enhancements that depend on the system’s physiological state. Even though the enhancement is modest due to the low concentrations expected for biological radicals, the ≈15-fold enhancements we report here could make MAS NMR observation possible in complex biochemical preparations and perhaps even some organelles and photosynthetic membranes, allowing MAS NMR observation of minimally perturbed functional biological systems. This could enable exciting new applications of MAS NMR. Thus, the enhancement from endogenous radicals could provide more and different sorts of information than are accessible now via conventional MAS NMR.
A few examples of systems in which a flavin semiquinone occurs naturally include nitric oxide synthase76,77 the cytochrome P450 reductases78, electron transfer flavoprotein79 and photolyase80. Moreover our method is applicable to other types of radicals as well. The larger enhancements and more favorable B0 dependence of the CE could also be exploited in conjunction with the use of an endogenous radical by addition of a second, exogenous radical whose resonant frequency is offset from that of the endogenous radical by the NMR frequency57. Similarly, recent work demonstrating DNP from a Mn2+ complex raises the possibility of DNP from endogenous metal sites with favourable properties81.
Conclusions
To our knowledge, this is the first demonstration of the use of a stable, endogenous biological paramagnetic cofactor as a basis for high-field DNP. We have demonstrated 1H-MAS NMR enhancement by a factor of 15, consistent with the low concentration of the radical that is expected of biological systems. DNP based on the endogenous radical conforms to the theoretical and experimental expectations for SE DNP. We found that deuteration of the protein yields a larger enhancement reflecting the longer T1n, likely because methyl rotors that can act as polarization sinks are eliminated and the 1H-1H dipolar interaction is attenuated. The observed enhancements are significant and could be used to increase MAS NMR signal intensities in 13C/15N-CPMAS spectra of biologically interesting samples. We hope that the enhancements provided by DNP in combination with the resolution provided by MAS NMR and selective isotopic labeling may make new experiments practical in minimally perturbed biochemical systems.
Supplementary Material
Acknowledgments
AFM acknowledges the hospitality of the Aspen Center for Physics. We thank an anonymous reviewer for alerting us to the work of Demco et al. and B. Corzilius and A. Smith for helpful discussions. This research was supported by the National Institutes of Health through grants EB002804, EB002026 to RGG and by the Petroleum Research Foundation via 44321-AC4 to AFM. AFM also acknowledges support from the office of the Vice President for Research and the College of Arts and Sciences (University of Kentucky) and TM acknowledges receipt of a postdoctoral fellowship of the Deutsche Forschungs Gemeinschaft.
Footnotes
Supporting Information Available
Seven figures showing, respectively, the X-band EPR spectrum of the FD SQ, simulation of the 140 GHz EPR spectrum of the FD SQ based on parameters obtained for photolyase, the dependence of SQ-based DNP enhancement on microwave power, locations of crystallographic water molecules on the surface of FD, DNP of 1H detected via CP to 13C, the buildup rates of 1H and 13C-detected DNP and comparison of a glassy and a non-glassy sample. A table providing an inventory of the H atoms in FD and details of calculations of average distances and distances from each H atom in FD to the nearest site at which TOTAPOL could approach or to the center of the bound flavin’s central ring. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Maly T, Debelouchina GT, Bajaj VS, Hu K-N, Joo C-G, MakJurkauskas ML, Sirigiri JR, van der Wel PCA, Herzfeld J, Temkin RJ, et al. J Chem Phys. 2008;128:052211–052219. doi: 10.1063/1.2833582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barnes AB, De Paëpe G, van der Wel PCA, Hu KN, Joo CG, Bajaj VS, Mak-Jurkauskas ML, Sirigiri JR, Herzfeld J, Temkin RJ, et al. Appl Magn Reson. 2008;34:237–263. doi: 10.1007/s00723-008-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linden AH, Lange S, Franks WT, Akbey ú, Specker E, van Rossum B-J, Oschkinat H. J Am Chem Soc. 2011;133:19266–19269. doi: 10.1021/ja206999c. [DOI] [PubMed] [Google Scholar]
- 4.Reggie L, Lopez JJ, Collinson I, Glaubitz C, Lorch M. J Am Chem Soc. 2011;133:19084–19086. doi: 10.1021/ja209378h. [DOI] [PubMed] [Google Scholar]
- 5.Günther, U. 2011, 1-47.
- 6.Atsarkin VA. Sov Phys Usp (english translation) 1978;21:725–745. [Google Scholar]
- 7.Hovav Y, Feintuch A, Vega S. J Magn Reson. 2010;207:176–189. doi: 10.1016/j.jmr.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 8.Hovav Y, Feintuch A, Vega S. J Magn Reson. 2012;214:29–41. doi: 10.1016/j.jmr.2011.09.047. [DOI] [PubMed] [Google Scholar]
- 9.Smith AA, Corzilius B, Barnes AB, Maly T, Griffin RG. J Chem Phys. 2012;136:015101–015116. doi: 10.1063/1.3670019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu K, Yu H, Swager T, Griffin R. J Am Chem Soc. 2004;126:10844–10845. doi: 10.1021/ja039749a. [DOI] [PubMed] [Google Scholar]
- 11.Song C, Hu K, Joo C, Swager T, Griffin R. J Am Chem Soc. 2006;128:11385–11390. doi: 10.1021/ja061284b. [DOI] [PubMed] [Google Scholar]
- 12.Barnes A, Corzilius B, Mak-Jurkauskas ML, Andreas LB, Bajaj VS, Matsuki Y, Belenky M, Lugtenburg J, Sirigiri JR, Temkin RJ, et al. Phys Chem Chem Phys. 2010;12:5861–5867. doi: 10.1039/c003763j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Wel P, Hu K, Lewandowski J, Griffin R. J Am Chem Soc. 2006;128:10840–10846. doi: 10.1021/ja0626685. [DOI] [PubMed] [Google Scholar]
- 14.Andreini C, Bertini I, Cavallaro G, Holliday G, Thornton J. J Biol Inorg Chem. 2008;13:1205–1218. doi: 10.1007/s00775-008-0404-5. [DOI] [PubMed] [Google Scholar]
- 15.Calle C, Sreekanth A, Fedin MV, Forrer J, Garcia-Rubio I, Gromov I, Hinderberger D, Kasumaj B, Leger P, Mancosu B, et al. Helvetica Chimica Acta. 2006;89:2495–2521. [Google Scholar]
- 16.Halkides CJ, Farrar CT, Larsen RG, Redfield AG, Singel DJ. Biochemistry. 1994;33:4019–4035. doi: 10.1021/bi00179a031. [DOI] [PubMed] [Google Scholar]
- 17.Hoffman BM. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3575–3578. doi: 10.1073/pnas.0636464100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyubenova S, Maly T, Zwicker K, Brandt U, Ludwig B, Prisner T. Accounts of Chemical Research. 2009;43:181–189. doi: 10.1021/ar900050d. [DOI] [PubMed] [Google Scholar]
- 19.Hore J, Broadhurst RW. Progress in Nuclear Magnetic Resonance Spectroscopy. 1993;25:345–402. [Google Scholar]
- 20.Van Schagen CG, Mueller F, Kaptein R. Biochemistry. 1982;21:402–407. doi: 10.1021/bi00531a030. [DOI] [PubMed] [Google Scholar]
- 21.Lee JH, Sekhar A, Cavagnero S. Journal of the American Chemical Society. 2011;133:8062–8065. doi: 10.1021/ja111613c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zysmilich MG, McDermott A. J Am Chem Soc. 1994;116:8362–8363. [Google Scholar]
- 23.Jeschke G, Matysik J. Chemical Physics. 2003;294:239–255. [Google Scholar]
- 24.Polenova T, McDermott AE. The Journal of Physical Chemistry B. 1998;103:535–548. [Google Scholar]
- 25.Thamarath SS, Heberle J, Hore PJ, Kottke T, Matysik J. Journal of the American Chemical Society. 2010;132:15542–15543. doi: 10.1021/ja1082969. [DOI] [PubMed] [Google Scholar]
- 26.Zech SG, Kurreck J, Eckert H-J, Renger G, Lubitz W, Bittl R. FEBS Lett. 1997;414:454–456. doi: 10.1016/s0014-5793(97)01054-5. [DOI] [PubMed] [Google Scholar]
- 27.Bennati M, Robblee J, Mugnaini V, Stubbe J, Freed J, Borbat P. J Am Chem Soc. 2005;127:15014–15015. doi: 10.1021/ja054991y. [DOI] [PubMed] [Google Scholar]
- 28.Hirsh DJ, Beck WF, Innes JB, Brudvig GW. Biochemistry. 1992;31:532–541. doi: 10.1021/bi00117a033. [DOI] [PubMed] [Google Scholar]
- 29.Swanson MA, Kathirvelu V, Majtan T, Frerman FE, Eaton GR, Eaton SS. J Am Chem Soc. 2009;131:15978–15979. doi: 10.1021/ja9059816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Curley GP, Carr MC, Mayhew SG, Voordouw G. Eur J Biochem. 1991;202:1091–1100. doi: 10.1111/j.1432-1033.1991.tb16475.x. [DOI] [PubMed] [Google Scholar]
- 31.Stanley RJ. Antioxidants & Redox Signaling. 2004;3:847–866. doi: 10.1089/15230860152665028. [DOI] [PubMed] [Google Scholar]
- 32.O’Farrell PA, Walsh MA, McCarthy AA, Higgins TM, Voordouw G, Mayhew SG. Biochemistry. 1998;37:8405–8416. doi: 10.1021/bi973193k. [DOI] [PubMed] [Google Scholar]
- 33.Lostao A, Gomez-Moreno C, Mayhew SG, Sancho J. Biochemistry. 1997;36:14334–14344. doi: 10.1021/bi971384h. [DOI] [PubMed] [Google Scholar]
- 34.Chang FC, Swenson RP. Biochemistry. 1999;38:7168–7176. doi: 10.1021/bi982203u. [DOI] [PubMed] [Google Scholar]
- 35.Watt W, Tulinsky A, Swenson RP, Watenpaugh KD. J Mol Bio. 1991;218:195–208. doi: 10.1016/0022-2836(91)90884-9. [DOI] [PubMed] [Google Scholar]
- 36.Ludwig ML, Pattridge KA, Metzger AL, Dixon MM, Eren M, Feng Y, Swenson RP. Biochemistry. 1997;36:1259–1280. doi: 10.1021/bi962180o. [DOI] [PubMed] [Google Scholar]
- 37.Stockman BJ, Euvrard A, Kloosterman DA, Scahill TA, Swenson RP. J Biomol NMR. 1993;3:133–149. doi: 10.1007/BF00178258. [DOI] [PubMed] [Google Scholar]
- 38.Yalloway GN, Lohr F, Wienk HL, Mayhew SG, Hrovat A, Knauf MA, Ruterjans H. J Biomol NMR. 2003;25:257–258. doi: 10.1023/a:1022814324367. [DOI] [PubMed] [Google Scholar]
- 39.Peelen S, Vervoort J. Arch Biochem Biophys. 1994;314:291–300. doi: 10.1006/abbi.1994.1445. [DOI] [PubMed] [Google Scholar]
- 40.Schnegg A, Kay C, Schleicher E, Hitomi K, Todo T, Mobius K, Weber A. Mol Phys. 2006;104:1627–1633. [Google Scholar]
- 41.Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.3r1. 2010 [Google Scholar]
- 42.Whitby LG. Biochemical Journal. 1953;54:437–442. doi: 10.1042/bj0540437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Becerra L, Gerfen G, Temkin R, Singel D, Griffin R. Phys Rev Lett. 1993;71:3561–3564. doi: 10.1103/PhysRevLett.71.3561. [DOI] [PubMed] [Google Scholar]
- 44.Granatstein VL, Parker RK, Armstrong CM. Proc IEEE. 1999;87:702–716. [Google Scholar]
- 45.Maly T, Bryant J, Ruben D, Griffin R. J Magn Reson. 2006;183:303–307. doi: 10.1016/j.jmr.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pines A, Gibby MG, Waugh JS. J Chem Phys. 1973;59:569–590. [Google Scholar]
- 47.Bennati M, Farrar C, Bryant J, Inati S, Weis V, Gerfen G, Riggs-Gelasco P, Stubbe J, Griffin R. J Magn Reson. 1999;138:232–243. doi: 10.1006/jmre.1999.1727. [DOI] [PubMed] [Google Scholar]
- 48.Becerra LR, Gerfen GJ, Bellew BF, Bryant JA, Hall DA, Inati SJ, Weber RT, Un S, Prisner TF, McDermott AE, Fishbein KW, Kreischer K, Temkin RJ, Singel DJ, Griffin RG. Journal of Magnetic Resonance. 1995;A117:28–40. [Google Scholar]
- 49.Hyde J, Pasenkiewicz-Gierula M, Jesmanowicz A, Antholine W. Appl Magn Reson. 1990;1:483–496. [Google Scholar]
- 50.Fuchs MR, Schleicher E, Schnegg A, Kay CWM, Torring JT, Bittl R, Bacher A, Richter G, Mobius K, Weber S. J Phys Chem B. 2002;106:8885–8890. [Google Scholar]
- 51.Kay C, Schleicher E, Hitomi K, Todo T, Bittl R, Weber S. Magn Reson Chem. 2005;43:S96–102. doi: 10.1002/mrc.1667. Spec no. [DOI] [PubMed] [Google Scholar]
- 52.Abragam A, Goldman M. Rep Prog Phys. 1978;41:395–467. [Google Scholar]
- 53.Jeffries CD. Physical Review Phys Rev PR. 1960;117:1056. [Google Scholar]
- 54.Weis V, Bennati M, Rosay M, Griffin RG. J Chem Phys. 2000;113:6795–6802. [Google Scholar]
- 55.Wind RA, Duijvestijn MJ, van der Lugt C, Manenschijn A, Vriend J. Prog NMR Spec. 1985;17:33–67. [Google Scholar]
- 56.Maly T, Miller A-F, Griffin RG. ChemPhysChem. 2010;11:999–1001. doi: 10.1002/cphc.200900908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu K, Bajaj V, Rosay M, Griffin R. J Chem Phys. 2007;126:044512. doi: 10.1063/1.2429658. [DOI] [PubMed] [Google Scholar]
- 58.Mak-Jurkauskas ML, Bajaj VS, Hornstein MK, Belenky M, Griffin RG, Herzfeld J. Proc Nat Aca Sci USA. 2008;105:883–888. doi: 10.1073/pnas.0706156105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu K-N, Song C, Yu H-h, Swager TM, Griffin RG. J Chem Phys. 2008;128:052302–052317. doi: 10.1063/1.2816783. [DOI] [PubMed] [Google Scholar]
- 60.Rosay MM. Sensitivity-enhanced nuclear magnetic resonance of biological solids. Massachusetts Institute of Technology; 2001. [Google Scholar]
- 61.Kagawa A, Murokawa Y, Takeda K, Kitagawa M. J Magn Reson. 2009;197:9–13. doi: 10.1016/j.jmr.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 62.Akbey Ü, Franks WT, Linden A, Lange S, Griffin RG, van Rossum B-J, Oschkinat H. Angew Chem Int Ed. 2010;49:7803–7806. doi: 10.1002/anie.201002044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nanni EA, Barnes AB, Matsuki Y, Woskov PP, Corzilius B, Griffin RG, Temkin RJ. J Magn Reson. 2011;210:16–23. doi: 10.1016/j.jmr.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bloembergen N. Physica. 1949;15:386–426. [Google Scholar]
- 65.Hovav Y, Feintuch A, Vega S. J Chem Phys. 2011;134:074509–074520. doi: 10.1063/1.3526486. [DOI] [PubMed] [Google Scholar]
- 66.Smith AA, Corzilius B, Barnes A, Maly T, Griffin RG. In preparation. 2011 [Google Scholar]
- 67.Demco DE, Johansson A, Tegenfeldt J. Solid State Nuclear Magnetic Resonance. 1995;4:13–38. doi: 10.1016/0926-2040(94)00036-c. [DOI] [PubMed] [Google Scholar]
- 68.Clauss J, Schmidt-Rohr K, Spiess HW. Acta Polymerica. 1993;44:1–17. [Google Scholar]
- 69.Blumberg WE. Phys Rev. 1960;119:79. [Google Scholar]
- 70.Solomon I. Physical Review Phys Rev PR. 1955;99:559. [Google Scholar]
- 71.Bertini I, Luchinat C, Parigi G. Current methods in inorganic chemistry. Elsevier; 2001. Solution NMR of paramagnetic molecules : applications to metallobiomolecules and models; pp. 336–366. [Google Scholar]
- 72.Bertini I, Luchinat C, Parigi G. Current methods in inorganic chemistry. Elsevier; 2001. Solution NMR of paramagnetic molecules : applications to metallobiomolecules and models; pp. 75–118. [Google Scholar]
- 73.Eaton SS, Eaton GR, Berliner LJ. Biomedical EPR / edited by Sandra R[ie S] Eaton, Gareth R Eaton, and Lawrence J Berliner. Kluwer Academic/Plenum Publishers; New York: 2005. Vol. Biological magnetic resonance, v. 23-24. [Google Scholar]
- 74.Matsuki Y, Maly T, Ouari O, Karoui H, Moigne Le F, Rizzato E, Lyubenova S, Herzfeld J, Prisner TF, Tordo P, et al. Angew Chem Int Ed. 2009;48:4996–5000. doi: 10.1002/anie.200805940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rosay M, Tometich L, Pawsey S, Bader R, Schauwecker R, Blank M, Borchard PM, Cauffman SR, Felch KL, Weber RT, et al. Phys Chem Chem Phys. 2010;12:5850–5860. doi: 10.1039/c003685b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stuehr DJ, Tejero J, Haque MM. FEBS J. 2009;276:3959–3974. doi: 10.1111/j.1742-4658.2009.07120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Noble MA, Munro AW, Rivers SL, Robledo L, Daff SN, Yellowlees LJ, Shimizu T, Sagami I, Guillemette JG, Chapman SK. Biochemistry. 1999;38:16413–16418. doi: 10.1021/bi992150w. [DOI] [PubMed] [Google Scholar]
- 78.Das A, Sligar SG. Biochemistry. 2009;48:12104–12112. doi: 10.1021/bi9011435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Swanson MA, Kathirvelu V, Majtan T, Frerman FE, Eaton GR, Eaton SS. Protein Sci. 2011;20:610–620. doi: 10.1002/pro.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Damiani MJ, Yalloway GN, Lu J, McLeod NR, O’Neill MA. Biochemistry. 2009;48:11399–11411. doi: 10.1021/bi901371s. [DOI] [PubMed] [Google Scholar]
- 81.Corzilius B, Smith AA, Barnes AB, Luchinat C, Bertini I, Griffin RG. J Am Chem Soc. 2011;133:5648–5651. doi: 10.1021/ja1109002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.