

Abstract
Opioid peptides and opiate drugs such as morphine, mediate their analgesic effects, but also undesired side effects, mostly through activation of the mu opioid receptor. However, delta- and kappa-opioid receptors can also contribute to the analgesic effects of opioids. Recent findings showed that simultaneous activation of multiple opioid receptors may result in additional analgesia with fewer side effects. Here, we evaluated the pharmacological profile of our formerly developed mixed mu/kappa-opioid receptor ligands, Dmt-c[D-Lys-Phe-Phe-Asp]NH2 (C-36) and Dmt-c[D-Lys-Phe-p-CF3-Phe-Asp]NH2 (F-81). The ability of these peptides to cross the blood–brain barrier was tested in the parallel artificial membrane permeability (PAMPA) assay. On the basis of the hot-plate test in mice after central and peripheral administration, analog F-81 was selected for the anti-nociceptive and anti-inflammatory activity assessment after peripheral administration.
Keywords: Blood–brain barrier permeability, hot-plate test, anti-nociceptive and anti-inflammatory activity, colitis induction, myeloperoxidase activity
Introduction
Treatment of pain, especially chronic, is a common problem of all societies and one of the biggest challenges for modern medicine. Approximately 20% of adults suffer from the chronic pain syndrome (CPS)1. Opioids are the most effective drugs in chronic pain treatment but centrally mediated analgesia of opioids is often associated with some dangerous side effects such as respiratory depression, constipation, and also with tolerance and dependence2,3. The majority of clinically available opioid analgesics acting centrally are mu opioid receptor agonists derived mostly from morphine, with progressive simplification of its structure4.
The data collected so far suggest a strong contribution of peripheral mechanisms in pain suppression5–7. Opioid receptor ligands acting peripherally show less side effects as compared with those acting in the central nervous system (CNS)5,8,9. Peripheral mechanisms of pain control are especially effective in case of local inflammation processes when an increased number of opioid receptors on primary afferent neurons is observed10–12.
Endogenous opioid peptides were thought to be an alternative for morphine-based drugs, as they produced less side effects13,14. Unfortunately, their use as drugs is hampered by their short half-life in biological fluids. Additionally, degradation of these peptides increases in the microenvironment of the inflamed tissue, due to the elevated temperature, acidosis, and enhanced proteolytic activity of some enzymes15–17. Administration of more stable opioid peptide analogs produces significant peripherally-mediated analgesia in human18–21 and animal models22–25.
The anti-nociceptive and anti-inflammatory effects are mediated mostly by activation of the peripheral mu- and kappa-opioid receptors26. Thus, the use of multifunctional opioid analogs, which simultaneously activate more than one receptor, may generate a better drug profile, with improved potency and/or reduced side effects27. Therefore, extensive structure–activity relationship studies of opioid peptides are focused on the development of analogs with various activity profiles and enhanced efficacy28–30.
Here, we characterized the artificial membrane permeability and anti-nociceptive activity in the mouse hot-plate test of our two formerly developed analogs Dmt-c[D-Lys-Phe-Phe-Asp]NH2 (C-36)31 and Dmt-c[D-Lys-Phe-p-CF3-Phe-Asp]NH2 (F-81)32, with the mu/kappa-opioid receptor profile. On the bases of the obtained results, F-81 was chosen for further evaluation as a peripherally restricted peptide with possible anti-nociceptive and anti-inflammatory activity.
Materials and methods
Kinetic solubility
Kinetic solubility assay was performed using Multiscreen HTS Vacuum Manifold (Merck Millipore, Warsaw, Poland). Compounds for this test were prepared as 10 mM stock solutions in DMSO and diluted in a standard PBS buffer (pH = 7.4), to the final concentrations of 0.5 mM. The solutions were applied into a 96-well filter plate and incubated for 90 min at room temperature, while shaking at 40 g and then filtrated. The concentration of each compound was determined on the basis of the prepared calibration curve using the UV–VIS spectrophotometric method. According to the absorption spectra of the compounds the wavelength used for the measurements was 280 nm for both, C-36 and F-81.
Parallel Artificial Membrane Permeability Assay
Parallel Artificial Membrane Permeability Assay (PAMPA) was performed using Multiscreen Filter Plate and Multiscreen Transport Receiver Plate (Merck Millipore, Warsaw, Poland). Tested, as well as reference compounds, were dissolved in 0.1 M PBS buffer containing 20% of ethanol, to the final concentration of 200 µM. A 300 µl aliquots of 0.1 M PBS solution (pH = 7.4) was dispensed into the wells of the donor chamber. Then 1% n-dodecane solution of the brain polar lipid extract (Avanti Polar Lipids; Sigma Aldrich, Poznan, Poland) was prepared to investigate the blood–brain barrier (BBB) permeability. Aliquots of the obtained lipid solutions (5 µl) were dispensed on the membranes of wells of the donor plate and left to dry. Then, solutions of the tested and reference (verapamil and theophylline) compounds (150 µl) were added into the wells of the donor plate. A complete system, consisting of the receiver plate with inserted donor plate, was incubated at room temperature for 4 h, shaking at 40 g, to guarantee good solvent mixing. Then, the samples were collected, concentration of the tested compounds in both compartments was determined using UV–VIS spectrophotometer (wavelength 280 nm for verapamil, C-36 and F-81, and 270 nm for theophylline, respectively) and the permeability coefficients (Pe) were calculated.
The ability of the tested compounds to permeate the artificial membranes was classified according to the literature33 as:
Pe > 4.0 – high permeation,
Pe < 2.0 – low permeation,
Pe from 2.0 to 4.0 – permeation uncertain.
Animals
Mice were housed for at least 1 week before the experimental sessions in sawdust-lined plastic cages (7–10 mice in each cage). Animals were housed at a constant temperature (21 ± 1 °C) and maintained under a 12 h light/dark cycle with food and water available ad libitum.
Male CD-1 mice (Harlan, Italy) weighing 25–30 g were used to determine the anti-nociceptive activity of the cyclopeptides in the hot-plate test after i.c.v. injection. Animal studies and research protocols were approved by the Service for Biotechnology and Animal Welfare of the Istituto Superiore di Sanità and authorized by the Italian Ministry of Health, according to Legislative Decree 26/14 (protocol number: 198/2013-B, 14 August 2013), which implemented the European Directive 2010/63/EEC on laboratory animal protection. Animal welfare was routinely checked by veterinarians from the Service for Biotechnology and Animal Welfare.
Male balb/c mice (Animal Facility of Nofer Institute of Occupational Medicine, Lodz, Poland) weighing 22–25 g were used to determine the anti-inflammatory and anti-nociceptive activity of cyclopeptides after i.p. administration. All procedures and protocols used in this study were approved by the Local Ethical Committee for Animal Research (protocol number: 20/ŁB708/2014, 26 May 2014). The study was carried out in accordance with the recommendations described in the Guide for the Care and Use of Laboratory Animals of the Medical University of Lodz, Poland. All experimental procedures were performed in accordance with the current guidelines for the care of laboratory animals (including the use of the 3Rs procedures) and in accordance with the ARRIVE guidelines34.
Assessment of anti-nociception after i.c.v. administration (hot-plate test)
The hot-plate test was performed as described earlier35. The intracerebroventricular (i.c.v.) injections were performed in a volume of 10 µl/animal, in the left brain ventricle of manually immobilized mice with a Hamilton microsyringe (50 µl) connected to a needle (diameter 0.5 mm). Peptides were also administrated in mice intravenously (i.v.) in a volume of 1 ml/kg. All compounds used for i.c.v. and i.v. administration were dissolved in saline. A transparent plastic cylinder (14 cm diameter, 31 cm height) was used to confine the mouse on the heated (55 ± 0.5 °C) surface of the plate. The animals were placed on the hot plate 5 min after the i.c.v. injection of saline (control) or peptides and the latencies to paw licking, rearing and jumping were measured at 5, 10, 20, 30, 45, 60, 90, and 120 min after administration of a peptide. A cut-off time of 240 s was used to avoid tissue injury. The percentage of the maximal possible effect (%MPE) was calculated as: %MPE = (t1 − t0)/(t2 − t0) × 100, where t0: control latency, t1: test latency, and t2: cut-off time. The median antinociceptive dose (ED50) was calculated according to the method of Litchfield and Wilcox36.
Induction of colitis and assessment of colonic damage
The experimental colitis was induced by intracolonic (i.c.) administration of 2,4,6-trinitrobenzenesulfonic acid (TNBS). Mice were anesthetized with isoflurane (Aerrane, Baxter, Deerfield, IL) and TNBS at the dose of 4 mg per mouse (dissolved in 30% EtOH in saline) in the final volume of 150 µl, was injected into the colon through a catheter inserted 3 cm proximally from the anus. Mice were then maintained in an inclined position for 1 min to ensure the proper distribution of the inductor in the colon. Next, recovery was allowed with food and water supplied. Control animals received vehicle alone (30% ethanol in saline; TNBS replaced with equivolume water).
The preventive effect of F-81 on the colitis development as well as its anti-inflammatory properties in the semi-chronic colitis model were characterized, allowing to determine therapeutic activity of the tested compound37.
In the prevention mouse model of colitis, F-81 was injected intraperitoneally (i.p.) at the dose of 1 mg/kg in saline, in the volume of 100 µl, once daily, for 3 days, 15 min prior to TNBS administration. Mice were weighted daily and monitored for clinical symptoms of colitis, including diarrhoea and bloody stool. On day 4, mice were sacrificed by rapid cervical dislocation and the macroscopic damage score was assessed.
In the semi-chronic model, the curative potential of F-81 was tested. TNBS was administered on day 0 and from day 3 to day 6 the F-81 treatment (1 mg/kg in saline, 100 µl, i.p) was performed. On day 7, mice were sacrificed and the evaluation of colonic damage was performed.
To perform the macroscopic damage score, the colon was removed immediately after euthanasia, opened longitudinally, rinsed with phosphate buffered saline (PBS), and examined using an established semiquantitative scoring system by adding individual scores for ulcer, colonic shortening, wall thickness, and the presence of haemorrhage, faecal blood, and diarrhoea, as described before37. For scoring ulcer and colonic shortening the following scale was used: ulcer: 0.5 points for each 0.5 cm; shortening of the colon: 1 point for >15%, 2 points for >25% (based on a mean length of the colon in untreated mice). The wall thickness was measured in millimetres, a thickness of n mm corresponded to n scoring points. The presence of haemorrhage, faecal blood, or diarrhoea increased the score by 1 point for each additional feature.
Histology
Distal colon sections from the preventive mouse model of colitis were stapled flat, mucosal side-up, onto cardboard strips and fixed in 10% formalin for 24 h at 4 °C. Samples were dehydrated, embedded in paraffin, sectioned at 5 µm, and mounted onto slides. The sections were then stained with haematoxylin and eosin and Motic AE31 microscope (Ted Pella, Sweden) photographs were taken using a digital imaging system consisting of a digital camera (Moticam 2300, Ted Pella, Sweden) and image analysis software (Motic Images Plus 2.0, Germany).
The microscopic total damage score was assessed using the following parameters: the goblet cell depletion (presence = 1, absence = 0), crypt abscesses (presence = 1, absence = 0), the destruction of mucosal architecture (normal = 1, moderate = 2, extensive = 3), the extent of muscle thickening (normal =1, moderate = 2, extensive = 3), and the presence and degree of cellular infiltration (normal = 1, moderate = 2, transmural = 3).
Determination of tissue myeloperoxidase (MPO) activity
The colon fragments (20–30 mg) were gently isolated from mice after total macroscopic damage score assessment, immediately washed with PBS, and homogenized in hexadecyltrimethylammonium bromide (HTAB) buffer (0.5% HTAB in 50 mM potassium phosphate buffer, pH 6.0; 50 mg of tissue/ml) using Ika Ultra Turrax Disperser T25 Digital 2 (Sigma Aldrich, Poznan, Poland). The homogenates were centrifuged (20 min, 13,200g, 4 °C) and the supernatants were transferred to the new test tubes. Then, 7 µl portions of supernatants were added on a 96-well plate, followed by 200 µl of 50 mM potassium phosphate buffer (pH = 6.0), containing 0.167 mg/ml of O-dianisidine hydrochloride and 0.05 µl of 1% H2O2. The absorbance was measured at 450 nm after 30 and 60 s (iMARK Microplate Reader, Biorad, UK). All measurements were done in triplicates.
The MPO activity was expressed in units/g of a wet tissue. One unit is determined as a quantity of enzyme able to convert 1 µmol of H2O2 to water in 1 min at room temperature. MPO activity units were calculated from the standard curve using purified peroxidase enzyme.
Mustard oil induced pain
To assess the anti-nociceptive effect of F-81 in mice with acute colitis, intestinal inflammation was induced by TNBS instillation three days prior to the experiment. Abdominal pain was induced in inflamed mice by the i.c. injection of 1% mustard oil (MO, allyl isothiocyanate) in 70% EtOH in saline. Mice were separated into clear plastic boxes (20 × 20 × 15 cm) and allowed a 5 min recovery after MO administration. Spontaneous behaviours (licking and stretching the abdomen, squashing of lower abdomen against the floor, and abdominal retraction) were observed and counted for 20 min.
Data analysis and terminology
Artificial membrane permeability was assessed in the PAMPA assay and the permeability coefficient (Pe) was calculated from the following equation:
where
Membrane retention was analysed using mass balance between a compound left in the donor and acceptor compartments and calculated using the following equation:
where
Va – volume of acceptor compartment,
Ca – concentration in acceptor compartment (after incubation),
Vd – volume of donor compartment,
Cd – concentration in donor compartment (after incubation),
Cinitial – initial concentration in donor compartment,
Cr – concentration in equilibrium,
p – membrane surface area (0.28 cm2),
t – time (s).
Results
Kinetic solubility
Kinetic solubility allows for determination of a compound solubility, based on the precipitation process. It is widely used at the early stages of drug discovery to assess the solubility of compounds in in vitro conditions in pharmacological assays.
In 0.1 M PBS (pH 7.4), C-36 and F-81 were both highly soluble (500 and 345 µM, respectively) (Table 1). The solubility of the tested peptides was sufficient to perform the PAMPA assay.
Table 1.
The kinetic solubility test results.
| Solubility |
Solubility |
||||
|---|---|---|---|---|---|
| Peptide | MW (g/mol) | Mean (µM) | SD | Mean (mg/ml) | SD |
| C-36 | 727 | ≥500 | – | ≥0.36 | – |
| F-81 | 795 | 345 | 15 | 0.27 | 0.01 |
Membrane permeability assay
PAMPA was used to assess passive permeability of cyclopeptides across the artificial BBB. Both tested compounds showed low permeation across this barrier (Table 2).
Table 2.
Blood–brain barrier permeability (PAMPA) results for cyclic pentapeptide analogs.
| Compound | BBB permeability Pe (10−6 cm−1) ± SD | Mean (%) Mass balance ± SD | Classification |
|---|---|---|---|
| Verapamil | 29.9 ± 6.5 | 89.28 ± 2.35 | High permeation |
| Theophylline | ≤0.58 | 98.21 ± 3.65 | Low permeation |
| C-36 | ≤0.58 | 97.87 ± 1.69 | Low permeation |
| F-81 | ≤0.58 | 95.75 ± 1.43 | Low permeation |
Assessment of anti-nociceptive activity
Anti-nociception was studied in the hot-plate test in mice after i.c.v. or i.v. administration of peptides. The results obtained in the dose-response studies after i.c.v. administration are shown in Figure 1(A). Both tested compounds showed dose-dependent anti-nociceptive activity, significantly stronger than that of endomorphin-2 (EM-2). The ED50 values (jumping response) for C-36 and F-81 were 57.78 and 17.27 ng, respectively, indicating that F-81 was approximately threefold more potent than C-36 (Figure 1(A)). In order to investigate if these peptides are able to cross the BBB, peripheral i.v. administration of the peptides was performed, and the results are reported in Figure 1(B). After i.v. administration at the dose of 20 mg/kg, only a negligible anti-nociceptive activity was observed for both compounds (Figure 1(B)). To characterize the involvement of opioid receptors in the anti-nociceptive action of analog F-81, co-administration studies with opioid receptor antagonists were performed. The anti-nociceptive effect of F-81 (10 ng/animal, i.c.v.) was blocked by β-funaltrexamine (β-FNA, 1 µg/animal), showing the involvement of the mu opioid receptors. The delta-opioid receptor antagonist, naltrindole (NTL, 1 µg/animal), and kappa-opioid receptor antagonist, norbinaltorphimine (nor-BNI, 5 µg/animal, i.c.v.), did not modify the anti-nociceptive action of F-81 (Figure 1(C)). Even though F-81 and C-36 showed significant kappa-affinity, the obtained results are in agreement with a generally accepted fact that the anti-nociceptive effects are mainly mediated by the mu opioid receptor32,38.
Figure 1.

Dose–response curves determined in the hot-plate test for the inhibition of jumping induced by i.c.v. injection of EM-2 and cyclic analogs C-36 and F-81 (Panel A). Effects induced by peptides after i.v. administration at the dose of 20 mg/kg (Panel B). The antagonist effect of β-funaltrexamine (β-FNA, 1 µg/animal, i.c.v.), naltrindole (NTL, 1 µg/animal, i.c.v.), and nor-binaltorphimine (nor-BNI, 5 µg/animal, i.c.v.), on the inhibition of jumping induced by F-81 (10 ng/animal, i.c.v.) (Panel C). *** is for p <.001 for agonist + β-FNA versus agonist alone using two-way ANOVA, followed by the Student–Newman– Keuls test. The data represent mean ± SEM of 10 mice per group.
Assessment of anti-inflammatory and anti-nociceptive activity of F-81 after peripheral administration
The i.c. administration of TNBS induced colonic inflammation in mice, as shown by the increased macroscopic damage score, ulceration of the intestinal wall and inflammation area which were significantly enhanced in the TNBS-treated group, as compared with the control group. The colon thickness and width were increased and colon length was decreased in the TNBS treated mice. Additionally, the MPO activity, which is an indicator of immune cell infiltration, was significantly enhanced in inflamed mice (Figures 2 and 3).
Figure 2.

Anti-inflammatory activity of F-81, administered i.p. once a day over 3 days, in the prevention mouse model of the TNBS-induced colitis. Macroscopic score (Panel A), inflammation area (Panel B), ulcer score (Panel C), colon thickness (Panel D), MPO activity (Panel E), colon width (Panel F), and colon length (Panel G). Statistical significance was assessed using one-way ANOVA and post hoc multiple comparison Student–Newman–Keuls test. ***p < .001, as compared with controls. #p < .05, ###p < .001, vs. TNBS-treated animals. Data represent mean ± SEM of eight mice per group.
Figure 3.

Anti-inflammatory activity of F-81, injected i.p. ameliorated experimental colitis in the semi-chronic TNBS-induced model. Macroscopic score (Panel A), inflammation area (Panel B), ulcer score (Panel C), colon thickness (Panel D) MPO activity (Panel E), colon width (Panel F), and colon length (Panel G). Statistical significance was assessed using one-way ANOVA and post hoc multiple comparison Student–Newman–Keuls test. ***p < .001, as compared with controls. #p < .05, ###p < .001, vs. TNBS-treated animals. Data represent mean ± SEM of eight mice per group.
In the prevention model of TNBS-induced colitis, F-81 (injected i.p. at the dose of 1 mg/kg, once daily) decreased macroscopic damage score (4.07 vs. 5.83 for F-81 and TNBS-treated group, respectively), the inflammation area (19.56 ± 2.16 vs. 24.42 ± 2.92), improved the ulcer score (1.43 ± 0.17 vs. 2.06 ± 0.22) and improved the colon width (6.09 ± 0.53 vs. 7.91 ± 0.23) as compared with the TNBS-treated animals. There were no differences in the intestinal wall thickness (0.95 ± 0.07 vs. 1.08 ± 0.12) and the colon length 8.23 ± 0.24 vs. 8.01 ± 0.39 (in comparison with inflamed mice). The activity of MPO was significantly decreased in F-81-treated mice as compared with the TNBS-treated group (3.65 ± 0.87 vs. 11.82 ± 1.72, respectively) (Figure 2(A–G)).
In the semi-chronic model of TNBS-induced colitis (Figure 3(A–G)), the anti-inflammatory effect of F-81 was shown by the decrease in the macroscopic damage score (2.18 ± 0.40 vs. 4.15 ± 0.54 for F-81- and TNBS-treated group, respectively) and inflammation area (12.23 ± 1.56 vs. 18.49 ± 3.95) and by improved ulcer score (0.92 ± 0.83 vs. 1.30 ± 0.12). No differences were observed in the colon thickness, width, or length between the F-81-treated and TNBS-treated groups. The MPO activity was also decreased in this model in the F-81-treated mice (7.88 ± 0.24 vs. 8.26 ± 0.15) (Figure 3(E)).
Microscopically, the induction of inflammation by TNBS was associated with a destruction of the intestinal wall (including architecture of mucosa and the presence of goblet cells and crypts), infiltration of immune cells and thickening of the muscle layer, as compared with naive animals (Figure 4(A–D)). The administration of F-81 (1 mg/kg) significantly improved the microscopic score (Figure 4(D)).
Figure 4.

Representative images of haematoxylin- and eosin-stained sections of distal colon obtained from the prevention mouse model of colitis for control (Panel A), TNBS-treated group (Panel B), TNBS- and F-81-treated group (Panel C). Microscopic damage score (Panel D). Statistical significance was assessed using one-way ANOVA and post hoc multiple comparison Student–Newman–Keuls test. ***p < .001, as compared with controls. #p < .05, ###p < .001, vs. TNBS-treated animals. Data represent mean ± SEM of eight mice per group.
The anti-nociceptive effect of F-81 was also characterized in the mouse model of abdominal pain induced by mustard oil instillation. Mice with TNBS-induced colitis were injected with F-81 (1 mg/kg, i.p.). This cyclopeptide significantly decreased the number of spontaneous pain behaviours; a strong anti-nociceptive activity was observed as compared with the control group (6.36 ± 1.98 vs. 29.28 ± 2.52, respectively) (Figure 5).
Figure 5.
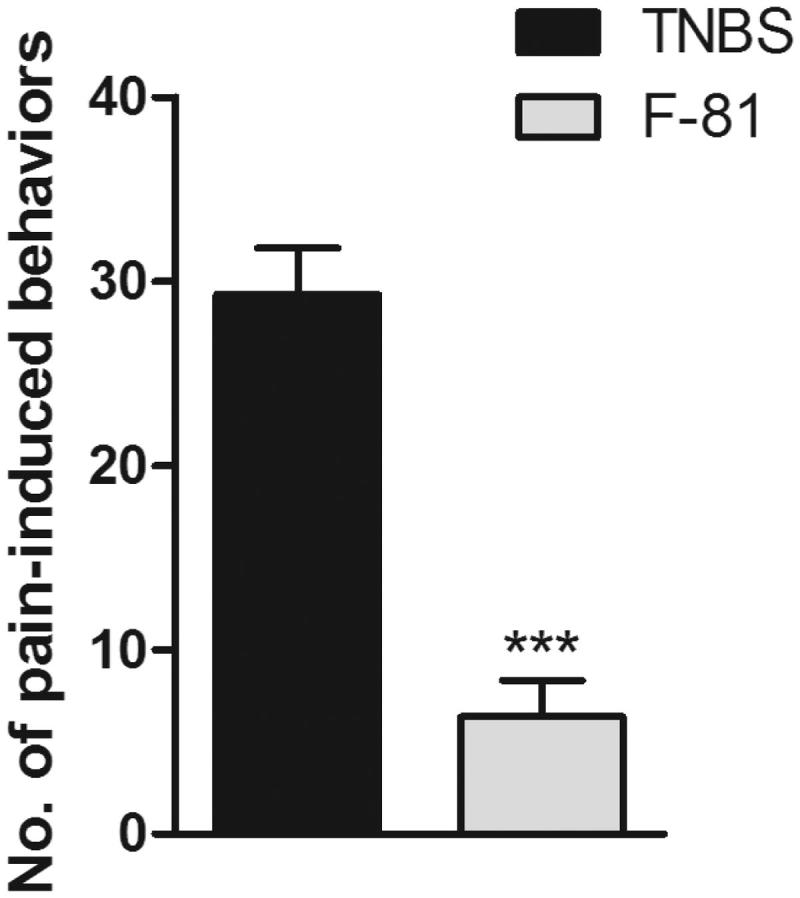
The anti-nociceptive effect of F-81 in the mustard oil-induced pain in mice with acute TNBS-induced colitis. Data represent mean ± SEM of eight mice per group. Statistical significance was assessed using t-test. ***p < .001, as compared with the control group.
Discussion
Opioid receptors are localized in the CNS and also in many peripheral tissues. The anti-nociceptive effects of opioid ligands given peripherally depends on their ability to cross the BBB. Ligands that can penetrate this barrier produce both, central and peripheral effects, while the action of those unable to reach the CNS is limited to the peripheral tissues. Peripherally restricted opioid analgesics, devoid of centrally mediated side effects can be a safer alternative for treating inflammatory painful disorders of gastrointestinal tract, skin, and joints39,40.
Endogenous opioid peptides elicit strong analgesic effect, however their usefulness after peripheral administration is very limited due to the rapid degradation in biological fluids41.
For several years, we have been engaged in the development of new, more stable opioid analogs with improved pharmacological profile. In this study, we tested the ability of cyclopeptides C-36 and F-81 (both with Dmt1) to cross the artificial BBB, using PAMPA. The PAMPA assay was previously shown to closely match the BBB membrane in such characteristics as hydrophobicity, rigidity, and fluidity, and is considered as an accurate passive diffusion permeability model for BBB42.
In the PAMPA experiment, both compounds showed low permeation through this artificial barrier. These results were confirmed in vivo in the mouse hot-plate test, since no anti-nociceptive activity was observed after peripheral (i.v.) administration of the tested compounds.
C-36 differs from our formerly published analog Tyr-c[D-Lys-Phe-Phe-Asp]NH2 [35] only by the presence of Dmt1 instead of Tyr1. As opposed to C-36, Tyr1-containing analog could reach the brain after i.v. administration. Recently, Zadina et al.30 reported several cyclopeptides of a similar structure also containing Tyr1 and those analogs were able to produce strong analgesia after peripheral administration. Therefore, the presence of Dmt1 in cyclic peptides may be a structural element responsible for lower permeability of such analogs through biological barriers.
However, the observed effect can be caused by other factors. Weltrowska et al.43 studied brain uptake of linear and cyclic DALDA (Tyr-D-Arg-Phe-Lys-NH2) analogs, both containing Dmt1. They observed a lower brain uptake and faster efflux of the more rigid cyclic peptide, indicating that structural flexibility strongly influences BBB permeability.
Since peripheral opioid receptors are involved in analgesia, especially in the presence of inflammation process, we decided to test the anti-nociceptive and anti-inflammatory activity of one of the analogs in abdominal pain models after peripheral administration. For these experiments F-81 was chosen, as its anti-nociceptive activity was threefold higher than that of C-36 in the mouse hot-plate test after central administration. The obtained data indicated that F-81 significantly attenuated inflammation and evoked strong anti-nociception in the mouse models of TNBS-induced colitis.
The observed effect of F-81 in the gastrointestinal tract is an interesting finding and can be useful in the further development of peripherally restricted agents.
Funding Statement
This work was supported by a grant from the National Science Center (Poland) to JP-C, No. DEC-2013/09/N/NZ7/00646, and a Grant from the Medical University of Lodz No. 502-03/1-156-02/502-14-301.
Disclosure statement
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Breivik H, Collett B, Ventafridda V, et al. . Survey of chronic pain in Europe: prevalence, impact on daily life, and treatment. Eur. J Pain 2006;10:287–333. [DOI] [PubMed] [Google Scholar]
- 2.Chu GH, Gu M, Cassel JA, et al. . Novel malonamide derivatives as potent kappa opioid receptor agonists. Bioorg Med Chem Lett 2007;17:1951–5. [DOI] [PubMed] [Google Scholar]
- 3.Brock C, Olesen SS, Olesen AE, et al. . Opioid-induced bowel dysfunction: pathophysiology and management. Drugs 2012;72:1847–65. [DOI] [PubMed] [Google Scholar]
- 4.Corbett AD, Henderson G, McKnight AT, Paterson SJ.. 75 years of opioid research: the exciting but vain quest for the Holy Grail. Br J Pharmacol 2006;147:S153–S62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Labuz D, Berger S, Mousa SA, et al. . Peripheral antinociceptive effects of exogenous and immune cell-derived endomorphins in prolonged inflammatory pain. J Neurosci 2006;26:4350–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Labuz D, Mousa SA, Schäfer M, et al. . Relative contribution of peripheral versus central opioid receptors to antinociception. Brain Res 2007;1160:30–8. [DOI] [PubMed] [Google Scholar]
- 7.Luster AD, Alon R, von Andrian UH.. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol 2005;6:1182–90. [DOI] [PubMed] [Google Scholar]
- 8.Hua S, Cabot PJ.. Mechanisms of peripheral immune-cell-mediated analgesia in inflammation: clinical and therapeutic implications. Trends Pharmacol Sci 2010;31:427–33. [DOI] [PubMed] [Google Scholar]
- 9.Iwaszkiewicz KS, Schneider JJ, Hua S.. Targeting peripheral opioid receptors to promote analgesic and anti-inflammatory actions. Front Pharmacol 2013;4:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Connor TM, O'Connell J, O'Brien DI, et al. . The role of substance P in inflammatory disease. J Cell Physiol 2004;201:167–80. [DOI] [PubMed] [Google Scholar]
- 11.Rittner HL, Labuz D, Schaefer M, et al. . Pain control by CXCR2 ligands through Ca2+-regulated release of opioid peptides from polymorphonuclear cells. Faseb J 2006;20:2627–9. [DOI] [PubMed] [Google Scholar]
- 12.Walker JS.Anti-inflammatory effects of opioids. Adv Exp Med Biol 2003;521:148–60. [PubMed] [Google Scholar]
- 13.Czapla MA, Gozal D, Alea OA, et al. . Differential cardiorespiratory effects of endomorphin 1, endomorphin 2, DAMGO, and morphine. Am J Respir Crit Care Med 2000;162:994–9. [DOI] [PubMed] [Google Scholar]
- 14.Wilson AM, Soignier RD, Zadina JE, et al. . Dissociation of analgesic and rewarding effects of endomorphin-1 in rats. Peptides 2000;21:1871–4. [DOI] [PubMed] [Google Scholar]
- 15.Babst R, Bongiorno L, Marini M, et al. . Trauma decreases leucine enkephalin hydrolysis in human plasma. J Pharmacol Exp Ther 1999;288:766–73. [PubMed] [Google Scholar]
- 16.Roques BP.Novel approaches to targeting neuropeptide systems. Trends Pharmacol Sci 2000;21:475–83. [DOI] [PubMed] [Google Scholar]
- 17.Schäfer M, Carter L, Stein C.. Interleukin 1 beta and corticotropin-releasing factor inhibit pain by releasing opioids from immune cells in inflamed tissue. Proc Natl Acad Sci USA 1994;91:4219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eisenach JC, Carpenter R, Curry R.. Analgesia from a peripherally active kappa-opioid receptor agonist in patients with chronic pancreatitis. Pain 2003;101:89–95. [DOI] [PubMed] [Google Scholar]
- 19.Stein C, Comisel K, Haimerl E, et al. . Analgesic effect of intraarticular morphine after arthroscopic knee surgery. N Engl J Med 1991;325:1123–6. [DOI] [PubMed] [Google Scholar]
- 20.Stein C, Küchler S.. Targeting inflammation and wound healing by opioids. Trends Pharmacol Sci 2013;34:303–12. [DOI] [PubMed] [Google Scholar]
- 21.Tegeder I, Meier S, Burian M, et al. . Peripheral opioid analgesia in experimental human pain models. Brain 2003;126:1092–102. [DOI] [PubMed] [Google Scholar]
- 22.Guan Y, Johanek LM, Hartke TV, et al. . Peripherally acting mu-opioid receptor agonist attenuates neuropathic pain in rats after L5 spinal nerve injury. Pain 2008;138:318–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mambretti EM, Kistner K, Mayer S, et al. . Functional and structural characterization of axonal opioid receptors as targets for analgesia. Mol Pain 2016;12. DOI: 10.1177/1744806916628734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moon SW, Park EH, Suh HR, et al. . The contribution of activated peripheral kappa opioid receptors (KORs) in the inflamed knee joint to anti-nociception. Brain Res 2016;1648:11–8. [DOI] [PubMed] [Google Scholar]
- 25.Vanderah TW, Largent-Milnes T, Lai J, et al. . amino acid tetrapeptides produce potent antinociception by selectively acting at peripheral kappa-opioid receptors. Eur J Pharmacol 2008;583:62–72. [DOI] [PubMed] [Google Scholar]
- 26.Jeanjean AP, Moussaoui SM, Maloteaux JM, Laduron PM.. Interleukin-1 beta induces long-term increase of axonally transported opiate receptors and substance P. Neuroscience 1995;68:151–7. [DOI] [PubMed] [Google Scholar]
- 27.Schiller PW.Bi- or multifunctional opioid peptide drugs. Life Sci 2010;86:598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis MP.Opioid receptor targeting ligands for pain management: a review and update. Expert Opin Drug Discov 2010;5:1007–22. [DOI] [PubMed] [Google Scholar]
- 29.Varamini P, Toth I.. Lipid- and sugar-modified endomorphins: novel targets for the treatment of neuropathic pain. Front Pharmacol 2013;4:155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zadina JE, Nilges MR, Morgenweck J, et al. . Endomorphin analog analgesics with reduced abuse liability, respiratory depression, motor impairment, tolerance, and glial activation relative to morphine. Neuropharmacology 2016;105:215–27. [DOI] [PubMed] [Google Scholar]
- 31.Perlikowska R, Malfacini D, Cerlesi MC, et al. . Pharmacological characterization of endomorphin-2-based cyclic pentapeptides with methylated phenylalanine residues. Peptides 2014;55:145–50. [DOI] [PubMed] [Google Scholar]
- 32.Piekielna J, Perlikowska R, do-Rego JC, et al. . Synthesis of mixed opioid affinity cyclic endomorphin-2 analogues with fluorinated phenylalanines. ACS Med Chem Lett 2015;6:579–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di L, Kerns EH, Fan K, et al. . High throughput artificial membrane permeability assay for blood-brain barrier. Eur J Med Chem 2003;38:223–32. [DOI] [PubMed] [Google Scholar]
- 34.Kilkenny C, Browne W, Cuthill IC, et al. . Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 2010;160:1577–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perlikowska R, do-Rego JC, Cravezic A, et al. . Synthesis and biological evaluation of cyclic endomorphin-2 analogs. Peptides 2010;31:339–45. [DOI] [PubMed] [Google Scholar]
- 36.Litchfield JT Jr, Wilcox F.. A simplified method of evaluating dose-effect experiments. J Pharmacol Exp Ther 1949;96:99–113. [PubMed] [Google Scholar]
- 37.Zielińska M, Lewandowska U, Podsędek A, et al. . Orally available extract from Brassica oleracea var. capitata rubra attenuates experimental colitis in mouse models of inflammatory bowel diseases. J Funct Foods 2015;17:587–99. [Google Scholar]
- 38.Perlikowska R, Piekielna J, Mazur M, et al. . Antinociceptive and antidepressant-like action of endomorphin-2 analogs with proline surrogates in position 2. Bioorg Med Chem 2014;22:4803–9. [DOI] [PubMed] [Google Scholar]
- 39.Lesniak A, Lipkowski AW.. Opioid peptides in peripheral pain control. Acta Neurobiol Exp 2011;71:129–38. [DOI] [PubMed] [Google Scholar]
- 40.Sehgal N, Smith HS, Manchikanti L.. Peripherally acting opioids and clinical implications for pain control. Pain Physician 2011;14:249–58. [PubMed] [Google Scholar]
- 41.Janecka A, Staniszewska R, Gach K, Fichna J.. Enzymatic degradation of endomorphins. Peptides 2008;29:2066–73. [DOI] [PubMed] [Google Scholar]
- 42.Di L, Kerns EH, Bezar IF, et al. . Comparison of blood-brain barrier permeability assays: in situ brain perfusion, MDR1-MDCKII and it was stated there that PAMPA PAMPA-BBB. J Pharm Sci 2009;98:1980–91. [DOI] [PubMed] [Google Scholar]
- 43.Weltrowska G, Nguyen TM, Chung NN, et al. . A cyclic tetrapeptide (“cyclodal”) and its mirror-image isomer are both high-affinity μ opioid receptor antagonists. J Med Chem 2016;59:9243–54. [DOI] [PMC free article] [PubMed] [Google Scholar]