

Abstract
Discovery of novel tool compounds and drug leads against a range of unorthodox protein targets has pushed both experimental screening methodologies as well as the field of structure based design to the limit in recent years. Increasingly, it has been recognized that some of the most desirable targets for the development of small molecule effectors are actually protein-protein and protein-nucleic acid interactions. There are numerous nontrivial challenges to pursuing small molecule lead compounds directed toward PPIs and PNIs: relatively shallow cavities, large surface areas that are natively complexed to macromolecules, complex patterns of interstitial waters, a paucity of “hot spots”, large conformational changes upon ligand binding, etc. Although there have been some notable successes targeting PPIs in the last decade, there has been distinctly less success in the realm of targeting PNIs. This chapter focuses on an approach, successfully applied by our group to address the challenge of gaining traction on the PPI target RAD52, which is a protein that binds both single stranded and double stranded DNA, and is an anti-cancer target for certain types of cancer. There are many approaches to tackling the difficult problems of finding effective small molecules that disrupt PPIs and PNIs, but the methods presented here offer a series of elegant solutions, which integrate experimental HTS, biophysical methods, docking and molecular dynamics in a powerful way. Additionally, the structural knowledge gained from these studies provides a means for rationally understanding what features lead to ligand affinity in these fascinating and highly unorthodox pockets.
1. The Challenge of Ligand Discovery and Design Targeting Protein-Nucleic Acid (and Protein-Protein) Interactions
It has become increasingly desirable to develop drug lead compounds against protein-macromolecular complexes. This is not surprising, as these unorthodox drug targets make up the bulk of the human proteome, and are involved in an array of pharmaceutically attractive phenomena, including cellular movement, assembly and disassembly of protein-complexes, chaperon activity, subcellular transport, nucleic acid binding, signal transduction, transcription, and other functions (Makley and Gestwicki, 2013, Venter et al., 2001). However, regardless of their medical importance, these unorthodox targets have been very recalcitrant to the facile identification of small molecule inhibitors of their native activities. Unlike classic drug targets, such as enzymes, the native ligand in PPIs or PNIs, even when known (which may often not be the case) does not usually provide a clear picture about how to proceed with small molecule scaffold discovery and/or development. Additionally, PPIs and PNIs often lack clearly distinguishable binding pockets, or have highly diffuse binding interfaces. The nature of these interfaces is complicated by the presence of “hot spots” that largely control the magnitude of the ligand binding energy (Yap et al., 2012, Makley and Gestwicki, 2013, Scott et al., 2016). Furthermore, additional complications arise when considering protein flexibility and the ligand associated changes pocket distribution (Dean et al., 2015, Eyrisch and Helms, 2007, Sperandio et al., 2008).
Most of what we know about the ability to successfully identify drug lead molecules against protein-macromolecular targets comes from the field of PPIs. There are certain types of PPIs that have been more amenable to drug discovery than others (Scott et al., 2016). Particular success has emerged from strategies that attempt to mimic a peptide or protein ligand, such that small molecule leads directly compete with the native macromolecular ligand (de Vega et al., 2007). In the field of PPI research, structural and biophysical approaches play a key role, often in iterative cycles of ligand development (Scott et al., 2016). These discovery campaigns have often revealed the fascinating, but complicated role that water plays in the shape, accessibility and binding activity of the PPI protein target. A number of key lessons should be noted from PPIs, which are likely to be at least partially relevant to PNIs. The buried regions in PPIs that have been amenable to inhibitor development are usually between 1,000 to 6,000 Å2 (Scott et al., 2016), with regions larger than 2,000 Å2 often consisting of a patchwork of buried and solvent accessible groups. Often there are increases in Trp and Tyr residues around the PPI binding pocket, as well as polar residues (e.g., Arg, Asp and His). The classical PPI possesses some hydrophobic core region flanked by a patchwork of polar regions, often utilizing interstitial waters. Generally, PPI inhibition strategies are more successful when the binding hot spots are relatively contiguous. Experimental approaches that have yielded results include classical HTS, fragment based drug discovery (FBDD), design of targeted peptides and peptidomimetics and computational approaches (i.e., structure based discovery and design). Traditional computational approaches to identification of PPI inhibitors are extremely challenging, due to the often ambiguous role of water and the inherent ligand-associated protein conformational changes in most PPIs (Huggins et al., 2011, Scott et al., 2016).
Although there are a vast array of difficulties in executing drug discovery campaigns against PPIs, it is potentially more challenging to attempt to disrupt PNIs, as much less is known about them (Yap et al., 2012). Nevertheless, our research group was interested in designing a campaign targeting the DNA repair protein RAD52, by specifically disrupting its ssDNA binding activity. The essence of our approach was to first carry out an experimental (in vitro) HTS campaign targeting the RAD52-ssDNA PNI, and to then employ computational and structural methods to learn about how certain chemotypes were disrupting the PNIs. A statistical approach was used to test the strength of our structural hypotheses about PNI disruption. Finally, this knowledge was then put to the test in an independent in silico HTS campaign against a database of interesting natural product compounds. Amazingly, a single natural product was selected for detailed biophysical study, and found to be in the high nM to low μM range (depending on the assay employed). A rational strategy for targeting the RAD52 PNI is, ostensibly, a nightmarish challenge for structure based design, in that only an unliganded crystal structure of RAD52 was available (PDB 1KN0), which suggests an exotic circular continuous ssDNA binding epitope (Hengel et al., 2016, Kagawa et al., 2002), with a patchwork of alternating hydrophobic and hydrophilic regions and complex water networks in the biding groove. Given the uncertainty of PDB 1KN0 representing the liganded conformation of the protein in solution, the lack of any known ligands that bind in this groove and the complex role of water, we sought to build an integrated workflow for PNI inhibitor discovery which capitalized on in vitro HTS initially, followed by computational studies built on a rigorous statistical framework for understanding why certain chemotypes disrupt the RAD52 PNI. This validated computational approach is then used to identify completely novel scaffolds in an in silico screening. Finally, we take great care in experimentally validating these PNI inhibitors via biophysical methods that confirm the projected activity of preventing ssDNA binding to the RAD52 target.
This chapter focuses largely on how to parlay the experimental HTS data into models that can be meaningfully tested for accuracy, and how to extrapolate these validated models into in silico screens against the challenging RAD52 target. Not only are these approaches relevant to discovering inhibitors against other DNA repair proteins, such as RAD51 recombinase and RPA ss-DNA binding proteins, whose binding may be assayed in a similar fashion(Grimme and Spies, 2011, Subramanyam et al., 2013), but to many types of PPI targets as well. The sections described below focus on I) how experimental HTS data was obtained for disrupting the RAD52 PPI, and an analysis of the most promising inhibitors, II) employing docking and molecular dynamics simulations to obtain information about the locus of action of the new inhibitors obtained from HTS, III) utilizing the Receiver Operating Characteristic (ROC) (Yap et al., 2012, Metz, 1978) statistical method to determine the accuracy of various computational workflow models, IV) application of the ROC-validated model in an in silico screening campaign against a database of natural products and a discussion of the role of biophysical validation using NMR WaterLOGSY.
2. Employing experimental HTS as a tool for learning about the nature of PNI lead compounds that disrupt the RAD52 PNI
To identify compounds that disrupt the RAD52-ssDNA interactions, a previously established Förster Resonance Energy Transfer (FRET)-based assay was adapted to the HTS format (Grimme and Spies, 2011, Grimme et al., 2010). The assay took advantage of certain known characteristics of RAD52; the ssDNA-binding region of RAD52 is continuous around the circumference of the ring with a shallow nucleic acid interactive groove that repeats in each monomer. Our FRET-based assay relied on the ability of ssDNA to bind and wrap around the narrow groove spanning the protein ring (Grimme and Spies, 2011, Grimme et al., 2010). FRET donor (Cy3) and acceptor (Cy5) were incorporated at the opposite ends of a 30-mer ssDNA to form Cy3-dT30-Cy5 (which can be synthesized commercially from IDT (Integrated DNA Technologies, INC)). An increase in the FRET signal occurs when the two fluorophores Cy3 and Cy5 were brought into proximity to one another during the process in which Cy3-dT30-Cy5 forms a 1:1 stoichiometric complex with RAD52. The positive control for disrupting the RAD52-ssDNA interaction was achieved using a stoichiometric complex of RAD52 with Cy3-dT30-Cy5 substrate competing with an excess amount of unlabeled ssDNA (Poly dT100); while the negative control consisted of an unperturbed stoichiometric complex of RAD52 with Cy3-dT30-Cy5 (Figure1A). A Multiflo dispenser (Biotek) was used for dispensing control mixtures into the plate, and compounds of interest were dispensed and mixed three times using a Microlab Star liquid handling robot (Hamilton). It is noted that from our experiment, the appropriate reaction volume along with the right plate type are valuable in producing an acceptable single/noise ratio. The quality of the HTS assay was quantified with the Z-factor, which accesses the assay’s performance in terms of separation between the positive and negative controls. A more detailed derivation of the Z-factor is described in Zhang et al (Zhang et al., 1999). For the evaluation of any assays, a Z-factor of 1 is ideal. The Z-factor from our RAD52-ssDNA FRET HTS assay was calculated to be 0.94, which fell between the range of 1 and 0.5, indicating an excellent assay with a large separation band for clear signal detection and superb reliability. Using this assay, the MicroSource SPECTRUM collection library (Microsource, Gaylordsville, CT), which contains a wide structural diversity, including 2320 drug, drug-like synthetic compounds and natural products with a range of known biological activities. Of the 2320 examined compounds, 96 preliminary hits were screened further for signal reproducibility and compound concentration dependent signal detection (Figure 1B). Based on the reported FRET signal and biochemical validation, 12 compounds were purchased with chemical structure confirmed by 1D NMR (Table 1).
Figure 1. FRET-based high throughput screening of the MicroSource Spectrum library.

(A) Control lanes from a 384-well plate. The positive control (blue) containing stoichiometric RAD52 with Cy3-dT30-Cy5 substrate challenged with an excess amount of unlabeled ssDNA (Poly dT100); while the negative control (red) consisted of unperturbed stoichiometric complex of RAD52 with Cy3-dT30-Cy5. Red and blue solid lines represent the average and the standard deviation is shown with error bars respectively. (B) 12 identified hits (green) from cherry-picked rescreening of the initial screen of the MicroSource SPECTRUM collection, with several false positives shown in blue. Originally published as a part of Figure 1(DOI: 10.7554/eLife.14740) in Hengel et al, 2016 (Hengel et al., 2016); used under a CC-BY license.
Table 1.
The twelve hits from the FRET-based HTS assay designed for identifying inhibitors of the RAD52-ssDNA interaction. NI=No inhibition. Originally published as a part of Table 1(DOI: 10.7554/eLife.14740) in Hengel et al, 2016 (Hengel et al., 2016) ; used under a CC-BY license.
| # | Small molecule name; CAS # | Small molecule structure | IC50 (DNA binding); FRET value at saturation | IC50 (annealing extent) | SAEM Δ G (kcal/mol) |
|---|---|---|---|---|---|
| ‘1’ | (−)-Epigallocatechin; 970-74-1 |
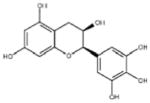
|
ssDNA: 1.8 ± 0.1 μM; 0.45 ± 0.004 ssDNA-RPA: 1.6 ± 0.1 μM; |
ssDNA: 4.9 ± 0.4 μM ssDNA-RPA 4.8 ± 1.8 μM; |
−8.60 |
|
| |||||
| ‘3’ | Methacycline hydrochloride; 3963-95-9 |
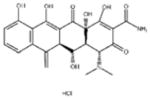
|
2.0 ± 0.17 μM; 0.47± 0.01 |
3.8 ± 0.2 μM | −4.61 |
|
|
|
||||
| ‘4’ | Rolitetracycline; 751-97-3 |
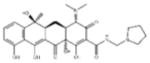
|
29 ± 8.2 μM; 0.56 ± 0.04 |
NI† | −10.50 |
|
| |||||
| ‘5’ | (−)-Epicatechin gallate; 1257-08-5 |
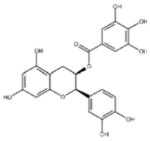
|
255 ± 16 μM; 0.41 ± 0.004 |
20 ± 0.7 μM | −9.87 |
|
| |||||
| ‘6’ | Epigallocatechin-3-monogallate; 989-51-5 |
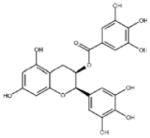
|
ssDNA: 277 ± 22 μM; 0.46 ± 0.01 ssDNA-RPA: 1.6 ± 0.5 μM; |
ssDNA: 6.7 ± 2.1 μM ssDNA-RPA: 3.7 ± 0.5 μM; |
−10.69 |
|
|
|
||||
| ‘7’ | (−)-Epicatechin; 490-46-0 |
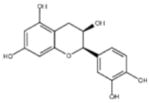
|
1.45 ± 0.11 μM; 0.51 ± 0.01 |
NI | −9.03 |
|
| |||||
| ‘14’ | Oxidopamine; 28094-15-7 1199-18-4 |
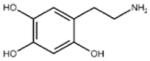
|
779 ± 51 μM; 0.50 ± 0.01 |
NI | −5.71 |
|
| |||||
| ‘15’ | Quinalizarin; 81-61-8 |
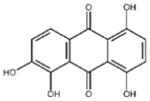
|
563 ± 40 μM; 0.51 ± 0.01 |
5.6 ± 0.6 μM | −9.17 |
|
|
|
||||
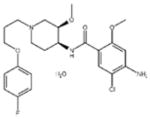
| ‘16’ | Cisapride monohydrate; 260779-88-2 81098-60-4 |
|
1.06 ± 0.05 μM; 0.50 ± 0.01 |
NI | −8.39 |
|
| |||||
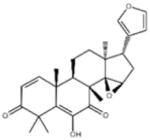
| ‘17’ | Cedrelone; 1254-85-9 |
|
>300 μM | NI | −10.0 |
|
| |||||
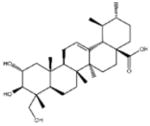
| ‘18’ | Asiatic acid; 464-92-6 18449-41-7 |
|
>800 μM | >100 μM | −11.33 |
|
| |||||
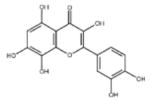
| ‘19’ | Gossypetin; 489-35-0 |
|
913 ± 58 nM; 0.49 ± 0.01 |
6.0 ± 2.3 μM | −9.30 |
In addition, similar FRET-based biochemical studies investigated the specificity of inhibition achieved by compound ‘1’ and ‘6’ to show that, compound ‘6’ interfered with the RAD52-dsDNA interaction while compound ‘1’ had no effect on the interaction between RAD52 and dsDNA. Furthermore, unlike the known RAD52 inhibitor 6-hydroxy-DL-dopa (Chandramouly et al., 2015), the inhibition exhibited by our hit compounds ‘1’ and ‘6’ did not disrupt the ring structural assembly of RAD52 as no tertiary structural changes were observed from our dynamic light scattering (DLS) experiments. Specifically, through performing WaterLOGSY experiments (see section 3 for more detailed descriptions), direct binding interactions were detected between RAD52 and hit compound ‘1’ and ‘6’ respectively (Figure 2).
Figure 2. WaterLOGSY experiments indicate direct interactions between HTS hit compounds and RAD52.

A. Aromatic region of the 1D 1H NMR spectrum of compound “1” (black) and the WaterLOGSY spectrum of 20 μM compound “1”in the presence of 3.3 μM RAD52 (red). The nonexchangeable proton peaks are labeled using atom names (blue) on the compound structure. B. Same as A, except representing the aromatic region of the 1D 1H NMR spectrum of compound “6” (black) and the WaterLOGSY spectrum of 40 μM compound “6” in the presence of 3.3 μM RAD52 (red). The nonexchangeable proton peaks are labeled using atom names (blue) as indicated on the compound structure. Originally published as a part of Figure 2a and 3a (DOI: 10.7554/eLife.14740) in Hengel et al, 2016 (Hengel et al., 2016); used under a CC-BY license.
3. Using docking and molecular dynamics simulations to make hypotheses about RAD52 PNI pharmacophores
Understanding small molecule placement in the ssDNA binding groove
Our next task was to find, with very high confidence, the features that are necessary for molecular recognition of our known (active) ligands with RAD52 (i.e., the binding orientation, ligand confirmations, and eventually the atomistic level structure of the RAD52-small molecule complexes). This information defines the RAD52 pharmacophore for our initial hits. Our primary interests focused on the compounds ‘1’ and ‘6’ from the HTS screen. For orthodox pockets, this would be an almost routine task using any number of docking approaches. However, due to the many challenges in targeting PNIs listed above, we required a multilayer approach that eventually led to a robust in silico screening. As the overall workflow in Figure 3 shows, our overarching goal is to confidently use in silico screening to identify novel chemical scaffolds that target RAD52. In order to accomplish this, we must first grapple with several stark problems. Firstly, a direct in silico campaign against this particular target is likely to generate too many false positives to be truly useful. Briefly, docking is an excellent tool for identifying the binding loci and orientation of small molecules, but is not good at making meaningful predictions of affinity rank ordering across ligand classes (Head, 2010). Additionally, although the technique can be useful in in silico screening, the large number of false positives is only amplified by increasing the size of the docking library. In addition to these classical problems, our lack of structural knowledge about the native liganded complex of RAD52 and the role of interstitial waters is concerning. Nevertheless, we believed that, given the current apo-structure and our HTS hits, particularly, ‘1’ and ‘6’, that we could employ various integrated docking and molecular dynamics strategies to inevitably determine the locus and orientation of these RAD52-ssDNA inhibitors. An increasing body of literature has shown that when docking and MD are employed together that meaningful binding affinity rank ordering can be made, and that the many complex roles of water can often be inferred from these simulations (Whalen et al., 2011, Whalen et al., 2013).
Figure 3. Docking workflow for screening small-molecule drugs against RAD52.

Originally published as a part of Figure 8a (DOI: 10.7554/eLife.14740) in Hengel et al, 2016 (Hengel et al., 2016); used under a CC-BY license.
Generally, the docking of small molecules involves positioning of the ligand with respect to a receptor structure, and determining the most likely binding mode, with regard to the geometries and projected affinities using scoring functions (Kitchen et al., 2004). The primary goal of a typical in silico screen would be to maximize the detection of known ligand as actives (i.e., true positives) while minimizing the errors of false positives (inactives wrongly assigned as actives) and false negative (actives wrongly assigned as inactives)(Malo et al., 2006). These classic approaches have been successfully applied to numerous receptor types for over three decades now. However, due to the many challenges associated with unorthodox targets, as mentioned above, very few in silico campaigns have succeeded in identifying leads against such target classes involved in PPI and PNI, and the prognosis for classical in silico screening approaches is not good (Scott et al., 2016).
The campaign described in this chapter is fundamentally different from the classical approach in that molecular dynamics approaches are used in the early phases to scrutinize both the positioning and the global conformation of the receptor-small molecule complex, such that we are explicitly and globally dealing with protein flexibility and solvation. The utility of performing MD simulations in the early phase of our workflow was to have the highest possible confidence for establishing a set of “true positives” that could be used in a statistical approach for assessing the degree of success in our in silico screening procedure, using the method of Receiver Operating Characteristic (Metz, 1978), which is described in detail below (Figure 4).
Figure 4. Implementation of the ROC analysis in our docking studies against the RAD52 target.

Originally published as a part of Figure 8a (DOI: 10.7554/eLife.14740) in Hengel et al, 2016 (Hengel et al., 2016) ; used under a CC-BY license.
There are an enormous array of potential docking approaches, and there are many factors that can affect the quality (accuracy and selectivity) of in silico screening approaches (Head, 2010). There is no one best method or docking method across target classes (Head, 2010), and certainly in the realm of attempting to disrupt a PPI or PNI, there is no clear indication about which docking algorithms are optimal. It was our contention that it is best to directly empirically assess any in silico screening workflow using quantitative metrics before attempting an actual production screen. Sections 3.2 to 3.4 detail I) the features used in the initial docking of the HTS lead compounds II) how molecular dynamics using a knowledge based force field approach was used to parse the docking results and to provide a much deeper level of detail in terms dealing with water III) how the ROC statistical approach was used to quantify the accuracy of an in silico screening approach.
I) Docking of HTS leads to the ssDNA binding grove of RAD52
As described above, RAD52 forms an oligomeric ring (Kagawa et al., 2002, Lloyd et al., 2002, Singleton et al., 2002); in which ssDNA binding site is located in a narrow groove, which is a continuous epitope that spans the ring circumference (Lloyd et al., 2005, Mortensen et al., 2002). There are repeated sub-pockets within each monomer, with alternating hydrophobic and hydrophilic regions, as well as numerous crystal waters. Our docking workflow employed a combination of sequences which initially used the Triangle Matcher approach for pose determination, followed by the London dG scoring function implemented in Molecular Operating Environment (MOE) 2013.08 (Chemical Computing Group Inc.). London dG is an empirical scoring function which attempts to approximate the binding energy of a small molecule to a protein (Chemical Computing Group Inc.). We then employed an additional layer of rigor by performing an all atom force field energy minimization of each docked pose, while the receptor was fixed, using the MMFF94x force field in MOE 2013.08, followed by rescoring the low energy pose with a physics based forcefield scoring function, GBVI/WSA dG; this approach is a highly parameterized version of the general MM/PBSA (or MM/GBSA) methodologies (Steinbrecher and Labahn, 2010, Wang and Kollman, 2000), in that it employs a forcefield interaction energy between ligand and receptor, and also calculates the difference in solvation energy using the GB/VI solvation model, and determines the change in the solvent accessible surface area (Naim et al., 2007). Not surprisingly, there were very large discrepancies between the top ranked poses when comparing the two different approaches. Additionally, there were some dissimilar poses, even within a single metric, which were different from one another as well. We addressed these challenges by using all atom molecular dynamics simulations with a Knowledge-Based Force Field (KBFF) to guide the selection process, which is described in detail below.
All of the lead inhibitors identified by HTS were subjected to the two docking protocols described above, using MOE 2013.08 to a portion of the ssDNA-binding groove of RAD52 spanning nearly a quarter of its circumference, which consists of three adjacent monomers. The receptor was built using PDB 1KN0, and subjected to preprocessing, including adding hydrogens, optimizing hydrogen bond networks, and assigning charges and protonation states. Additionally, ligands were preprocessed to assign protonation states, assign charges, and energy minimized using the MMFF94x force field in MOE (Chemical Computing Group Inc.). The ligands were then docked into the large (quarter circumference) pocket described above, using the systematic Triangle Matcher search algorithm in MOE (Chemical Computing Group Inc.). One then has a database of potential poses with docking scores. We kept the top 30 poses (based on the London dG scoring function) and these were further subjected to the ligand energy minimization with a rigid RAD52 receptor using the MMFF94x force field, followed by a free energy scoring using the GBIV/WSB dG approach described above.
II) Use of Molecular Dynamics Simulations to Expand on Docking Pose Selection
Following the docking workflow outlined above, we subjected all docked poses to a much more physically rigorous treatment, employing an all atom molecular dynamics simulations with simulated annealing, using the Yamber03 KBFF (Krieger et al., 2004), using fully explicit solvent conditions (Dean et al., 2015). Briefly, each docked complex (i.e., each pose structure) was placed into a simulation cell and solvated, and charge-neutralized with physiological salt concentration, followed by optimizations of the solvent and the hydrogen bond network of the receptor-ligand complex, and finally a phased simulated annealing minimization was performed (the full technical details of a very similar MD simulation are described in more detail in Whalen et al., 2011 (Whalen et al., 2011) and Dean et al., 2015(Dean et al., 2015) No restraints or fixed atoms were employed (i.e., all atoms in the ligand and the entire RAD52 complex, ions and solvent were free). When the convergence criteria for the simulated annealing was met, the VINA (Trott and Olson, 2010) docking utility in YASARA(Krieger et al., 2002) was invoked to perform a “local docking”, which means that the VINA simulation cell is placed only in an area just around the ligand, so that the docking score reflects the position via molecular dynamics. Additionally, interstitial water molecules were determined and retained to be used by the VINA scoring function. Our workflow (Hengel et al., 2016), featured a user-specified parameter for defining what constituted an interstitial water. This allowed us to parse all the docking poses from the docking workflow outlined above. In these studies, we found that interstitial water structure was a major factor contributing to ligand pose and the quality of the docking scores. In Hengel et al., we referred to this automated process as Simulated Annealing Energy Minimization (SEAM) (Hengel et al., 2016).
The nature of the many sub-pockets along this three monomer continuous epitope provided a large number of potential poses for both compounds ‘1’ and ‘6’. The automated SEAM approach was applied to 34 unique docked poses, respectively, along the large quarter circumference ssDNA binding groove (Figure 5A). There was a clear lack of consensus in the docking, which was clarified by the SEAM. Generally, what one finds is that regardless of the docking approach used, the vast majority of docking poses that yield high scores result in poor performance when subjected to SEAM, since introduction of both realistic water structure and full ligand and receptor flexibility expose these poses as weak binders.
Figure 5. Spatial arrangement of inhibitors ‘1’ and ‘6’ with RAD52-ssDNA binding groove.

A. Binding positions of inhibitors ‘1’ and ‘6’ along the sub-pockets of three individual monomers (yellow, green and blue) of the RAD52 ring (PDB 1KN0). Dotted lines indicate the approximate boundaries of ssDNA-binding groove. B. Ligand interactive maps of inhibitors ‘1’ and ‘6’ inside the sub-pockets of RAD52. Originally published as Figure 4 (DOI: 10.7554/eLife.14740) in Hengel et al, 2016 (Hengel et al., 2016) ; used under a CC-BY license.
Interestingly, the top scoring poses of compounds ‘1’ and ‘6’ using the SEAM approach yielded complexes with unique binding sub-pockets along the RAD52 binding groove, suggesting that they may have distinct activities with regard to disrupting complexation with ssDNA (Figure 5). Both compounds occupy complex pockets lying at the interface of the two RAD52 monomers. ‘1’ mediates its interactions through an array of residues, including R55, V128, E140, as well as through water contacts via G59, M56, and K141. ‘6’ binds via hydrogen bonding with D149 and I166, and acts via water mediated contacts with E140, K144, and R153 (Figure. 5B). It was very interesting and encouraging for our SEAM approach to note that all final compound placements include interactions, directly or through interstitial waters, with key RAD52 residues, which have previously been reported to be involved in ssDNA binding (Lloyd et al., 2005) (Figure. 5B). Particular key residues in the locus of the docked compounds include R55, Y65, K152, R153 and R156, which have been shown to be critical to ssDNA binding to RAD52 (Lloyd et al., 2005) (Figure. 5B). Additionally, K141 and K144, also involved in the SEAM pharmacophore, have been linked to particular RAD52 activities via mutation studies (Mortensen et al., 2002).
Importantly, all of the lead compounds subjected to the SEAM approach yielded complexes in which interstitial water plays a role in the binding of the ligand. In retrospect, it is clear that the unorthodox nature of the RAD52 ssDNA binding groove demands the use of some type of explicit solvent treatment to accurately capture small molecule binding. Interestingly, many of the interstitial waters identified in the SEAM method are not involved in high quality hydrogen bonds that one may frequently see in very buried enzyme active sites, but rather, represent a van der Waals binding surface. This suggests that it may be possible to design better ligands, which both displaces these waters, and gain binding energy by filling these potential hydrogen binding positions with the receptor.
III) Use of the ROC method for assessing docking workflow accuracy
It should be kept in mind that the overall goal of this project was to build a workflow that would lead to a high accuracy in silico screening method for RAD52 (i.e., a structure based approach for identification of novel inhibitors). In essence, the SEAM method outlined above has provided us with what will be used as “actual positives” in a statistical assessment of docking workflow accuracy. That raises the question about what approach one may use for “actual negatives” in such a docking workflow. An excellent choice in lieu of using experimental negatives, is to employ decoy compounds that possess a number of similar properties as true hits, but that are topologically distinct. We employed the Database of Useful Decoys-Enhanced (DUD-E) website to generate a series of 50 “DUDS” for each true positive. The general utility of employing DUDS is discussed in Huang et al (Huang et al., 2006). The idea is to use property matching to compounds of interest, such that compounds with similar molecular weight, estimated water-octanol partitioning coefficient (cLogP), number of rotatable bonds, number of hydrogen bond acceptors, number of hydrogen bond donors and net charge are selected from among a data base.
Having a set of true positives and true negatives allows one to apply quantitative tests of accuracy to docking workflows. We used the statistical method of analyzing ROC curves to optimize the balance of true positives, false positives, true negatives and false negatives (Alexandre Varnek, 2008, Metz, 1978). The ROC approach allows one to make informed decisions about the type of accuracy desired. For screening purposes, accuracy itself is not the goal. It helps to understand that the performance of a classifier can be defined by the following indices:
One way to view accuracy is that it can be represented by these two different indices:
We can also examine the number of incorrect classifications as well:
If we have only two classifications, positive or negative, then the total fraction of correct and incorrect decisions must be unity, thus:
Due to these inherent constraints, it is only necessary to express one of the fractions from each of the above equations in order to capture the necessary information for quantifying accuracy. The central point is that Accuracy may be achieved in different ways, and that it would be ideal to test how a given workflow performs before actually employing it. Given overlapping populations of true negatives and true positives, a classifier threshold value must be defined, which optimizes these benefits and drawbacks (Figure 6). Importantly, there is no absolute best classifier threshold; it is a subjective decision that depends on many factors that the researcher must weigh. The best way to make the key decision about what threshold value to employ is to vary its value and see how this affects the fraction of true positives and fraction of false negatives, respectively. ROC analysis is usually performed by changing the classifier threshold, and determining how the fraction of true positives and true negatives change (since determination of only these two values reports on all four classifier fractions).
Figure 6. Decision threshold selection is not absolute.

Two simulated distribution (black and blue) of a quantity with one possible decision threshold (red) shown.
An example of a typical ROC curve is shown in Figure 7, in which the fraction of true positives is plotted on the ordinate and the fraction of false positives is plotted on the abscissa; the range of values will always be from zero to one, since these are limits of the fractions. As we vary the classifier threshold, the form that the function takes in the ROC plot is, of course, dictated by the nature and the shape of the overlap of the two distributions (as in Figure 6). Importantly, we see that all possible permutations of the fraction of true positives and fraction of true negatives are represented on the ROC curve. This is the real power of the technique, in that it affords the researcher the opportunity see the performance of the workflow over the complete range of classifier values, and choose what value is most conducive to the current study. Figure 7 indicates that a very high classifier threshold yields the portion of the ROC curve in the lower left quadrant of the plot, and that very low classifier thresholds would be associated with the upper right quadrant. An ideal ROC curve would reach the upper left corner of the plot, while a classifier that did no better than random would cross the diagonal of the graph (Figure 7). In other words, the closer to the diagonal an ROC curve is, the worse is the protocol used to distinguish the actives from the decoys (i.e., DUDS in this case), and the closer to the ideal curve, the more the docking protocol gets to fully distinguishing actives from decoys.
Figure 7.

A typical conventional ROC curve with decreasing threshold strictness.
The nature of our particular in silico screening campaign is that we would like to focus on screening natural product compounds, since they have a long history of producing excellent lead compounds in challenging ligand discovery campaigns. However, these compounds are usually relatively precious, and significant labor may be expended in their preparation. Thus, in terms of our particular workflow, we were very interested in a high classifier threshold, which would come close to completely separating the populations, even if this means that we get a good deal of false negatives. In other words, we very much wanted to avoid false positives (the usual bane of in silico screening). Fortunately, our particular workflow, employing SEAM, provided a near perfect ROC curve, which allowed us to confidently employ the use of natural product library, as discussed below.
Docking scores were used for determining the ROC optimal classifier threshold values based on the cost-benefit analysis described above. The original HTS and its poses were determined to be the only “actual positives”, and the DUDS and their respective poses were treated as “actual negatives”; any poses above the classifier threshold value were True positives, and values below were designated as True negatives. The ROC curves were analyzed using the metric of the area under the curve (AUC) (DeLong et al., 1988). The AUC reports on the probability that, when we compare the scores for a randomly selected active compound and a randomly selected decoy, that the in silico screen will rank the active compound ahead of the decoy (DeLong et al., 1988).
The docking scores for the poses with the most active compounds exhibited bimodal frequency distribution (Figure 8A), and the docking protocols’ ability to distinguish between active compounds and decoys was quantified (Figure 8B). As mentioned above, our goal was to be able to employ a high classifier threshold, in order to reduce the cost of false positives. The ROC curve for ‘1’ illustrates how the optimized in silico classification process is nearly ideal in distinguishing false positives from true positives, since it assigns more negative docking scores (i.e., more favorable predicted binding energies) for active conformations than for those of the decoy compounds. Thus, up to this point we have used retrospective information gathered from HTS, and used molecular dynamics and docking to expand our understanding of the complexation of these hits with the ssDNA binding groove of RAD52. We now pivot our focus to a completely prospective study. This represents a much more significant, and risky challenge, since we are no longer retrospectively analyzing HTS results. Indeed, true prospective structure based discovery of specific low μM to high nM hits using strictly in silico screening, especially against challenging or novel targets, is quite rare (Head, 2010). Our philosophy for the campaign described in Hengel et al.(Hengel et al., 2016), was to use ROC curve analysis in such a way as to be able to focus on screening of interesting and relatively precious natural products. Natural products clearly have an unrivaled history in drug discovery, and often represent the primary hits against targets. We employed the AnalytiCon MEGx Natural Products Screen Library (AnalytiCon Discovery GmbH, Postsdam, Germany) - the in silico version of an actual library, consisting of highly purified plant, fungal and microbial derived compounds, which are available for purchase.
Figure 8. Application of ROC in combination with binding energy based docking scores for novel small molecule inhibitors for RAD52-ssDNA interaction.

A. Calculated docking scores (kcal/mol) for the binding configurations of compound ‘1’ in comparison to decoys compounds. As the low docking scores indicate low binding energies, which correlate to favorable interactions, the histogram shows a clear distinction between true positives from compound ‘1’ and true negatives from ‘decoys’. B. Receiving-operating characteristics (Metz, 1978) curve used with the threshold shown yielding an AUC value of 0.9973. Originally published as a part of Figure 8b and 8c (DOI: 10.7554/eLife.14740) in Hengel et al, 2016 (Hengel et al., 2016); used under a CC-BY license.
To summarize, the overall workflow is shown in Figure 3. A database composed of the natural products and a control selected from the HTS hits were created and preprocessed for virtual screening, as described above. The Dock utility of MOE was used to generate a database of the top 30 scoring poses from each compound, followed by overall ranking of poses for the entire database. The top scoring compounds were then selected for further analysis. Poses with more negative scores than the best scoring of our control (i.e., an actual positive in the ROC curve context) were then subjected to the SEAM docking procedure detailed above. Following SEAM, all of the poses were ranked and evaluated. This resulted in nine compounds from the AnalytiCon database that had poses with scores better than those of the internal control. The best scoring structure was ordered from AnalytiCon Discovery GmbH (Postsdam, Germany) for in vitro inhibition studies. This compound, NP-004255, is a macrocyle ester, consisting of three trihydroxylated phenolic moieties; it is called corilagin, which belongs to a family of secondary plant metabolites called ellagitannins. Importantly, the hit was validated by both NMR WaterLOGSY and the FRET-based method described above, as fully described in Hengel et al., 2016 (Hengel et al., 2016), as shown in Figure 9. NP-004255 was shown to bind to RAD52, and compete for ssDNA binding. Two assays were performed to obtain IC50 values, competition with ssDNA only and competition in the presence of the ssDNA binding protein RPA, yielding values of 1.5 ű 0.2 uM and 0.5 ± 0.1 μM, respectively. The importance of multiple forms of orthogonal validation of any hit, whether from an in vitro or in silico HTS campaign, cannot be overemphasized. It is especially important to have at least one assay that establishes direct binding to the target (i.e., not a functional assay, but an assay that shows direct interaction between the proposed hit and the target). Methods for direct binding that are frequently employed include surface plamon resonance (SPR), NMR WaterLOGSY and crystallography. WaterLOGSY was an excellent approach in this case, since the method derives from the magnetization transfer from solvent to a bound ligand, which often proceeds through interstitial water contacts. Figure 10 illustrates the placement of NP-004255 within the RAD52-ssDNA binding groove, as a comparison to the HTS hits shown (Figure 5).
Figure 9. Biochemical validation of NP-004255.

A. Aromatic region of the 1D 1H NMR spectrum of compound NP-004255 alone (black) and the WaterLOGSY spectrum of 40 μM compound NP-004255 in the presence of 3.3 μM RAD52 (red). The nonexchangeable proton peaks (blue) using atom names are shown on the structure of compound NP-004255. (B) IC50 values for inhibition of ssDNA binding and wrapping were determined using FRET-based assays that follow the change in geometry of a Cy3-dT30-Cy5 substrate (black circles). The computed IC50 value is shown above the curve. Titration of the RAD52–dsDNA with NP-004255 (gray boxes) shows that this inhibitor does not perturb the RAD52–dsDNA interaction. (C) Aromatic region of the 1D 1H NMR spectrum of compound NP-004255 alone (black) and the WaterLOGSY spectrum of 40 μM compound NP-004255 in the presence of 3.3 μM RPA (red). (D)Titration of the RAD52-RPA-Cy3-dT30-Cy5 complex with NP-004255 (black circles). The computed IC50 value is shown below the curve. Green squares show titration of the RPA-Cy3-dT30-Cy5 complex with NP-004255 indicating NP-004255 does not perturb the RPA-ssDNA. Originally published as Figure 9 (DOI: 10.7554/eLife.14740) in Hengel et al, 2016 (Hengel et al., 2016); used under a CC-BY license.
Figure 10. Placement of RAD52 NP-004255 within the ssDNA binding groove.

A. Electrostatic potential surface of three monomers of the RAD52-NTD (PDB 1KN0) positioning NP-004255 within the ssDNA binding groove. B. MOE ligand map of NP-004255 when binding RAD-52. Originally published as Figure 8d and 8e (DOI: 10.7554/eLife.14740) in Hengel et al, 2016 (Hengel et al., 2016); used under a CC-BY license.
NP-004255 binds to RAD52 in an analogous way as ‘1’ and ‘6’, employing a buried interstitial water network, and adopts a helical-like conformation that fits nicely into the ssDNA binding groove (Figure 10). Its complexation produces hydrogen bonding patterns ideally located to prevent ssDNA access to residues that are important for complexation(Hengel et al., 2016, Lloyd et al., 2005).
4. Implications for employing ROC and MD-informed workflows in the discovery of novel PNI inhibitors
The workflow described in this chapter was highly successful in the challenging domain of prospectively discovering the pharmacophore of the PNI of RAD52. The data show that choice of workflow, employing experimental, docking, and MD simulations can be designed to almost perfectly separate hits from negatives. The use of ROC curves, allowed us to optimize the threshold to meet our very low risk tolerance (for false positives), such that we could focus on screening natural product libraries. We believe that this approach should work well for generally disrupting challenging macromolecular-protein targets, as long as the system under investigation is amenable to some of the biophysical validation methods described above. However, our choice of a very strict cutoff threshold raises some potential caveats. Namely, we may have generated several false negatives in the natural product screen, which might have been excellent inhibitors. This could be alleviated by employing cheaper, standard synthetic diversity lead libraries, and using a moderate cutoff threshold. Nevertheless, it is our belief that natural product macrocycles, such as NP-004255 provide a unique opportunity to target PNIs and PPIs. Their stereochemical complexity and pre-organized ring structures provides a functionality that is difficult to replicate in classical synthetic leads (Driggers et al., 2008). As evidenced by Hengel et al (Hengel et al., 2016), and the data provided in this chapter, the extended nature of the PNI binding site is complementary to the pre-organized macrocycle ligand. It is almost certainly the case that a significant contributing factor to its efficacy is reduction in the entropic penalty in protein binding, due to this pre-organization. While these features often provide excellent complementarity to protein targets, they are also associated with the, surprisingly, sufficient bioavailability of macrocycles. Interestingly, natural products, especially larger ones, are known to have complex conformational ensembles in solution, which may contribute to their unique properties. The nature of the workflow described in this chapter, and in Hengel et al., is not optimized to capture these natural product properties. This is largely because, although it employs MD simulations of the protein-ligand complex, it still relies in part on docking; for classic drug like compounds this should not be a problem, but for large macrocycles it would fail to capture true breadth their solution conformational ensembles. One possible solution to this is to use a free energy calculation that is amenable to medium throughput analysis, such as Extended Linear Response (ELR), which has been highly successful in explicitly capturing the contributions of interstitial waters to ligand binding (Whalen et al., 2013).
Acknowledgments
This work was supported by National Institutes of Health (R01-GM097373). The funders had no role in study design, data collection, analysis, decision to publish or preparation of the chapter.
References
- Chemical Computing Group Inc. Molecular Operating Environment. 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2013.08 ed. [Google Scholar]
- ALEXANDRE VARNEK AT. Chemoinformatics Approaches to Virtual Screening. Royal Society of Chemistry; 2008. [Google Scholar]
- CHANDRAMOULY G, MCDEVITT S, SULLIVAN K, KENT T, LUZ A, GLICKMAN JF, ANDRAKE M, SKORSKI T, POMERANTZ RT. Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem Biol. 2015;22:1491–1504. doi: 10.1016/j.chembiol.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE VEGA MJ, MARTIN-MARTINEZ M, GONZALEZ-MUNIZ R. Modulation of protein-protein interactions by stabilizing/mimicking protein secondary structure elements. Curr Top Med Chem. 2007;7:33–62. doi: 10.2174/156802607779318325. [DOI] [PubMed] [Google Scholar]
- DEAN SF, WHALEN KL, SPIES MA. Biosynthesis of a Novel Glutamate Racemase Containing a Site-Specific 7-Hydroxycoumarin Amino Acid: Enzyme-Ligand Promiscuity Revealed at the Atomistic Level. ACS Cent Sci. 2015;1:364–373. doi: 10.1021/acscentsci.5b00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DELONG ER, DELONG DM, CLARKE-PEARSON DL. Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics. 1988;44:837–45. [PubMed] [Google Scholar]
- DRIGGERS EM, HALE SP, LEE J, TERRETT NK. The exploration of macrocycles for drug discovery--an underexploited structural class. Nat Rev Drug Discov. 2008;7:608–24. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- EYRISCH S, HELMS V. Transient pockets on protein surfaces involved in protein-protein interaction. J Med Chem. 2007;50:3457–64. doi: 10.1021/jm070095g. [DOI] [PubMed] [Google Scholar]
- GRIMME JM, HONDA M, WRIGHT R, OKUNO Y, ROTHENBERG E, MAZIN AV, HA T, SPIES M. Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic Acids Res. 2010;38:2917–30. doi: 10.1093/nar/gkp1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIMME JM, SPIES M. FRET-based assays to monitor DNA binding and annealing by Rad52 recombination mediator protein. Methods Mol Biol. 2011;745:463–83. doi: 10.1007/978-1-61779-129-1_27. [DOI] [PubMed] [Google Scholar]
- HEAD M. Docking: a domesday report. Cambridge University Press; 2010. [Google Scholar]
- HENGEL SR, MALACARIA E, FOLLY DA SILVA CONSTANTINO L, BAIN FE, DIAZ A, KOCH BG, YU L, WU M, PICHIERRI P, SPIES MA, SPIES M. Small-molecule inhibitors identify the RAD52-ssDNA interaction as critical for recovery from replication stress and for survival of BRCA2 deficient cells. Elife. 2016;5:e14740. doi: 10.7554/eLife.14740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG N, SHOICHET BK, IRWIN JJ. Benchmarking sets for molecular docking. Journal of Medicinal Chemistry. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUGGINS DJ, MARSH M, PAYNE MC. Thermodynamic Properties of Water Molecules at a Protein-Protein Interaction Surface. J Chem Theory Comput. 2011;7:3514–3522. doi: 10.1021/ct200465z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAGAWA W, KURUMIZAKA H, ISHITANI R, FUKAI S, NUREKI O, SHIBATA T, YOKOYAMA S. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol Cell. 2002;10:359–71. doi: 10.1016/s1097-2765(02)00587-7. [DOI] [PubMed] [Google Scholar]
- KITCHEN DB, DECORNEZ H, FURR JR, BAJORATH J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3:935–49. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- KRIEGER E, DARDEN T, NABUURS SB, FINKELSTEIN A, VRIEND G. Making optimal use of empirical energy functions: force-field parameterization in crystal space. Proteins. 2004;57:678–83. doi: 10.1002/prot.20251. [DOI] [PubMed] [Google Scholar]
- KRIEGER E, KORAIMANN G, VRIEND G. Increasing the precision of comparative models with YASARA NOVA--a self-parameterizing force field. Proteins. 2002;47:393–402. doi: 10.1002/prot.10104. [DOI] [PubMed] [Google Scholar]
- LLOYD JA, FORGET AL, KNIGHT KL. Correlation of biochemical properties with the oligomeric state of human rad52 protein. J Biol Chem. 2002;277:46172–8. doi: 10.1074/jbc.M207262200. [DOI] [PubMed] [Google Scholar]
- LLOYD JA, MCGREW DA, KNIGHT KL. Identification of residues important for DNA binding in the full-length human Rad52 protein. J Mol Biol. 2005;345:239–49. doi: 10.1016/j.jmb.2004.10.065. [DOI] [PubMed] [Google Scholar]
- MAKLEY LN, GESTWICKI JE. Expanding the Number of ‘Druggable’ Targets: Non-Enzymes and Protein–Protein Interactions. Chemical Biology & Drug Design. 2013;81:22–32. doi: 10.1111/cbdd.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MALO N, HANLEY JA, CERQUOZZI S, PELLETIER J, NADON R. Statistical practice in high-throughput screening data analysis. Nature Biotechnology. 2006;24:167–175. doi: 10.1038/nbt1186. [DOI] [PubMed] [Google Scholar]
- METZ CE. Basic principles of ROC analysis. Semin Nucl Med. 1978;8:283–98. doi: 10.1016/s0001-2998(78)80014-2. [DOI] [PubMed] [Google Scholar]
- MORTENSEN UH, ERDENIZ N, FENG Q, ROTHSTEIN R. A molecular genetic dissection of the evolutionarily conserved N terminus of yeast Rad52. Genetics. 2002;161:549–62. doi: 10.1093/genetics/161.2.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAIM M, BHAT S, RANKIN KN, DENNIS S, CHOWDHURY SF, SIDDIQI I, DRABIK P, SULEA T, BAYLY CI, JAKALIAN A, PURISIMA EO. Solvated interaction energy (SIE) for scoring protein-ligand binding affinities. 1. Exploring the parameter space. J Chem Inf Model. 2007;47:122–33. doi: 10.1021/ci600406v. [DOI] [PubMed] [Google Scholar]
- SCOTT DE, BAYLY AR, ABELL C, SKIDMORE J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov. 2016;15:533–50. doi: 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- SINGLETON MR, WENTZELL LM, LIU Y, WEST SC, WIGLEY DB. Structure of the single-strand annealing domain of human RAD52 protein. Proc Natl Acad Sci U S A. 2002;99:13492–7. doi: 10.1073/pnas.212449899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPERANDIO O, MITEVA MA, SEGERS K, NICOLAES GA, VILLOUTREIX BO. Screening Outside the Catalytic Site: Inhibition of Macromolecular Inter-actions Through Structure-Based Virtual Ligand Screening Experiments. Open Biochem J. 2008;2:29–37. doi: 10.2174/1874091X00802010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEINBRECHER T, LABAHN A. Towards accurate free energy calculations in ligand protein-binding studies. Curr Med Chem. 2010;17:767–85. doi: 10.2174/092986710790514453. [DOI] [PubMed] [Google Scholar]
- SUBRAMANYAM S, JONES WT, SPIES M, SPIES MA. Contributions of the RAD51 N-terminal domain to BRCA2-RAD51 interaction. Nucleic Acids Res. 2013;41:9020–32. doi: 10.1093/nar/gkt691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TROTT O, OLSON AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VENTER JC, ADAMS MD, MYERS EW, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- WANG W, KOLLMAN PA. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J Mol Biol. 2000;303:567–82. doi: 10.1006/jmbi.2000.4057. [DOI] [PubMed] [Google Scholar]
- WHALEN KL, CHAU AC, SPIES MA. In silico optimization of a fragment-based hit yields biologically active, high-efficiency inhibitors for glutamate racemase. ChemMedChem. 2013;8:1681–9. doi: 10.1002/cmdc.201300271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHALEN KL, TUSSEY KB, BLANKE SR, SPIES MA. Nature of allosteric inhibition in glutamate racemase: discovery and characterization of a cryptic inhibitory pocket using atomistic MD simulations and pKa calculations. J Phys Chem B. 2011;115:3416–24. doi: 10.1021/jp201037t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAP JL, CHAUHAN J, JUNG KY, CHEN LJ, PROCHOWNIK EV, FLETCHER S. Small-molecule inhibitors of dimeric transcription factors: Antagonism of protein-protein and protein-DNA interactions. Medchemcomm. 2012;3:541–551. [Google Scholar]
- ZHANG JH, CHUNG TDY, OLDENBURG KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. Journal of Biomolecular Screening. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]