

Abstract
Nonsense mutations are present in 10% of patients with CF, produce a premature termination codon in CFTR mRNA causing early termination of translation, and lead to lack of CFTR function. There are no currently available animal models which contain a nonsense mutation in the endogenous Cftr locus that can be utilized to test nonsense mutation therapies. In this study, we create a CF mouse model carrying the G542X nonsense mutation in Cftr using CRISPR/Cas9 gene editing. The G542X mouse model has reduced Cftr mRNA levels, demonstrates absence of CFTR function, and displays characteristic manifestations of CF mice such as reduced growth and intestinal obstruction. Importantly, CFTR restoration is observed in G542X intestinal organoids treated with G418, an aminoglycoside with translational readthrough capabilities. The G542X mouse model provides an invaluable resource for the identification of potential therapies of CF nonsense mutations as well as the assessment of in vivo effectiveness of these potential therapies targeting nonsense mutations.
Introduction
Cystic Fibrosis (CF) is an autosomal recessive genetic disorder caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. CFTR is an anion channel expressed throughout the body with highest expression in epithelial tissues. Absence of CFTR function impairs transepithelial fluid and electrolyte permeation and results in viscous mucus along the epithelial lining of organs leading to a wide range of disease manifestations. Common manifestations of CF include lung failure [1–3], pancreatic insufficiency [4], intestinal obstruction [5–8], and reduced growth [9–11]. Over 2000 unique CFTR variants have been identified, with ~300 categorized as definitively CF-causing mutations [12, 13]. CF-causing nonsense mutations are the second most common CF mutation type, and are found in approximately 10% of patients with CF [14, 15]. Nonsense mutations create a premature termination codon (PTC) in CFTR, which leads to premature termination of CFTR translation [16].
Recently, there has been progress in the development of CFTR modulators that restore CFTR function in patients with specific CFTR mutations. Examples include VX-770 (ivacaftor) for gating mutations like G551D [17, 18] and VX-809 (lumacaftor) for the misfolding mutation F508del [19, 20]. However, no therapies are available to CF patients that restore function to nonsense mutations in CFTR. Certain drugs, like gentamicin and PTC124 (ataluren), have been shown to interact with the ribosome to induce readthrough of PTCs by allowing insertion of a near-cognate aminoacyl tRNA and translation of a full-length protein [21–23]. Both gentamicin and PTC124 have been used in clinical trials for patients with nonsense mutations [24–27]. However, gentamicin treatment is associated with nephrotoxicity and ototoxicity [28] and PTC124 has produced no significant improvements in adult CF patients with nonsense mutations in Phase 3 clinical trials [29]. Therefore, no currently available treatment for CF nonsense mutations is both safe and clinically effective.
Because of the safety issues surrounding PTC readthrough strategies, animal models of CFTR nonsense mutations are essential for in vivo validation of new therapies. Current CF mouse models with nonsense mutations were created in a way that either does not allow for correction of the nonsense mutation or [30, 31] or utilizes non-endogenous CFTR with tissue specific expression [14]. Therefore, a model with global expression of the G542X mutation in the endogenous mouse Cftr will have a greater value for examining the efficacy of nonsense mutation therapies on Cftr in multiple tissue types with native expression levels. In this manuscript, we describe the generation of a mouse model with expression of the G542X mutation, the most common nonsense mutation in CF [32], using CRISPR/Cas9 gene editing technology. Mice homozygous for the G542X mutation have reduced Cftr expression and absence of CFTR function in the airway and intestine. These mice display typical cystic fibrosis manifestations such as poor growth and reduced survival due to intestinal obstruction. Importantly, we demonstrate that pharmacological readthrough of the G542X nonsense mutation in this model allows production of functional CFTR. The G542X mouse will be a valuable model for the examination of nonsense mutation therapies for CF and other genetic diseases caused by nonsense mutations.
Materials and methods
Generation of the G542X allele
To produce the G542X mouse Cftr allele (CftrG542X) using the gene editing system CRISPR/Cas9, guide RNAs (gRNA) were selected in exon 12 of mouse Cftr using CRISPR design software that identifies optimal gRNAs based on proximity to target as well as off-target cutting predictions [33]. Four of these gRNAs were tested in vitro using guide-it gRNA in vitro transcription and screening kit (Clontech) for the ability to guide Cas9 nuclease activity to the desired DNA sequence. One gRNA (25 ng/ul; PNABio), a 120 bp single stranded oligonucleotide (ssODN) containing the G542X mutation (25 ng/ul; IDT) centered on the cut site and either Cas9 mRNA (25–50 ng/ul; PNABio) or Cas9 protein (25 ng/ul; PNABio) were injected in the pronucleus of C57BL/6 one-cell embryos. Embryos were then placed in pseudo-pregnant females to develop. Ear punches from the 22 resulting pups were sequenced using next generation sequencing. The region of interest was PCR amplified (~300 bp) with flanking primers that were tailed with a universal sequence. A secondary PCR amplification was then performed on the primary PCR product using a set of primers specific for the universal sequence and tailed with I5,I7 Illumina index and specific barcode sequences allowing for the sequencing of multiple samples at the same time. The mixture of barcoded PCR products were analyzed on an Illumina MiSeq instrument using a paired-end 300 base pair kit allowing for 25 million reads per run. Data were de-convoluted and variant analysis in comparison to the original sequence was performed using the Outknocker software program [34].
Mice
In some experiments, the G542X mouse model was compared to a previously published CFTR null model carrying a S489X mutation (Cftrtm1Unc) which is congenic on the C57BL/6J background [31]. Mice homozygous for these mutations were created by breeding heterozygous males and females and wild type littermates were used as controls. Genotyping was completed by PCR analysis using DNA extracts from ear biopsies. To detect the G542X allele (319 bp) primers P1 (5’- ACAAGACAACACAGTTCTCT -3’) and P2 (5’ TCCATGCACCATAACAACAAGT -3’) were used. To detect the wildtype (WT) allele (319 bp) P2 and P3 (5’- ACAAGACAACACAGTTCTTG -3’) were used in a separate reaction. PCR reactions were completed for 40 cycles of 95°C for 30 seconds, 58°C for 30 seconds and 72°C for 30 seconds and products were run out on 2% agarose gels. All mice were allowed unrestricted access to water and solid chow (Harlan Teklad 7960; Harlan Teklad Global Diets). All animals were maintained on a 12-h light, 12-h dark schedule at a mean ambient temperature of 22°C and were housed in standard polysulfone microisolator cages in ventilated units with corncob bedding. Animals were monitored on a daily basis, and weight was assessed every 5 days from 10 to 40 days of age. Length of 6-wk-old euthanized mice was assessed from nose to anus by use of digital calipers. For G418 treatment, mice were intraperitoneal injected on three consecutive days (25mg/kg b.w.). Twenty four hours after last injection, mice were sacrificed and lungs were flash frozen and RNA prepared as described below. The Institutional Animal Care and Use Committee of Case Western Reserve University approved all animal protocols.
Expression analysis
One μg of RNA was reversed transcribed in cDNA using QScript cDNA synthesis kit (VWR). Real-time quantitative PCR was performed on a StepOne PCR system (Applied Biosystems). Cftr expression was assessed using a TaqMan expression assay which used primers spanning exon 17 and 18 (Mm00445197; Applied Biosystems). Expression was normalized to β-actin as the endogenous control. Each RNA sample was used to make cDNA in duplicate and the expression results were then averaged to yield the final value. The average of each sample was then expressed as a percentage of wildtype expression.
Bioelectric measurements
Nasal potential difference (NPD) measurements were obtained as previously described [35, 36]. NPD (mV) was assessed after the addition of chloride-free HEPES-buffered saline containing 10 μM forskolin. Short circuit measurements on intestinal sections were obtained as previously described [36]. The change in short-circuit current was calculated after the addition of 10 μM forskolin and 100 μM IBMX to the basolateral side of the intestinal sections.
Intestinal organoid harvesting and culture
Intestinal organoids were harvested from adult mice as described by others [37]. Briefly, the mouse was sacrificed, the small intestine removed and flushed using PBS. The intestine was cut longitudinally, villi removed with a razor blade and the remaining intestine was cut into ~0.5 cm segments and suspended in Gentle Cell Dissociation Reagent (STEMCELL Technologies) in a 50 mL conical tube for 30 minutes under gentle agitation with a shaker. In a sterile tissue culture hood, the intestinal segments were vigorously shaken by hand for 30 seconds, and the supernatant was deposited in a 10 cm dish. The segments were resuspended in ice-cold PBS lacking Mg++ and Ca++, and vigorously shaken again for 30 seconds. This process was repeated until four fractions of supernatant were produced. The fraction which was most enriched for intestinal crypts was filtered using a 100 μm cell strainer, and pelleted at 450xg for 10 minutes. The supernatant was discarded, and the pellet was resuspended in 500 μl of a 1:1 mixture of MatriGel (Corning) and Intesticult Organoid Growth Media (OGM; STEMCELL Technologies). Crypts were diluted to a concentration of 10 crypts/μl, and plated into non-tissue culture treated 24-well plates, with 35 μl of MatriGel:OGM mixture deposited in each well. The MatriGel:OGM was solidified at 37°C for 15 minutes, then 500 μl of OGM was gently deposited in each well. The organoids were stored in a 37°C incubator with 5% CO2. The OGM was changed once every three days, and the organoids were passaged once every 7 days.
Measurement of intestinal organoid swelling
Forskolin-induced swelling (FIS) of intestinal organoids was performed similar to previously described methods [38, 39]. Briefly, intestinal organoids which had been cultured for 6–8 days were split into non-tissue culture treated 48-well plates (Genesee Scientific) and allowed to grow for 24 hours before drug treatment. Organoids were treated with G418 (20–100 μM) or PTC124 (1–20 μM) for 72 hours prior to FIS measurements. FIS was assessed by treating organoids with 10 μM forskolin and imaging for 5 hours under live cell conditions with brightfield microscopy on a Lionheart FX Automated Microscope (BioTek). FIS was quantified by identifying and normalizing organoid area to T = 0 with Gen5 ImagePrime software (BioTek). Area under the curve (AUC) at T = 300 minutes was calculated to compare statistical significance between treatment groups. All organoids used for swelling quantifications were between passages 1–4.
Statistics
Results are expressed as the mean +/- S.E.M. Differences between groups were determined using either an ANOVA with post-hoc Tukey test or an unpaired t test. A P value of <0.05 was considered significant.
Results
Generation of the G542X mutation
To generate the G542X mutation in mouse Cftr, the CRISPR/Cas9 gene editing system was utilized. Guide RNAs (gRNAs) in proximity to the DNA sequence corresponding to glycine at position 542 in mouse Cftr exon 12 were selected and tested in vitro for their ability to support efficient Cas9 nuclease activity to the DNA region as indicated by the reduction of full length DNA amplicon (Fig 1A). The gRNA that supported the highest Cas9 nuclease activity was injected in one-cell mouse embryos along with a ssODN containing the G542X mutation and either Cas9 mRNA or protein (Fig 1B). DNA from all mice originating from these injected embryos was sequenced. Of 22 mice, 9 (40.9%) had at least one Cftr allele containing the G542X mutation due to homology-directed repair (HDR) with the ssODN, 5 (22.7%) did not have the G542X mutation but had other mutations (insertions/deletions) due to nonhomologous end joining and 8 (34.4%) had no mutation of Cftr in exon 12. One of the founder mice containing the G542X allele was selected to establish and expand the G542X colony.
Fig 1. Generation of the G542X mutation.

(A) To validate gRNA efficiency in guiding Cas9 nuclease to the desired site, an in vitro assay was performed. PCR amplified DNA containing exon 12 and surrounding region of mouse Cftr (1056 bp) is displayed on an agarose gel with no gRNAs (-) or with 1 of 4 different gRNAs (1–4). Cas9 nuclease activity results in the cleavage of the DNA into fragments of ~750 bp and ~300 bp. (B) Normal Cftr mouse DNA and amino acid sequence around the desired mutation site is shown with the gRNA sequence (in box) and the protospacer adjacent motif sequence recognized by Cas9 (in blue). A portion of the sequence for the G542X oligo is also shown with the substitution change shown in red. The G to T mutation corresponds to the glycine to stop mutation. A silent T to C mutation was also inserted to assist with genotyping and verify HDR had occurred.
Cftr expression is reduced and CFTR function is absent in G542X mice
Cftr expression was assessed using qRTPCR from various tissues of G542X and WT littermates. Cftr expression levels in tissues from G542X mice were significantly reduced compared to WT expression levels ranging from 2.5±0.5% in the ileum to 28.4±8.6% of WT levels in the lung (Fig 2). Assessment of CFTR function in the airway was performed using NPD measurements in the nasal lumen. Activation of CFTR and the subsequent Cl- secretion into the nasal lumen causes a decrease in voltage across the nasal epithelium. Mice with functional CFTR display a decrease in NPD (-14.6±1.6 mV) while G542X mice have a slightly positive NPD (4.38±2.1 mV; Fig 3A) indicating no functional CFTR similar to other CF mouse models [35, 36, 40, 41]. CFTR function in the intestinal epithelium was examined by measuring short-circuit current (ΔIsc) in intestinal segments from G542X and WT mice before and after activation of CFTR. In WT intestinal segments, a significant increase in short-circuit current is observed following CFTR stimulation. Corresponding segments from G542X mice displayed significantly reduced changes in short-circuit current (Fig 3B), similar to other CF mouse models [36, 42, 43].
Fig 2. Cftr expression in tissues from G542X mice.

Cftr expression in tissues from G542X and WT littermates were evaluated using qRTPCR. Airway (nasal epithelium, lung), intestine (duodenum, jejunum, ileum), and kidney Cftr expression as a percentage of WT Cftr expression is displayed. G542X expression was significantly reduced in all tissues compared to WT. (P<0.05 vs. WT. n≥5 per group).
Fig 3. CFTR function in the airway and intestine of G542X mice.

(A) NPD measurements and (B) change in intestinal short circuit current (ΔIsc) measurements from the duodenum, jejunum, and ileum from WT and G542X mice are shown. (*P<0.05 vs. WT. n≥4 per group).
G542X mice display characteristic CF manifestations
The most common cause of morbidity in CF mouse models is intestinal obstruction [5, 36, 44, 45]. G542X homozygous mice displayed a reduced survival rate compared to WT littermates (33.3% vs. 97.9%; Fig 4A). Intestinal obstruction was observed in all G542X mice post-mortem but not in WT littermates. Another common disease manifestation in CF mouse models is reduced growth, or failure to thrive [31, 36, 43, 46]. G542X homozygous mice demonstrated significantly reduced length compared to sex-matched 6-week old WT littermates (in cm: males 7.6±0.2 vs. 8.4±0.1; females 6.0±0.3 vs. 7.8±0.1; Fig 4B). To assess growth, offspring from G542X heterozygote crosses were weighed at 5 day intervals from 10–40 days of age. G542X homozygous mice displayed significantly reduced weight at all time points compared to sex-matched WT littermates (Fig 4C and 4D).
Fig 4. Survival and growth characteristics of G542X mice.
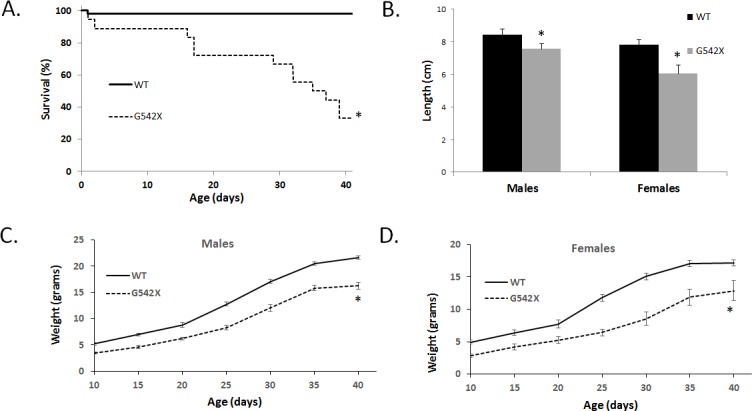
(A) Survival of G542X and WT mice up to 40 days of age. (B) Length of G542X and WT mice at 6 weeks of age. (C, D) Weight of G542X and WT mice sex-matched mice up to 40 days of age. Weight at every age was significantly different between G542X and WT littermates. (n≥10 per group. *P<0.05 vs. WT).
G542X CFTR function is restored following pharmacological nonsense mutation readthrough by G418 but not by PTC124
The ability to produce functional CFTR following treatment with potential therapies is crucial for the G542X mouse model to have full utility as a model of CF nonsense mutations. To examine CFTR function following readthrough, we generated intestinal organoids from G542X homozygous mice. Intestinal organoids are a three-dimensional cell culture model produced from LGR5+ stem cells in the intestinal crypt with budding outgrowths and a hollow lumen [39]. Activation of CFTR by forskolin allows flow of Cl- anions into the intestinal organoid lumen, increasing lumen osmolality, drawing in fluid, and causing organoid FIS. FIS is absent in organoids lacking CFTR function, allowing sensitive detection of CFTR activity. G542X organoids were treated for 72 hours with the aminoglycoside G418, a suppressor of nonsense mutations [47, 48]. FIS was examined by imaging organoid swelling over 300 minutes under live cell conditions with brightfield microscopy (Fig 5A). We observed a dose-dependent FIS response to G418-induced readthrough indicating restored CFTR function (normalized percent area at 300 minutes: No G418: 114±1.7; 20 μM: 135±2.3; 50 μM: 160±3.0; 100 μM: 181±1.7, Fig 5B). Statistical significance between treatment groups was compared by calculating area under the curve (AUC) at 300 minutes for each treatment groups (Fig 5C). AUC of organoids treated with G418 were significantly increased compared to non-treated organoids (Fig 5C). S489X organoids, which are non-correctable Cftr-null mutants, did not display FIS following 72 hour treatment with 100 μM G418, confirming that FIS in G542X organoids is due to readthrough of nonsense mutations (Normalized percent area at 300 minutes: 113±0.7, Fig 5B and 5C). Similar to tissues from G542X homozygous mice, cultured intestinal organoids have a reduced amount of Cftr expression compared to those from WT littermates (24.0±1.0%) (Fig 5D). However, concordant with the increase in CFTR function in G418 treated organoids from G542X mice, G418 treatment also significantly increased the amount of detectable Cftr mRNA (Fig 5D). Short term treatment of G542X mice with G418 also resulted in a significant increase in the amount of detectable Cftr mRNA in tissue (Fig 5E). Interestingly, PTC124 treatment of the G542X organoid model did not stimulate detectable FIS at any of the concentrations tested and thus resulted in no significant difference of AUC between PTC124 treated and non-treated organoids (1–20 μM; Fig 6A–6C).
Fig 5. Intestinal organoids and tissue from G542X mice are used to test G418-mediated nonsense mutation readthrough.
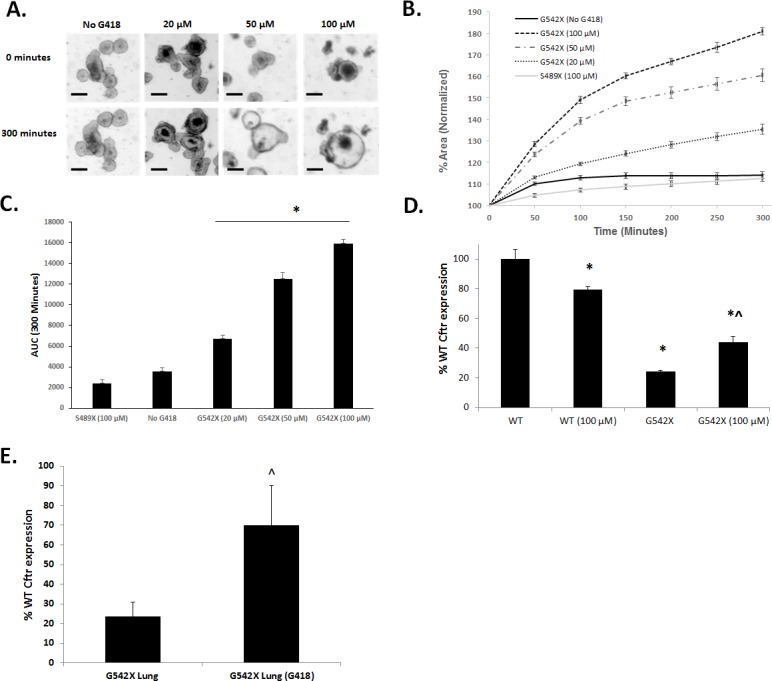
Intestinal organoids were incubated with indicated doses of G418, or vehicle for 72 hours prior to swelling with 10 μM forskolin. (A) Representative images at T = 0 and T = 300 minutes of organoid swelling, conditions as indicated. (Scale bar = 100 μm) (B) Total change in area was measured over 300 minutes, and FIS was quantified by normalizing the total organoid area to T = 0. (n = 5 wells of organoids for each treatment, ± SE). (C) AUC at T = 300 for indicated treatment groups. (*P<0.05 compared to untreated G542X). (D) Expression of Cftr in WT and G542X organoids with and without G418 treatment as a percentage of untreated WT organoids. (E) Expression of Cftr in lung from untreated G542X mice or treated with G418 compared to expression from WT mice. (*P<0.05 compared to untreated WT. ^P<0.05 compared to untreated G542X.).
Fig 6. Intestinal organoids from G542X mice are used to test PTC124 mediated readthrough.
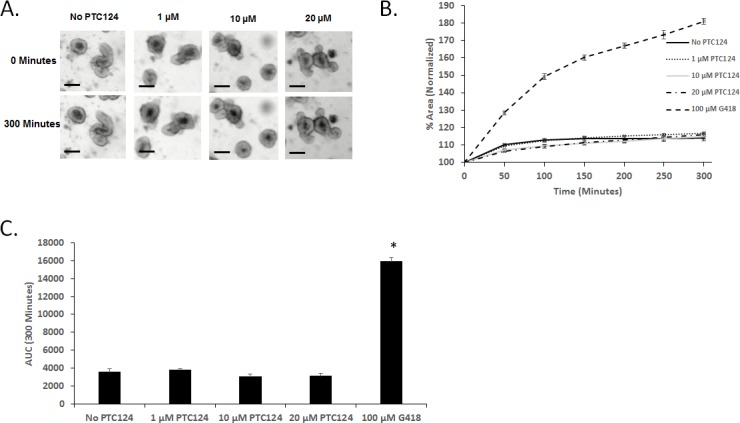
G524X organoids were treated with the indicated doses of PTC124 for 72 hours prior to the FIS assay. (A) Representative images at T = 0 and T = 300 minutes of organoid swelling, conditions as indicated. (Scale bar = 100 μm). (B) Normalized areas for PTC124 treated organoids are shown, compared to vehicle and 100 μM G418-treated organoids. (C) AUC measurements at 300 minutes. (*P<0.05 compared to untreated G542X.).
Discussion
Currently there are only two FDA approved therapies that directly target CFTR dysfunction, the primary defect of CF [17, 20]. These therapies are approved to modulate aberrant protein that is produced from the CFTR gene in patients with specific mutations that do not include nonsense mutations. Nonsense mutations typically result in little to no protein function. Reduced mRNA levels, through nonsense-mediated decay (NMD), dramatically decreases the amount of protein produced. In addition, any protein that is synthesized is truncated due to the PTC and, depending on severity of the truncation, leads to significantly limited protein function [49, 50]. Current research efforts to increase the amount of full length protein from nonsense mutations include both inducing readthrough of the PTC and targeting the NMD pathway to increase mRNA levels [49, 50].
Readthrough of PTCs through nonsense suppression can be achieved with aminoglycosides that reduce the efficiency of termination and allow for the insertion of near-cognate tRNA [21]. Promising results in cell and mouse models of nonsense mutations have led to the use of the aminoglycoside gentamicin in patients [14, 21, 24, 51]. In CF, functional improvements in CFTR have been described in CF patients receiving topical application to the nasal mucosa [26] but renal and otic toxicities are a concern with long term treatment. Interestingly, a minor component B1 of gentamicin has been shown to provide effective readthrough activity [52], suggesting that an enrichment of this component of gentamicin or modified aminoglycosides with reduced toxicity may hold promise for the future [23]. A high-throughput screen identified PTC124 as a candidate compound with readthrough ability without the toxicity concerns of aminoglycosides [22]. Despite a lack of readthrough efficacy of PTC124 in specific in vitro reporter systems [53], PTC124 treatment of preclinical models of CF have displayed functional improvements [54]. While some initial patient studies suggested PTC124 efficacy in CF [25, 55], larger studies failed to show significant improvements in CF patients with nonsense mutations in Phase 3 clinical trials [29]. While PTC124 will not be used as a therapy for CF, investigation of the compound is still underway for other nonsense mutation genetic disorders such as Duchenne muscular dystrophy.
There has been recent success in the identification of possible nonsense mutation suppressors in CFTR using high-throughput screens [56–59]. The in vitro tools created in each of these screens made the identification of these hits possible. However, a CF animal model to carry out in vivo studies to test the efficacy of these hits is not available to the research community. The first Cftr mutation made in the mouse was a nonsense mutation created by replacing the normal exon sequence with the premature stop codon, S489X along with selectable markers. The integration of the selectable markers in the exon precludes the production of functional protein [30, 31] and thus serves as a negative control for readthrough strategies. The only current animal model shown to produce functional CFTR following readthrough is a transgenic mouse that expresses human CFTR cDNA containing the G542X mutation driven by a rat Fatty Acid Binding Protein (FABP) promoter that results in intestinal-specific expression [14]. The conditions of CFTR expression in this model differ significantly from endogenous expression and readthrough of G542X can only be assessed in the intestine. In addition, because cDNA was used and there is no intron splicing, the mRNA produced is not susceptible to NMD which limits the interpretation of any in vivo studies. Non-endogenous expression levels and/or the absence of NMD of the resulting mRNA may explain why PTC124 displayed functional improvement in the intestine of hCFTR-G542X [54] model but failed to show similar improvements in patients.
In this study, we created a mouse model of CF containing the G542X mutation in the endogenous Cftr gene. We utilized the CRISPR/Cas9 gene editing system to create the G542X mutation in one-cell mouse embryos. This is the first report of Cftr CRISPR/Cas9 editing to create a mouse model. As previously reported, the use of CRISPR/Cas9 significantly reduces the time of gene editing in embryos compared to embryonic stem cells [60]. In addition, this gene editing system is highly efficient in the creation of the desired mutation. Traditional gene editing in embryonic stem (ES) cells typically produced 1–5% of ES colonies harboring the correctly edited gene and could take 1–2 years to produce the targeting construct and correctly targeted founder mice. With CRISPR/Cas9, 40.9% of founder mice contained the G542X mutation and were produced in as few as 3 months. One disadvantage in utilizing CRISPR/Cas9 gene editing is the possible creation of double-strand breaks in similar sequences to the target which can create off-target effects [61]. A low incidence of off-target mutations has been identified in mice created using CRISPR/Cas9 [62, 63]. While off-target mutations can occur, subsequent backcrosses to a parental strain (e.g., C57Bl/6) can remove any unintended consequences from off-target mutations.
Mice homozygous for the G542X mutation demonstrate reduced Cftr expression throughout the body. While our analysis cannot rule out that a truncated form of CFTR protein is produced, there is a clear absence of CFTR function in the airway and intestine of these mice. The G542X mice also display severe CF manifestations, including reduced growth, and reduced survival due to intestinal obstruction. In this model, mRNA levels can be increased and CFTR function can be corrected through readthrough by G418. Interestingly, the utilization of PTC124 in the intestinal organoid model did not demonstrate any improvement in CFTR function. This is consistent with studies that show no significant improvement in CF patients chronically administered PTC124. All of these findings confirm that this G542X mouse model can be utilized to test the efficacy of nonsense mutation therapies.
The G542X mouse model also provides an unlimited source of primary cells to potentially identify new therapies for nonsense mutations. For example, the prominent intestinal organoid FIS phenotype, which indicates CFTR function, has the potential to be used for high-throughput screens similar to those currently utilizing CF patient samples [39, 64]. The high degree of similarity between organoids and tissue in vivo may increase the odds of identifying translationally beneficial compounds in a high-throughput screen compared to current screens using transfected cell lines or in vitro reporter assays.
In conclusion, this is the first study to generate a mouse model containing CRISPR/Cas9 edited Cftr and the first CF animal model that can be utilized to assess whole body efficacy of nonsense mutation directed therapies. With several recent screens identifying potential PTC readthrough compounds, the G542X mouse model can be utilized to prioritize which compounds to test in a clinical trial that may be beneficial to CF patients. The ability to analyze in vivo effectiveness of current and future potential nonsense mutation therapies will not only increase our understanding of how these therapies work on a basic level but lead to treatments for CF patients with nonsense mutations.
Acknowledgments
We thank Alma Wilson, Molly Schneider and Amanda Barabas of the CF Mouse Models Core at Case Western Reserve University for their work in maintaining the mouse colony. The work was supported by the Cystic Fibrosis Foundation (Hodges15XX0).
Data Availability
All relevant data are within the paper.
Funding Statement
This work was supported by grants from the Cystic Fibrosis Foundation Therapeutics (Hodges15XX0 to CAH and Conlon15XX0 to RAC, https://www.cff.org/ CFF). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Livraghi A, Randell SH. Cystic fibrosis and other respiratory diseases of impaired mucus clearance. Toxicologic pathology. 2007;35(1):116–29. Epub 2007/02/28. doi: 10.1080/01926230601060025 . [DOI] [PubMed] [Google Scholar]
- 2.Hobbs CA, Da Tan C, Tarran R. Does epithelial sodium channel hyperactivity contribute to cystic fibrosis lung disease? The Journal of physiology. 2013;591(18):4377–87. Epub 2013/07/24. doi: 10.1113/jphysiol.2012.240861 ; PubMed Central PMCID: PMCPMC3784186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pillarisetti N, Williamson E, Linnane B, Skoric B, Robertson CF, Robinson P, et al. Infection, inflammation, and lung function decline in infants with cystic fibrosis. American journal of respiratory and critical care medicine. 2011;184(1):75–81. Epub 2011/04/16. doi: 10.1164/rccm.201011-1892OC . [DOI] [PubMed] [Google Scholar]
- 4.Singh VK, Schwarzenberg SJ. Pancreatic insufficiency in Cystic Fibrosis. J Cyst Fibros. 2017;16 Suppl 2:S70–S8. doi: 10.1016/j.jcf.2017.06.011 . [DOI] [PubMed] [Google Scholar]
- 5.De Lisle RC, Borowitz D. The cystic fibrosis intestine. Cold Spring Harbor perspectives in medicine. 2013;3(9):a009753 doi: 10.1101/cshperspect.a009753 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colombo C, Ellemunter H, Houwen R, Munck A, Taylor C, Wilschanski M. Guidelines for the diagnosis and management of distal intestinal obstruction syndrome in cystic fibrosis patients. J Cyst Fibros. 2011;10 Suppl 2:S24–8. Epub 2011/06/18. doi: 10.1016/s1569-1993(11)60005-2 . [DOI] [PubMed] [Google Scholar]
- 7.Cleghorn GJ, Stringer DA, Forstner GG, Durie PR. Treatment of distal intestinal obstruction syndrome in cystic fibrosis with a balanced intestinal lavage solution. Lancet (London, England). 1986;1(8471):8–11. Epub 1986/01/04. . [DOI] [PubMed] [Google Scholar]
- 8.Sun L, Rommens JM, Corvol H, Li W, Li X, Chiang TA, et al. Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nature genetics. 2012;44(5):562–9. doi: 10.1038/ng.2221 ; PubMed Central PMCID: PMC3371103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lai HC, Corey M, FitzSimmons S, Kosorok MR, Farrell PM. Comparison of growth status of patients with cystic fibrosis between the United States and Canada. Am J Clin Nutr. 1999;69(3):531–8. doi: 10.1093/ajcn/69.3.531 . [DOI] [PubMed] [Google Scholar]
- 10.Scaparrotta A, Di Pillo S, Attanasi M, Consilvio NP, Cingolani A, Rapino D, et al. Growth failure in children with cystic fibrosis. Journal of pediatric endocrinology & metabolism: JPEM. 2012;25(5–6):393–405. Epub 2012/08/11. . [DOI] [PubMed] [Google Scholar]
- 11.Kyle UG, Shekerdemian LS, Coss-Bu JA. Growth failure and nutrition considerations in chronic childhood wasting diseases. Nutrition in clinical practice: official publication of the American Society for Parenteral and Enteral Nutrition. 2015;30(2):227–38. Epub 2014/11/08. doi: 10.1177/0884533614555234 . [DOI] [PubMed] [Google Scholar]
- 12.Hasty P, O'Neal WK, Liu KQ, Morris AP, Bebok Z, Shumyatsky GB, et al. Severe phenotype in mice with termination mutation in exon 2 of cystic fibrosis gene. Somat Cell Mol Genet. 1995;21(3):177–87. . [DOI] [PubMed] [Google Scholar]
- 13.O'Neal WK, Hasty P, McCray PB Jr., Casey B, Rivera-Perez J, Welsh MJ, et al. A severe phenotype in mice with a duplication of exon 3 in the cystic fibrosis locus. Hum Mol Genet. 1993;2(10):1561–9. . [DOI] [PubMed] [Google Scholar]
- 14.Du M, Jones JR, Lanier J, Keeling KM, Lindsey JR, Tousson A, et al. Aminoglycoside suppression of a premature stop mutation in a Cftr-/- mouse carrying a human CFTR-G542X transgene. Journal of molecular medicine (Berlin, Germany). 2002;80(9):595–604. Epub 2002/09/13. doi: 10.1007/s00109-002-0363-1 . [DOI] [PubMed] [Google Scholar]
- 15.Bidou L, Allamand V, Rousset JP, Namy O. Sense from nonsense: therapies for premature stop codon diseases. Trends in molecular medicine. 2012;18(11):679–88. Epub 2012/10/23. doi: 10.1016/j.molmed.2012.09.008 . [DOI] [PubMed] [Google Scholar]
- 16.Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Annals of human genetics. 2003;67(Pt 5):471–85. Epub 2003/08/28. . [DOI] [PubMed] [Google Scholar]
- 17.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–72. Epub 2011/11/04. doi: 10.1056/NEJMoa1105185 ; PubMed Central PMCID: PMC3230303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(44):18825–30. Epub 2009/10/23. doi: 10.1073/pnas.0904709106 ; PubMed Central PMCID: PMCPmc2773991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ren HY, Grove DE, De La Rosa O, Houck SA, Sopha P, Van Goor F, et al. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Molecular biology of the cell. 2013;24(19):3016–24. Epub 2013/08/09. doi: 10.1091/mbc.E13-05-0240 ; PubMed Central PMCID: PMCPMC3784376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, et al. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–31. doi: 10.1056/NEJMoa1409547 ; PubMed Central PMCID: PMCPMC4764353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2(4):467–9. . [DOI] [PubMed] [Google Scholar]
- 22.Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87–91. Epub 2007/04/24. doi: 10.1038/nature05756 . [DOI] [PubMed] [Google Scholar]
- 23.Xue X, Mutyam V, Tang L, Biswas S, Du M, Jackson LA, et al. Synthetic aminoglycosides efficiently suppress cystic fibrosis transmembrane conductance regulator nonsense mutations and are enhanced by ivacaftor. American journal of respiratory cell and molecular biology. 2014;50(4):805–16. doi: 10.1165/rcmb.2013-0282OC ; PubMed Central PMCID: PMC4068923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malik V, Rodino-Klapac LR, Viollet L, Wall C, King W, Al-Dahhak R, et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Annals of neurology. 2010;67(6):771–80. Epub 2010/06/03. doi: 10.1002/ana.22024 . [DOI] [PubMed] [Google Scholar]
- 25.Sermet-Gaudelus I, Boeck KD, Casimir GJ, Vermeulen F, Leal T, Mogenet A, et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. American journal of respiratory and critical care medicine. 2010;182(10):1262–72. Epub 2010/07/14. doi: 10.1164/rccm.201001-0137OC . [DOI] [PubMed] [Google Scholar]
- 26.Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349(15):1433–41. doi: 10.1056/NEJMoa022170 . [DOI] [PubMed] [Google Scholar]
- 27.McDonald CM, Campbell C, Torricelli RE, Finkel RS, Flanigan KM, Goemans N, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet (London, England). 2017;390(10101):1489–98. doi: 10.1016/S0140-6736(17)31611-2 . [DOI] [PubMed] [Google Scholar]
- 28.Sandoval RM, Reilly JP, Running W, Campos SB, Santos JR, Phillips CL, et al. A non-nephrotoxic gentamicin congener that retains antimicrobial efficacy. Journal of the American Society of Nephrology: JASN. 2006;17(10):2697–705. Epub 2006/09/15. doi: 10.1681/ASN.2005101124 . [DOI] [PubMed] [Google Scholar]
- 29.Kerem E, Konstan MW, De Boeck K, Accurso FJ, Sermet-Gaudelus I, Wilschanski M, et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. The Lancet Respiratory medicine. 2014;2(7):539–47. Epub 2014/05/20. doi: 10.1016/S2213-2600(14)70100-6 ; PubMed Central PMCID: PMCPmc4154311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koller BH, Kim HS, Latour AM, Brigman K, Boucher RC Jr., Scambler P, et al. Toward an animal model of cystic fibrosis: targeted interruption of exon 10 of the cystic fibrosis transmembrane regulator gene in embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(23):10730–4. ; PubMed Central PMCID: PMC53004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, et al. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257(5073):1083–8. . [DOI] [PubMed] [Google Scholar]
- 32.Schloesser M, Arleth S, Lenz U, Bertele RM, Reiss J. A cystic fibrosis patient with the nonsense mutation G542X and the splice site mutation 1717–1. Journal of medical genetics. 1991;28(12):878–80. Epub 1991/12/01. ; PubMed Central PMCID: PMCPMC1017168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–32. doi: 10.1038/nbt.2647 ; PubMed Central PMCID: PMCPMC3969858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmid-Burgk JL, Schmidt T, Gaidt MM, Pelka K, Latz E, Ebert TS, et al. OutKnocker: a web tool for rapid and simple genotyping of designer nuclease edited cell lines. Genome Res. 2014;24(10):1719–23. doi: 10.1101/gr.176701.114 ; PubMed Central PMCID: PMCPMC4199374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brady KG, Kelley TJ, Drumm ML. Examining basal chloride transport using the nasal potential difference response in a murine model. Am J Physiol Lung Cell Mol Physiol. 2001;281(5):L1173–9. doi: 10.1152/ajplung.2001.281.5.L1173 . [DOI] [PubMed] [Google Scholar]
- 36.Hodges CA, Cotton CU, Palmert MR, Drumm ML. Generation of a conditional null allele for Cftr in mice. Genesis. 2008;46(10):546–52. doi: 10.1002/dvg.20433 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141(5):1762–72. doi: 10.1053/j.gastro.2011.07.050 . [DOI] [PubMed] [Google Scholar]
- 38.Pattison AM, Blomain ES, Merlino DJ, Wang F, Crissey MA, Kraft CL, et al. Intestinal Enteroids Model Guanylate Cyclase C-Dependent Secretion Induced by Heat-Stable Enterotoxins. Infection and immunity. 2016;84(10):3083–91. Epub 2016/08/03. doi: 10.1128/IAI.00639-16 ; PubMed Central PMCID: PMCPMC5038068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dekkers JF, Wiegerinck CL, de Jonge HR, Bronsveld I, Janssens HM, de Winter-de Groot KM, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19(7):939–45. doi: 10.1038/nm.3201 . [DOI] [PubMed] [Google Scholar]
- 40.Kelley TJ, Cotton CU, Drumm ML. In vivo activation of CFTR-dependent chloride transport in murine airway epithelium by CNP. The American journal of physiology. 1997;273(5 Pt 1):L1065–72. . [DOI] [PubMed] [Google Scholar]
- 41.van Heeckeren AM, Schluchter MD, Drumm ML, Davis PB. Role of Cftr genotype in the response to chronic Pseudomonas aeruginosa lung infection in mice. Am J Physiol Lung Cell Mol Physiol. 2004;287(5):L944–52. Epub 2004/07/13. doi: 10.1152/ajplung.00387.2003 [pii]. . [DOI] [PubMed] [Google Scholar]
- 42.Clarke LL, Grubb BR, Gabriel SE, Smithies O, Koller BH, Boucher RC. Defective epithelial chloride transport in a gene-targeted mouse model of cystic fibrosis. Science. 1992;257(5073):1125–8. . [DOI] [PubMed] [Google Scholar]
- 43.Zeiher BG, Eichwald E, Zabner J, Smith JJ, Puga AP, McCray PB Jr., et al. A mouse model for the delta F508 allele of cystic fibrosis. J Clin Invest. 1995;96(4):2051–64. doi: 10.1172/JCI118253 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilke M, Buijs-Offerman RM, Aarbiou J, Colledge WH, Sheppard DN, Touqui L, et al. Mouse models of cystic fibrosis: phenotypic analysis and research applications. J Cyst Fibros. 2011;10 Suppl 2:S152–71. doi: 10.1016/S1569-1993(11)60020-9 . [DOI] [PubMed] [Google Scholar]
- 45.Hodges CA, Grady BR, Mishra K, Cotton CU, Drumm ML. Cystic fibrosis growth retardation is not correlated with loss of Cftr in the intestinal epithelium. Am J Physiol Gastrointest Liver Physiol. 2011;301(3):G528–36. Epub 2011/06/11. doi: ajpgi.00052.2011 [pii] doi: 10.1152/ajpgi.00052.2011 ; PubMed Central PMCID: PMC3174541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Delaney SJ, Alton EW, Smith SN, Lunn DP, Farley R, Lovelock PK, et al. Cystic fibrosis mice carrying the missense mutation G551D replicate human genotype-phenotype correlations. Embo J. 1996;15(5):955–63. . [PMC free article] [PubMed] [Google Scholar]
- 47.Nudelman I, Glikin D, Smolkin B, Hainrichson M, Belakhov V, Baasov T. Repairing faulty genes by aminoglycosides: development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorganic & medicinal chemistry. 2010;18(11):3735–46. Epub 2010/04/23. doi: 10.1016/j.bmc.2010.03.060 . [DOI] [PubMed] [Google Scholar]
- 48.Heier CR, DiDonato CJ. Translational readthrough by the aminoglycoside geneticin (G418) modulates SMN stability in vitro and improves motor function in SMA mice in vivo. Hum Mol Genet. 2009;18(7):1310–22. Epub 2009/01/20. doi: 10.1093/hmg/ddp030 ; PubMed Central PMCID: PMCPmc2655772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keeling KM, Xue X, Gunn G, Bedwell DM. Therapeutics based on stop codon readthrough. Annu Rev Genomics Hum Genet. 2014;15:371–94. doi: 10.1146/annurev-genom-091212-153527 ; PubMed Central PMCID: PMCPMC5304456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez B, Rodriguez-Pombo P, Ugarte M, Desviat LR. Readthrough strategies for therapeutic suppression of nonsense mutations in inherited metabolic disease. Mol Syndromol. 2012;3(5):230–6. doi: 10.1159/000343086 ; PubMed Central PMCID: PMCPMC3531923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sermet-Gaudelus I, Renouil M, Fajac A, Bidou L, Parbaille B, Pierrot S, et al. In vitro prediction of stop-codon suppression by intravenous gentamicin in patients with cystic fibrosis: a pilot study. BMC Med. 2007;5:5 doi: 10.1186/1741-7015-5-5 ; PubMed Central PMCID: PMCPMC1852113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baradaran-Heravi A, Niesser J, Balgi AD, Choi K, Zimmerman C, South AP, et al. Gentamicin B1 is a minor gentamicin component with major nonsense mutation suppression activity. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(13):3479–84. doi: 10.1073/pnas.1620982114 ; PubMed Central PMCID: PMCPMC5380045. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.McElroy SP, Nomura T, Torrie LS, Warbrick E, Gartner U, Wood G, et al. A lack of premature termination codon read-through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biol. 2013;11(6):e1001593 doi: 10.1371/journal.pbio.1001593 ; PubMed Central PMCID: PMCPMC3692445 the lack of efficacy of PTC124 by conducting our own drug discovery programme to discover and develop nonsense mutation read-through agents. Subsequent to the completion of this work, WHIM initiated a partnership with Glaxo SmithKline (GSK) on a closely related project. The work described herein was conducted prior to, and independently of, any interaction with GSK. No materials or support were received from GSK and no agreements are in place with GSK concerning the execution or publication of this work, although they were at one point involved in discussions about its publication. The collaboration with GSK has now ended. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(6):2064–9. Epub 2008/02/15. doi: 10.1073/pnas.0711795105 ; PubMed Central PMCID: PMCPmc2538881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet (London, England). 2008;372(9640):719–27. doi: 10.1016/S0140-6736(08)61168-X . [DOI] [PubMed] [Google Scholar]
- 56.Benhabiles H, Gonzalez-Hilarion S, Amand S, Bailly C, Prevotat A, Reix P, et al. Optimized approach for the identification of highly efficient correctors of nonsense mutations in human diseases. PLoS One. 2017;12(11):e0187930 doi: 10.1371/journal.pone.0187930 ; PubMed Central PMCID: PMCPMC5683606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gonzalez-Hilarion S, Beghyn T, Jia J, Debreuck N, Berte G, Mamchaoui K, et al. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J Rare Dis. 2012;7:58 doi: 10.1186/1750-1172-7-58 ; PubMed Central PMCID: PMCPMC3562214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang F, Shang H, Jordan NJ, Wong E, Mercadante D, Saltz J, et al. High-Throughput Screening for Readthrough Modulators of CFTR PTC Mutations. SLAS Technol. 2017;22(3):315–24. doi: 10.1177/2472630317692561 . [DOI] [PubMed] [Google Scholar]
- 59.Mutyam V, Du M, Xue X, Keeling KM, White EL, Bostwick JR, et al. Discovery of Clinically Approved Agents That Promote Suppression of Cystic Fibrosis Transmembrane Conductance Regulator Nonsense Mutations. American journal of respiratory and critical care medicine. 2016;194(9):1092–103. doi: 10.1164/rccm.201601-0154OC ; PubMed Central PMCID: PMCPMC5114449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153(4):910–8. doi: 10.1016/j.cell.2013.04.025 ; PubMed Central PMCID: PMCPMC3969854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31(9):822–6. doi: 10.1038/nbt.2623 ; PubMed Central PMCID: PMCPMC3773023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iyer V, Shen B, Zhang W, Hodgkins A, Keane T, Huang X, et al. Off-target mutations are rare in Cas9-modified mice. Nat Methods. 2015;12(6):479 doi: 10.1038/nmeth.3408 . [DOI] [PubMed] [Google Scholar]
- 63.Nakajima K, Kazuno AA, Kelsoe J, Nakanishi M, Takumi T, Kato T. Exome sequencing in the knockin mice generated using the CRISPR/Cas system. Sci Rep. 2016;6:34703 doi: 10.1038/srep34703 ; PubMed Central PMCID: PMCPMC5048150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hagemeijer MC, Siegwart DJ, Strug LJ, Cebotaru L, Torres MJ, Sofoluwe A, et al. Translational research to enable personalized treatment of cystic fibrosis. J Cyst Fibros. 2017. doi: 10.1016/j.jcf.2017.10.017 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.