

Abstract
AIM
To identify the effect and regulatory mechanism of amyloid β (Aβ) protein on retinal pigment epithelial (RPE) cells in cell proliferation and apoptosis, and clarify Aβ role in the pathogenesis of age-related macular degeneration (AMD).
METHODS
The model of Aβ25-35 protein cytotoxicity in RPE cell was successfully established to investigate the effect of Aβ protein on RPE cells in vitro. Based on Aβ protein, the specific inhibitors (HY-50682 or BAY11-7082) or activating agent (lipopolysaccharide) was used to analyze the regulatory mechanism of Aβ protein to RPE cells on cell proliferation and apoptosis by flow cytometry, real-time polymerase chain reaction, Western blotting, enzyme-linked immunosorbent assay and dual-luciferase reporter gene assay.
RESULTS
The number of RPE cells, treated with Aβ25-35 from 0.3 to 60 µmol/L, significantly reduce (P<0.01), and had the dose-dependent effect. Aβ protein 60 µmol/L inhibits the G1/S phase transition (P<0.01) and down-regulated cyclin E mRNA level (P<0.01). Similarly, Aβ25-35 induced a significant increase of cell apoptosis, accompanied by the significantly higher level of activated caspase 3 protein. Furthermore, nuclear factor-kappaB (NF-κB) activity and phosphorylated Iκ-Ba level would significantly lower in treated RPE cells. Using specific inhibitors or activating agent based on the Aβ, the cell numbers, NF-κB activity, phosphorylated Iκ-Ba level, receptor for advanced glycation endproducts (RAGE) gene expression levels, cyclin E mRNA level and activated caspase 3 level had accordingly changed by different methods, confirming that RAGE/NF-κB signaling pathway involved in the regulation of Aβ protein on RPE cell apoptosis and proliferation.
CONCLUSION
Aβ protein inhibits cell proliferation and activates apoptosis via inactivation of the RAGE/NF-κB signaling pathway in RPE cell.
Keywords: amyloid β protein, retinal pigment epithelial cells, proliferation, apoptosis, receptor for advanced glycation endproducts, nuclear factor-kappaB, age-related macular degeneration
INTRODUCTION
Age-related macular degeneration (AMD) is the global third major reason of vision loss, and, in many countries, it is the principal cause of nonreversible loss of vision in people over 60 years old[1]. Retinal pigment epithelial (RPE) cell is the pigmented cell layer between photoreceptors and choroidal vessels and maintains the structural and functional integrity of the retina. Early RPE dysfunction, which is an early and crucial event in AMD, progresses to cell damage and death, potentially leading to geographic atrophy, scotopic sensitivity, and photoreceptor cell death[2]–[3].
Amyloid β (Aβ), a peptide commonly associated with Alzheimer's disease (AD)[4], is the major element of drusen[5]–[6]. There is an evidence in primary neurons and the brains of AD patients that Aβ can activate signaling molecules and pathways, including nuclear factor-kappaB (NF-κB), which is well known for its wide roles in inflammation, immune responses, and regulation of cell division and apoptosis[7]–[10]. The eye and brain from one embryonic germ layer similar to pathogenesis and morphological manifestations of AMD and AD, mutual pathway of Aβ precursor protein were associated with the pathological aggregation of fibrillar Aβ protein pathological aggression and the β-amyloidopathy development in structural elements of the eye and brain[11]–[12]. The researchers demonstrated the keynote of AMD and AD is β-amyloidopathy. The formation of intracellular Aβ would induce the development of AMD or AD by leading to cytotoxicity, neurodegeneration and activating the pathological apoptosis.
Previous study has confirmed that an Aβ-mediated inflammatory response in retinal cells may be responsible for the disorder of early-stage AMD[13], and results in acute intoxication in RPE cells which is also associated with the pernicious onset of early-stage AMD[14]–[15]. Thus, it is believed that Aβ-induced chronic inflammation is a crucial pathogenic process in early-stage AMD. However, the detailed molecular mechanism by which Aβ contributes to the pathogenesis of AMD is not fully understood. Here, we tried to establish an in vitro RPE cell model with Aβ25-35 protein cytotoxicity to investigate the effects of Aβ on RPE cells and illuminate the molecular mechanisms underlying the pathogenesis of AMD.
MATERIALS AND METHODS
Cell Culture
Human RPE cells [also known as acute retinal pegment epitheliitis-19 (ARPE)] were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% fetal bovine serum and 100 U penicillin-streptomycin at 37°C in a 5% CO2 atmosphere.
Retinal Pigment Epithelial Cell Amyloid β25-35 Cytotoxicity Model
Of 1 mg Aβ25-35 protein (Sigma-Aldrich, St Louis, MO, USA) was dissolved in 0.36 mL RPMI 1640 medium to create a stock solution of 2.5 mmol/L Aβ25-35. The solution was incubated in a CO2 incubator for 5d and mixed daily to obtain the Aβ25-35 oligomer. ARPE cells were grown to 20% confluence and treated with Aβ25-35 oligomer at concentrations of 0 (medium), 0.3, 1, 5, 20, and 60 µmol/L for 48h. Aβ25-35 cytotoxicity was determined by quantifying cell numbers applying MTT assay. For some experiments, RPE cells were treated for 48h with 60 µmol/L Aβ25-35 in the presence or absence of 100 ng/mL HY-50682 and/or 5 µmol/L BAY11-7082 to specifically inhibit receptor for advanced glycation endproducts (RAGE) and Iκ-Bα, respectively, and the cells were then collected for analysis.
Cell Proliferation Assay
Cells were cultured in 96-well plates with a density of 1.0×104 cells/well and treated with Aβ25-35 in the presence or lack of HY-50682, BAY11-7082 or lipopolysaccharide (LPS) (5 µg/mL). Cell proliferation was measured by MTT reagent (5 mg/mL) and 180 µL medium to each well and incubating for 4h. Cell viability was identified by detecting absorption at 490 nm.
Flow Cytometric Analysis of the Cell Cycle
Cells were treated with Aβ25-35 in the presence or absence of BAY11-7082 for 36h. Cells were collected, mixed with 0.25% Triton X-100 and 5 µL propidium iodide (PI), and incubated for 30min at room temperature in the dark. Cells were resuspended in 0.5 mL of phosphate-buffered saline and analyzed immediately using a FACSCanto II flow cytometer.
Flow Cytometric Analysis of Apoptosis Cell
Apoptosis was analyzed by double staining of the cells with PI and annexin V. Cells were treated by Aβ25-35 in the presence or lack of BAY11-7082 for 36h, collected, and washed twice with ice-cold phosphate-buffered saline. The pellets were resuspended in 100 µL annexin V-binding, transferred to a 5-mL culture tube containing 5 µL annexin V-FITC, and mixed with 10 µL PI. The tube was vortexed gently and then incubated for 15min at room temperature in the dark. Next, 300 µL binding buffer was added, and the cells were analyzed instantly using a FACSCanto II flow cytometer.
Quantitative Reverse Transcription-polymerase Chain Reaction
Total ribonucleic acid (RNA) was extracted from the cells by TRIzol (Invitrogen; Carlsbad, CA, USA) according to instructions of the manufacturer. First strand cDNA was synthesized employing PrimeScript RT Master kit (Takara; Dalian, China). Quantitative reverse transcription-polymerase chain reaction (RT-Q-PCR) was carried out applying SYBR Green reagents on a 7500 real-time PCR system (Applied Biosystems; Foster City, CA, USA) as described previously. The specific primers that were utilized are listed in Table 1. For quantification, gene expression was normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression using the 2−ΔΔCt method.
Table 1. The specific primers for RT-Q-PCR in this study.
| Gene | Accession No. | Sequence |
| GAPDH | NM_001289746 | F: 5′-GGTCACCAGGGCTGCTTTTA-3′ |
| R: 5′-TTCCCGTTCTCAGCCTTGAC-3′ | ||
| CCNE | NM_001322262 | F: 5′-ATACTTGCTGCTTCGGCCTT-3′ |
| R: 5′-TCAGTTTTGAGCTCCCCGTC-3′ | ||
| RAGE | NM_001136 | F: 5′-GAAACTGAACACAGGCCGGA-3′ |
| R: 5′-ACACGGACTCGGTAGTTGGA-3′ |
GAPDH: Glyceraldehyde-3-phosphate dehydrogenase; CCNE: Cyclin E; RAGE: Receptor for advanced glycation endproducts.
Western Blotting Analysis
Antibodies against caspase 3 (CASP3) were purchased from Santa Cruz Biotechnology. Proteins were separated by SDS-PAGE applying a NuPAGE 12% Bis-Tris gel and shifted to a nitrocellulose membrane. The membrane was probed with anti-CASP3 primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies.
Enzyme-linked Immunosorbent Assay
Cells were treated by Aβ25-35 plus HY-50682 or BAY11-7082 for 36h, collected, and detected the expression level of RAGE, and the activity Iκ-Bα using enzyme-linked immunosorbent assay (ELISA) kits (R&D Systems, Minneapolis, MN, USA) according to the instructions of manufacturer.
Dual-luciferase Reporter Assay
The pNF-κB-Luc reporter plasmid expressing firefly luciferase was purchased from Beyotime Biotechnology (Shanghai, China). RPE cells were transiently transfected with pNF-κB-Luc or control plasmids. A Renilla luciferase plasmid was co-transfected to normalize expression, and IL-6 gene expression was used as the positive control. After 36h incubation, firefly and Renilla luciferase activities were detected applying the dual-luciferase reporter assay kit (Promega, USA) according to the instructions of manufacturer.
Statistical Analysis
Data were presented as mean±standard error (SE) of at least three independent experiments. Differences between groups were analyzed applying Student's t-test. A P-value of less than 0.05 was considered statistically significant.
RESULTS
Effect of Amyloid β Protein on Retinal Pigment Epithelial Cells
RPE cells were treated with Aβ25-35 (0.3, 1, 5, 20, or 60 µmol/L) for 48h, and the proliferation was detected by MTT assay. The results showed that Aβ25-35 significantly and dose-dependently inhibited number of RPE cells compared with medium-treated cells (P<0.05 or P<0.01; Figure 1A) from concentrations 0.3 to 60 µmol/L. These experiments successfully established the Aβ cytotoxicity model of RPE cells. And all subsequent experiments were performed with the optimal concentration of 60 µmol/L Aβ25-35.
Figure 1. Effect of Aβ on cell proliferation or apoptosis in RPE cells.

A: The number of RPE cells by MTT in different groups gradually and significantly reduced from 0.3 to 60 µmol/L Aβ25-35 compared to that without Aβ25-35; B, C: After treated with 60 µmol/L Aβ25-35 for 36h, the cell cycle significantly inhibited, and the cell apoptosis significantly prompted; D: After treated with 60 µmol/L Aβ25-35 for 36h, the CCNE mRNA level by RT-Q-PCR significantly down-regulated; E: The level of activated CASP3 by Western blotting in RPE cells with or without 60 µmol/L Aβ25-35 for 36h obviously increased. Data were means±SE (n=8 in MTT; or n=3 in others). aP<0.05, bP<0.01 with t-test.
The flow cytometric (FCM) assay also was performed to detect the phases of cell cycle. After treatment with 60 µmol/L Aβ25-35 compared with control cells without Aβ25-35, the percentage of RPE cells in S phase was significantly lower (P<0.05), but in G1 phase was substantially higher (P<0.05; Figure 1B), suggesting that Aβ25-35 inhibits the G1/S phase transition. Genetically, cyclin E (CCNE) mRNA level (measured by RT-Q-PCR analysis) were significantly down-regulated (P<0.01) in cells treated with 60 µmol/L Aβ25-35 for 36h compared with control cells (Figure 1C).
The observed inhibition of RPE cell number by Aβ also might reflect the induction of apoptosis. We detected the cell apoptosis by FCM assay. In RPE cells treated with 60 µmol/L Aβ25-35 compared with control cells, Aβ25-35 induced a significant increase in PI-negative and annexin V-positive cells (P<0.05; Figure 1D). Treatment with the cytotoxic agent etoposide was used as a positive control for apoptosis detection. The level of activated CASP3 protein (measured by Western blotting) was markedly increased in cells treated with 60 µmol/L Aβ25-35 for 36h compared with control cells (Figure 1E).
Mediation of Nuclear Factor-kappaB Pathway in the Regulation of Amyloid β25-35
It was verified if NF-κB signaling involved in the effects of Aβ25-35 on RPE cells in this study. The dual-luciferase reporter assay result has provided the direct evidence as shown in Figure 2A, NF-κB activity in RPE cells significantly declined (P<0.01) after treated with Aβ25-35. To confirm the above view, the detection of phosphorylated Iκ-Bα was further performed. The level of phosphorylated Iκ-Bα was significantly lower (P<0.05) in cells treated with Aβ25-35 than in control cells, and this decrease was further significantly enhanced (P<0.05) in cells with the co-treatment of Aβ25-35 plus BAY11-7082 (Figure 2B). After treated with Aβ25-35 plus BAY11-7082 or LPS for 48h, the cell number was detected. Compared to the cells only treated with Aβ25-35, the number was significantly lower (P<0.05) in cell treated by Aβ25-35 plus BAY11-7082 (Figure 2C), but significantly higher (P<0.05) in cell treated by Aβ25-35 plus LPS (Figure 2D).
Figure 2. NF-κB pathway medicating the effect of Aβ on RPE cells.

A: The level of NF-κB activity by ELISA had significantly decreased in RPE cells treated with 60 µmol/L Aβ25-35 for 36h; B: The level of Iκ-Bα phosphorylation was significantly decreased in RPE cells treated with 60 µmol/L Aβ25-35, and greatly decreased with 60 µmol/L Aβ25-35 plus 5 µg/mL LPS treatment; C, D: The number of RPE cells by MTT reduced after treated with 60 µmol/L Aβ25-35 plus 5 µmol/L BAY11-7082, but increase after treated with 60 µmol/L Aβ25-35 plus 5 µg/mL LPS; E: The CCNE mRNA level greatly down-regulated by 60 µmol/L Aβ25-35 plus 5 µmol/L BAY11-7082 for 36h, but significantly up-regulated by 60 µmol/L Aβ25-35 plus 5 µg/mL LPS for 36h; F, G: The level of activated CASP3 by Western blotting in RPE cells with 60 µmol/L Aβ25-35 plus 5 µmol/L BAY11-7082, obviously increased, but obviously decreased on the basis of 60 µmol/L Aβ25-35 addition plus 5 µg/mL LPS for 36h. Data are means±SE (n=8 in MTT; or n=3 in others). aP<0.05; bP<0.01 with t-test.
Genetically, using RPE cells with Aβ25-35 in the presence of BAY11-7082 treated for 36h, the changes in CCNE mRNA and activated CASP3 protein levels were further detected by RT-Q-PCR and Western blotting. Compared with cells treated with Aβ25-35 alone, cells co-treated with Aβ25-35 plus BAY11-7082 had significantly lower level of CCNE mRNA (P<0.05; Figure 2E), and activated CASP3 protein level had obviously increased (Figure 2F). However, using RPE cells with Aβ25-35 in the presence of LPS treated for 36h, compared with cells treated with Aβ25-35 alone, cells co-treated with Aβ25-35 plus LPS had significantly higher levels of CCNE mRNA (P<0.05; Figure 2E), and activated CASP3 protein levels had obviously decreased (Figure 2G).
Effect of Amyloid β on Retinal Pigment Epithelial Cell Through Receptor for Advanced Glycation Endproducts/nuclear Factor-kappaB Pathway
To determine whether RAGE involved in the regulation of Aβ25-35 in RPE cells, the levels of RAGE mRNA and protein were analyzed. RPE cells treated with 60 µmol/L Aβ25-35 for 36h, the expression of RAGE mRNA and protein had significantly down-regulated (P<0.01) compared with control cells. In addition, RAGE mRNA and protein levels showed greatly down-regulated (P<0.05) in cells co-treated for 36h with Aβ25-35 plus HY-50682 compared with cells treated with Aβ25-35. Similarly, the expression levels of RAGE mRNA and protein were significantly down-regulated (P<0.01, P<0.05) in RPE cells treated with Aβ25-35 and BAY11-7082 to inhibit NF-κB signaling pathuay (Figure 3A, 3B).
Figure 3. RAGE interacting with the NF-κB pathway to medicate the effect of Aβ on RPE cells.

A, B: Both of mRNA and protein expression levels of RAGE gene by RT-Q-PCR and ELISA had the different reduction in RPE cells with no treatment and other different treatments (60 µmol/L Aβ25-35, 60 µmol/L Aβ25-35 plus 100 ng/mL HY-50682, 60 µmol/L Aβ25-35 plus 5 µmol/L BAY11-7082); C: The activity of NF-κB by the dual-luciferase reporter assay reduced in RPE cells treated with 60 µmol/L Aβ25-35 plus 100 ng/mL HY-50682 and only with 60 µmol/L Aβ25-35; D: The level of phosphorylated Iκ-Bα by ELISA in RPE cells decreased in RPE cells treated with 100 ng/mL HY-50682 on the basis of 60 µmol/L Aβ25-35; E: The number of RPE cells by MTT, which treated with 100 ng/mL HY-50682 on the basis of 60 µmol/L Aβ25-35, was greatly lower. Data were means±SE (n=8 in MTT; or n=3 in others). aP<0.05; bP<0.01 with t-test.
Further, we found that the results by the dual-luciferase reporter assay confirmed that NF-κB activity was greatly reduced after RAGE inhibition based on the treatment of Aβ25-35 (P<0.05; Figure 3C). Similarly, the phosphorylation of Iκ-Bα was significantly lower (P<0.01) in cells co-treated with Aβ25-35 and HY-50682 than in cells treated with Aβ25-35 alone (P<0.01; Figure 3D). Finally, RPE cell number was significantly lower (P<0.05) after combined treatment with Aβ25-35 plus the RAGE inhibitor HY-50682 than only treatment with Aβ25-35 (P<0.05; Figure 3E). Considered to all results, the proposed molecular regulatory network is presented in Figure 4.
Figure 4. The underlying mechanism of Aβ protein induction AMD in RPE cell.
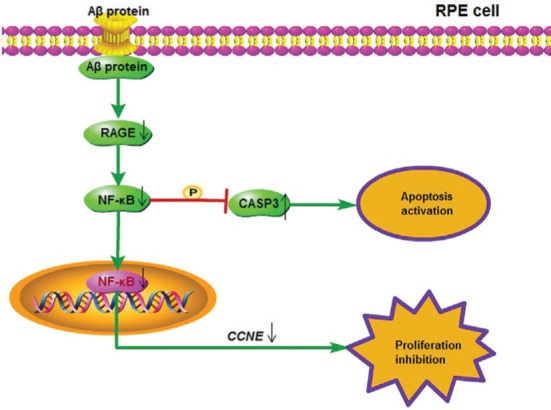
Aβ protein inhibited cell proliferation and activated apoptosis of RPE cells via the RAGE/NF-κB signaling pathway. Aβ protein could down-regulate the mRNA expression of CCNE to hinder the cell cycle, and activate CASP3 to enhance the apoptosis by the decrease of the RAGE/NF-κB activities.
DISCUSSION
AMD is the principal cause of irreversible vision loss in people over the age of 60y[1]. Aβ has the acute intoxication in RPE cells which is associated with the early-stage AMD[14]–[15], but the detailed molecular mechanism by which Aβ contributes to AMD's pathogenesis is not fully understood.
In present study, we successfully established an RPE cell toxicity model by adding different concentrations of Aβ25-35 protein, and Aβ25-35 would induce the significant reduction of cell number with a dose-dependent manner from 0.3 to 60 µmol/L. The result by FCM revealed that Aβ25-35 protein would inhibit the proliferation and activate apoptosis of RPE cell. CCNE and activated CASP3, respectively as the key factor in cell proliferation or apoptosis[16]–[17], were detected to verify the effect of Aβ. The changes of CCNE mRNA and activated CASP3 levels in treated cells with 60 µmol/L Aβ25-35 had supported the view on Aβ protein to RPE cells.
Considering the important role of NF-κB signaling pathway in AD induced by Aβ[7]–[10], the NF-κB pathway also was analyzed. Here, NF-κB level and Iκ-Bα activity were significantly lower in RPE cells after treated with 60 µmol/L Aβ25-35, suggesting that Aβ inactivated NF-κB signaling pathway to contribute to the effect of cell proliferation or apoptosis in RPE cells. Further, RPE cells were treated with BAY11-7082 (a specific NF-κB pathway inhibitor) on the basis of Aβ25-35 (60 µmol/L), the cell number and CCNE mRNA level would decrease with a greater extent, and the activated CASP3 level would significant increase. However, RPE cells were treated with LPS (the NF-κB pathway activating agent) on the basis of Aβ25-35 (60 µmol/L), the cell number, CCNE mRNA level and activated CASP3 level had the opposite change. That is to say, LPS could relieve the cytotoxicity of RPE cells by activating the NF-κB pathway. Generally speaking, it was confirmed that Aβ could inhibit the proliferation and promote the apoptosis of RPE cell by inactivating the NF-κB signaling pathway.
As previous reported, there is a close association between RAGE and NF-κB signaling[18]–[20]. Therefore, we also investigated whether RAGE involved in the effects of Aβ25-35 on RPE cells. For RPE cells were treated with 60 µmol/L Aβ25-35, RAGE mRNA and protein levels would significantly down-regulate, hinting Aβ would regulate the expression of RAGE gene in RPE cells. In RPE cells treated with HY-50682 (a specific RAGE inhibitor) on the basis of 60 µmol/L Aβ25-35, accompanied by blocking of RAGE, the phosphorylation of Iκ-Bα was significantly lower, and NF-κB activity was greatly reduced. On the contrary, the levels of RAGE mRNA and protein also significantly down-regulated after NF-κB pathway blocked. In addition, after RPE cells treated with Aβ25-35 plus HY-50682 for 48h, the cell number was significantly lower, and greatly reduced by treatment with Aβ25-35 plus HY-50682 and BAY11-7082. These results fully confirmed that RAGE involved in the effects of Aβ25-35 on RPE cells, and had a interaction cooperation relationship with NF-κB pathway in RPE cells.
In conclusion, it is the first to illustrate the underlying regulatory mechanism that Aβ inhibited cell proliferation and activated apoptosis through RAGE/NF-κB signaling pathway in RPE cells (Figure 4). These results suggest that Aβ may be an important factor in the induction of AMD. Further work will be required to determine the pathways and mechanisms that mediate and regulate the toxic effect of Aβ on RPE cells.
Acknowledgments
Conflicts of Interest: Ye Z, None; He SZ, None; Li ZH, None.
REFERENCES
- 1.Mehta S. Age-related macular degeneration. Prim Care. 2015;42(3):377–391. doi: 10.1016/j.pop.2015.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Shaw PX, Stiles T, Douglas C, Ho D, Fan W, Du H, Xiao X. Oxidative stress, innate immunity, and age-related macular degeneration. AIMS Mol Sci. 2016;3(2):196–221. doi: 10.3934/molsci.2016.2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Algvere PV, Kvanta A, Seregard S. Drusen maculopathy: a risk factor for visual deterioration. Acta Ophthalmol. 2016;94(5):427–433. doi: 10.1111/aos.13011. [DOI] [PubMed] [Google Scholar]
- 4.Tonnies E, Trushina E. Oxidative stress, synaptic dysfunction, and Alzheimer's disease. J Alzheimers Dis. 2017;57(4):1105–1121. doi: 10.3233/JAD-161088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao T, Gao J, Van J, To E, Wang A, Cao S, Cui JZ, Guo JP, Lee M, McGeer PL, Matsubara JA. Age-related increases in amyloid beta and membrane attack complex: evidence of inflammasome activation in the rodent eye. J Neuroinflammation. 2015;12:121. doi: 10.1186/s12974-015-0337-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson DH, Talaga KC, Rivest AJ, Barron E, Hageman GS, Johnson LV. Characterization of beta amyloid assemblies in drusen: the deposits associated with aging and age-related macular degeneration. Exp Eye Res. 2004;78(2):243–256. doi: 10.1016/j.exer.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 7.Snow WM, Albensi BC. Neuronal gene targets of NF-κB and their dysregulation in Alzheimer's disease. Front Mol Neurosci. 2016;9:118. doi: 10.3389/fnmol.2016.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi S, Liang D, Chen Y, Xie Y, Wang Y, Wang L, Wang Z, Qiao Z. Gx-50 reduces β-amyloid-induced TNF-α, IL-1β, NO, and PGE2 expression and inhibits NF-κB signaling in a mouse model of Alzheimer's disease. Eur J Immunol. 2016;46(3):665–676. doi: 10.1002/eji.201545855. [DOI] [PubMed] [Google Scholar]
- 9.Srinivasan M, Lahiri DK. Significance of NF-κB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer's disease and multiple sclerosis. Expert Opin Ther Targets. 2015;19(4):471–487. doi: 10.1517/14728222.2014.989834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi ZM, Han YW, Han XH, Zhang K, Chang YN, Hu ZM, Qi HX, Ting C, Zhen Z, Hong W. Upstream regulators and downstream effectors of NF-κB in Alzheimer's disease. J Neurol Sci. 2016;366:127–134. doi: 10.1016/j.jns.2016.05.022. [DOI] [PubMed] [Google Scholar]
- 11.Ermilov VV, Nesterova AA. β-amyloidopathy in the pathogenesis of age-related macular degeneration in correlation with neurodegenerative diseases. Adv Exp Med Biol. 2016;854:119–125. doi: 10.1007/978-3-319-17121-0_17. [DOI] [PubMed] [Google Scholar]
- 12.Zhao Y, Bhattacharjee S, Jones BM, Hill JM, Clement C, Sambamurti K, Dua P, Lukiw WJ. Beta-amyloid precursor protein (βAPP) processing in Alzheimer's disease (AD) and age-related macular degeneration (AMD) Mol Neurobiol. 2015;52(1):533–544. doi: 10.1007/s12035-014-8886-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu RT, Gao J, Cao S, Sandhu N, Cui JZ, Chou CL, Fang E, Matsubara JA. Inflammatory mediators induced by amyloid-beta in the retinaand RPE in vivo: implications for inflammasome activation in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2013;54(3):2225–2237. doi: 10.1167/iovs.12-10849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tesco G, Koh YH, Tanzi RE. Caspase activation increases beta-amyloid generation independently of caspase cleavage of the beta-amyloid precursor protein (APP) J Biol Chem. 2003;278(46):46074–46080. doi: 10.1074/jbc.M307809200. [DOI] [PubMed] [Google Scholar]
- 15.Ding JD, Johnson LV, Herrmann R, Farsiu S, Smith SG, Groelle M, Mace BE, Sullivan P, Jamison JA, Kelly U, Harrabi O, Bollini SS, Dilley J, Kobayashi D, Kuang B, Li W, Pons J, Lin JC, Bowes Rickman C. Anti-amyloid therapy protects against retinal pigmented epithelium damage and vision loss in a model of age-related macular degeneration. Proc Natl Acad Sci U S A. 2011;108(28):E279–E287. doi: 10.1073/pnas.1100901108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M, Ouyang L, Zheng Z, Xiang D, Ti A, Li L, Dan Y, Yu C, Li W. E3 ubiquitin ligase FBW7α inhibits cholangiocarcinoma cell proliferation by downregulating c-Myc and cyclin E. Oncol Rep. 2017;37(3):1627–1636. doi: 10.3892/or.2017.5432. [DOI] [PubMed] [Google Scholar]
- 17.Pu X, Storr SJ, Zhang Y, Rakha EA, Green AR, Ellis IO, Martin SG. Caspase-3 and caspase-8 expression in breast cancer:caspase-3 is associated with survival. Apoptosis. 2017;22(3):357–368. doi: 10.1007/s10495-016-1323-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park R, Kook SY, Park JC, Mook-Jung I. Aβ1-42 reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-κB signaling. Cell Death Dis. 2014;5:e1299. doi: 10.1038/cddis.2014.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan X, Gu J, Zhao B, Wang S, Yuan J, Wang C, Chen J, Liu J, Feng L, Jia X. Ginseng improves cognitive deficit via the RAGE/NF-κB pathway in advanced glycation end product-induced rats. J Ginseng Res. 2015;39(2):116–124. doi: 10.1016/j.jgr.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bortolotto V, Grilli M. Every cloud has a silver lining: proneurogenic effects of Aβ oligomers and HMGB-1 via activation of the RAGE-NF-κB axis. CNS Neurol Disord Drug Targets. 2017;16(10):1066–1079. doi: 10.2174/1871527315666160803153459. [DOI] [PubMed] [Google Scholar]