

Abstract
The cell division cycle 25 phosphatases (CDC25A, B, and C; E.C. 3.1.3.48) are key regulator of the cell cycle in human cells. Their aberrant expression has been associated with the insurgence and development of various types of cancer, and with a poor clinical prognosis. Therefore, CDC25 phosphatases are a valuable target for the development of small molecule inhibitors of therapeutic relevance. Here, we used an integrated strategy mixing organic chemistry with biological investigation and molecular modeling to study novel quinonoid derivatives as CDC25 inhibitors. The most promising molecules proved to inhibit CDC25 isoforms at single digit micromolar concentration, becoming valuable tools in chemical biology investigations and profitable leads for further optimization.
Keywords: CDC25, enzyme inhibitors, quinonoid, organic synthesis, molecular modeling
Introduction
Protein tyrosine phosphatases (E.C. 3.1.3.48) are a large family of enzymes that catalyze the removal of the phosphate group from tyrosine residues, and are widely expressed in mammals and other organisms1,2. Among them, cell division cycle 25 phosphatases (CDC25A, B and C isoforms) are central regulators of the cell cycle in human cells, driving and tuning each phase of cell cycle progression2–5. Expression and activity of these enzymes are tightly regulated in physiological conditions, whereas abnormal expression of CDC25 has been detected in many high-grade tumors, such as breast, prostate, ovarian, endometrial, colorectal, esophageal, thyroid, gastric and hepatocellular cancers, glioma, neuroblastoma, non-Hodgkin lymphoma and acute myeloid leukemia5–7. Overexpression of CDC25 induces a bypass of cell cycle phases controls, allowing malignant cells to get through the phases and to divide, and has been correlated with tumor aggressiveness, high grade tumors and low vital prognosis for patients8–10. Taken together, these evidences suggest that CDC25 phosphatases are promising targets for the development of anti-cancer therapies. To date, a number of CDC25 inhibitors have been described in the literature11–13, and some of them proved to be rather efficient14. These compounds belong to various chemical classes, such as quinonoids, electrophilic inhibitors, thiophenic derivatives, phosphate surrogates and coumarins15,16) and were obtained by multiple sources, including organic synthesis, or marine organisms extraction17. In this context, it is worth mentioning that Georgantea et al. have recently identified two CDC25 inhibitors among 21 sesquiterpenes isolated from the Caribbean soft coral Pseudopterogogia rigida18.
Some molecules have been evaluated on mice-xenografted human tumors, showing a decrease of tumor size through tumor growth arrest. However, none of these compounds has been selected for further development, due to cytotoxicity at bioactive concentrations19–21. Accordingly, there is still a critical need for novel and efficient CDC25 lead inhibitors from various chemical classes, to be further developed as anticancer agents.
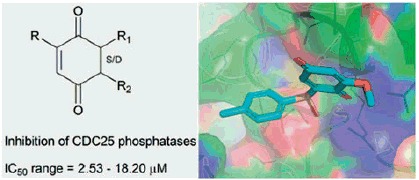
From a medicinal chemistry standpoint, quinones and quinone-like compounds seem to be very promising candidates for CDC25 inhibition, also considering that minor changes in the chemical composition of the side chain of quinone structures can lead to a significant variation in cytotoxicity22–24. Therefore, the design of various quinonoid derivatives with strong inhibitory activity and low cytotoxicity is definitely conceivable. Here, we synthesized the quinonoid derivatives 1–7 (Figure 1), which were evaluated as inhibitors of CDC25 phosphatases and could be potentially of therapeutic benefit. The binding mode of the most potent compounds to CDC25 isoforms was investigated by molecular docking simulations, thus suggesting a rational explanation for the observed biological activity.
Figure 1.

Quinones and quinone-like compounds 1–7.
Materials and methods
Production and purification of recombinant human CDC25
Human glutathione-S-transferase (GST)-Cdc25 recombinant enzymes were used to evaluate the inhibitory potential of compounds.
Recombinant human GST-CDC25 proteins were produced as previously described by Brault et al.25 Briefly, the GST-tagged Cdc25s were expressed in the bacterial expression system Escherichia coli BL21-DE3 pLys S, transformed by a plasmidic vector (pGEX 2T) containing the sequences encoding full length CDC25. Production of recombinant proteins was induced via an IPTG induction system. Then, cells were lysed and centrifuged to recover the supernatant which was purified with a GSH-agarose column system, and recombinant GST-CDC25 proteins were eluted and collected in fractions. Activity, purity and protein concentration of the fractions were evaluated.
CDC25 enzymatic activity was measured by a dephosphorylation assay with 3-O methyl fluorescein phosphate as described26. Briefly, the assay was performed in 96-well plates in buffer [50 mM Tris–HCl, 50 mM NaCl, 1 mM EDTA and 0.1% SAB, pH 8.1], 3-O-methylfluorescein phosphate was used as substrate. After 2 h at 30 °C, 3-O-methylfluorescein fluorescent emission was measured with a CytoFluor system (Perspective Applied Biosystems, Villebon-sur-Yvette, France; excitation filter: 475 nm; emission filter: 510 nm).
Statistics and analytical models
Assays were performed in triplicate, and the experiment was independently performed three times. The results are expressed as percentage of inhibition of CDC25 phosphatase activity in presence of the tested compounds (and compared to DMSO control). All compounds were tested at a 100 µM concentration. Naphtoquinone (20 µM) was used as positive reference inhibitor.
IC50 values for CDC25 inhibition were evaluated by in vitro fluorimetric assays and were determined with sigmoid curves plotted by using a non-linear approximation model based on the least square method (GraphPad Prism software, La Jolla, CA).
Molecular modeling
Ligand conformational analysis was carried out with Omega2, version 2.5.1.4 (OpenEye, Santa Fe, NM)27,28, allowing the storage of the 600 most favorable conformations. Molecular docking was then performed with the FRED docking program, version 3.2.1 (OpenEye, Santa Fe, NM)29–31, while rescoring of docking poses was performed with the XSCORE program32 and with the molecular mechanics generalized-Born surface area (MM-GBSA) method33, using a procedure described elsewhere34. To perform molecular docking, the crystallographic structures coded by PDB IDs 1C2535, 1CWS36, and 3OP3 were selected as representative for CDC25A, CDC25B and CDC25C, respectively. For homology modeling purposes, sequences of human CDC25A, CDC25B and CDC25C were retrieved from the UniProt Knowledgebase (UniProtKB – http://www.uniprot.org/) under the accession codes P30304, P30305 and P30307, respectively37, and were aligned by Clustal38. The full structure of catalytic domain of the CDC25C was generated by Modeller 9v539. The best protein model was chosen based on the DOPE score.
Results and discussion
Chemistry
Compound 1 was prepared from vanillin according to Noland procedure40 with slight modifications (Scheme 1). The Noland procedure used MOM (methoxymethyl-) as protecting group for the hydroquinone 1a. While MOM chloride used for this protection is quite expensive and highly toxic, we preferred to protect the hydroxyquinone as the ethoxyethyl ether 1b. Furthermore, CAN oxidation of the MOM-protected hydroquinone 2a lead in our hands to lower isolated yields (50%) of the sulfinylquinone 1 compared to EE-protected hydroquinone 2b (70%). Details on the synthetic procedure to compound 1, chemical and spectroscopic characterizations are described in the Supporting Information.
Scheme 1.
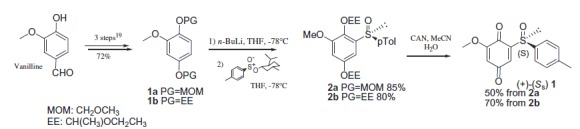
Preparation of the quinone 1 from vanillin.
Compounds 2 and 3 were prepared according to our previous work starting from commercially available 2-methylhydroquinone (Scheme 2)41. Details on the synthesis and chemical characterization of 2 and 3 are reported in the Supporting Information.
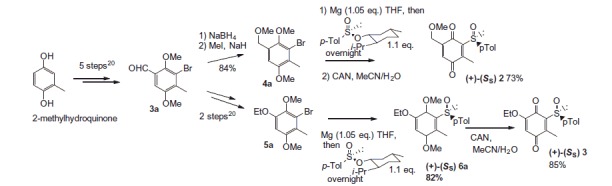
Scheme 2. Preparation of the quinones 2 and 3 from 2-methylhydroquinone.
Quinonoids 4–7 were described as synthetic intermediates in our previous work towards the total synthesis of salvinorin A and analogs42.
Inhibition of CDC25A, B, and C by 1–7
A preliminary evaluation of the inhibitory activity of 1–7 against CDC25 isoforms A, B and C was performed at 100 µM concentration of each compound, in order to remove low-potency inhibitors and to focus further efforts on most promising molecules. Results showed that four compounds, namely 1, and 3–5, were potent inhibitors of the three CDC25 isoforms (Figure 2), whereas 2, 6 and 7 inhibited the CDC25 isoforms to a lesser extent (residual activity of the CDC25 enzymes was higher than 10% at 100 µM). For this reason, these molecules were discarded, while 1, and 3–5 were selected for further investigations.
Figure 2.

Preliminary screening of the test-set. The inhibition of CDC25A (left/blue bars), CDC25B (middle/green bars), and CDC25C (right/red bars) isoforms by 100 μM of 1–7 was evaluated. DMSO served as negative inhibition control (100% residual CDC25 activity), while the reference inhibitor naphtoquinone at 20 μM serve as positive control.
The half-maximal inhibitory concentration (IC50) of compounds 1, and 3–5 was evaluated against each CDC25 isoform. Notably, all values were below 20 µM, and the small molecules proved to inhibit more potently CDC25A with respect to CDC25B and CDC25C. Moreover, as reported in Table 1, compounds 1 and 5 were the most potent inhibitors of CDC25A (IC50 = 2.64 and 2.53 µM, respectively). These values are of particular interest, especially in comparison to literature data (best IC50 usually between 0.1 and 5 µM)43. CDC25B was inhibited by 1 and 3–5 with lower potency among the CDC25 isoforms, with 3 and 4 being the weakest inhibitors of the test set showing IC50 in the double digit micromolar concentration. Finally, CDC25C was inhibited with IC50 from 5.41 and 9.43 µM.
Table 1.
Inhibition of human CDC25 isoforms by quinones and quinone-like compounds 1, and 3–5.
| IC50 (μM)* |
|||
|---|---|---|---|
| Mol | CDC25A | CDC25B | CDC25C |
| 1 | 2.64 ± 0.62 | 6.99 ± 0.21 | 5.72 ± 0.27 |
| 3 | 3.19 ± 1.02 | 11.13 ± 1.01 | 5.41 ± 0.52 |
| 4 | 3.73 ± 1.53 | 18.20 ± 1.68 | 9.43 ± 1.25 |
| 5 | 2.53 ± 0.23 | 7.46 ± 2.17 | 5.76 ± 0.42 |
Values are expressed as mean of triplicates ± standard deviation (SD).
Molecular modeling
The crystallographic structures of CDC25A, CDC25B and CDC25C isoforms were selected as described in the experimental section, and used as rigid receptors in molecular docking simulations. Whereas multiple structures are available in the protein data bank (rcsb.org/pdb) to describe the CDC25A and CDC25B catalytic domain2,35,36, the only structure available of CDC25C has a residues gap within the active site, which may hamper structure-based molecular simulations. Therefore, the full structure of CDC25C catalytic domain was obtained by homology modeling. Molecular docking was performed with FRED (OpenEye, Santa Fe, NM)29–31. The best docking-base complex of each molecule was then submitted to energy minimization in explicit water solvent, before the ligand binding affinity was recalculated by means of two different rescoring methods (see below).
Overall, molecular docking simulations showed that molecules 1, and 3–5 are able to fit within the active site of CDC25A, CDC25B and CDC25C (Figure 3) and to occlude the accessibility to the catalytic cysteine, thus providing a structural explanation to the inhibitory effect observed in vitro. In detail, the sulfoxide and the quinone moieties establish H-bond interactions with the side chain of polar residues flanking the catalytic cysteine in CDC25A (Figure 3A) such as Arg436 and Arg439. The hydrophobic portion of 1 and 3 establishes hydrophobic interactions with Phe432 or Tyr386, whereas 4 and 5 share a very similar binding mode and establish a “parallel displaced” π–π interaction with the aromatic side chain of His490. Notably, the same pharmacophores were engaged in docking towards CDC25B (Figure 3B). Indeed, the quinone and sulfoxide moieties establish H-bond interactions with Arg482, Tyr428, Arg479 and Arg544 of CDC25B that surround the catalytic cysteine residue within the active site. In CDC25C, 1 and 3 share a common binding mode and establish H-bonds with Arg383 and Ser380 of the catalytic loop by means of the quinone moiety. Similarly, 4 and 5 establish H-bonds with Arg383 and His437 and occupy the catalytic site by occluding the accessibility to the catalytic Cys377 from the solvent area (Figure 3C).
Figure 3.

Predicted binding mode of active molecules 1, 3, 4 and 5 towards the crystallographic structure of CDC25A (Panel A), CDC25B (Panel B), and the homology model of CDC25C catalytic domain (Panel C). Small molecules are shown as sticks. The protein is shown as cartoon. Side chains of residues within 5 Å from small molecules are showed as lines. H-bond interactions are highlighted by dashed lines, and residues contacted by H-bonds are labeled. For the sake of representation, H atoms were omitted. The catalytic cysteine residue is shown as sticks and is labeled.
Theoretical binding affinity of active molecules was computed by means of MM-GBSA and XSCORE. The MM-GBSA approach calculates the free energy of binding of a ligand towards a protein and is also used to rescore docking or virtual screening results33,34,44–46; the XSCORE function performs a precise and reliable estimation of the ligand’s pKd32. Notably, results in Table 2 reflect the trend of potency observed in vitro, showing that compounds are expected to bind to CDC25 enzymes with a micromolar affinity, further reinforcing the consistency of predicted binding modes.
Table 2.
Predicted binding affinity of active molecules towards CDC25A, CDC25B and CDC25C.
| CDC25A |
CDC25B |
CDC25C |
||||
|---|---|---|---|---|---|---|
| Mol | MM-GBSA (kcal/mol) | XSCORE (pKd) | MM-GBSA (kcal/mol) | XSCORE (pKd) | MM-GBSA (kcal/mol) | XSCORE (pKd) |
| 1 | −25.70 | 5.24 | −24.98 | 5.17 | −29.09 | 5.84 |
| 3 | −23.81 | 5.12 | −19.25 | 5.14 | −27.30 | 5.87 |
| 4 | −24.02 | 5.11 | −17.62 | 4.51 | −19.70 | 4.55 |
| 5 | −27.56 | 5.57 | −24.30 | 5.08 | −27.12 | 5.61 |
Conclusion
In this work, we synthesized and tested in vitro a number of novel quinonoid derivatives as inhibitors of CDC25 phosphatases. Due to the implication of these enzymes in the fine regulation of the cell cycle, as well as in the origin and progression of various types of cancer, these molecules may be profitable starting points for further investigations. The most promising leads showed inhibition of CDC25 isoforms at low micromolar concentration (single digit). Their binding mode to CDC25 isoforms was investigated by molecular modeling, showing that these inhibitors are able to fit the catalytic active site and to sterically occlude the access to the catalytic cysteine residue from the bulk solvent.
In summary, these low molecular weight quinonoid derivatives are tools for chemical biology studies, as well as profitable leads for further investigation as anticancer candidates.
Supplementary Material
Acknowledgements
Authors are sincerely grateful to Dr. Nadine Martinet for her networking efforts and profitable discussions. The authors wish to acknowledge networking contribution by the COST Actions CM1106 “Chemical Approaches to Targeting Drug Resistance in Cancer Stem Cells” and CM1407 “Challenging organic syntheses inspired by nature – from natural products chemistry to drug discovery”. The authors wish to thank the OpenEye Free Academic Licensing Program for providing a free academic license for molecular modeling and chemoinformatics software.
Disclosure statement
The authors report no conflicts of interest.
References
- 1.Mascarello A, Chiaradia-Delatorre LD, Mori M, et al. Mycobacterium tuberculosis-secreted tyrosine phosphatases as targets against tuberculosis: exploring natural sources in searching for new drugs. Curr Pharm Des 2016;22:1561–9. [DOI] [PubMed] [Google Scholar]
- 2.Rudolph J.Cdc25 phosphatases: structure, specificity, and mechanism. Biochemistry 2007;46:3595–604. [DOI] [PubMed] [Google Scholar]
- 3.Ducommun B, Draetta G, Young P, Beach D.. Fission yeast cdc25 is a cell-cycle regulated protein. Biochem Biophys Res Commun 1990;167:301–9. [DOI] [PubMed] [Google Scholar]
- 4.Boutros R, Lobjois V, Ducommun B.. CDC25B involvement in the centrosome duplication cycle and in microtubule nucleation. Cancer Res 2007;67:11557–64. [DOI] [PubMed] [Google Scholar]
- 5.Boutros R, Lobjois V, Ducommun B.. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer 2007;7:495–507. [DOI] [PubMed] [Google Scholar]
- 6.Kristjansdottir K, Rudolph J.. Cdc25 phosphatases and cancer. Chem Biol 2004;11:1043–51. [DOI] [PubMed] [Google Scholar]
- 7.Brenner AK, Reikvam H, Lavecchia A, Bruserud O.. Therapeutic Targeting the Cell Division Cycle 25 (CDC25) phosphatases in human acute myeloid leukemia – the possibility to target several kinases through inhibition of the various CDC25 isoforms. Molecules 2014;19:18414–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boutros R, Dozier C, Ducommun B.. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol 2006;18:185–91. [DOI] [PubMed] [Google Scholar]
- 9.Cangi MG, Cukor B, Soung P, et al. Role of the Cdc25A phosphatase in human breast cancer. J Clin Invest 2000;106:753–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonin S, Brunetti D, Benedetti E, et al. Expression of cyclin-dependent kinases and CDC25a phosphatase is related with recurrences and survival in women with peri- and post-menopausal breast cancer. Virchows Arch 2006;448:539–44. [DOI] [PubMed] [Google Scholar]
- 11.Lavecchia A, Di Giovanni C, Novellino E.. CDC25A and B dual-specificity phosphatase inhibitors: potential agents for cancer therapy. Curr Med Chem 2009;16:1831–49. [DOI] [PubMed] [Google Scholar]
- 12.Brault L, Bagrel D.. Activity of novel Cdc25 inhibitors and preliminary evaluation of their potentiation of chemotherapeutic drugs in human breast cancer cells. Life Sci 2008;82:315–23. [DOI] [PubMed] [Google Scholar]
- 13.Lazo JS, Aslan DC, Southwick EC, et al. Discovery and biological evaluation of a new family of potent inhibitors of the dual specificity protein phosphatase Cdc25. J Med Chem 2001;44:4042–9. [DOI] [PubMed] [Google Scholar]
- 14.Lavecchia A, Di Giovanni C, Novellino E.. Inhibitors of Cdc25 phosphatases as anticancer agents: a patent review. Expert Opin Ther Pat 2010;20:405–25. [DOI] [PubMed] [Google Scholar]
- 15.Contour-Galcera MO, Lavergne O, Brezak MC, et al. Synthesis of small molecule CDC25 phosphatases inhibitors. Bioorg Med Chem Lett 2004;14:5809–12. [DOI] [PubMed] [Google Scholar]
- 16.He R, Zeng LF, He Y, et al. Small molecule tools for functional interrogation of protein tyrosine phosphatases. FEBS J 2013;280:731–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brisson M, Foster C, Wipf P, et al. Independent mechanistic inhibition of cdc25 phosphatases by a natural product caulibugulone. Mol Pharmacol 2007;71:184–92. [DOI] [PubMed] [Google Scholar]
- 18.Georgantea P, Ioannou E, Evain-Bana E, et al. Sesquiterpenes with inhibitory activity against CDC25 phosphatases from the soft coral Pseudopterogorgia rigida. Tetrahedron 2016;72:3262–9. [Google Scholar]
- 19.Brezak MC, Valette A, Quaranta M, et al. IRC-083864, a novel bis quinone inhibitor of CDC25 phosphatases active against human cancer cells. Int J Cancer 2009;124:1449–56. [DOI] [PubMed] [Google Scholar]
- 20.Brezak MC, Quaranta M, Contour-Galcera MO, et al. Inhibition of human tumor cell growth in vivo by an orally bioavailable inhibitor of CDC25 phosphatases. Mol Cancer Ther 2005;4:1378–87. [DOI] [PubMed] [Google Scholar]
- 21.Brezak MC, Quaranta M, Mondesert O, et al. A novel synthetic inhibitor of CDC25 phosphatases: BN82002. Cancer Res 2004;64:3320–5. [DOI] [PubMed] [Google Scholar]
- 22.Monks TJ, Hanzlik RP, Cohen GM, et al. Quinone chemistry and toxicity. Toxicol Appl Pharmacol 1992;112:2–16. [DOI] [PubMed] [Google Scholar]
- 23.Monks TJ, Jones DC.. The metabolism and toxicity of quinones, quinonimines, quinone methides, and quinone-thioethers. Curr Drug Metab 2002;3:425–38. [DOI] [PubMed] [Google Scholar]
- 24.Ollinger K, Brunmark A.. Effect of hydroxy substituent position on 1,4-naphthoquinone toxicity to rat hepatocytes. J Biol Chem 1991;266:21496–503. [PubMed] [Google Scholar]
- 25.Brault L, Denance M, Banaszak E, et al. Synthesis and biological evaluation of dialkylsubstituted maleic anhydrides as novel inhibitors of Cdc25 dual specificity phosphatases. Eur J Med Chem 2007;42:243–7. [DOI] [PubMed] [Google Scholar]
- 26.Valente S, Bana E, Viry E, et al. Synthesis and biological evaluation of novel coumarin-based inhibitors of Cdc25 phosphatases. Bioorg Med Chem Lett 2010;20:5827–30. [DOI] [PubMed] [Google Scholar]
- 27.Hawkins PC, Skillman AG, Warren GL, et al. Conformer generation with OMEGA: algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J Chem Inf Model 2010;50:572–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.OpenEye OMEGA 2.5.1.4: OpenEye Scientific Software, Santa Fe, NM. Available from: http://www.eyesopen.com. [Google Scholar]
- 29.McGann M.FRED pose prediction and virtual screening accuracy. J Chem Inf Model 2011;51:578–96. [DOI] [PubMed] [Google Scholar]
- 30.McGann M.FRED and HYBRID docking performance on standardized datasets. J Comput Aided Mol Des 2012;26:897–906. [DOI] [PubMed] [Google Scholar]
- 31.OpenEye FRED 3.0.1 OpenEye Scientific Software, Santa Fe, NM. Available from: http://www.eyesopen.com. [Google Scholar]
- 32.Wang RX, Lai LH, Wang SM.. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput-Aided Mol Des 2002;16:11–26. [DOI] [PubMed] [Google Scholar]
- 33.Miller BR, McGee TD, Swails JM, et al. MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput 2012;8:3314–21. [DOI] [PubMed] [Google Scholar]
- 34.Mori M, Manetti F, Botta M.. Predicting the binding mode of known NCp7 inhibitors to facilitate the design of novel modulators. J Chem Inf Model 2011;51:446–54. [DOI] [PubMed] [Google Scholar]
- 35.Fauman EB, Cogswell JP, Lovejoy B, et al. Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell 1998;93:617–25. [DOI] [PubMed] [Google Scholar]
- 36.Reynolds RA, Yem AW, Wolfe CL, et al. Crystal structure of the catalytic subunit of Cdc25B required for G2/M phase transition of the cell cycle. J Mol Biol 1999;293:559–68. [DOI] [PubMed] [Google Scholar]
- 37.UniProt C UniProt: a hub for protein information. Nucleic Acids Res 2015;43:D204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007;23:2947–8. [DOI] [PubMed] [Google Scholar]
- 39.Sali A, Blundell TL. Comparative Protein modeling by satisfaction of spatial restraints. J Mol Biol 1993;234:779–815. [DOI] [PubMed] [Google Scholar]
- 40.Noland WE, Kedrowski BL.. Quinone approaches toward the synthesis of aflatoxin B(2). Org Lett 2000;2:2109–11. [DOI] [PubMed] [Google Scholar]
- 41.Lanfranchi DA, Hanquet G.. Asymmetric Diels-Alder reactions of a new enantiomerically pure sulfinylquinone: a straightforward access to functionalized Wieland-Miescher ketone analogues with (R) absolute configuration. J Org Chem 2006;71:4854–61. [DOI] [PubMed] [Google Scholar]
- 42.Lanfranchi DA, Bour C, Hanquet G.. Enantioselective access to key intermediates for salvinorin A and analogues. Eur J Org Chem 2011;2818–26. [Google Scholar]
- 43.Lavecchia A, Di Giovanni C, Novellino E.. CDC25 phosphatase inhibitors: an update. Mini Rev Med Chem 2012;12:62–73. [DOI] [PubMed] [Google Scholar]
- 44.Hou T, Wang J, Li Y, Wang W.. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 2011;51:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mori M, Botta M.. Drug design and screening by in silico approaches. Trypanosomat Dis Mol Routes Drug Disc 2013;57–79. [Google Scholar]
- 46.Infante P, Mori M, Alfonsi R, et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J 2015;34:200–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.