

Abstract
Short-term synaptic plasticity contributes to many computations in the brain and allows synapses to keep a finite record of recent activity. Here we have investigated the mechanisms underlying an intriguing form of short-term plasticity termed labile LTP, at hippocampal and PFC synapses in male rats and male and female mice. In the hippocampus, labile LTP is triggered by high-frequency activation of presynaptic axons and is rapidly discharged with further activation of those axons. However, if the synapses are quiescent, they remain potentiated until further presynaptic activation. To distinguish labile LTP from NMDAR-dependent forms of potentiation, we blocked NMDARs in all experiments. Labile LTP was synapse-specific and was accompanied by a decreased paired pulse ratio, consistent with an increased release probability. Presynaptic Ca2+ and protein kinase activation during the tetanus appeared to be required for its initiation. Labile LTP was not reversed by a PKC inhibitor and did not require either RIM1α or synaptotagmin-7, proteins implicated in other forms of presynaptic short-term plasticity. Similar NMDAR-independent potentiation could be elicited at synapses in mPFC. Labile LTP allows for rapid information storage that is erased under controlled circumstances and could have a role in a variety of hippocampal and prefrontal cortical computations related to short-term memory.
SIGNIFICANCE STATEMENT Changes in synaptic strength are thought to represent information storage relevant to particular nervous system tasks. A single synapse can exhibit multiple overlapping forms of plasticity that shape information transfer from presynaptic to postsynaptic neurons. Here we investigate the mechanisms underlying labile LTP, an NMDAR-independent form of plasticity induced at hippocampal synapses. The potentiation is maintained for long periods as long as the synapses are infrequently active, but with regular activation, the synapses are depotentiated. Similar NMDAR-independent potentiation can also be induced at L2/3-to-L5 synapses in mPFC. Labile LTP requires a rise in presynaptic Ca2+ and protein kinase activation but is unaffected in RIM1α or synaptotagmin-7 mutant mice. Labile LTP may contribute to short-term or working memory in hippocampus and mPFC.
Keywords: hippocampus, LTP, memory, PFC, synaptic plasticity, working memory
Introduction
Changes in synaptic strength are thought to represent information storage relevant to particular nervous system tasks. Evidence accumulated over many years supports an important role for NMDAR-dependent LTP in learning and memory, as well as in other neuroadaptations to external stimuli (brain development, addiction, nociception) (Malenka and Bear, 2004; Nicoll, 2017). Perhaps the most widely studied synapse in the vertebrate brain is that between hippocampal CA3 and CA1 pyramidal cells. Specific deletion of NMDARs at these synapses produces deficits in spatial learning tasks and disruption of place fields (Bannerman et al., 1995; Tsien et al., 1996; Nakazawa et al., 2004; Place et al., 2012; Morris, 2013; Morris et al., 2013).
A single synapse can exhibit multiple overlapping forms of plasticity that shape information transfer from presynaptic to postsynaptic neuron. Short-term plasticity occurs at nearly all synapses, even after as few as two presynaptic action potentials. Facilitation, short-term depression, endocannabinoid-mediated short-term depression, and post-tetanic potentiation (PTP) modulate responses based on the recent history of activation of those synapses (Zucker and Regehr, 2002; Chevaleyre et al., 2006; Fioravante and Regehr, 2011; Jackman and Regehr, 2017), and specific synapses often have a characteristic short-term plasticity signature (e.g., rapidly developing depression during repeated action potentials). On a short time scale, many cognitive tasks require that a neural ensemble be persistently active or primed to retain an item of information on a seconds to tens of seconds time scale. Pattern completion, maintenance of temporal order, and working memory all require information to be retained in response to transient sensory input (Funahashi et al., 1989; Goldman-Rakic, 1995; Abbott and Regehr, 2004; Major and Tank, 2004; Kaminśki et al., 2017; Wolff et al., 2017). It has been theorized that this is achieved by persistent firing of relevant neurons, but recent work in rodents and humans suggests that synaptic potentiation may also contribute in an “activity-silent” manner (Funahashi et al., 1989; Barak and Tsodyks, 2007; Hansel and Mato, 2013; Wolff et al., 2015; Rose et al., 2016).
CA3-to-CA1 hippocampal synapses exhibit short-term facilitation, PTP, endocannabinoid depression, and NMDAR-dependent LTP (Ohno-Shosaku et al., 2002; Fioravante and Regehr, 2011; Herring and Nicoll, 2016). At the same synapses, we have now characterized a form of synaptic potentiation that is initiated by high-frequency activity of CA3 pyramidal cell axons and is apparently stored for seconds, or for up to hours as long as the synapse is stimulated infrequently. However, the synapses are depotentiated when activation at higher frequency is resumed. This plasticity, which has been called transient LTP or labile LTP, has been noted previously (Xiao et al., 1996; Volianskis and Jensen, 2003), but is often not distinguished in LTP experiments because typical test stimulus frequency causes relatively rapid depotentiation. Labile LTP has properties that might be useful in retaining information for short periods, but also allows synaptic strength to reset rapidly after use. Here we have probed the frequency dependence, synapse specificity, and molecular and pharmacological properties of labile LTP.
Materials and Methods
Slice preparation.
All experiments were performed strictly adhering to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and as approved by the Brown Institutional Animal Care and Use Committee. Male Sprague Dawley rats were used; in some experiments, RIM1α knock-out mice (JAX B6;129P2Rims1tm1Sud/J) and synaptotagmin-7 (syt-7) knock-out mice (JAX B6.129.S1-Syt7tm1Nan/J) were used. RIM1α knock-out mice were bred as heterozygotes crossed to heterozygotes, syt-7 knock-out mice were bred as homozygotes to homozygotes, and homozygote results are reported here. Each strain had been backcrossed onto a C57BL/6J background for at least 7 generations. Both male and female mice were used. Mice (p28–40) or male Sprague Dawley rats (Charles River, p28–40), were deeply anesthetized with isoflurane and decapitated. Coronal brain slices were prepared as described previously (Kauer, 1999; Mair and Kauer, 2007). The 400-μm-thick slices (300 μm for whole-cell recordings) were placed into ice-cold ACSF containing the following (in mm): 126 NaCl, 26 NaHCO3, 2.5 KCl, 1.3 NaH2PO4, 2.5 CaCl2, 1.2 MgSO4, or 124 NaCl, 3.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 2 MgSO4, 26 NaHCO3, saturated with 95% O2-5%CO2 (Volianskis and Jensen, 2003). No differences were observed in the two ACSF compositions. For prefrontal cortical slices, two slices per animal ∼200–300 μm caudal to the olfactory bulb and just rostral to the basal ganglia were prepared (plates 8–10, Paxinos and Watson, 2013). Slices were stored for 1–6 h submerged in oxygenated ACSF at room temperature.
Electrophysiological recordings.
Slices were placed in a submerged recording chamber constantly perfused at ∼2–3 ml/min with bubbled ACSF and heated to 29°C–31°C. In all experiments, 50 μm d-APV was included in the ACSF to block NMDARs. Field potentials were evoked and recorded from area CA1 as described previously (Kauer, 1999); and in mPFC slices, the stimulating electrode was placed in layer 2/3 and the recording electrode in layer 5 (Mair and Kauer, 2007). The recording electrode was filled with 2N NaCl and placed in stratum radiatum. In experiments for Figure 2, stimulating electrodes were positioned on either side of the recording electrode (all in stratum radiatum), and stimulation was delivered alternately to each input. Paired pulses were evoked at 50 ms interstimulus interval. Field potentials were recorded with an Axopatch or AM Systems amplifier and Brownlee postamplifier and filtered at 3 kHz for storage on a PC. The initial slope of the field potential was measured with custom software written for Labview and kindly donated by Dr. Daniel Madison. Amplitudes rather than slopes of mPFC fields were measured because of their rapid time course. Baseline stimulation frequency was 1/45 s (0.022 Hz) except as noted. High-frequency stimulation (HFS) was delivered at 100 Hz for 1 s at 1.5 times the test current intensity; this train was repeated twice, 20 s apart except for data in Figure 2 in which the train was repeated 4 times. After HFS in most experiments, stimulation was not resumed for at least 5 min and was then resumed at once per 5 min (0.00334 Hz) except as noted.
Figure 2.

Labile LTP is synapse-specific and is maintained if post-HFS stimuli are delivered at low frequency. a, Example experiment in which the baseline fEPSPs were evoked every 45 s. Post-HFS after a 10 min pause, stimulation was resumed at once every 5 min. Top, fEPSP slope. Bottom, PPR. Inset, Five averaged fEPSPs before (1, black) and at the end of the recording (2, red). Calibration: 10 ms, 0.5 mV. b, Top, Diagram of recording arrangement with a stimulating electrode on either side of the extracellular recording site. Bottom, Experiments using two stimulation pathways. Pathway 1, fEPSP slope/PPR from the pathway that received HFS (arrow); Pathway 2, fEPSP slope/PPR from the control pathway in the same slice that did not receive HFS (n = 9, fEPSP in HFS vs control pathway, p = 0.021; PPR in HFS vs control pathway, p = 0.0394, unpaired t tests). Experiments that had potentiation of >10% in the control pathway (perhaps when the two pathways were not truly independent) were excluded from the dataset to emphasize that labile LTP is still observed in the HFS pathway even without potentiation of the control pathway.
For extracellular EGTA-AM experiments, slices were either incubated for 1 h before and during recording in 200 μm EGTA-AM or 200 μm EGTA-AM was added to the bath solution after recording began. For intracellular EGTA experiments, whole-cell recordings were made from visually identified CA1 pyramidal cells as described previously (Gibson et al., 2008). The recording solution was identical to ACSF except that 50 μm picrotoxin was included to block GABAA receptors. Patch pipettes were filled with internal recording solution containing the following (in mm): 117 K gluconate, 2.8 NaCl, 5 MgCl2, 20 HEPES, 2 ATP-Na+, 0.3 GTP-Na+, and 15 EGTA. Neurons were held at −70 mV throughout the recording period, and for at least 20 min before HFS to allow EGTA to diffuse to synaptic sites, a protocol known to attenuate various forms of LTP dependent upon postsynaptic Ca2+ (Lenz and Alger, 1999; Cho et al., 2001; Lapointe et al., 2004). The cell input resistance and series resistance were monitored throughout each experiment; cells were discarded if these values changed by >10% during the experiment. Recordings were amplified and low-pass filtered at 3 kHz and digitally sampled using pCLAMP software (Molecular Devices). EPSC amplitudes were measured using pClamp.
Experimental design and statistical analysis.
Because of the low stimulation frequencies, paired pulse ratios (PPRs) were calculated for each time point as slope fEPSP2/fEPSC1 (hippocampus), fEPSP2/fEPSP1 (PFC), or amplitude EPSC2/EPSC1. All results are expressed as the mean ± SEM. The slope or amplitude of the average of 15 fEPSPs/EPSCs before HFS was compared with all fEPSPs/EPSCs recorded after tetanus for statistical testing (7–14 points), unless otherwise noted. For Figure 2 experiments using alternately stimulated pathways, experiments in which the untetanized pathway potentiated >10% were excluded to demonstrate that even when no significant potentiation occurs on the untetanized pathway, significant labile LTP is still observed. Statistical significance was determined using paired t tests on raw data before and after a manipulation except where noted, and p values <0.05 were considered significant and are denoted with an asterisk. N values were based on previous experience and represent the number of slices used, except for experiments with knock-out mice for which n equals number of mice. Experiments using RIM1α knock-out mice were performed blind to genotype; CA3-CA1 synapses from syt-7 mice lack paired pulse facilitation, making blinding imperfect.
Materials.
GF109203X (Tocris Bioscience) and EGTA-AM (Invitrogen) were dissolved in DMSO and diluted 1:1000 for use; 0.1% DMSO had no effect on labile LTP (data not shown). All other reagents were from Sigma or Tocris Bioscience and were dissolved in water or ACSF.
Results
HFS of CA3 afferents in the hippocampal slice is followed by transient potentiation (labile LTP) (Volianskis and Jensen, 2003). Although NMDAR antagonists were reported to prevent labile LTP induction (Volianskis and Jensen, 2003; Volianskis et al., 2013b, 2015), we find instead that, even with NMDARs blocked using 50 μm d-APV, robust labile LTP was induced (Fig. 1). This d-APV-resistant transient potentiation was still observed even with a pause in stimulation after HFS for 10–20 min, a time point when short-term facilitation, depression, and PTP have recovered to control levels, confirming previous observations (Volianskis and Jensen, 2003). The onset and subsequent decrement of labile LTP after tetanus were paralleled by a decreased PPR that gradually returned to baseline values, consistent with the idea that labile LTP results from a transient increase in presynaptic transmitter release (compare Volianskis and Jensen, 2003). Blocking NMDARs distinguishes labile LTP from common forms of LTP and NMDAR-dependent short-term potentiation (Collingridge et al., 1983; Kauer et al., 1988; Malenka, 1991; Park et al., 2014). All of the subsequent experiments were performed in the presence of 50 μm d-APV to isolate NMDAR-independent synaptic plasticity.
Figure 1.

Labile LTP can be triggered by HFS of CA3-CA1 synapses in the presence of 50 μm d-APV. a, fEPSPs in hippocampal area CA1 were evoked every 10 s. HFS (arrow; 100 Hz, 1 s repeated twice) elicited labile potentiation observable even after a 10 min pause without stimulation. Example from one hippocampal slice: Top (black), fEPSP; Bottom (blue), PPR. Inset, Average of 5 fEPSPs before (1, black) and just after resumption of stimulation after tetanus (2, red). Calibration: 10 ms, 0.5 mV. b, Top, Average of 9 similar experiments showing mean ± SEM. Bottom, Raw data showing fEPSP slopes and PPR from each experiment (n = 9); fEPSPs, p = 0.014 for the first 2 min after resuming stimulation (paired t test). PPR, p = 0.0041 at 2 min after tetanus (paired t test). *p < 0.05. In this and all experiments, 50 μm d-APV was present for at least 10 min before HFS.
After the tetanus, if the stimulation frequency was reduced to a very low rate (once per 5 min), labile LTP was maintained, no longer exhibiting any decrement over periods of ≥50 min after tetanus (Fig. 2a). Again, the potentiation was accompanied by a significant drop in the PPR. Control pathways recorded simultaneously in the same slices did not exhibit these changes in EPSP slope or PPR (Fig. 2b), demonstrating that labile LTP is synapse-specific, apparently not induced at neighboring synapses that do not receive HFS. Together, these data suggest that brief repetitive activation of presynaptic afferents allows these activated synapses to retain information for long periods, but only if the synapses are activated infrequently after the triggering event.
Do other excitatory synapses with a likely role in memory formation also exhibit labile LTP? We next tested excitatory synapses in slices of mPFC, a brain region strongly implicated in working memory. In the presence of d-APV, fEPSPs recorded in layer 5 while stimulating layer 2–3 exhibited modest potentiation 2 min after HFS when stimulated at 0.1 Hz (Fig. 3a–c). If instead, these synapses were stimulated infrequently 10 min after HFS, they remained potentiated (Fig. 3d–f). Similarly to hippocampal synapses, in the presence of d-APV, HFS triggered stabile potentiation accompanied by decreased PPR. These data suggest that a form of potentiation similar to labile LTP also exists at mPFC synapses.
Figure 3.

In PFC, HFS in d-APV elicits potentiation similar to hippocampal labile LTP. fEPSPs evoked in layer 2/3 and recorded in layer 5 of rat mPFC. a, Diagram of electrode placements. b, Average of 5 experiments illustrating transient potentiation of PFC fEPSPs after HFS (arrow) followed by a 10 min pause in stimulation; stimulation was at 0.1 Hz throughout. c, Raw data for these experiments (fEPSPs, baseline vs first 2 min after HFS, p = 0.031; PPR, baseline vs first 2 min after HFS, p = 0.313). d, Example experiment showing that HFS triggers potentiation when stimulation is resumed at once per 5 min following a 10 min pause. Inset, Average of 5 fEPSPs before (1, black) and during potentiation (2, red). Calibration: 10 ms, 0.5 mV. e, Average of 18 similar experiments. f, Raw data for each experiment in e (fEPSP amplitude, p = 0.007; PPR, p = 0.009; paired t tests). *p < 0.05.
A remarkable feature of the persistent potentiation in hippocampus is that, once induced and monitored using very low-frequency stimulation, increasing the stimulus frequency returned the EPSPs and PPRs toward control levels (Fig. 4). One hypothesis to explain the origin of labile LTP is that the tetanus causes nascent synapses to form and become functional; these newly functional synapses might be more susceptible to presynaptic depression caused by vesicle depletion, and thus might seem to disappear upon resumption of higher stimulus frequencies (Xiao et al., 2004). To test this idea, we established labile LTP and then increased the stimulus frequency to produce depotentiation; we then halted stimulation for 10 min. Upon resuming stimulation at very low frequency, however, we did not recover potentiation (Fig. 4a–c). Our results suggest that nascent weak synapses or transient vesicle depletion cannot account for the depotentiation of synapses expressing labile LTP, but indicate instead that, once presynaptic action potential frequency reaches a certain threshold, the synapses are truly depotentiated.
Figure 4.

Synapses with labile LTP are depotentiated by stimulation at a higher frequency. CA3-CA1 synapses were stimulated once per 45 s before delivering HFS (arrow). After a 20 min pause, stimuli were resumed at 1 per 5 min for 50 min (blue bar); next, stimulus frequency was increased to 0.1 Hz for 10 min (magenta bar), and then after a 10 min pause, stimulus frequency was returned to once every 5 min (blue bar). a, Example experiment demonstrating that labile LTP triggered by HFS returns to near baseline values after stimulation at 0.1 Hz. Top, Black symbols represent fEPSP. Bottom, Blue symbols represent PPR. Inset left, Average of 3 fEPSPs before (1, black) and during labile LTP (2, red). Inset right, Average of 3 fEPSPs during labile LTP and after 0.1 Hz stimulation (3, green, “depotentiation”). b, Average of 7 similar experiments. Top, Black symbols represent fEPSP slope. Bottom, Blue symbols represent PPR. c, Raw data showing fEPSP slopes (F(1,8) = 8.0, p = 0.014; baseline vs labile LTP: p = 0.0104; baseline vs after 0.1 Hz: p = 0.95) and PPR from each experiment (F(1,8) = 9.41, p = 0.0105; baseline vs labile LTP p = 0.025; baseline vs after 0.1 Hz, p = 0.039; repeated-measures one-way ANOVA followed by Dunnett's multiple-comparisons test). *p < 0.05.
The decreased PPR accompanying labile LTP indicates persistently increased presynaptic release (Felmy and von Gersdorff, 2006). Similarly, facilitation and PTP are also triggered by presynaptic trains of action potentials, are accompanied by a decreased PPR, and require elevated Ca2+ in the presynaptic terminal (Fioravante and Regehr, 2011). To test whether Ca2+ during the train is necessary for labile LTP, we next chelated Ca2+ using the membrane-permeant compound, EGTA-AM. As reported previously for hippocampal synapses (Ouanounou et al., 1996; Salin et al., 1996b; Nanou et al., 2016), bath application of EGTA-AM (200 μm) only modestly depressed the initial fEPSP but strongly attenuated paired pulse facilitation (Fig. 5a), presumably by decreasing the presynaptic Ca2+ required for facilitation (Atluri and Regehr, 1996). In the presence of EGTA-AM, labile LTP was blocked; and in some experiments, synaptic depression was observed after HFS (Fig. 5b, top). To ensure that EGTA-AM had reached a functional steady state, we instead preincubated slices for 1 h before the start of recording; in the continued presence of EGTA-AM, labile LTP was not induced (Fig. 5b, bottom). EGTA-AM will chelate Ca2+ in both presynaptic and postsynaptic neurons in these experiments. We next selectively chelated Ca2+ in the postsynaptic neuron by including 15 mm EGTA in the recording pipette during whole-cell recordings from CA1 pyramidal neurons before HFS. Labile LTP was robustly induced in these experiments (Fig. 5c,d), demonstrating that postsynaptic Ca2+ is not required for labile LTP induction, and instead suggesting that the block of labile LTP by EGTA-AM may result from chelation of presynaptic Ca2+.
Figure 5.
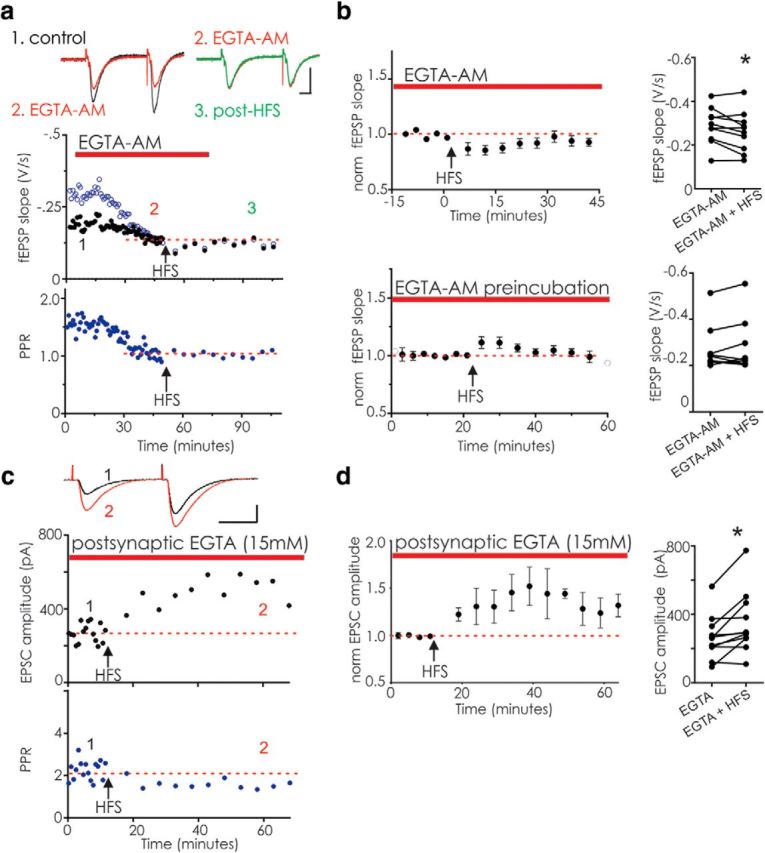
Labile LTP is blocked by bath-applied EGTA-AM but not by postsynaptically delivered EGTA. a, Example experiment showing acute bath application of 200 μm EGTA-AM to a slice. Top, fEPSP slope. Black symbols represent fEPSP1. Open blue symbols represent fEPSP2. Bottom, PPR for the same experiment. EGTA-AM decreased the second fEPSP with little effect on fEPSP1 (1, control; 2, EGTA-AM). Subsequent HFS did not elicit labile LTP nor a decrease in the PPR (3, post-HFS). Inset, Average of 5 fEPSPs before EGTA-AM application (black), as well as before (red) and after HFS (green). Calibration: 10 ms, 0.5 mV. b, Top left, Average of 10 similar experiments, illustrating the last 15 min of baseline recording in EGTA-AM; EGTA-AM was applied for at least 30 min before HFS. Right, Raw data from each experiment. On average, HFS did not elicit labile LTP but instead elicited a significant depression (all points after HFS over the next 45 min; p = 0.043, paired t test). Bottom left, Averaged data for 9 experiments for which slices were incubated for 60 min in EGTA-AM before being recorded from in the continued presence of EGTA-AM. Right, Raw data from each experiment. HFS did not induce labile LTP (p = 0.44, paired t test). Potentiation following HFS was significantly reduced in both acutely applied EGTA-AM and preincubation experiments compared with labile LTP in the 9 slices in Figure 2 that had HFS (acute, p = 0.0003; preincubated, p = 0.039; unpaired t test). c, Example whole-cell voltage-clamp recording from a CA1 pyramidal cell with EGTA (15 mm) included in the intracellular pipette solution. HFS was delivered at least 20 min after break-in to ensure diffusion of EGTA. Inset, Average of 5 EPSCs comparing baseline (1, black) with labile LTP (2, red). Calibration: 20 ms, 200 pA. Top, Black symbols represent EPSC. Bottom, Blue symbols represent PPR. d, Left, Average of 9 similar experiments with 15 mm EGTA in the pipette (EPSC after HFS vs baseline, p = 0.010, paired t test). Right, Raw data from each experiment. *p < 0.05.
While chelation of Ca2+ by EGTA-AM may have multiple effects, potentially inhibiting the release of most neurotransmitters and modulators, as well as other signaling molecules, the decreased PPR and block of labile LTP by EGTA-AM are consistent with the idea that a Ca2+-dependent process in the presynaptic terminal underlies labile LTP. We next explored the involvement of several candidate presynaptic proteins implicated in short-term synaptic plasticity.
RIM1α is a central component of the release machinery in nerve terminals, interacting with multiple proteins, including the synaptic vesicle proteins Rab3 and synaptotagmins as well as munc-13, ELKS, and liprins (Schoch et al., 2002). RIM1α knock-out mice lack LTP at several synapses in which LTP is known to require a rise in presynaptic Ca2+ and is maintained by presynaptic increases in glutamate release (Salin et al., 1996a; Castillo et al., 2002; Fourcaudot et al., 2008). Because of these similarities with labile LTP, we next examined slices from RIM1α knock-out mice. Robust labile LTP was triggered in synapses from RIM1α knock-out mice (Fig. 6a), suggesting that this form of potentiation is mechanistically different from other presynaptically maintained forms of LTP.
Figure 6.

Labile LTP is intact in RIM1α or syt-7 knock-out mice but blocked by a protein kinase inhibitor. a, Hippocampal slices from RIM1α KO mice express labile LTP after tetanus (n = 5, p = 0.019, paired t test). Inset, fEPSPs before (1, black) and after HFS (2, red). b, Hippocampal slices from syt-7 KO mice express labile LTP after HFS (n = 7, p = 0.002, paired t test). Inset, fEPSPs before (1, black) and after HFS (2, red). c, The PKC inhibitor GF109203X (10 μm) was added to the bathing medium once labile LTP was established in rat hippocampal slices (n = 11, labile LTP before and after 30 min GF, p = 0.20, paired t test). Inset, fEPSPs before (1, black), during labile LTP (2, red), and after 10 min GF109203X (3, blue). d, The broad-spectrum kinase inhibitor H-9 (200 μm) was added to the bathing medium at least 20 min before induction of labile LTP in rat hippocampal slices (n = 6, p = 0.91, paired t test). Inset, fEPSPs before HFS and (1, black) and after HFS in H-9 (2, red). Calibration: 0.5 mV, 10 ms. a–d, Right, Raw data for each experiment. *p < 0.05.
Syt-7 is a Ca2+-binding protein found at many central synapses that contributes to presynaptic asynchronous transmitter release as well as postsynaptic AMPAR trafficking (Sugita et al., 2001; Li et al., 2017; Wu et al., 2017). It was recently shown that several different synapses in the syt 7 knock-out mouse have markedly attenuated facilitation, among these the CA3-CA1 synapse that supports labile LTP (Jackman et al., 2016; Turecek et al., 2017), and we therefore next measured labile LTP in the syt-7 knock-out mouse. As reported, paired pulse facilitation was nearly absent at the CA3-CA1 synapse in slices from these animals; however, labile LTP appeared unaffected (Fig. 6b). Despite their shared roles in plasticity involving release probability and presynaptic Ca2+, neither RIM1α nor syt-7 is necessary for labile LTP.
The long period following the HFS before resuming stimulation in our experiments makes it unlikely that accumulated Ca2+ remains in the terminal, but persistent activation of a Ca2+-dependent enzyme, such as PKC, could account for the maintained potentiation. PTP can be blocked by the PKC inhibitor, GF109203X, and this compound blocks PTP at CA3-CA1 synapses (Brager et al., 2003; Korogod et al., 2007; Wierda et al., 2007; Wang et al., 2016). However, once labile LTP was established, 10 μm GF109203X did not significantly reverse the potentiation (Fig. 6c). To further test the role of protein kinases in the induction of labile LTP, we next bath-applied the broad-spectrum kinase inhibitor, H-9. At 200 μm, this kinase is expected to block PKA, PKG, PKC, and CaMKII. H-9 effectively blocked the induction of labile LTP (Fig. 6d).
Discussion
We have characterized a novel form of synaptic potentiation at hippocampal synapses that differs from other forms of potentiation. Like other examples of LTP, labile LTP is maintained after it is triggered without further synaptic activity, even after a long silent period. However, unlike LTP, if the synapses are active after its induction, even at 0.1 Hz, depotentiation occurs within a few minutes (Volianskis and Jensen, 2003), but labile LTP will remain stable with stimulation at 0.0033 Hz. Labile LTP is synapse-specific, as it is not observed at neighboring unstimulated synapses.
Mechanisms underlying labile LTP
Mechanistically, labile LTP does not require NMDAR activation, and thus is distinct from short-term potentiation (Kauer et al., 1988; Volianskis et al., 2013a) or NMDAR-dependent LTP (Collingridge et al., 1983). Despite indications that labile LTP may involve GluN2B (Volianskis et al., 2013b), receptors including this subunit are largely blocked by 50 μm d-APV (Buller et al., 1994); we thus conclude that NMDARs are not required for the labile LTP we characterized. If NMDARs are present on presynaptic terminals at a given synapse (Berretta and Jones, 1996; McGuinness et al., 2010), they could theoretically boost labile LTP magnitude by increasing presynaptic Ca2+, and thus increase the magnitude of labile LTP (sometimes called short-term potentiation) observed in earlier studies (Volianskis and Jensen, 2003; Volianskis et al., 2013b). This would not have been seen in our study as d-APV was present throughout all experiments. Furthermore, labile LTP is sustained in the presence of d-APV when synapses are stimulated infrequently (once per 5 min, 0.00334 Hz); in earlier experiments, more frequent stimulation after induction may have contributed to synapse depotentiation (Volianskis and Jensen, 2003).
The correlated reduction in the PPR suggests a presynaptic locus for labile LTP maintenance. Bath-applied EGTA-AM, but not postsynaptically delivered EGTA, prevented labile LTP induction, consistent with a requirement for elevated presynaptic Ca2+. Labile LTP was still induced under conditions in which PTP and facilitation are attenuated or absent, demonstrating that it is mechanistically distinct from these presynaptic processes and at least some of their underlying proteins. RIM1α and syt-7, presynaptic proteins implicated in the control of neurotransmitter release, are not required for labile LTP, and PKC is not essential for labile LTP maintenance. Our results using a broad-spectrum kinase inhibitor, H-9, suggest that kinase activation is necessary for labile LTP. We speculate that a Ca2+-sensitive enzyme is the most likely candidate to mediate labile LTP induction. Multiple Ca2+-dependent protein kinases, including CaMKII, PKC, and myosin light-chain kinase, can increase release probability (de Jong and Verhage, 2009). Mice lacking synapsins exhibit attenuated short-term synaptic plasticity notable after HFS in hippocampal slices (Rosahl et al., 1993, 1995), hinting at a potential role for these vesicle-associated phosphoproteins in labile LTP. Depotentiation after labile LTP also appears to be a Ca2+-dependent process; in hippocampal synapses, 0.133 Hz stimulation depotentiated labile LTP, even with glutamate receptors blocked by kynurenic acid and LY341495; however, the same stimulation in zero-Ca2+ solution did not permit depotentiation (Volianskis and Jensen, 2003). Together with the block of labile LTP by EGTA-AM reported here, this observation indicates a requirement for Ca2+ both to initiate labile LTP and for depotentiation. Further work will be needed to identify the proteins that control both processes.
Role of labile LTP in hippocampal and prefrontal circuitry
Labile LTP was originally described using field potential recordings from rat hippocampal slices, and we now demonstrate that labile LTP can also be observed in mouse hippocampus and in whole-cell recordings, and a labile potentiation with similar features can be elicited in prefrontal cortical synapses. Its intriguing properties may allow labile LTP to contribute to a variety of network processes. Labile LTP would be expected to tag synapses that have recently been highly active, increasing their probability of release for a defined period. At hippocampal and cortical synapses with relatively low release probability (Rosenmund et al., 1993; Dobrunz and Stevens, 1997), such a transient tag may permit specific inputs to increase their typical signal-to-noise ratio. This in turn is expected to increase the likelihood of coincident glutamate release and postsynaptic depolarization, promoting induction of NMDAR-dependent LTP. In the hippocampus, labile LTP could contribute to retention of information at specific synapses for short periods. This might be useful for hippocampal function during episodic learning, maintaining temporal order information, or perhaps could contribute to pattern completion or trace conditioning (Cohen and Eichenbaum, 1991; Shors, 2004; Kesner and Hopkins, 2006; Cowan, 2008). CA3 pyramidal cells fire in vivo in bursts at ∼200 Hz, often with ≥6 action potentials per bursts, repeated every ∼1–2 s (Kowalski et al., 2016). The 100 Hz HFS protocol used here to induce labile LTP is of longer duration than likely occurs in vivo, but the frequency is in the physiological range. Labile LTP can also be elicited with 30 Hz afferent stimulation or a theta burst protocol, demonstrating that it can be triggered by various patterns of presynaptic activity at relatively high frequency (Volianskis and Jensen, 2003).
The PFC is generally thought to have a key role in working memory (Fuster, 2009; D'Esposito and Postle, 2015). Recently, there has been considerable discussion of the possibility that working memory may not result solely from persistent neuronal firing, and memory may remain even after pauses in the persistent activity (LaRocque et al., 2013; Stokes, 2015). We observed potentiation with some features reminiscent of hippocampal labile LTP at layer 2–3 to layer 5 PFC synapses, where it could contribute to working memory, retaining information at recently active synapses without the expense of persistently active firing in cells of the relevant network.
Although not yet tested explicitly, labile LTP may enable high-frequency activation of a single axon to elevate the release probability for a period of time dependent on subsequent presynaptic activity of that axon. This nonassociative arrangement is different from NMDAR-dependent forms of plasticity in its independence of the postsynaptic neuron, instead depending in theory only on presynaptic afferent activity. Labile LTP cannot on its own account for acquisition of novel associations, but instead would simply retain the information of recent local network activity and could contribute to nonassociative retention of information over short time periods.
In many electrophysiological experiments, labile LTP can be masked by other short- and long-term plasticity phenomena, and so may have easily been overlooked. The observation of labile LTP at synapses in both hippocampal and cortical circuits raises the question of how widespread this phenomenon is. For example, do CA3 or layer 2/3 afferents synapsing on GABAergic interneurons also exhibit labile LTP upon HFS? Do GABAergic nerve terminals exhibit labile LTP? Our results suggest a requirement for presynaptic Ca2+ and activation of kinases to trigger labile LTP, but beyond that our experiments have only ruled out several likely participants. Understanding the mechanisms that underlie labile LTP should clarify its behavioral functions and how it may be used by neurons embedded in distinct circuits.
Footnotes
This work was supported by National Institutes of Health Grants NS050570 and NS088453 to J.A.K., Brown Institute for Brain Science to B.P., and a Deutsche Forschungsgemeinschaft Grant PR1719/1-1 to B.P. We thank Rachel Stevenson, other J.A.K. laboratory members, and Drs. Carlos Aizenman, Barry Connors, Chris Moore, David Badre, and Theresa Desrochers for helpful suggestions; and Ayumi Tsuda for technical assistance.
The authors declare no competing financial interests.
References
- Abbott LF, Regehr WG (2004) Synaptic computation. Nature 431:796–803. 10.1038/nature03010 [DOI] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG (1996) Determinants of the time course of facilitation at the granule cell to purkinje cell synapse. J Neurosci 16:5661–5671. 10.1523/JNEUROSCI.16-18-05661.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannerman DM, Good MA, Butcher SP, Ramsay M, Morris RG (1995) Distinct components of spatial learning revealed by prior training and NMDA blockade. Nature 378:182–186. 10.1038/378182a0 [DOI] [PubMed] [Google Scholar]
- Barak O, Tsodyks M (2007) Persistent activity in neural networks with dynamic synapses. PLoS Comput Biol 3:e35. 10.1371/journal.pcbi.0030035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta N, Jones RS (1996) Tonic facilitation of glutamate release by presynaptic N-methyl-D-aspartate autoreceptors in the entorhinal cortex. Neuroscience 75:339–344. 10.1016/0306-4522(96)00301-6 [DOI] [PubMed] [Google Scholar]
- Brager DH, Cai X, Thompson SM (2003) Activity-dependent activation of presynaptic protein kinase C mediates post-tetanic potentiation. Nat Neurosci 6:551–552. 10.1038/nn1067 [DOI] [PubMed] [Google Scholar]
- Buller AL, Larson HC, Schneider BE, Beaton JA, Morrisett RA, Monaghan DT (1994) The molecular basis of NMDA receptor subtypes: native receptor diversity is predicted by subunit composition. J Neurosci 14:5471–5484. 10.1523/JNEUROSCI.14-09-05471.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC (2002) RIM1alpha is required for presynaptic long-term potentiation. Nature 415:327–330. 10.1038/415327a [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE (2006) Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci 29:37–76. 10.1146/annurev.neuro.29.051605.112834 [DOI] [PubMed] [Google Scholar]
- Cho K, Aggleton JP, Brown MW, Bashir ZI (2001) An experimental test of the role of postsynaptic calcium levels in determining synaptic strength using perirhinal cortex of rat. J Physiol 532:459–466. 10.1111/j.1469-7793.2001.0459f.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen NJ, Eichenbaum H (1991) The theory that wouldn't die: a critical look at the spatial mapping theory of hippocampal function. Hippocampus 1:265–268. 10.1002/hipo.450010312 [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Kehl SJ, McLennan H (1983) Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J Physiol 334:33–46. 10.1113/jphysiol.1983.sp014478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan N. (2008) What are the differences between long-term, short-term, and working memory? Prog Brain Res 169:323–338. 10.1016/S0079-6123(07)00020-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong AP, Verhage M (2009) Presynaptic signal transduction pathways that modulate synaptic transmission. Curr Opin Neurobiol 19:245–253. 10.1016/j.conb.2009.06.005 [DOI] [PubMed] [Google Scholar]
- D'Esposito M, Postle BR (2015) The cognitive neuroscience of working memory. Annu Rev Psychol 66:115–142. 10.1146/annurev-psych-010814-015031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF (1997) Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron 18:995–1008. 10.1016/S0896-6273(00)80338-4 [DOI] [PubMed] [Google Scholar]
- Felmy F, von Gersdorff H (2006) Late switch for post-tetanic potentiation: once again it's Ca2+. Focus on “An increase in calcium influx contributes to post-tetanic potentiation at the rat calyx of held synapse.” J Neurophysiol 96:2840–2841. 10.1152/jn.00783.2006 [DOI] [PubMed] [Google Scholar]
- Fioravante D, Regehr WG (2011) Short-term forms of presynaptic plasticity. Curr Opin Neurobiol 21:269–274. 10.1016/j.conb.2011.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourcaudot E, Gambino F, Humeau Y, Casassus G, Shaban H, Poulain B, Lüthi A (2008) cAMP/PKA signaling and RIM1alpha mediate presynaptic LTP in the lateral amygdala. Proc Natl Acad Sci U S A 105:15130–15135. 10.1073/pnas.0806938105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funahashi S, Bruce CJ, Goldman-Rakic PS (1989) Mnemonic coding of visual space in the monkey's dorsolateral prefrontal cortex. J Neurophysiol 61:331–349. 10.1152/jn.1989.61.2.331 [DOI] [PubMed] [Google Scholar]
- Fuster JM. (2009) Cortex and memory: emergence of a new paradigm. J Cogn Neurosci 21:2047–2072. 10.1162/jocn.2009.21280 [DOI] [PubMed] [Google Scholar]
- Gibson HE, Edwards JG, Page RS, Van Hook MJ, Kauer JA (2008) TRPV1 channels mediate long-term depression at synapses on hippocampal interneurons. Neuron 57:746–759. 10.1016/j.neuron.2007.12.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS. (1995) Cellular basis of working memory. Neuron 14:477–485. 10.1016/0896-6273(95)90304-6 [DOI] [PubMed] [Google Scholar]
- Hansel D, Mato G (2013) Short-term plasticity explains irregular persistent activity in working memory tasks. J Neurosci 33:133–149. 10.1523/JNEUROSCI.3455-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring BE, Nicoll RA (2016) Long-term potentiation: from CaMKII to AMPA receptor trafficking. Annu Rev Physiol 78:351–365. 10.1146/annurev-physiol-021014-071753 [DOI] [PubMed] [Google Scholar]
- Jackman SL, Regehr WG (2017) The mechanisms and functions of synaptic facilitation. Neuron 94:447–464. 10.1016/j.neuron.2017.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman SL, Turecek J, Belinsky JE, Regehr WG (2016) The calcium sensor synaptotagmin-7 is required for synaptic facilitation. Nature 529:88–91. 10.1038/nature16507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminśki J, Sullivan S, Chung JM, Ross IB, Mamelak AN, Rutishauser U (2017) Persistently active neurons in human medial frontal and medial temporal lobe support working memory. Nat Neurosci 20:590–601. 10.1038/nn.4509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA. (1999) Blockade of hippocampal long-term potentiation by sustained tetanic stimulation near the recording site. J Neurophysiol 81:940–944. 10.1152/jn.1999.81.2.940 [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC, Nicoll RA (1988) NMDA application potentiates synaptic transmission in the hippocampus. Nature 334:250–252. 10.1038/334250a0 [DOI] [PubMed] [Google Scholar]
- Kesner RP, Hopkins RO (2006) Mnemonic functions of the hippocampus: a comparison between animals and humans. Biol Psychol 73:3–18. 10.1016/j.biopsycho.2006.01.004 [DOI] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R (2007) Posttetanic potentiation critically depends on an enhanced Ca(2+) sensitivity of vesicle fusion mediated by presynaptic PKC. Proc Natl Acad Sci U S A 104:15923–15928. 10.1073/pnas.0704603104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski J, Gan J, Jonas P, Pernía-Andrade AJ (2016) Intrinsic membrane properties determine hippocampal differential firing pattern in vivo in anesthetized rats. Hippocampus 26:668–682. 10.1002/hipo.22550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe V, Morin F, Ratte S, Croce A, Conquet F, Lacaille JC (2004) Synapse-specific mGluR1-dependent long-term potentiation in interneurones regulates mouse hippocampal inhibition. J Physiol 555:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRocque JJ, Lewis-Peacock JA, Drysdale AT, Oberauer K, Postle BR (2013) Decoding attended information in short-term memory: an EEG study. J Cogn Neurosci 25:127–142. 10.1162/jocn_a_00305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz RA, Alger BE (1999) Calcium dependence of depolarization-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J Physiol 521:147–157. 10.1111/j.1469-7793.1999.00147.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Chanaday NL, Xu W, Kavalali ET (2017) Synaptotagmin-1- and synaptotagmin-7-dependent fusion mechanisms target synaptic vesicles to kinetically distinct endocytic pathways. Neuron 93:616–631.e3. 10.1016/j.neuron.2016.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair RD, Kauer JA (2007) Amphetamine depresses excitatory synaptic transmission at prefrontal cortical layer V synapses. Neuropharmacol 52:193–199. [DOI] [PubMed] [Google Scholar]
- Major G, Tank D (2004) Persistent neural activity: prevalence and mechanisms. Curr Opin Neurobiol 14:675–684. 10.1016/j.conb.2004.10.017 [DOI] [PubMed] [Google Scholar]
- Malenka RC. (1991) Postsynaptic factors control the duration of synaptic enhancement in area CA1 of the hippocampus. Neuron 6:53–60. 10.1016/0896-6273(91)90121-F [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF (2004) LTP and LTD: an embarrassment of riches. Neuron 44:5–21. 10.1016/j.neuron.2004.09.012 [DOI] [PubMed] [Google Scholar]
- McGuinness L, Taylor C, Taylor RD, Yau C, Langenhan T, Hart ML, Christian H, Tynan PW, Donnelly P, Emptage NJ (2010) Presynaptic NMDARs in the hippocampus facilitate transmitter release at theta frequency. Neuron 68:1109–1127. 10.1016/j.neuron.2010.11.023 [DOI] [PubMed] [Google Scholar]
- Morris RG. (2013) NMDA receptors and memory encoding. Neuropharmacology 74:32–40. 10.1016/j.neuropharm.2013.04.014 [DOI] [PubMed] [Google Scholar]
- Morris RG, Steele RJ, Bell JE, Martin SJ (2013) N-Methyl-d-aspartate receptors, learning and memory: chronic intraventricular infusion of the NMDA receptor antagonist d-AP5 interacts directly with the neural mechanisms of spatial learning. Eur J Neurosci 37:700–717. 10.1111/ejn.12086 [DOI] [PubMed] [Google Scholar]
- Nakazawa K, McHugh TJ, Wilson MA, Tonegawa S (2004) NMDA receptors, place cells and hippocampal spatial memory. Nat Rev Neurosci 5:361–372. 10.1038/nrn1385 [DOI] [PubMed] [Google Scholar]
- Nanou E, Sullivan JM, Scheuer T, Catterall WA (2016) Calcium sensor regulation of the CaV2.1 Ca2+ channel contributes to short-term synaptic plasticity in hippocampal neurons. Proc Natl Acad Sci U S A 113:1062–1067. 10.1073/pnas.1524636113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA. (2017) A brief history of long-term potentiation. Neuron 93:281–290. 10.1016/j.neuron.2016.12.015 [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Tsubokawa H, Mizushima I, Yoneda N, Zimmer A, Kano M (2002) Presynaptic cannabinoid sensitivity is a major determinant of depolarization-induced retrograde suppression at hippocampal synapses. J Neurosci 22:3864–3872. 10.1523/JNEUROSCI.22-10-03864.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouanounou A, Zhang L, Tymianski M, Charlton MP, Wallace MC, Carlen PL (1996) Accumulation and extrusion of permeant Ca2+ chelators in attenuation of synaptic transmission at hippocampal CA1 neurons. Neuroscience 75:99–109. 10.1016/0306-4522(96)00319-3 [DOI] [PubMed] [Google Scholar]
- Park P, Volianskis A, Sanderson TM, Bortolotto ZA, Jane DE, Zhuo M, Kaang BK, Collingridge GL (2014) NMDA receptor-dependent long-term potentiation comprises a family of temporally overlapping forms of synaptic plasticity that are induced by different patterns of stimulation. Philos Trans R Soc Lond B Biol Sci 369:20130131. 10.1098/rstb.2013.0131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C (2013) The rat brain in stereotaxic coordinates. San Diego, CA: Academic Press. [Google Scholar]
- Place R, Lykken C, Beer Z, Suh J, McHugh TJ, Tonegawa S, Eichenbaum H, Sauvage MM (2012) NMDA signaling in CA1 mediates selectively the spatial component of episodic memory. Learn Mem 19:164–169. 10.1101/lm.025254.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosahl TW, Geppert M, Spillane D, Herz J, Hammer RE, Malenka RC, Südhof TC (1993) Short-term synaptic plasticity is altered in mice lacking synapsin 1. Cell 75:661–670. 10.1016/0092-8674(93)90487-B [DOI] [PubMed] [Google Scholar]
- Rosahl TW, Spillane D, Missler M, Herz J, Selig DK, Wolff JR, Hammer RE, Malenka RC, Südhof TC (1995) Essential functions of synapsins I and II in synaptic vesicle regulation. Nature 375:488–493. 10.1038/375488a0 [DOI] [PubMed] [Google Scholar]
- Rose NS, LaRocque JJ, Riggall AC, Gosseries O, Starrett MJ, Meyering EE, Postle BR (2016) Reactivation of latent working memories with transcranial magnetic stimulation. Science 354:1136–1139. 10.1126/science.aah7011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL (1993) Nonuniform probability of glutamate release at a hippocampal synapse. Science 262:754–757. 10.1126/science.7901909 [DOI] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA (1996a) Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron 16:797–803. 10.1016/S0896-6273(00)80099-9 [DOI] [PubMed] [Google Scholar]
- Salin PA, Scanziani M, Malenka RC, Nicoll RA (1996b) Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci U S A 93:13304–13309. 10.1073/pnas.93.23.13304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Südhof TC (2002) RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature 415:321–326. 10.1038/415321a [DOI] [PubMed] [Google Scholar]
- Shors TJ. (2004) Memory traces of trace memories: neurogenesis, synaptogenesis and awareness. Trends Neurosci 27:250–256. 10.1016/j.tins.2004.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes MG. (2015) ‘Activity-silent’ working memory in prefrontal cortex: a dynamic coding framework. Trends Cogn Sci 19:394–405. 10.1016/j.tics.2015.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Han W, Butz S, Liu X, Fernández-Chacón R, Lao Y, Südhof TC (2001) Synaptotagmin VII as a plasma membrane Ca(2+) sensor in exocytosis. Neuron 30:459–473. 10.1016/S0896-6273(01)00290-2 [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S (1996) The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87:1327–1338. 10.1016/S0092-8674(00)81827-9 [DOI] [PubMed] [Google Scholar]
- Turecek J, Jackman SL, Regehr WG (2017) Synaptotagmin-7 confers frequency invariance onto specialized depressing synapses. Nature 551:503–506. 10.1038/nature24474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volianskis A, Jensen MS (2003) Transient and sustained types of long-term potentiation in the CA1 area of the rat hippocampus. J Physiol 550:459–492. 10.1113/jphysiol.2003.044214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volianskis A, Collingridge GL, Jensen MS (2013a) The roles of STP and LTP in synaptic encoding. PeerJ 1:e3. 10.7717/peerj.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volianskis A, Bannister N, Collett VJ, Irvine MW, Monaghan DT, Fitzjohn SM, Jensen MS, Jane DE, Collingridge GL (2013b) Different NMDA receptor subtypes mediate induction of long-term potentiation and two forms of short-term potentiation at CA1 synapses in rat hippocampus in vitro. J Physiol 591:955–972. 10.1113/jphysiol.2012.247296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volianskis A, France G, Jensen MS, Bortolotto ZA, Jane DE, Collingridge GL (2015) Long-term potentiation and the role of N-methyl-d-aspartate receptors. Brain Res 1621:5–16. 10.1016/j.brainres.2015.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CC, Weyrer C, Paturu M, Fioravante D, Regehr WG (2016) Calcium-dependent protein kinase C is not required for post-tetanic potentiation at the hippocampal CA3 to CA1 synapse. J Neurosci 36:6393–6402. 10.1523/JNEUROSCI.0708-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda KD, Toonen RF, de Wit H, Brussaard AB, Verhage M (2007) Interdependence of PKC-dependent and PKC-independent pathways for presynaptic plasticity. Neuron 54:275–290. 10.1016/j.neuron.2007.04.001 [DOI] [PubMed] [Google Scholar]
- Wolff MJ, Ding J, Myers NE, Stokes MG (2015) Revealing hidden states in visual working memory using electroencephalography. Front Syst Neurosci 9:123. 10.3389/fnsys.2015.00123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff MJ, Jochim J, Akyürek EG, Stokes MG (2017) Dynamic hidden states underlying working-memory-guided behavior. Nat Neurosci 20:864–871. 10.1038/nn.4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Bacaj T, Morishita W, Goswami D, Arendt KL, Xu W, Chen L, Malenka RC, Südhof TC (2017) Postsynaptic synaptotagmins mediate AMPA receptor exocytosis during LTP. Nature 544:316–321. 10.1038/nature21720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao MY, Niu YP, Wigström H (1996) Activity-dependent decay of early LTP revealed by dual EPSP recording in hippocampal slices from young rats. Eur J Neurosci 8:1916–1923. 10.1111/j.1460-9568.1996.tb01335.x [DOI] [PubMed] [Google Scholar]
- Xiao MY, Wasling P, Hanse E, Gustafsson B (2004) Creation of AMPA-silent synapses in the neonatal hippocampus. Nat Neurosci 7:236–243. 10.1038/nn1196 [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64:355–405. 10.1146/annurev.physiol.64.092501.114547 [DOI] [PubMed] [Google Scholar]