

Summary
Exhausted CD8 T cells (Tex) are immunotherapy targets in chronic infection and cancer, but a comprehensive assessment of Tex cell diversity in human disease is lacking. Here we developed a transcriptomic- and epigenetic-guided mass cytometry approach to define core exhaustion-specific genes and disease-induced changes in Tex cells in HIV and human cancer. Single-cell proteomic profiling identified 9 distinct Tex cell clusters using phenotypic, functional, transcription factor and inhibitory receptor co-expression patterns. An exhaustion severity metric was developed and integrated with high-dimensional phenotypes to define Tex cell clusters that were: present in healthy subjects; common across chronic infection and cancer or enriched in either disease; linked to disease severity; and changed with HIV therapy. Combinatorial patterns of immunotherapy targets on different Tex cell clusters were also defined. This approach and associated datasets present a resource for investigating human Tex cell biology, with implications for immune-monitoring and immunomodulation in chronic infections, autoimmunity and cancer.
Keywords: CD8 T cell, T cell exhaustion, mass cytometry, chronic infection, HIV, cancer immunology, lung cancer, systems immunology, immune checkpoint
ETOC BLURB
Exhausted T cells (Tex) have poor function in chronic infections and cancer but can be therapeutically re-invigorated. Bengsch et al. use genes modified epigenetically during exhaustion and high-dimensional CyTOF profiling to define Tex cell heterogeneity in humans with HIV or lung cancer, and link Tex cell features to disease progression and response to immunotherapy.
Introduction
Exhausted CD8 T cells (Tex cells) often develop during chronic infections and cancer and prevent optimal control of disease. They have functional defects, co-express multiple inhibitory receptors (IRs) and develop an altered transcriptional, epigenetic, metabolic and differentiation program (Wherry and Kurachi, 2015). Novel immunotherapies target IRs expressed by Tex cells such as PD-1 or CTLA-4 and are having dramatic effects in cancer patients with potential applications in other settings (Callahan et al., 2016). Tex cells have been implicated in the response to checkpoint blockade, but the underlying immunological mechanisms of therapeutic response or failure in humans remain poorly understood. Recent epigenetic studies in mice and humans indicate that Tex cells represent a unique T cell lineage, compared to functional effector T cells (TEFF) and memory T cells (TMEM) (Pauken et al., 2016; Philip et al., 2017; Sen et al., 2016). The Tex cells S epigenetic landscape changes relatively little after PD-1 pathway blockade mediated re-invigoration (Pauken et al., 2016), suggesting that Tex cells are a stable, distinct and disease relevant cell type.
Specific characteristics of Tex cells can vary in different disease settings (Wherry and Kurachi, 2015). It has been unclear whether the diverse descriptions of exhaustion represent technical differences (e.g. choice of IRs examined), conceptual differences (e.g., about the degree of dysfunction) or disease-related features of Tex cells. Identifying and monitoring changes of Tex cells in humans has been challenging since many molecules expressed by Tex cells are also identified in other T cell subsets or expressed by recently activated cells. Moreover, different subsets of Tex cells have been defined that include progenitor and more terminally exhausted populations. These Tex cell subsets can be identified by T-bet, Eomes, TCF-1 and PD-1 expression, and additional heterogeneity may exist based on follicular homing molecules such as CXCR5 (Blackburn et al., 2008; He et al., 2016; Im et al., 2016; Paley et al., 2012; Utzschneider et al., 2016; Wu et al., 2016). Tex cells have also been implicated in autoimmune diseases where transcriptional signatures of Tex cells are enriched in patients with less severe disease (McKinney et al., 2015). These observations suggest the possibility of a core program of exhaustion that is tuned differently to the precise disease setting. An approach that identifies and interrogates the heterogeneity of Tex cells and relates this information to disease status could provide new insights and opportunities for intervention based on manipulating Tex cells.
In this study, we developed a systems immunology approach to identify and deeply interrogate Tex cells in human disease. At the center of this approach is the development of a core exhaustion signature that integrates transcriptomic and epigenomic profiling of Tex cells in the chronic LCMV system and then distills this mouse epi-genomic signature into a robust and stable Tex cell gene expression signature that translates to human biology in HIV disease and human lung cancer. Deep mass cytometry profiling combined with bioinformatic analysis of the high-dimensional data allowed the identification of a set of multi-parametric exhaustion phenotypes. These data then distinguished 9-12 Tex cell clusters as well as subsets reflecting canonical T cell differentiation states, such as TN, TEFF, TMEM, and tissue resident memory T cells (TRM). Using this framework we defined an “exhaustion score” for each cluster based on functional features. Linking changes in these Tex cell clusters to disease parameters in HIV and lung cancer defined subtypes of Tex cells more prominently impacted by antigen (viral load) versus environment (i.e. low CD4/CD8 ratio, CD4 counts) and revealed a conserved Tex cell program, but also changes in the diversity of exhausted subpopulations in HIV compared to lung cancer. These data now provide a resource to identify Tex cell populations with distinct patterns of expression of IRs, cytotoxic molecules and transcription factors that may be relevant for designing checkpoint blockade therapies, but they also suggest possible relationships between Tex cell subtypes. These data provide a novel resource that should aid future studies and improve our understanding of exhaustion in chronic infection, autoimmunity and cancer.
Results
Identification of genes with unique expression patterns in Tex cells
Exhausted CD8 T cells (Tex cells) undergo major transcriptional changes that distinguish them from naïve (TN), effector (TEFF) and memory (TMEM) T cells (Figure 1A) (Wherry and Kurachi, 2015). Functional and transcriptional features of exhaustion arise after ~2 weeks of chronic infection (Angelosanto et al., 2012; Doering et al., 2012). We hypothesized that a core signature of genes specifically regulated in Tex cells during chronic LCMV infection could be used to identify and monitor Tex cells in other settings. We aimed to identify a focused set of highly exhaustion-biased genes, validate it against data from other settings of exhaustion and then use epigenetic information for individual genes to further refine the signature. This signature could then enable the development of a comprehensive single-cell protein-based method for interrogating the biology of human Tex cells by mass cytometry. Thus, we first identified genes that specifically displayed increased- or decreased expression in virus-specific Tex cells during chronic LCMV infection compared to TN, TEFF, and TMEM in acute resolving LCMV Armstrong infection (Figures 1A, 1B). These criteria identified genes highly biased to Tex cells compared to activation-related genes found in TEFF, such as Cd38, but not Havcr1 encoding Tim-3. We validated the selection of genes by Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) comparing Tex cells isolated after 30d of clone 13 infection to TMEM, TEFF and TN (Figure 1C). We also investigated whether this signature would enrich in subsets of Tex cells (Blackburn et al., 2008; Im et al., 2016; Paley et al., 2012). GSEA showed strong enrichment in signatures of the more terminally exhausted Tex cell subset expressing high levels of PD-1 or Tim-3 compared to the progenitor subset of Tex cells expressing lower levels of PD-1 or CXCR5 (Figure 1D) (Blackburn et al., 2008; Im et al., 2016). However, the genes selected also enriched in the less terminal subsets of Tex cells if these cells were compared to TEFF rather than terminal Tex cells (data not shown) suggesting high sensitivity of this signature. Moreover, this exhaustion signature strongly enriched in tumor infiltrating lymphocytes (TIL) from melanoma patients versus peripheral blood and in HIV-specific T cells from HIV progressor patients versus elite controllers (Figure 1E), in agreement with previous reports (Baitsch et al., 2011; Quigley et al., 2010). We also noted that a number of exhaustion genes were enriched in elite controllers indicating that the signature also included genes that might be useful for discriminating less dysfunctional exhaustion states (Figure 1E). Extending these analyses to other transcriptomic datasets also identified more exhausted human T cell populations in silico such as CD39+ HCV-specific CD8 T cells (Gupta et al., 2015) (Figure 1F, Supplementary table 1). Leading edge analysis identified genes strongly contributing to the enrichment, including ENTPD1 (encoding CD39), CTLA4, PDCD1 and CD38 that were common to enrichment for TILs from melanoma and chronic HCV infection (Figures 1G, H). In sum, these analyses identified a transcriptomic signature of Tex cells in chronic LCMV infection that was shared across species and disease types. Moreover, the patterns of enrichment suggested that elements of this signature might be capable of distinguishing different features of exhaustion in distinct human diseases.
Figure 1. Mouse-derived transcriptomic exhaustion signature translates to human exhaustion.
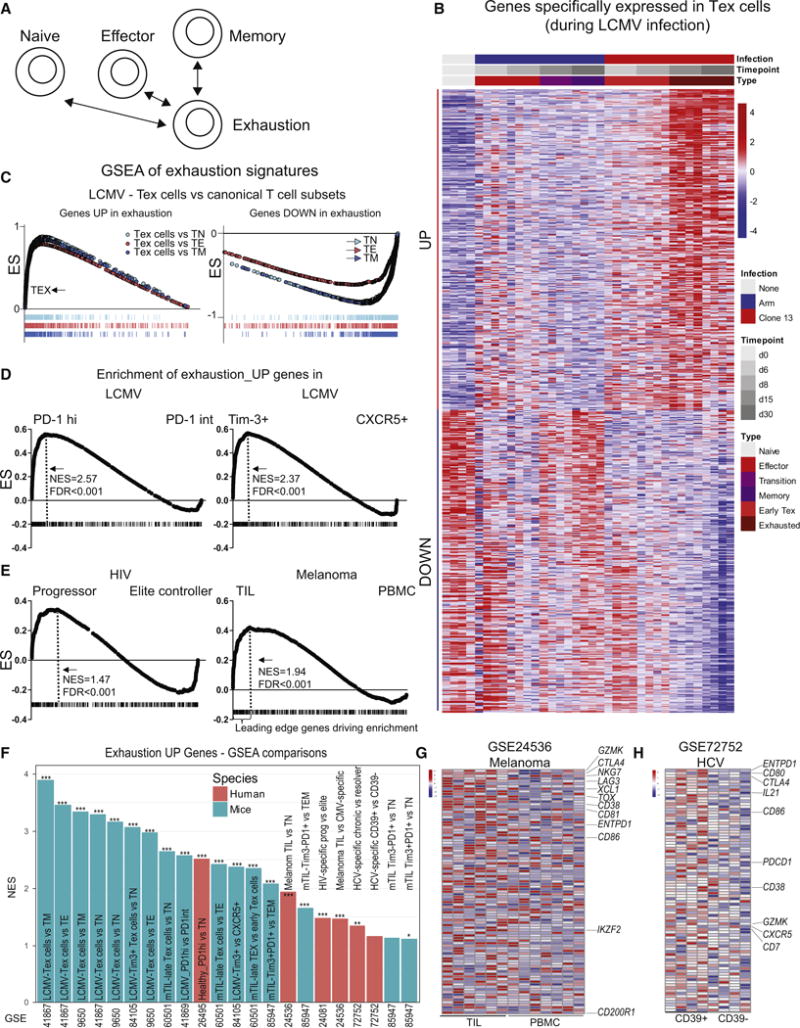
(A) Genes transcriptionally increased or decreased in virus-specific CD8 T cells from d15 and d30 of LCMV clone 13 infection (Tex cells) were compared to TN, TEFF, TMEM from LCMV Arm infection (GSE41867) and exhaustion-specific genes defined based on moderated Bayesian statistics. (B) Heatmap of transcriptomic data (see also Supplementary Table 2). (C) Exhaustion genesets validated for enrichment in Tex cells versus TN, TEFF, or TMEM in LCMV infection via GSEA. (D) Enrichment of gene signature was analyzed in transcriptomic data from Tex cell subpopulations (PD-1Hi versus PD-1Int, Tim-3+ versus CXCR5+) from LCMV clone 13 infection (GSE41869; GSE84105) or (E) human HIV-specific CD8 T cells from HIV elite controller versus progressor patients (GSE24081) or PBMC versus TIL from melanoma patients (GSE 24536). FDR and normalized enrichment score (NES) are indicated. Dashed lines in D and E indicate leading edge genes driving the NES. (F) The exhaustion gene signature was analyzed in multiple mouse and human datasets of Tex cell populations (detailed in Supplementary Table 1) and NES plotted for each comparison. *** FDR<0.001, ** <0.01, * <0.05. (G and H) Heatmap for leading edge genes driving enrichment for genes with increased expression in exhaustion in melanoma (PBMC versus TIL) (GSE 24536), and HCV (CD39+ versus CD39− cells) (GSE 72752).
Uniquely regulated Tex cell genes identified by epigenetic changes
Epigenetic patterns may be more faithful indicators of cell identity than gene expression. We hypothesized that genes uniquely regulated in Tex cells that also displayed specific epigenetic changes (i.e. at open chromatin regions (OCR: e.g. enhancers) would provide a more robust and stable signature of exhaustion. To test this hypothesis, we identified enhancers in Tex cells from chronic LCMV infection compared to TN, TEFF and TMEM using epigenomic profiling by Assay for Transposase-Accessible Chromatin with high throughput sequencing (ATAC-Seq) in published datasets (Pauken et al., 2016; Sen et al., 2016).
Starting with the differentially expressed genes identified in Figure 1, 313 and 182 exhaustion specific genes (with increased or decreased expression in Tex cells, respectively) also contained associated Tex cell -related epigenetic (e.g. enhancer) changes (Figures 2A, 2B, Supplementary Table 2). These genes included those with more accessible OCR close to genes encoding IRs (e.g., Pdcd1, Tigit, Ctla4), ectoenzymes implicated in metabolic regulation (e.g. Cd38, Entpd1), chemokines and cytokines (e.g. Xcl1) and transcription factors (e.g. Eomes, Ikzf2, Tox) (Figure 2B and Supplementary Table 2). Genes with reduced accessibility of OCR linked to decreased expression in Tex cells (e.g., Ccr7, Il7r, Nt5e, Tcf7, Lef1) were also identified (Supplementary Table 2). Tex cell genes with associated OCR changes contributed significantly more frequently to the enrichments observed in the comparisons of Tex cell populations across diseases in Figure 1 compared to genes without a Tex cell-related epigenetic change (Figures 2C). This feature was manifest by significantly higher GSEA leading edge contributions (Figures 2C, Supplementary Table 3), but different Tex cell genes often contributed to the leading edge depending on the comparison (Figures 2D, 2E). GO term analysis indicated that “epigenomically selected” exhaustion genes with increased expression and enhancer accessibility were enriched in immune activation and regulation of phosphorylation pathways, whereas genes with decreased expression and reduced enhancer accessibility enriched for metabolic processes, among others (Figure 2F). Thus, key genes distinguishing Tex cells from canonical T cell subsets are revealed by a combination of unique transcriptomic expression patterns and associated epigenetic changes. These uniquely regulated genes are strong candidates for biomarkers of exhaustion across diverse disease types.
Figure 2. Uniquely regulated genes in exhaustion identified by epigenetic accessibility.
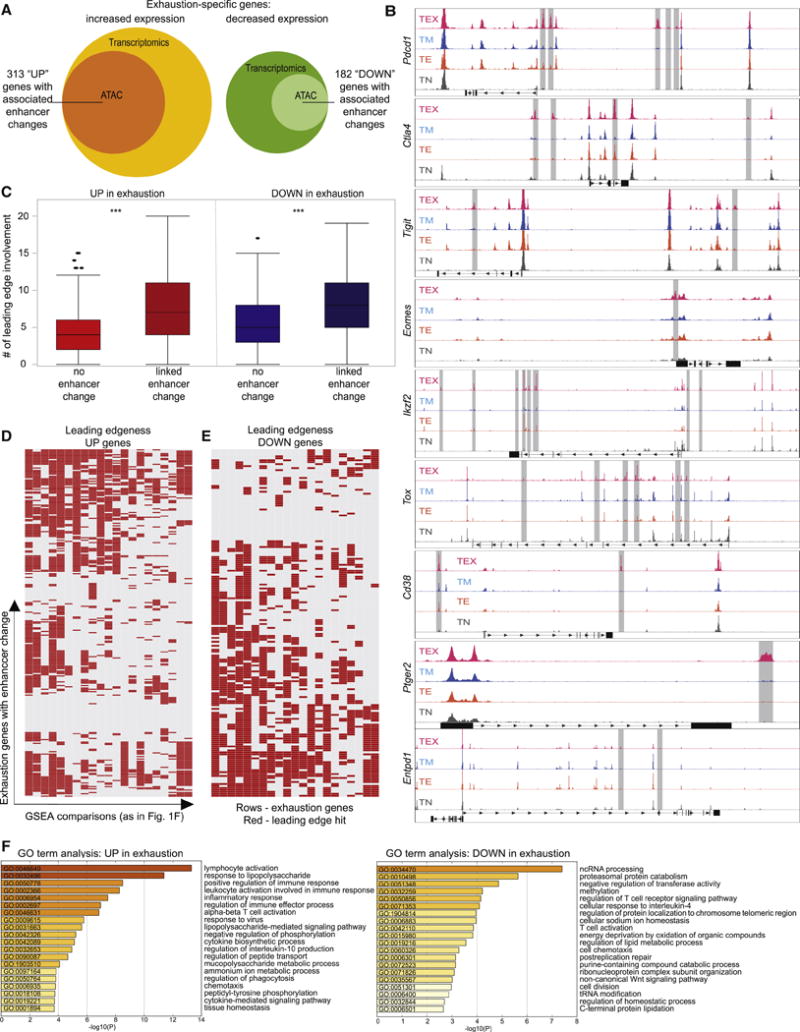
Genes specifically regulated in Tex cells from Figure 1, were analyzed for epigenetic changes in ATAC-seq datasets from LCMV infection (GSE86797, GSE87646). Genes are detailed in Supplementary Table 2. (A) The fraction of transcriptionally identified genes with associated epigenetic changes (increased accessibility of open chromatin regions (OCR) near exhaustion genes for UP-, decreased accessibility of OCRs in the vicinity of DOWN-exhaustion genes) is shown. (B) Exemplary ATAC-seq tracks indicating increased OCR (highlighted by grey bars) near exhaustion genes from GSE86797. (C) Exhaustion genes were analyzed for associated OCR changes and the role in driving the enrichment (“leading edge”) in the comparisons of Tex cells versus other T cell datasets, as detailed in Supplementary Tables 1 and 3. Genes with an associated OCR change displayed higher leading edge involvement. *** p<0.001. The leading edge contribution of exhaustion signature genes with an associated OCR change is shown as a binary heatmap for genes up- (D) and down-regulated in exhaustion (E) (rows indicate genes, columns individual GSEA comparisons, red denoting leading edge contribution (for details, see Supplementary Table 3). (F) GO Term analysis of the exhaustion-specific genesets with associated OCR changes. The 20 GO terms with the lowest p values are shown.
CD8 T cells expressing exhaustion genes are biomarkers of HIV disease progression
Tex cells are a hallmark of chronic HIV infection. We hypothesized that converting the population-based epigenomic exhaustion signature defined above to a single-cell profiling approach could provide insights into the diversity of Tex cells LS in HIV disease. We thus constructed a mass cytometry panel that integrated 16+ epigenomically-selected exhaustion-related gene products together with other T cell markers for defining lineage and other differentiation states (Supplementary Table 4). The genes selected for further analysis by CyTOF were chosen, in part, based on the availability of high quality antibodies for cytometry analysis. Other epigenomically-selected genes are also available for future analyses including ADAM19, BHLHE41, DUSP4, GLP1R, GPR65, GPR155, IFI27, IFI44, PRDM1, PTPN13, RGS16, and SLC22A15 (Supplementary Table 2 and 3). The exhaustion genes encoding proteins selected for this CyTOF panel had a high leading edge contribution to the enrichment of the exhaustion signature in different diseases (Figures 3A, 3B). To interrogate the discriminatory potential in single-cell datasets and to test how the 16 exhaustion markers selected for the CyTOF panel compared to the larger epigenomically selected list, we used Gene Set Variation Analysis (GSVA) of published CD8 T cell single-cell transcriptomic dataset from human melanoma tumor-infiltrating lymphocytes (TIL) (Tirosh et al., 2016). Both the total epigenomically selected genes, and the subset selected for CyTOF discriminated considerable variation in the scRNA-seq data, with similar discriminating potential (Supplementary Figure 1), indicating that this subset of genes was emblematic of key features of exhaustion.
Figure 3. Mass cytometry analysis of exhaustion molecule expression.
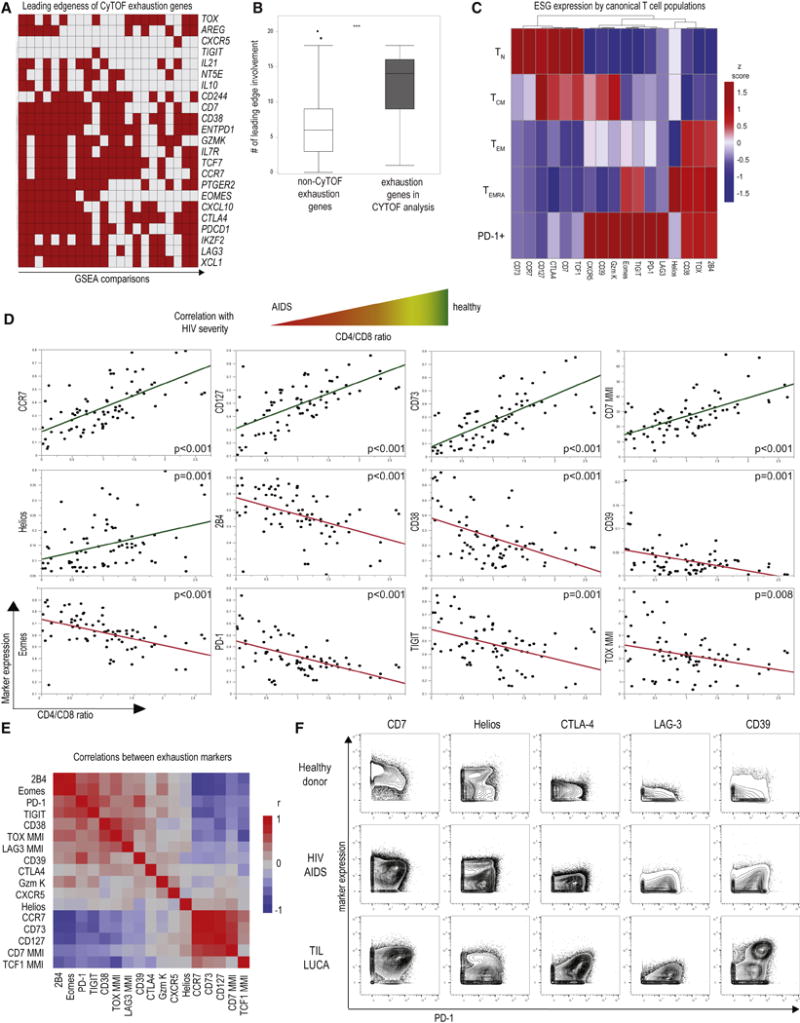
(A) Exhaustion genesets defined in Figure 1 and 2 were used to design an exhaustion-focused mass cytometry panel. The leading edge contribution of genes chosen for CyTOF is shown; rows indicate genes, columns individual GSEA comparisons. See also Supplementary Table 4. Cytokines and chemokines were analyzed using a dedicated panel (see Supplementary Table 6) (B) Genes selected for CyTOF had significantly higher leading edge contribution in the GSEA analyses of Tex cells compared to the remaining exhaustion genes (*** p <0.001) and showed similar ability to discriminate Tex cells in single-cell transcriptomic data (see Supplementary Figure 1). (C) Exhaustion markers were analyzed on canonical CD8 T cell populations (TN, TCM, TEM, TEMRA) and total PD-1+ CD8 T cells in HC and patients with HIV and lung cancer. Heatmap depicts exhaustion marker expression by median metal intensity (MMI) on concatenated CD8 T cell data from PBMC (n=35; see Supplementary Table 7). (D) Linear regression analysis versus CD4/CD8 ratio was performed for marker expression in patients with HIV infection and HC using percent positive or MMI as indicated. Each dot represents an individual patient CD8 T cells. (n=75 samples from 48 HIV patients and HC were analyzed, higher sample number indicates longitudinal samples when available, for details see Supplementary Table 7). Green - positive correlation; red - negative correlation. Similar results were obtained in a repeat analysis on a different mass cytometer (Supplementary Figure 2). (E) These data were further analyzed for cross-correlation of exhaustion marker expression estimated by pairwise method (see also Supplementary Figure 3). (F) The expression of indicated exhaustion markers on CD8 T cells is plotted versus PD-1 in a representative HC, an untreated HIV patient with a CD4/CD8 ratio of 0.06 typical of AIDS, and tumor-infiltrating lymphocytes isolated from a lung cancer patient.
We next applied this CyTOF panel to analysis of PBMC from healthy control subjects (HC) and patients with active HIV infection as well as HIV patients on anti-retroviral therapy (ART) (Supplementary Table 5). In subsequent analyses, we also included samples from lung cancer patients including PBMC, macroscopically uninvolved lung tissue and TIL. We first examined classically defined TN, TEFF, effector memory (TEM), central memory (TCM) and terminally differentiated effector memory RA (TEMRA) as well as PD-1+ CD8 T cells for the expression of epigenomically-selected exhaustion markers (Figures 3C, 3D). PD-1+ cells expressed more of these markers than any of the other phenotypically defined CD8 T cell populations, whereas TN and TCM expressed molecules linked to decreased expression by Tex cells (Figure 3C).
We next examined the correlation between expression of individual exhaustion molecules with the CD4/CD8 ratio, an established metric of severity of HIV disease (Figure 3D). Molecules predicted to be decreased in Tex cells correlated with health, mild disease, and a higher CD4/CD8 ratio (e.g. CCR7, CD73, CD127), whereas molecules predicted to be increased in Tex cells correlated with low CD4/CD8 ratios indicating advanced disease (e.g. 2B4, CD38, CD39, Eomes, PD-1, TIGIT, TOX) (Figure 3D; Supplementary Figure 2). Correlation matrix analysis identified sets of highly co-regulated exhaustion-related molecules in HIV, such as PD-1, Eomes, 2B4, TIGIT and CD38 (Figure 3E, Supplementary Figure 3). Several of these molecules, (i.e. CD38 and PD-1) are known indicators of immune activation and exhaustion in progressive HIV infection, though others such as TOX are less well understood. Some markers predicted to be highly expressed in exhaustion did, however, only display trends towards enrichment in severe HIV (LAG-3, CTLA-4), or were even associated with less severe disease (CD7, Helios), suggesting more complex relationships captured poorly by the analysis of single markers. Indeed, for individual patients with HIV-AIDS or lung cancer more extreme Tex cell phenotypes existed that were identified, for example, by co-expression of CD7 and PD-1 (Figure 3F). Moreover, other molecules that displayed no obvious, negative (CTLA-4, LAG-3, CD39) or a complex (Helios) co-expression pattern with PD-1 in HC became positively associated with PD-1 in disease pointing towards the need for high-dimensional analysis of Tex cells.
A high-dimensional single-cell map of exhaustion reveals distinct locations of virus-specific T cells and TILs in the Tex cell landscape
To visualize the complex Tex cell phenotypes defined by this CyTOF panel, we first used a tSNE-based dimensionality reduction approach integrating the information from exhaustion markers expression of non-naïve CD8 T cells into an “exhaustion map” (Figure 4A). Samples from different batches were quality controlled and displayed similar results in the high-dimensional analysis (Supplementary Figure 4). Cells with closely related high-dimensional phenotypes localized in neighboring areas of the map (Figure 4B). For example, PD-1 expression largely overlapped with Eomes, multiple other IRs and a paucity of CD127 expression (Figure 4B). Other regions displayed expression of other Tex cell genes (e.g. CD38, CD39, Helios and TOX) and/or different patterns of PD-1 co-expression (Figure 4B). This exhaustion map was used to interrogate differences in exhaustion states across patients and diseases (Figure 4C). Tetramer+ virus-specific CD8 T cells targeting CMV- and influenza virus (FLU) epitopes localized to distinct areas of the exhaustion map compared to HIV-specific CD8 T cells, confirming the ability of this approach to distinguish known differentiation patterns of virus-specific CD8 T cells (Figure 4D). Moreover, the HIV-specific CD8 T cells, but not FLU-specific and few CMV-specific populations overlapped with the PD-1+ part of the exhaustion map (Figures 4B, 4D). Thus, examination of virus-specific CD8 T cells validated the exhaustion map in the ability distinguish HIV-, CMV- and FLU-specific populations.
Figure 4. An exhaustion map allows comparison of Tex cells across HIV and lung cancer.
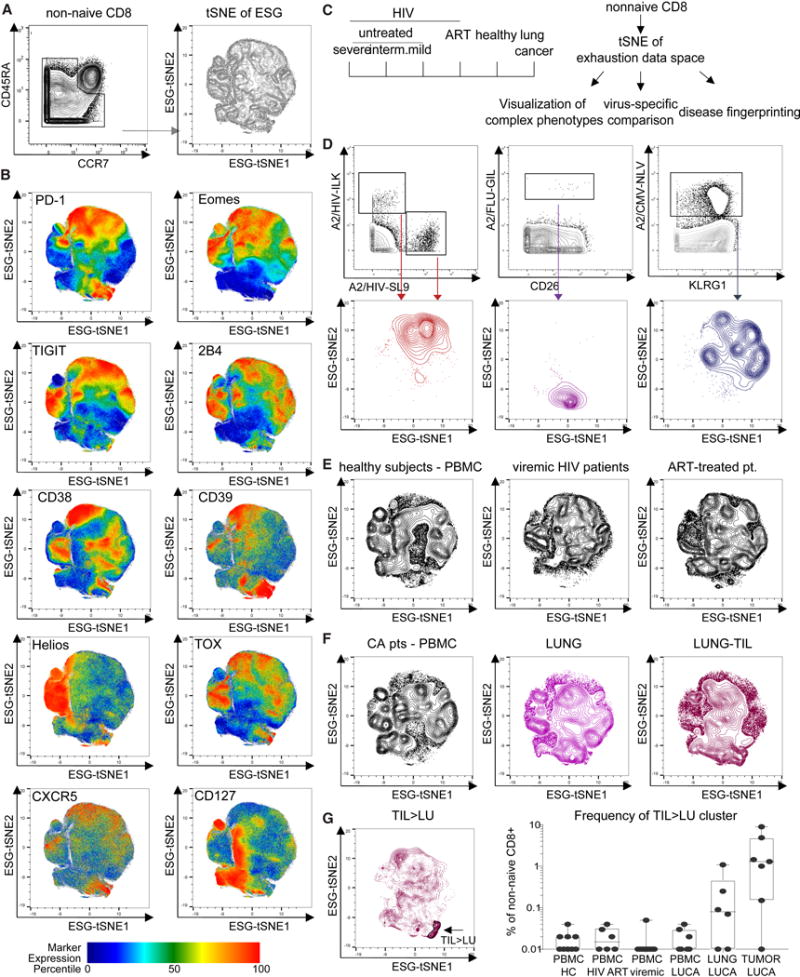
(A) An exhaustion map was generated by tSNE-based dimensionality reduction on 48 samples (see Supplementary Table 7) using information about expression of 16 exhaustion markers (see Supplementary Table 4) on nonnaive (CD45RA−CCR7−) CD8 T cells. (B) Expression of individual molecules (upper left corner of each panel) on the exhaustion map is visualized (color based on percentile of marker expression). (C) Schematic: The exhaustion map was generated for HIV patients with varying severity of untreated disease based on CD4/CD8 ratio and ART-treated patients and compared to HC and patients with lung cancer. (D) HIV-, FLU-, and CMV-specific CD8 T cells identified by tetramer staining were visualized on the exhaustion map. (E) Total CD8 T cells from HC and viremic and ART-treated HIV+ patients and from (F) lung cancer patient samples were mapped to the exhaustion landscape: PBMC (left), macroscopically uninvolved lung tissue (middle) or tumor-infiltrating lymphocytes (right). (G) Differential overlay of TILs compared to CD8 T cells from uninvolved lung on the exhaustion map. A TIL>LU cluster was gated (gate indicated by the arrow) and validated on a per-sample basis (right).
We then examined all non-naïve CD8 T cells from HC and HIV patients on and off therapy. Concatenated data shown in Figure 4E revealed distinct distributions of T cell populations in these cohorts, pointing to the possibility of using such “exhaustion fingerprints” for dissecting disease states. Although Tex cells have been reported in viral infections as well as cancer, it remains unclear whether there are common versus distinct features of Tex cells in different diseases. To address this question we next examined TIL isolated from lung cancer patients and T cells isolated from macroscopically unaffected adjacent tissue (Figure 4F). TIL mapped to regions with some overlap with HIV-specific T cells, but also displayed distinct features (Figure 4F). However, the lung tissue microenvironment might contribute to the TIL signature. After removal of the phenotypic signature of cells from the uninvolved lung tissue, TIL-enriched features became apparent that included regions with partial overlap with HIV-specific CD8 T cells and co-expression of PD-1, other IRs and Eomes (top of exhaustion map) but also other regions of the exhaustion landscape highly enriched in TIL with PD-1 and CD39 co-expression (Figures 4B, 4F, 4G). In sum, high-dimensional profiling of Tex cells identified distinct features of the differentiation landscape for HIV-specific CD8 T cells and TILs.
High-dimensional Tex cell cluster dynamics in HIV infection
We hypothesized that additional insights into these high-dimensional data and disease relevance could be achieved by a non-redundant high-dimensional analytical approach using phenograph (Figure 5A) (Levine et al., 2015) that enables high stability of cluster identification without downsampling or dimensionality-reduction (Melchiotti et al., 2017). Phenograph analysis identified 25 clusters based on expression of exhaustion markers (30 clusters were computed, but clusters c14, c20, c22, c24, c30 contained few events and were excluded from further analyses; see STAR Methods). Some clusters were identified that phenotypically represented TN and TCM (clusters such as c13, c15 or c21) or populations with features of TEM (e.g. clusters c10 and c11) or TEMRA-like cells (e.g., clusters c10, c9, c23) (Figures 5B, 5C). In contrast, the cluster contribution to PD-1+ CD8 T cells showed more diversity and no single cluster contributed to more than 15% of the population (Figure 5B), suggesting a high resolution of this approach for Tex cell subset discrimination. Approximately 9-12 clusters contained putative Tex cells based on co-expression of 3 or more (up to 9) IRs (c1, c2, c3, c4, c9, c16, c18, c19, c27, c28, c29), whereas 9 clusters were associated with TN and/or TCM phenotypes and contained cells with <3 IRs. TEM and TEMRA phenotype cells were contained in 3-8 distinct clusters. Notably, 3 of these clusters contained cells with <3 IRs, but the other clusters containing classically defined TEM and TEMRA also contained clusters expressing IRs (Figure 5C). This observation likely reflects the fact that phenotypically exhausted cells are mostly CD27+CD45RA− (Bengsch et al., 2010; Huang et al., 2017) and fall into the CD8 T cell subset defined by the classic definition of TEM (CCR7−CD45RA−). Thus, the high dimensional approach is necessary to parse these different CD8 T cell populations.
Figure 5. High-dimensional clustering identifies Tex cell phenotypes linked to HIV disease progression.
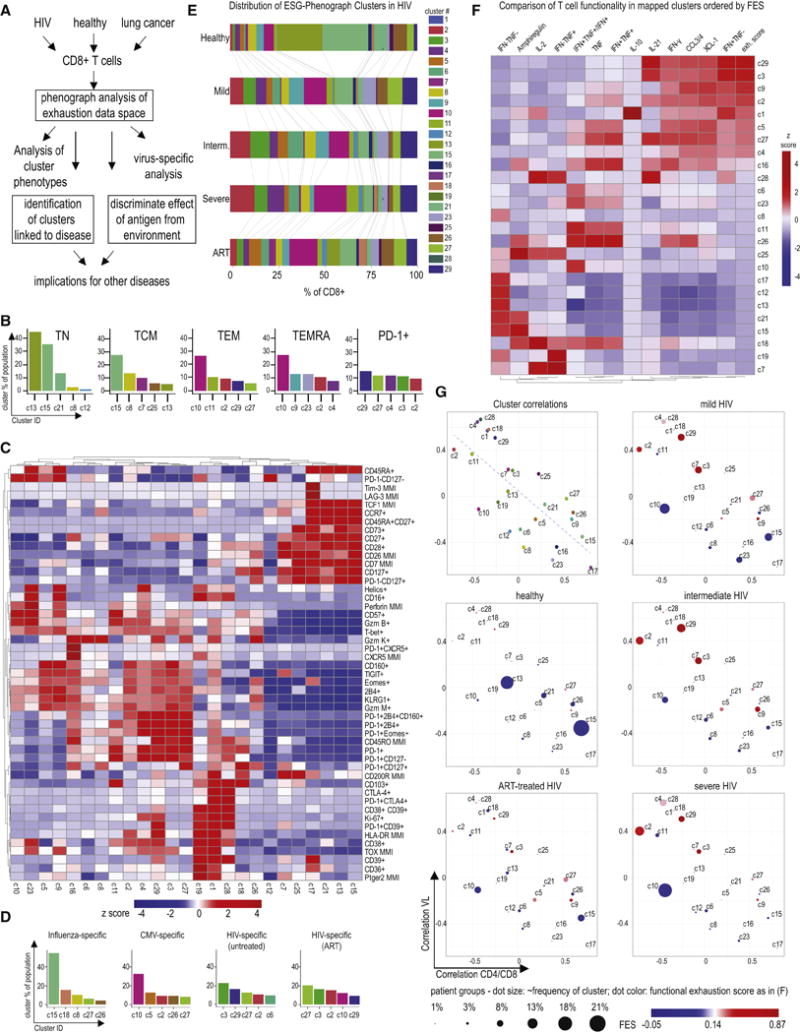
(A) Schematic of the pipeline for high-dimensional CD8 Tex cellcluster identification by phenograph and assessment in disease (B) Phenograph analysis of Tex cell markers was performed on live singlet CD45+CD3+CD8+ T cells (n=48; see Supplementary Table 7). Canonical CD8 T cell populations and total PD-1+ T cells were analyzed for their composition based on the phenograph analysis. The top 5 phenograph clusters within TN, TCM, TEM, TEMRA and PD-1+ CD8 T cells are shown. (C) Phenograph clusters were tested for expression of T cell markers using manual gating. Heatmap indicates expression of markers or marker combinations (using (+) or (−), as in PD-1+CD39+) or MMI (e.g., TOX). Row- and column-based clustering was performed using Pearson’s correlation. The heatmap coloring reflects z scores after row normalization, as indicated. (D) The contribution of phenograph clusters to virus-specific T cell responses from HIV patients and HC detected via tetramer staining was analyzed, the top 5 clusters are shown (n=24 tetramer responses; CMV n=4, FLU n=5, HIV n=15). Changes in phenograph cluster composition of HIV-specific T cells on or off antiretroviral therapy (ART) are displayed. (E) The distribution of phenograph clusters in HC and HIV patients (total n= 25) with differing disease states (CD4/CD8 ratio for viremic “Severe”: <0.2, “Intermediate”: 0.2-0.5, “Mild”: >0.5) is shown. The coloring reflects cluster assignment. The mean frequency of each cluster for each patient population is depicted by the size of the corresponding bar. (F) Viremic HIV and control samples were stimulated with PMA/Ionomycin and analyzed for cytokine expression by cluster mapping using phenograph classify function and the scaffold parameters detailed in Supplementary Table 6. Heatmap indicates gated expression of markers or marker combinations and the functional exhaustion score (FES) (see STAR Methods). Column-based clustering using Pearson’s correlation metric was performed. Rows are arranged by FES. Values displayed are column normalized. (G) The correlation of each phenograph cluster frequency with key parameters of HIV disease progression in viremic HIV patients (i.e. CD4/CD8 ratio and viral load) was plotted (upper left panel). This coordinate system displays the relative frequency of each cluster in HC, HIV patients with untreated disease and patients on ART therapy (remaining panels). The dot size corresponds linearly to cluster relative abundance, the color corresponds to the FES.
Applying this approach to virus-specific CD8 T cells revealed distinct cluster distribution patterns for HIV-, FLU-, and CMV-specific CD8 T cells (Figure 5D). Cluster c10 that contained cells with high expression of T-bet, Granzyme B, Perforin and CD57, a phenotype often associated with TEMRA and/or T cell senescence was strongly enriched in CMV-specific CD8 T cells (Figure 5D). In contrast, cluster c15 with hallmarks of TCM was enriched in FLU-specific CD8 T cells from the blood (Figures 5B, 5C, 5D) whereas FLU-specific CD8 T cells from lungs were enriched in clusters c18 and c28 that expressed CD103, a molecule often expressed by resident memory T cells (TRM) (Supplementary Figure 5) (Schenkel and Masopust, 2014). HIV-specific CD8 T cells enriched in clusters with high expression of exhaustion markers, and the distribution of these Tex cell clusters was altered in ART-treated HIV patients (Figures 5C, 5D). Notably, many cluster composition changes found in HIV-specific CD8+ T cells, such as the increase of c27 and decrease of c29 and c3 in ART-treated patients, were also observed on the global CD8 level (Figure 5E).
T cell exhaustion was originally defined by altered and often poor function (Zajac et al., 1998). However, Tex cells are not functionally inert. Rather, they are characterized by reduced IFN-γ production and a poor ability to simultaneously produce IFN-γ and TNF, lack of IL-2 and reduced cytotoxicity but often retain the ability to make some IFN-γ and also robustly produce chemokines (Betts et al., 2006; Crawford et al., 2011; Fuller and Zajac, 2003; Wherry et al., 2003). Our transcriptomic and epigenomic profiling also indicated up-regulation of the chemokines CCL3/4, XCL1 and cytokines IL10, IL-21 and Amphiregulin in Tex cells (Supplementary Table 2). It remained unclear whether the high dimensional clusters identified by phenotype also reflected functional characteristics of exhaustion. We thus interrogated functional features of exhaustion and their relationship to the clusters determined above by stimulating CD8 T cells from viremic HIV patients and examined functionality (Figure 3A, Supplementary Table 2) using a panel built on the framework of 13 phenotypic exhaustion markers (Supplementary Table 6). Mapping chemokine and cytokine production to phenotypic clusters identified distinct cluster function (Figure 5F). Expression of CCL3/4, XCL1, and IL-21 was identified in clusters such as c2, c16 and c29 with other features of exhaustion. In contrast, Amphiregulin, stained primarily in clusters with naïve or memory phenotypes (e.g. c13, c15, c17, c21; Figure 5F) rather than Tex cell clusters, though the reasons for this disparity between the genomic and protein data are currently unclear. Cells that retained IFN-γ production but lost TNF production (i.e. reduced polyfunctionality) were prominently associated with the clusters c3, c16, c29 that also co-expressed many IRs (Figures 5C, 5F) consistent with exhaustion. To enable comparisons, we developed a functional “exhaustion score” (FES) that increased for functional hallmarks of exhaustion (e.g. loss of TNF or IL-2 production, upregulation of chemokines) and decreased with the presence of effector or memory functionality (e.g. IL-2 production or IFN-γ and TNF coproduction) (see STAR Methods). The highest FES was observed for cluster c29 that phenotypically resembled highly exhausted T cells based on the co-expression of 6-7 IRs and other features of Tex cells (Figures 5C, 5F). This analysis identified 9 clusters with high FES that also mapped to clusters with high IR co-expression, such as the two clusters with the highest FES (“top 2”, c3, c29). However, 3 clusters (c18, c19, c28) that co-expressed 3-4 IRs had intermediate to low FES. As expected, TCM, TEM and TEMRA enriched for clusters with low FES (Figure 5F). Notably, even HC had detectable frequencies of Tex cell clusters including especially those with lower FES such as c5, c9, c16 and c27 (Figures 5D, 5E).
We next asked how these clusters changed with HIV disease severity and after control of viral replication by ART. Tex cell clusters such as c2 and c29 were expanded in advanced HIV infection, but were decreased in ART therapy (Figure 5E). Clusters c1, c2, c4, c11, c18, c28 and c29 were positively associated with both high viral load and low CD4/CD8 ratio (Figure 5G), left upper panel). Other clusters including the Tex cell clusters c5, c9 and c27 as well as TN/TCM clusters (e.g., c15 and c17) were associated with low viral load and higher CD4/CD8 ratio. In contrast, clusters such as c10, a cluster with characteristics of TEM and TEMRA and low FES and abundant in CMV-specific T cells (Figure 5D), were more strongly associated with low CD4/CD8 ratio than changes in viral load, and c10 in particular, was expanded in HIV infection suggesting a bystander effect (Figure 5G). Many Tex cell clusters were linked to severe HIV disease (e.g., c29, c2, c3, c4), displayed co-expression of IRs (e.g. PD-1, 2B4, CD160, and TIGIT) and high Eomes, a phenotype consistent with severe exhaustion (Paley et al., 2012) (Figures 5C, 5G). Interestingly, some clusters were enriched in mild HIV, expressed molecules consistent with progenitor Tex cells (e.g., CD127, some TCF1: c16, c27) in addition to IRs and other exhaustion markers, and c16 included CXCR5+ cells (Figures 5C, 5G). A link to mild disease was also observed for a cluster with low PD-1, but high 2B4, CD160 and TIGIT and high expression of cytotoxic molecules (c9), suggesting preserved cytotoxicity potential and features of exhaustion in the absence of high PD-1. Together, these results point to multiple subtypes of Tex cells differentially linked to HIV disease progression or therapy and identified features of Tex cells and other T cell subsets that are preferentially associated with changes in viral load, immune dysregulation (captured by CD4/CD8 ratio) and response to ART.
Distinct functional, phenotypic and transcriptional features of Tex cell clusters
To test whether these analyses could distinguish Tex cells from TEFF, we generated TEFF in vitro starting with total PBMC or sorted TN, TCM, TEM, or TEMRA and then examined the functional and phenotypic profile of the resulting TEFF. TEFF had high polyfunctionality, IFN-γ and TNF coproduction, and a low FES score (Figure 6A). In this analysis, CD39, LAG-3, Helios and CTLA-4 were higher on activated TEFF, whereas Tex cell clusters displayed higher expression of Eomes, TOX, 2B4 and TIGIT, among other molecules (Figure 6B). This high-dimensional approach clearly distinguished Tex cell from TEFF, a distinction that has been challenging using other approaches based on individual markers. As expected, Tex cell clusters did not represent typical TN, TCM, TEM, or TEMRA populations, instead, 7 of the top 9 clusters with highest FES showed high PD-1 expression (Figure 6C). To test how the FES related to the high-dimensional phenotypes, we plotted the phenograph-derived clusters using tSNE (Supplementary Figure 6), and projected the FES onto this simplified cluster map (Figure 6D). Clusters with positive exhaustion scores fell in close proximity (Figure 6D). Analysis of the transcription factor expression patterns in Tex cell clusters revealed high Eomes and TOX in clusters with a high FES (Figure 6E). In contrast, high T-bet and Helios were present in Tex cell cluster c9 that expressed many cytotoxic molecules. Finally, TCF1 was expressed by a subset of Tex cells, including clusters c1 and c16, but this transcription factor was highest in non-Tex cells (Figure 6E) consistent with the major role for TCF1 in TN and TMEM. These results showed that high dimensional Tex cell clusters display distinct phenotypic, transcriptional and functional properties.
Figure 6. Distinct exhausted high dimensional clusters are enriched in HIV patients and differentially linked to HIV progression.
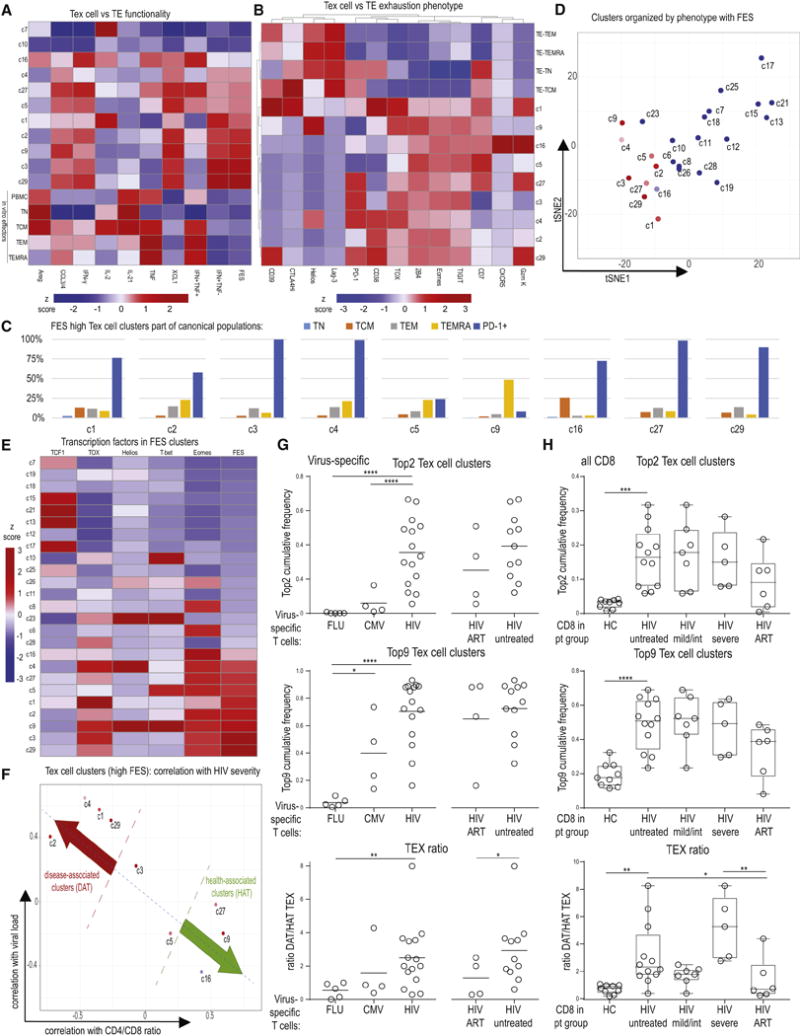
(A) The FES was calculated for in vitro differentiated TEFF (TEFF generated from total PBMC, or sorted TN, TCM, TEM, or TEMRA see STAR Methods) and compared to phenograph clusters. TCM and TEMRA-enriched clusters c7 and c10 are displayed for comparison in addition to phenograph clusters with high FES. (B) Comparison of in vitro differentiated TEFF (as in A) to the 9 Tex cell subsets with highest FES. Median frequencies of populations positive for given marker are displayed. Heatmap is clustered by row and column using Pearson’s correlation. (C) Tex cell clusters with high FES were analyzed for classical differentiation subsets. Depicted is the frequency of the indicated Tex cell clusters that fell into the classically gated TN, TCM, TEM, or TEMRA phenotypes or was PD-1+ (D) Phenograph clusters were plotted based on a tSNE analysis using exhaustion marker expression as outlined in Supplemental Figure 3 and colored by the FES. (E) Clusters were analyzed for transcription factor expression and arranged based on FES. Heatmap is clustered by rows using Pearson’s correlation. (F) Tex cell clusters with high FES were plotted versus correlation of cluster frequency with CD4/CD8 and viral load. (G) Virus-specific T cells identified in PBMCs from HC and HIV patients were analyzed for the prevalence of the Top 2 (upper graph) and Top 9 (middle graph) clusters with highest FES (sum of percentages for Top 2 and Top 9 clusters is displayed). The TEX ratio (lower graph) is shown as the sum of clusters defined to be disease associated (DAT; i.e. linked to severe HIV) divided by the sum of clusters defined to be health associated (HAT; i.e., linked to mild HIV), as in Figure 6F. (H) As in (G), Top 2, Top 9 and TEX ratio was determined for CD8+ T cells from PBMC of HC and HIV patients and displayed by HIV disease stage. Heatmap coloring in (A), (B), (E) reflects z scores after column normalization.
Use of Tex cell clusters to interrogate disease associations
We next asked whether the distribution of Tex cell clusters might inform about disease state. We thus identified the 9 Tex cell clusters with highest FES (c1, c2, c3, c4, c5, c9, c16, c27, c29; among which c3 and c29 were highest (“Top 2”)) and grouped them based on correlation with CD4/CD8 ratio and viral load in viremic untreated HIV patients into those linked to severe disease (“Disease Associated Tex cells”, (DAT) (c1, c2, c4, c29)) and those associated with mild disease (“Health Associated Tex cells”, (HAT) (c9, c16, c27)) (Figure 6F). We then analyzed FLU-, CMV- or HIV-specific T cells for the sum of the frequency of the Top 2 or Top 9 Tex cell clusters and for the ratio of clusters linked to severe (disease) versus mild (health) HIV infection (Tex ratio: DAT/HAT). HIV-specific T cells had higher frequencies of exhausted clusters and also a higher TEX ratio compared to FLU- and CMV-specific T cells (Figure 6G). The Tex ratio also revealed changes during ART therapy that was characterized by an increase in HAT (Figure 6G). These findings observed on HIV-specific CD8+ T cells could be extended to total CD8 T cells that showed an enrichment of the Top 2 and Top 9 Tex cell clusters and a higher DAT/HAT Tex ratio in viremic HIV patients compared to HC and some reduction of this ratio upon therapy (Figure 6H). Although the correlations associating clusters with severe or mild HIV were derived from viremic untreated patients (Figure 5), the correlations of Tex cellclusters with the CD4/CD8 ratio remained stable in ART-treated patients (Supplementary Figure 6). Thus, detailed analysis of Tex cell biology provided insight into changes in HIV disease and therapy and may serve as a framework to understand specific features of exhaustion involved in different stages of disease and guide novel therapeutic approaches.
Organ- and disease-specific changes in Tex cells in lung cancer
One unresolved question is whether key features of exhaustion are shared across different diseases and/or tissue sites. To interrogate this issue, we examined CD8 T cells from patients with newly diagnosed lung cancer using samples from peripheral blood, lung tumors and macroscopically unaffected lung tissue. Clusters with TN and TCM-like features (c13, c15, c21) were reduced in lung cancer patient PBMC compared to HC (Figures 7A). Larger changes in cluster distribution were observed between the blood, the lung tissue and TIL including an enrichment in Tex cell clusters c2 and c29, as well as the TEM/TEMRA cluster c10 in TIL (Figure 7A). Clusters expressing CD103 were enriched in the uninvolved lung tissue and TIL samples (e.g., c11, c18, c28) suggesting a lung tissue imprint on both Tex cells and non-exhausted populations (Figure 7A). The TRM-like populations also included FLU-specific cells (Supplementary Figure 5). Thus, cells with general features of Tex cells in the respiratory tract of lung cancer patients overlap with those observed in blood, but this anatomical location was also associated with alterations in Tex cells and non-exhausted cell populations that may relate to tissue-specific programming.
Figure 7. TIL dysfunction in lung cancer is linked to Tex cell phenotypes shared with severe HIV and tissue-associated features.
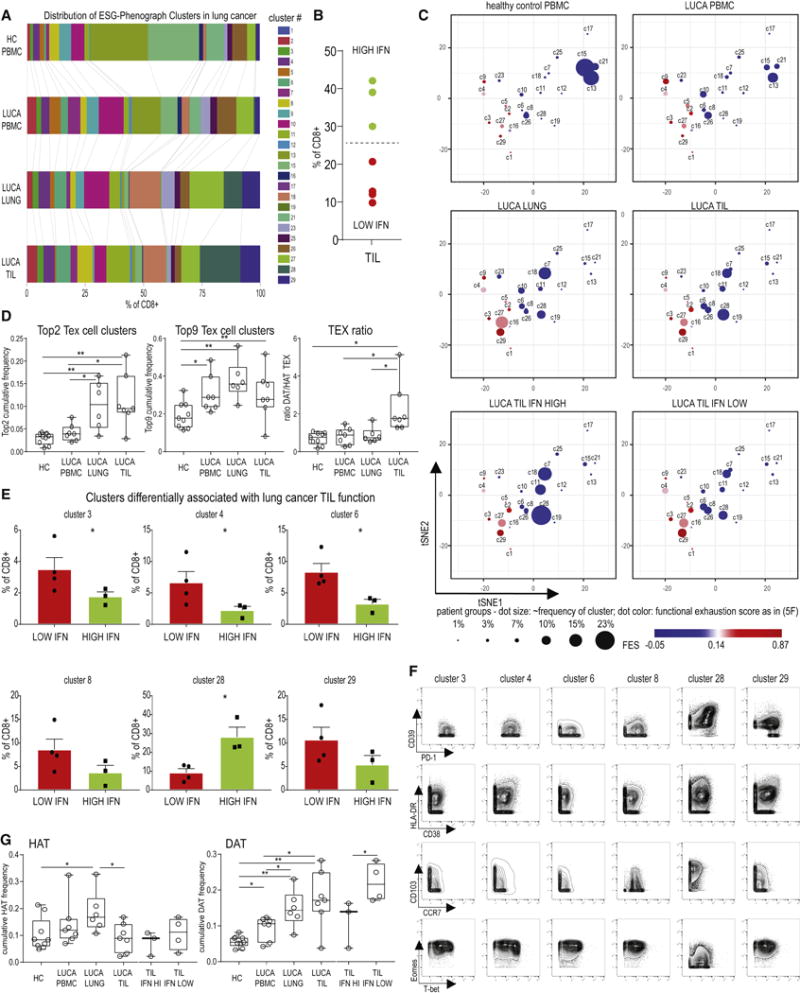
(A) Distribution of phenograph clusters in the blood, uninvolved lung tissue and tumor from 7 lung cancer patients and HC. The mean frequency of each cluster in each patient population is indicated by the size of the corresponding bar. (B) Tumors were evaluated based on CD8 TIL IFN-γ production following overnight anti-CD3 stimulation, and stratified into high and low TIL functionality. (C) The relative frequency of each cluster is shown on the same exhaustion coordinate system as in Figure 6D and Supplementary Figure 6. (D) Sum of the frequencies for the Top 2 and Top 9 Tex cell clusters and TEX ratio were determined as defined in Figure 6. (E) Clusters overrepresented in low or high functionality TIL are shown (for stacked bar analysis see Supplementary Figure 5). * indicates p <0.05. c8: p=0.07; c29: p=0.08. (F) Bivariate plots indicate expression of markers of exhaustion, activation, tissue residency and transcriptional programming for clusters differentially linked to tumor functionality. Plots display concatenated CD8 T cell data from lung cancer patients and HC as assigned by phenograph clustering. (G) The sum of the frequencies of HAT or DAT clusters linked to mild or severe HIV was determined in the lung cancer cohort. TIL data was analyzed both as total aggregate data and separating the high and low functionality samples as shown in (B). DAT clusters enrich in the dysfunctional tumor microenvironment in lung cancer.
Enrichment of Tex cell signatures in poorly functional lung cancer TIL populations
To interrogate how these TIL clusters related to function, IFN-γ production was examined after short-term in vitro stimulation (Figure 7B). Samples were then grouped into high and low IFN-γ producers. We plotted the changes in cluster distribution between CD8 T cells isolated from HC or lung cancer patients and also compared TIL with high versus low IFN-γ functionality (Figure 7C). Blood from lung cancer patients had a notable loss of TN and TCM clusters (c13, c15) and enrichment of Tex cell clusters c4 and c9. Lung tissue was enriched for clusters such as c11, c18 and c28 that expressed CD103 (Figure 7C). In more functional TIL populations the non-Tex cell clusters c11 and c18 as well as cluster c28 were enriched whereas in TIL with low functionality Tex cell clusters c4, c27 and c29 were overrepresented (Figure 7C). We investigated whether the results from HIV could be used to inform exhaustion and disease in the tumor context. Compared to PBMC, CD8 T cells from lung and TIL enriched in the sum of the Top 2 or Top 9 Tex cell clusters identified above (Figure 7D). Moreover, the DAT/HAT Tex ratio strongly increased in the TIL samples compared to the adjacent lung (Figure 7D). Examining specific clusters, c3, c4, and c6 were enriched in TIL with low IFN-γ production, with similar trends for c8 and c29 (Figure 7E). These clusters co-expressed PD-1 and Eomes, and many also had high co-expression of multiple IRs (Figures 5C, 7F). In contrast, c28 was overrepresented in tumors with higher IFN-γ production (Figure 7C, 7E). This cluster expressed CD103 as well as some PD-1 and other exhaustion-associated molecules (e.g., CD39, CTLA4, TOX), but lacked expression of other features of severe exhaustion, such as Eomes and or high expression of other IRs (e.g., 2B4, CD160, TIGIT) and also did not have high FES in the HIV data. Although high and low function TIL contained HAT clusters (as defined above), low function TIL were substantially enriched in “Disease Associated” Tex cells Figure 7G). These analyses revealed conserved Tex cell biology across HIV and cancer, with additional insights into disease specific enrichments. They demonstrate the ability to use an epigenomically-guided CyTOF approach to connect the differentiation landscape of Tex cells across tissues, disease type and disease severity. Moreover, this resource reveals common Tex cell biology and disease specific features.
Discussion
We developed a systems immunology pipeline to profile the heterogeneity of Tex cells in human disease. This pipeline integrated information from bulk transcriptomic and epigenomic datasets to develop a novel, focused single-cell proteomic profiling approach. The ability to identify Tex cells at the single cell level by cytometric approaches has remained challenging due to the lack of a distinct marker of Tex cells. For example, many individual proteins implicated in exhaustion, such as PD-1, are also expressed by other activated cells. Here, we used an epigenomic-driven mass cytometry approach to identify Tex cell populations in humans with chronic infection and cancer. Tex cells identified using this approach displayed differential expression of exhaustion markers compared to TEFF, TMEM and TN, as predicted. Notably, Tex cells were even detected in HC blood suggesting that exhaustion is a normal aspect of T cell biology that is amplified in specific disease settings. In HIV infection, more dysfunctional Tex cell subpopulations were enriched in more severe disease and similar Tex cell clusters were found in more dysfunctional TIL in lung cancer. Applying such approaches to distinct diseases will facilitate identification of Tex cell features associated with specific types of diseases, anatomical locations and will guide understanding of changes in Tex cell populations with immunotherapeutic interventions in chronic infections, cancer and even autoimmunity.
We identified 12 putative Tex cell clusters with considerable heterogeneity in the precise pattern of IR and transcription factor co-expression. Eomes was a prominent feature of Tex cell clusters linked to disease severity in HIV and TIL. Notably, however, c18, c19 and c28 expressed 4-5 IRs, but they lacked Eomes and had low FES. Among these, c19 and c28 (but not c18) expressed TOX. Thus, based on transcription factors and IRs, not only was it possible to identify phenotypically exhausted cells in human chronic infections and cancer (as predicted from previous work), but it was also possible to identify discrete types of Tex cells present in different diseases. Interrogating T cell function helped validate the nature of putative Tex cell clusters and also provided a framework to link changes in cluster distribution to disease. Often, changes in polyfunctionality, rather than production of IFN-γ alone, more accurately reflect T cell exhaustion (Betts et al., 2006; Fuller and Zajac, 2003; Wherry et al., 2003). The residual functions of Tex cells may be important in establishing a host-pathogen or host-tumor equilibrium. For example, in chronic SIV infection, virus-specific CD8 T cells become exhausted (Velu et al., 2009), but CD8 T cell depletion causes increases in viral load and progression to AIDS (Schmitz et al., 1999). Here, using high-dimensional cytometry, we developed a functional exhaustion score, FES, that was linked to high-dimensional T cell phenotypes. The FES validated the phenotypically defined Tex cell clusters since clusters with a high FES had high co-expression of many exhaustion markers, such as IRs (e.g., c1, c29) and high expression of Eomes in combination with PD-1 (e.g., c3, c29). In contrast, a lower FES identified Tex cells with lower IR expression and higher levels of CD127, TCF1 and/or CXCR5 expression (e.g. c16, c27), phenotypes previously associated with more functional Tex cells (Blackburn et al., 2008; Im et al., 2016; Paley et al., 2012; Utzschneider et al., 2016; Wieland et al., 2017). Thus, the combined high-dimensional epigenomically-guided phenotypic and functional analysis accurately identified Tex cells and provides a resource to interrogate how changes in the Tex cell landscape are associated with disease.
Overall, this approach revealed several novel concepts about the biology of Tex cells in humans. First, Tex cells exist in HC, albeit at low frequency, and are enriched in clusters with lower FES. Second, both HIV and lung cancer were enriched for Tex cell subtypes that appeared severely exhausted by phenotype and FES (e.g. c3, c29). These observations point to a common core biology of Tex cells present across diseases. Third, distinct clusters were overrepresented in HIV (e.g. c2) or lung cancer (e.g. c27 or c28), perhaps representing features of tissue location (e.g. CD103 in c28) or other disease specific effects. Fourth, Tex cells were identified that were associated with better health in chronic disease. Clusters c9, c16, c27 were associated with higher CD4/CD8 ratio and lower viral load in HIV infection and were preserved in HIV+ ART patients suggesting a potential benefit to the host or enhanced durability based on cytotoxic molecule expression (c9), CD127 and TCF1 expression (c16, c27) and/or CXCR5 coexpression with PD-1 (c16). Finally, many of the Tex cell clusters defined in HIV that associated with severe disease were also linked to poor TIL function (e.g. c3, c4, c29). In lung cancer a separate cluster with high CD103 expression, c28, was associated with more functional TIL consistent with a low FES for this cluster in the HIV setting.
Heterogeneity in Tex cells has been identified in animal models that relate to either terminal exhaustion or, conversely, persistence and the ability to be reinvigorated by PD-1 pathway blockade (Blackburn et al., 2008; Im et al., 2016; Paley et al., 2012; Utzschneider et al., 2016; Wu et al., 2016), but the role in human disease has remained unclear. Our approach resolved this heterogeneity in human Tex cell clusters. For example, c16 expressed Eomes and PD-1 but also moderate TCF-1 and high CXCR5, suggesting a similarity to the described TCF-1+ CXCR5+ subset (He et al., 2016; Im et al., 2016; Utzschneider et al., 2016; Wieland et al., 2017; Wu et al., 2016). Some CXCR5 and TCF1 expression was also observed in c1, and moderate CXCR5 expression was observed in c29, both clusters with higher FES than c16, suggesting different degrees of exhaustion and potential precursor-progeny relationships among CXCR5+ and TCF1+ Tex cells. An additional implication of this heterogeneity is the differential expression of immunoregulatory targets. Identifying IR co-expression patterns on subsets of Tex cells with suspected progenitor capacity or on Tex cells with disease specific enrichment might allow more effective immunotherapies. For example, clusters enriched in lung cancer TIL (e.g. c28) had prominent expression of CTLA-4 unlike those in the blood of HIV patients suggesting potentially more benefit of targeting this molecule in cancer. Better understanding of Tex cell heterogeneity should provide insights into disease progression, and can be used to identify target populations responding to therapy.
Recent epigenetic work indicates that Tex cells constitute a distinct T cell lineage and their fate appears largely epigenetically stable even after PD-1 pathway blockade (Pauken et al., 2016; Philip et al., 2017; Sen et al., 2016). The epigenetic information about Tex cells used to construct our CyTOF panel allowed this high dimensional single cell approach to be anchored on genes/proteins that were “fate-specific” for T cell exhaustion. Using that backbone, heterogeneity in combinatorial expression of these fate-selected activation and differentiation molecules allowed us to resolve patterns of Tex cell differentiation in human disease. Together, these observations suggest a model where within a T cell lineage or fate, cells can exist in multiple states of differentiation and/or activation. Comparing the patterns of Tex cell clusters across disease severity in HIV and between HIV and lung cancer allows speculation about possible relationships between Tex cell subpopulations in a model of T cell exhaustion in humans (Supplementary Figure 7). Notably, this analysis also clearly identified distinct populations with TN, TMEM, TEMRA, TRM and senescent phenotypes. Thorough understanding of T cell exhaustion is becoming increasingly relevant for improving immunotherapies. The deep phenotyping presented here implicates differential expression of checkpoint blockade targets on different exhaustion subsets and suggests approaches for high-dimensional profiling of Tex cells in checkpoint blockade and other therapeutic interventions. Moreover, this resource should provide an opportunity to further interrogate Tex cell biology in human disease. Thus, these results have implications for our understanding, diagnostics, immune-monitoring and immunomodulatory approaches in chronic infection, autoimmunity and cancer.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, E. John Wherry (wherry@pennmedicine.upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Blood was acquired with the written informed consent of all study participants and with the approval of the University of Pennsylvania Institutional Review Board. For HIV cohorts, blood samples were obtained (Penn Center for AIDS Research (CFAR) (IRB# 815056)), for lung cancer, blood and tissue samples were obtained (IRB# 813004) and for HC blood was obtained (IRB# 820151). PBMC and TIL were extracted as described (Huang et al., 2017). Cohort information is provided in Supplementary Table 5. Detailed information on which samples were used in which figures is provided in Supplementary Table 7.
METHOD DETAILS
Mass Cytometry
Mass cytometry reagents were obtained or generated by custom conjugation to isotope-loaded polymers using MAXPAR kit (Fluidigm). Mass cytometry antibodies used are shown in Supplementary Table 4. Exhaustion-specific markers were chosen for mass cytometry based on: 1) presence in the transcriptional signature of Tex cells; 2) the presence of unique, Tex cell-specific epigenetic changes in the gene locus (either gained or lost); and 3) the availability of suitable antibodies for CyTOF, either pre-conjugated or after in-house conjugation and validation. Staining was performed as described (Bengsch et al., 2017). Briefly, single-cell suspensions were pelleted, incubated with 20 μM Lanthanum-139 (Trace Sciences)-loaded maleimido-mono-amine-DOTA (Macrocyclics) in PBS for 10min at RT for live/dead discrimination (LD). Cells were washed in staining buffer and resuspended in surface antibody cocktail, incubated for 30min at RT, washed twice in staining buffer, fixed and permeabilized using FoxP3 staining buffer set (eBioscience), and stained intracellularly for 60min at RT. Cells were further washed twice before fixation in 1.6% PFA (Electron Microscopy Sciences) solution containing 125nM Iridium overnight at 4C. Prior to data acquisition on CyTOF2 (Fluidigm), and in a repeat cohort experiment on a CyTOF Helios (Fluidigm), cells were washed twice in PBS and once in dH2O. Mass cytometry data on samples from 57 patients were acquired in different batches. In particular, samples analyzed in Figures 4–7 were obtained in three batches detailed in Supplementary Table 7 using the same core antibodies, with similar CyTOF instrument performance. For batch control, we also used bead-based normalization and analyzed PBMC from a single control donor in every batch, displaying similar results in the high-dimensional analysis (Supplementary Figure 4). Later, repeat cohort analyses were performed by reanalysis of patient samples from the same original bleed date stained at a later time point and acquired on a different mass cytometer and resulted in similar conclusions as the original analysis (Figure 3, Supplementary Figure 2). For cytokine analysis, samples were split for phenotyping or stimulation with PMA/Ionomycin in complete media for 5h at 37C in the presence of Monensin and Brefeldin A and stained for mass cytometry analysis using the panel outlined in Supplementary Table 6.
Cell sorting and in vitro culture for TEFF activation
PBMC or sorted T cell populations were stimulated with anti-CD3/CD28 beads (Miltenyi Biotec) in the presence of 20 U/ml IL-2 (Stemcell) for 72 hours in supplemented culture media (RPMI 1640 (Gibco) supplemented with L-glutamine, 10% FCS and Penicillin/Streptomycin). Sorting was performed on a FACS Aria II (BD Biosciences) after staining for TN (CCR7+CD45RA+CD27+), TCM (CD27+CD45RA−CCR7+), TEM (CD27−CCR7−CD45RA−), TEMRA (CD27−CCR7−CD45RA+) and PD-1+ populations using anti-CD27-BV785 (clone O323), anti-CD45RA-BV605 (clone HI100), anti-PD-1-BV421 (clone E12.2.H7), anti-CD8 APC-Fire (clone RPA-T8) (Biolegend), anti-CCR7-FITC (clone 150503) (BD Biosciences), and after staining with life/dead reagent Ghost Violet (Tonbo).
QUANTIFICATION AND STATISTICAL ANALYSIS
High dimensional data analysis
Bead-based normalization of CyTOF data was performed using the Nolan lab normalizer available through https://github.com/nolanlab/bead-normalization/releases. FCS files were further analyzed by commercial software FlowJo v10 (TreeStar), FCSExpress 6 (DeNovo Software) and ViSNE (Cytobank). R based tSNE analysis was performed using Rtsne package. Phenograph was performed using RPhenograph package implemented via cytofkit package, described in (Chen et al., 2016; Levine et al., 2015). Analysis of exhaustion data space using ViSNE or Phenograph was performed on mass channels corresponding to exhaustion-specific molecules as defined through Figures 1 and 2 and detailed in Supplementary Table 4. Phenograph analysis of exhaustion data space on Iridium intercalator positive, singlet LD negative CD45+CD3+CD8 T cells identified 30 high-dimensional clusters, of which 5 (c14, c20, c22, c24, c30) represented cell frequencies <0.01% of CD8 T cells after quality control gating and were excluded from downstream analyses. After Phenograph and Visne analysis, data was integrated into fcs files and further processed by FlowJo or FCSExpress.
Transcriptomic and epigenomic data analysis
Transcriptional profiling of LCMV-specific T cells available through the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/gds) under the accession number GSE41867 was described in (Doering et al., 2012). ATAC-Seq data was described in (Pauken et al., 2016; Sen et al., 2016) (GSE97646, GSE86881). Transcriptional profiling data was downloaded from GEO and annotated using R 3.3.1 and GEOquery package. ATAC-Seq open chromatin region (OCR) analysis was done as in (Pauken et al., 2016; Sen et al., 2016) (GSE97646, GSE86881). Identification of exhaustion-specific transcriptomic and epigenomic expression patterns was performed using moderated Bayesian statistics calculated by the limma package. Specifically, genes with increased or decreased expression in virus-specific Tex cells were selected if their moderated T statistic was (>=2.9) compared to TN, TEFF and TMEM.
Gene set enrichment and variation analysis
Gene set enrichment analysis (GSEA) using Broad Institute software (http://www.broadinstitute.org/gsea/index.jsp) was performed on microarray data from GEO (indicated in Supplementary Table 1). Exhaustion-specific gene signatures were tested by GSEA. Normalized Enrichment scores (NES) and leading edge (LE) genes obtained by GSEA were used for comparison across different datasets in Figures 1, 2 and 3. In Supplementary Figure 1, Gene set variation analysis (GSVA) using GSVA R package (Hanzelmann et al., 2013) was performed to interrogate single cell transcriptomic data from (Tirosh et al., 2016) and assess different exhaustion gene sets in the tumor microenvironment. Briefly, CD8 T cell single-cell data was obtained from NIH GEO (GSE72056), and GSVA of single-cell data was performed using the full epigenomically-and transcriptomically defined exhaustion gene list or a subset of genes later analyzed by CyTOF. The results of the GSVA analysis using the exhaustion gene sets were used to calculate a GSVA exhaustion score (GSVA_score_UP – GSVA_score DN).
Exhaustion function mapping
Exhaustion-specific markers shared between the phenotyping and stimulation panel (“scaffold”, outlined in Supplementary Table 6) were used to map post-stimulation samples to pre-stimulation clusters by the “classify” mode of Phenograph (Levine et al., 2015). The training data was constructed by sampling equal amounts of cells (50000) from each of the samples with a stimulus. The exhaustion markers common to both the unstimulated and post stimulation data, CTLA4, CD7, CD127, Helios, PD-1, CCR7, Eomes, CD39, TOX, TIGIT, CXCR5, 2B4, LAG3 were used for these analyses. For each stimulated sample, a nearest neighbor graph using the Jaccard metric was constructed using the training data and cells from the stimulated sample. Random walk probabilities through the graph were used to assign clusters to each of the stimulated cells. See (Levine et al., 2015) for a more detailed description. The concordance between the mapped and pre-stim data is shown in Supplementary Figure 4. A functional exhaustion score (FES) was then calculated using the production of IL-2 and CCL3/4, as well as IFN-γ and TNF coproduction (2*(%IFN+TNF−)-(%IFN−TNF+)-(%IL-2+))*(%CCL3/4+).
Heatmap display
Heatmaps were generated using the Pheatmap R package (v. 1.0.8). Color is based on the z-score and indicated by a color palette in the figures next to the heatmaps.
Statistical analysis and data visualization
Statistical analysis was performed using JMP 12.2.0 (SAS), GraphPad Prism 7.02 and R 3.3.1 limma package. Group comparisons in Figures 5–7 were performed using unpaired t test with Welch’s correction. In Figure 5 and 6, simple regression analysis of phenograph cluster frequencies in viremic HIV patient samples was performed versus viral load and the CD4/CD8 ratio. The respective Pearson correlation was plotted using R ggplot2 package. The cluster dot size displayed in Figure 5G and 7C was scaled proportionally to the abundance of individual clusters (% of CD8). In Figure 6 and 7, sum of the percentage of phenograph clusters per sample was calculated for the top2 and top9 clusters with highest FES shown in Figure 5 and 6, the Disease- or Health Associated Tex cell clusters in HIV (DAT/HAT) indicated in Figure 6. In addition a TEX ratio was calculated using the sum of the frequency of DAT divided by the sum of the frequency of HAT to assess a skewing of the composition of different qualities of Tex cells across diseases.
DATA AND SOFTWARE AVAILABILITY
The high-dimensional mass cytometry phenotyping data is available through Cytobank. A summary of the transcriptomic and epigenomic datasets and relevant links to data repositories are provided in the Key Resource table.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-2B4, clone C1.7 | Biolegend | RRID:AB_1279194 Cat#329502 |
| Anti-Amphiregulin, polyclonal AB-1 | Thermo | RRID:AB_59522 Cat#RB-257-PABX |
| Anti-CCL3/4, clone 93342 | R&D | RRID:AB_2259652 Cat#MAB2701 |
| Anti-CCR7, clone G043H7, 159Tb | Fluidigm | RRID:AB_2714155 Cat#3159003A |
| Anti-CCR7-FITC (clone 150503) | BD Biosciences | RRID:AB_10561679 Cat# 561271 |
| Anti-CD103, clone Ber-ACT8 | Biolegend | RRID:AB_535945 Cat#121402 |
| Anti-CD127, clone HIL-7R-M21 | BD Biosciences | RRID:AB_394494 Cat#552853 |
| Anti-CD16, clone 3G8, 209Bi | Fluidigm | RRID:AB_2661791 Cat#3209002B |
| Anti-CD160, clone BY55 | Biolegend | RRID:AB_2074411 Cat#341202 |
| Anti-CD19, clone HIB19 | Biolegend | RRID:AB_314232 Cat#302202 |
| Anti-CD200R2, clone OX-108 | Biolegend | RRID:AB_1027731 Cat#329302 |
| Anti-CD26, clone BA5b | Biolegend | RRID:AB_314286 Cat#302702 |
| Anti-CD27, clone L128, 155Gd | Fluidigm | RRID:AB_2687645 Cat#3155001B |
| Anti-CD27-BV785 (clone O323) | Biolegend | RRID:AB_2562674 Cat#302832 |
| Anti-CD28, clone CD28.2 | Biolegend | RRID:AB_314304 Cat#302902 |
| Anti-CD3, clone UCHT1 | Biolegend | RRID: AB_314056 Cat#300402 |
| Anti-CD36, clone 5-271 | Biolegend | RRID:AB_1279228 Cat#336202 |
| Anti-CD38, clone HIT2, 167Er | Fluidigm | RRID:AB_2687640 Cat#3167001B |
| Anti-CD39, clone A1 | Biolegend | RRID:AB_940438 Cat#328202 |
| Anti-CD4, clone RPA-T4 | Biolegend | RRID:AB_314070 Cat#300502 |
| Anti-CD45, clone HI30, 89Y | Fluidigm | RRID: AB_2661851 Cat#3089003B |
| Anti-CD45RA, clone H100 | BD Biosciences | RRID:AB_395877 Cat#555486 |
| Anti-CD45RA-BV605 (clone HI100) | Biolegend | RRID:AB_2563814 Cat#304134 |
| Anti-CD45RO, clone UCHL1 | BD Biosciences | RRID: AB_395882 Cat#555491 |
| Anti-CD57, clone TB01 | Ebioscience | RRID: AB_1311193 Cat #16-0577-85 |
| Anti-CD7, clone eBio124-1 | Ebioscience | RRID:AB_823132 Cat#14-0079-82 |
| Anti-CD73, clone AD2 | Biolegend | RRID:AB_2154067 Cat#344002 |
| Anti-CD8, clone RPA-T8 | Biolegend | RRID:AB_314120 Cat#301002 |
| Anti-CD8-APC-Fire750 (clone RPA-T8) | Biolegend | RRID:AB_2572113 Cat#100766 |
| Anti-CTLA-4, clone BNI3 | BD Biosciences | RRID:AB_396173 Cat#555850 |
| Anti-CXCL10, clone J034D6 | Biolegend | RRID:AB_2563206 Cat#519502 |
| Anti-CXCR5, clone RF8B2 | BD Biosciences | RRID:AB_394324 Cat#552032 |
| Anti-Eomes, clone WD1928 | Ebioscience | RRID:AB_2572882 Cat#14-4877-82 |
| Anti-FoxP3, clone PCH101, 162Dy | Fluidigm | RRID:AB_2687650 Cat#3162011A |
| Anti-Granzyme B, clone CLB-GB11 | Novus | RRID:AB_10012261 Cat#NBP1-50071 |
| Anti-Granzyme K, clone GM6C3 | Santa Cruz | RRID:AB_2263772 Cat#sc-56125 |
| Anti-Granzyme M, clone 4B2G4 | Bovenschen lab | n/a |
| Anti-Helios, clone 22F6 | Biolegend | RRID:AB_10900638 Cat#137202 |
| Anti-HLA-DR, clone L243 | Biolegend | RRID:AB_314680 Cat#307602 |
| Anti-ICOS clone C398.4A | Biolegend | RRID:AB_416326 Cat#313502 |
| Anti-IFN-gamma, clone B27 | Biolegend | RRID:AB_315435 Cat#506502 |
| Anti-IL-10, clone JES3-9D7 | Biolegend | RRID:AB_315168 Cat#501402 |
| Anti-IL-2, clone MQ1-17H12 | Ebioscience | RRID:AB_468408 Cat#14-7029-85 |
| Anti-IL-21, clone 3A3-N2 | Biolegend | RRID:AB_1027621 Cat#513002 |
| Anti-Ki-67, clone B56 | BD Biosciences | RRID:AB_396287 Cat#556003 |
| Anti-KLRG1, clone 13F12F2 | Pircher lab | n/a |
| Anti-LAG-3, clone 17B4 | Enzo | RRID:AB_2133353 Cat#ALX-804-806 |
| Anti-PD-1, clone EH12.2H7 | Biolegend | RRID:AB_940488 Cat#329902 |
| Anti-PD-1-BV421 (clone E12.2.H7) | Biolegend | RRID:AB_10960742 Cat#329920 |
| Anti-Perforin, clone B-D48 | Abcam | RRID:AB_2169084 Cat#ab47225 |
| Anti-Ptger2, polyclonal AB9472 | Merck Millipore | RRID:AB_2174912 Cat#AB9472 |
| Anti-T-bet, clone 4B10, 160Gd | Fluidigm | RRID:AB_763634 Cat#3160010B |
| Anti-TCF1, clone 7F11A10 | Biolegend | RRID:AB_2562103 Cat#655202 |
| Anti-TIGIT, clone MBSA43 | Ebioscience | RRID:AB_10718831 Cat#16-9500-82 |
| Anti-Tim-3, clone F38-2E2, 153Eu | Fluidigm | RRID:AB_2687644 Cat#3153008B |
| Anti-TNF, clone MAb11 | Ebioscience | RRID:AB_468489 Cat#14-7349-85 |
| Anti-Tox, clone Rea473 | Miltenyi | n/a |
| Anti-XCL1, clone 109001 | R&D | RRID:AB_2217055 Cat#MAB6951 |
| Biological Samples | ||
| PBMC from HIV patients | Penn Center for AIDS Research (CFAR), University of Philadelphia | http://www.med.upenn.edu/cfar/ |
| PBMC from healthy individuals | University of Pennsylvania, Human Immunology Core and Institute for Immunology | https://pathbio.med.upenn.edu/hic/site/ |
| PBMC and lung, tumor tissue from lung cancer patients | Lung Cancer Immunobiology Translational Center of Excellence of the Abramson Cancer, University of Pennsylvania | https://www.pennmedicine.org/cancer/cancer-research/translating-research-to-practice/lung-cancer-tce |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Antibody Stabilizer | Candor | Cat#131050 |
| Gadolinium-157 | Trace Sciences | n/a |
| Ghost Violet 510 | Tonbo | Cat#13-0870 |
| GolgiPlug | BD Biosciences | Cat#555029 |
| GolgiStop | BD Biosciences | Cat#5554724 |
| Indium-113 | Trace Sciences | n/a |
| Indium-115 | Trace Sciences | n/a |
| Ionomycin | Sigma-Aldrich | Cat #I0634 |
| Iridium (Cell-ID Intercalator-Ir 500 μM) | Fluidigm | Cat #201192B |
| Lanthanum-139 | Trace Sciences | n/a |
| Maleimido-mono-amine-DOTA | Macrocyclics | Cat#B-272 |
| Paraformaldehyde 16% | FisherScientific | Cat#50-980-487 |
| pMHC Monomers | NIH tetramer core | n/a |
| Peptide CMV pp65 495-503 NLVPMVATV | Eunoia Biotech | n/a |
| Peptide HIV-1 gag 77-85 SLYNTVATL | Eunoia Biotech | n/a |
| Peptide HIV-1 pol 476-484 ILKEPVHGV | Eunoia Biotech | n/a |
| Peptide Influenza A M1 58-66 GILGFVFTL | Eunoia Biotech | n/a |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma-Aldrich | Cat#P8139 |
| Streptavidin | Newell lab | n/a |
| TCEP | ThermoFisher | Cat#PI77720 |
| Critical Commercial Assays | ||
| EQ Four Element Calibration Beads | Fluidigm | Cat#201078 |
| FoxP3 Transcription factor staining kit | ThermoFisher | Cat#A25866A |
| Maxpar DN3 Multimetal Labeling Kit | Fluidigm | n/a |
| Maxpar X8 Multimetal Labeling Kit | Fluidigm | Cat#201300 |
| Deposited Data | ||
| ATAC-Seq TEX LCMV data Pauken et al. | PMID: 27789795 | GEO: GSE86797 |
| ATAC-Seq TEX LCMV data Sen et al. | PMID: 27789799 | GEO: GSE87646 |
| CyTOF fcs file repository Cytobank | This paper | https://premium.cytobank.org/cytobank/experiments/154556/ and https://premium.cytobank.org/cytobank/experiments/154563/ |
| GEO datasets analyzed by GSEA (see table S1) | This paper | table S1 |
| Microarray TEX LCMV data Doering et al. | PMID: 23159438 | GEO: GSE41867 |
| Sc RNA-Seq MM data Tirosh et al. | PMID: 27124452 | GEO: GSE72056 |
| Software and Algorithms | ||
| Cytobank | Cytobank, Inc | https://www.cytobank.org/ |
| Cytofkit | PMID:27662185 | https://github.com/JinmiaoChenLab/cytofkit |
| FCS Express 6 | De Novo Software | https://www.denovosoftware.com/ |
| FlowJo v10 | Tree Star | https://www.flowjo.com/solutions/flowjo/downloads |
| GSEA | PMID:17644558 | https://software.broadinstitute.org/gsea/ |
| GSVA | PMID:23323831 | https://bioconductor.org/packages/release/bioc/html/GSVA.html |
| JMP 12.2.0 | SAS | https://www.jmp.com |
| Limma | PMID:25605792 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Mass cytometry bead based normalizer | PMID:23512433 | https://github.com/nolanlab/bead-normalization/releases |
| Pheatmap | PMID:23685480 | https://CRAN.R-project.org/package=pheatmap |
| Phenograph | PMID:26095251 | https://www.c2b2.columbia.edu/danapeerlab/html/phenograph.html |
| Prism 7.02 | Graph Pad Software | https://www.graphpad.com/scientific-software/prism/ |
| R | The R foundation | https://www.r-project.org/ |
| ViSNE | PMID:23685480 | https://www.c2b2.columbia.edu/danapeerlab/html/cyt.html |
| Other | ||
| Amicon 30kDa Ultrafiltraition spin columns | Merck Millipore | Cat #UFC503096 |
| Amicon 3kDa Ultrafiltraition spin columns | Merck Millipore | Cat #UFC500396 |
| EasySep™ Human CD8+ T Cell Isolation Kit | StemCell | Cat#17953 |
| T Cell Activation/Expansion Kit | Miltenyi | Cat#130-091-441 |
Supplementary Material
These supplementary tables exceeding 3 pages are provided: Supplementary Table 2 related to Figures 1 and 2: Transcriptomic and epigenomic defined exhaustion-specific gene list (mouse and corresponding human orthologs)
Table summarizing the transcriptomic-derived exhaustion-specific genes (mouse and human gene IDs), and whether these genes also had a matched epigenetic OCR change (1-YES, 0-NO).
Supplementary Table 3 related to Figures 2 and 3: Leading edge analysis of exhaustion genes in Tex cell subsets.
Table summarizing the leading edge contribution of exhaustion-specific genes with respect to the GSEA analysis performed. Genes are annotated by their summarized “leading edgeness” (LE); whether they showed an associated OCR change revealed by ATAC-seq and whether they were analyzed by CyTOF.
Highlights.
Unbiased identification of unique Tex genes using transcriptomics and epigenomics
High-dimensional CyTOF profiling of human Tex gene products reveals heterogeneity
Identification of key disease-relevant Tex cell populations in HIV and lung cancer
Development of exhaustion metrics applicable to human immune monitoring
Acknowledgments
We thank D. Gudonis, M. Cummins and the UPenn Center for AIDS Research (P30-AI-045008) for HIV patient samples, the UPenn Human Immunology Core for HC samples (P30-CA016520), the Corporal Michael J. Crescenz VA Medical Center Research Program for shared use of its CyTOF mass cytometer, the NIH tetramer core for HLA/peptide monomers and H. Pircher for the KLRG1 antibody. B.B. was supported by German Research Foundation (BE5496/1-1 and BE5496/1-2), O.K.by F30AI129263, A.H. by T32CA009615-26 and a Measey Research Fellowship, S.O.B. by T32-CA009140. This work was partially supported by the Lung Cancer Immunobiology Translational Center of Excellence of the Abramson Cancer Center (S.O., S.M.A., and E.J.W.). This work was supported by grants from the NIH (AI105343, AI082630, AI112521, AI115712, AI117718, AI108545, and AI117950) and the Parker Institute for Cancer Immunotherapy to E.J.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions “CRediT” taxonomy:
Conceptualization – B.B. and E.J.W.; Methodology – B.B.; Algorithmic analysis – B.B., M.S., S.M., P.G.; Validation – B.B., T.O., R.H., K.C.; Formal analysis – B.B., O.K., M.S., S.M., S.OB.; Investigation – Experiments and data acquisition B.B., T.O., S.OB.; Resources – A.H., K.C., E.N., N.B., D.P., S.A., E.J.W.; Writing – Original Draft - B.B., E.J.W.; Visualization - B.B., O.K., S.M., P.G.; Supervision – E.J.W.; Funding Acquisition – B.B., E.J.W.
Declaration of Interest:
E.J.W. has a patent licensing agreement on the PD-1 pathway. S.M.A. is the recipient of a sponsored research agreement from Janssen Pharmaceuticals. A provisional patent application has been filed based on work presented in this manuscript.
References
- Angelosanto JM, Blackburn SD, Crawford A, Wherry EJ. Progressive loss of memory T cell potential and commitment to exhaustion during chronic viral infection. J Virol. 2012;86:8161–8170. doi: 10.1128/JVI.00889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengsch B, Ohtani T, Herati RS, Bovenschen N, Chang KM, Wherry EJ. Deep immune profiling by mass cytometry links human T and NK cell differentiation and cytotoxic molecule expression patterns. J Immunol Methods. 2017 doi: 10.1016/j.jim.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengsch B, Seigel B, Ruhl M, Timm J, Kuntz M, Blum HE, Pircher H, Thimme R. Coexpression of PD-1, 2B4, CD160 and KLRG1 on exhausted HCV-specific CD8+ T cells is linked to antigen recognition and T cell differentiation. PLoS Pathog. 2010;6:e1000947. doi: 10.1371/journal.ppat.1000947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A. 2008;105:15016–15021. doi: 10.1073/pnas.0801497105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan MK, Postow MA, Wolchok JD. Targeting T Cell Co-receptors for Cancer Therapy. Immunity. 2016;44:1069–1078. doi: 10.1016/j.immuni.2016.04.023. [DOI] [PubMed] [Google Scholar]
- Chen H, Lau MC, Wong MT, Newell EW, Poidinger M, Chen J. Cytofkit: A Bioconductor Package for an Integrated Mass Cytometry Data Analysis Pipeline. PLoS Comput Biol. 2016;12:e1005112. doi: 10.1371/journal.pcbi.1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford A, Angelosanto JM, Nadwodny KL, Blackburn SD, Wherry EJ. A role for the chemokine RANTES in regulating CD8 T cell responses during chronic viral infection. PLoS Pathog. 2011;7:e1002098. doi: 10.1371/journal.ppat.1002098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37:1130–1144. doi: 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller MJ, Zajac AJ. Ablation of CD8 and CD4 T cell responses by high viral loads. J Immunol. 2003;170:477–486. doi: 10.4049/jimmunol.170.1.477. [DOI] [PubMed] [Google Scholar]
- Gupta PK, Godec J, Wolski D, Adland E, Yates K, Pauken KE, Cosgrove C, Ledderose C, Junger WG, Robson SC, et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog. 2015;11:e1005177. doi: 10.1371/journal.ppat.1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature. 2016;537:412–428. doi: 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016;537:417–421. doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amirel AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523:612–616. doi: 10.1038/nature14468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchiotti R, Gracio F, Kordasti S, Todd AK, de Rinaldis E. Cluster stability in the analysis of mass cytometry data. Cytometry A. 2017;91:73–84. doi: 10.1002/cyto.a.23001. [DOI] [PubMed] [Google Scholar]
- Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, Wherry EJ. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338:1220–1225. doi: 10.1126/science.1229620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen D, Kurachi M, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016 doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545:452–456. doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley M, Pereyra F, Nilsson B, Porichis F, Fonseca C, Eichbaum Q, Julg B, Jesneck JL, Brosnahan K, Imam S, et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat Med. 2010;16:1147–1151. doi: 10.1038/nm.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel JM, Masopust D. Tissue-resident memory T cells. Immunity. 2014;41:886–897. doi: 10.1016/j.immuni.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, et al. The epigenetic landscape of T cell exhaustion. Science. 2016 doi: 10.1126/science.aae0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity. 2016;45:415–427. doi: 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- Velu V, Titanji K, Zhu B, Husain S, Pladevega A, Lai L, Vanderford TH, Chennareddi L, Silvestri G, Freeman GJ, et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature. 2009;458:206–210. doi: 10.1038/nature07662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland D, Kemming J, Schuch A, Emmerich F, Knolle P, Neumann-Haefelin C, Held W, Zehn D, Hofmann M, Thimme R. TCF1+ hepatitis C virus-specific CD8+ T cells are maintained after cessation of chronic antigen stimulation. Nat Commun. 2017;8:15050. doi: 10.1038/ncomms15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol. 2016;1 doi: 10.1126/sciimmunol.aai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
These supplementary tables exceeding 3 pages are provided: Supplementary Table 2 related to Figures 1 and 2: Transcriptomic and epigenomic defined exhaustion-specific gene list (mouse and corresponding human orthologs)
Table summarizing the transcriptomic-derived exhaustion-specific genes (mouse and human gene IDs), and whether these genes also had a matched epigenetic OCR change (1-YES, 0-NO).
Supplementary Table 3 related to Figures 2 and 3: Leading edge analysis of exhaustion genes in Tex cell subsets.
Table summarizing the leading edge contribution of exhaustion-specific genes with respect to the GSEA analysis performed. Genes are annotated by their summarized “leading edgeness” (LE); whether they showed an associated OCR change revealed by ATAC-seq and whether they were analyzed by CyTOF.