

Abstract
Cutaneous somatosensory neurons convey innocuous and noxious mechanical, thermal, and chemical stimuli from peripheral tissues to the CNS. Among these are nociceptive neurons that express calcitonin gene-related peptide-α (CGRPα). The role of peripheral CGRPα neurons (CANs) in acute and injury-induced pain has been studied using diphtheria toxin ablation, but their functional roles remain controversial. Because ablation permanently deletes a neuronal population, compensatory changes may ensue that mask the physiological or pathophysiological roles of CANs, particularly for injuries that occur after ablation. Therefore, we sought to define the role of intact CANs in vivo under baseline and injury conditions by using noninvasive transient optogenetic inhibition. We assessed pain behavior longitudinally from acute to chronic time points. We generated adult male and female mice that selectively express the outward rectifying proton pump archaerhodopsin-3 (Arch) in CANs, and inhibited their peripheral cutaneous terminals in models of neuropathic (spared nerve injury) and inflammatory (skin-muscle incision) pain using transdermal light activation of Arch. After nerve injury, brief activation of Arch reversed the chronic mechanical, cold, and heat hypersensitivity, alleviated the spontaneous pain, and reversed the sensitized mechanical currents in primary afferent somata. In contrast, Arch inhibition of CANs did not alter incision-induced hypersensitivity. Instead, incision-induced mechanical and heat hypersensitivity was alleviated by peripheral blockade of CGRPα peptide-receptor signaling. These results reveal that CANs have distinct roles in the time course of pain during neuropathic and incisional injuries and suggest that targeting peripheral CANs or CGRPα peptide-receptor signaling could selectively treat neuropathic or postoperative pain, respectively.
SIGNIFICANCE STATEMENT The contribution of sensory afferent CGRPα neurons (CANs) to neuropathic and inflammatory pain is controversial. Here, we left CANs intact during neuropathic and perioperative incision injury by using transient transdermal optogenetic inhibition of CANs. We found that peripheral CANs are required for neuropathic mechanical, cold, and heat hypersensitivity, spontaneous pain, and sensitization of mechanical currents in afferent somata. However, they are dispensable for incisional pain transmission. In contrast, peripheral pharmacological inhibition of CGRPα peptide-receptor signaling alleviated the incisional mechanical and heat hypersensitivity, but had no effect on neuropathic pain. These results show that CANs have distinct roles in neuropathic and incisional pain and suggest that their targeting via novel peripheral treatments may selectively alleviate neuropathic versus incisional pain.
Keywords: archaerhodopsin, incision, inflammation, mechanotransduction, nerve injury, nociceptor
Introduction
Cutaneous nerve endings sense environmental events, including noxious mechanical, thermal, and chemical stimuli that are then conveyed to the spinal cord (body) or trigeminal tract (face) (Basbaum et al., 2009). Classically, pain-sensing neurons (nociceptors) are divided into two general subsets of nociceptors: peptidergic neurons, which express the neuropeptides calcitonin gene-related peptide-α (CGRPα) and substance P; and nonpeptidergic neurons, which express isolectin-B4 or MrgD (Basbaum et al., 2009; Cavanaugh et al., 2009; Ghitani et al., 2017). However, recent studies have revealed that CGRPα neurons (CANs) are more heterogeneous (Usoskin et al., 2015; Li et al., 2016; Nguyen et al., 2017). Approximately 20% of Aδ or C-fiber isolectin-B4+ neurons (classically identified as nonpeptidergic) express CGRP (Li et al., 2016) and are also TRKA+ and TRPV1+ (belonging to the NP2 subtype) (Usoskin et al., 2015). Furthermore, two populations exist within the classically defined CGRP peptidergic neurons: PEP1 and PEP2. Both subtypes express CGRP, TRKA, KIT, and Nav1.8/9. PEP1 and PEP2 neurons differ in that PEP1 neurons are unmyelinated, express TAC1 and TRPV1, and express low levels of PLXNC1, whereas PEP2 neurons are myelinated and express CNTNAP2, FAM19A1, and NEFH. Notably, CANs can respond to painful stimuli and directly transmit the signal to the CNS, in part, by releasing CGRPα from their central terminals (Basbaum et al., 2009; McCoy et al., 2012). At their peripheral endings, CANs can also release CGRPα that then acts on other non-neuronal cells or on CGRP− neuronal endings. When CGRPα is released from peripheral sensory endings, it causes vasodilation and inflammation by acting on CGRP1 receptors that are expressed by CGRP− neuronal, endothelial cells that line blood vessels, immune cells, and keratinocytes (Hagner et al., 2002; Eftekhari and Edvinsson, 2010; Assas et al., 2014; Russell et al., 2014). Exciting studies have conclusively shown a clear role for CAPS in headache pain, resulting in the development of clinically effective novel therapeutics for migraine and headache disorders (Goadsby and Edvinsson, 1993; Olesen et al., 2004; Deen et al., 2017; Tso and Goadsby, 2017).
CANs have also been implicated in nociceptive transmission and neurogenic inflammation in nonheadache persistent pain; however, their roles in pain with distinct etiologies (i.e., neuropathic vs inflammatory injuries) remain debated (McCoy et al., 2012, 2013; Iyer et al., 2014; Daou et al., 2016; Iyengar et al., 2017). Chemical ablation of CANs showed that they are essential for baseline cold and heat detection, as well as heat hypersensitivity after both neuropathic and inflammatory injuries (McCoy et al., 2012, 2013). However, mechanical detection was unaffected by chemical ablation of CANs in both normal baseline and injury conditions (McCoy et al., 2013). Nonetheless, reports on the role of CANs in injury-induced mechanical hypersensitivity are conflicting (Iyer et al., 2014; Daou et al., 2016; Iyengar et al., 2017). Both systemic CGRP antagonist administration (Iyengar et al., 2017) and optogenetic inhibition of broadly defined nociceptive neurons that include CANs (Iyer et al., 2014; Daou et al., 2016) decrease the injury-induced mechanical hypersensitivity. Because ablation and genetic deletion methods permanently remove neurons or proteins, they can potentially cause unrecognized compensatory molecular, synaptic, and circuitry changes in vivo that may underlie the discrepancies in CGRPα-related mechanical hypersensitivity after injury (Eisener-Dorman et al., 2009). Optogenetic inhibition overcomes these potential complications of ablation and genetic deletion approaches by leaving the neurons, their normal signaling mechanisms, and their anatomical circuits intact, and by allowing precise temporal control over neuronal activity (Boada et al., 2014; Daou et al., 2016; St John Smith, 2018).
Here, we selectively inhibited CANs in vivo and in vitro using a tamoxifen-inducible CGRPα Cre mouse line to express the light-sensitive proton pump arachaerhodopsin-3 (Arch) exclusively in the CANs of adult mice. We assessed the contributions of CANs longitudinally at acute and chronic time points following neuropathic and inflammatory (incision) injuries.
Materials and Methods
Animals.
All animal protocols were in accordance with the National Institute of Health guidelines and were approved by the Institutional Animal Care and Use Committee of the Medical College of Wisconsin (AUA #0383). Mice were housed in a 14:10 light/dark cycle with ad libitum access to food and water. Adult C57BL/6J (RRID:IMSR_JAX:000664) mice and homozygous CGRPα Cre+ Arch/Arch (CGRP-Arch Cre+) and CGRPα Cre− Arch/Arch littermates (CGRP-Arch Cre−) 8–24 weeks old were used. Approximately equal numbers of males and females were used for most experiments, except where noted. No differences were found between sexes for any of the assays, and as such, data from both sexes were combined. Selective expression of Arch in CANs was achieved using a CreER-LoxP system. Homozygous Ai35D (B6;129S-Gt(ROSA)26Sortm35.1(CAG-aop3/GFP)Hze/J mice, The Jackson Laboratory catalog #012735, RRID:IMSR_JAX:012735) have a stop codon flanked by two LoxP sites that prevents Arch-GFP transcription. These mice were mated with the Chuang CGRPCreER mouse line (Song et al., 2012) to produce CGRP-Arch Cre+ and CGRP-Arch Cre− mice. All animals, regardless of CreER expression, were intraperitoneally injected with 75 mg tamoxifen/kg body weight for 5 consecutive days. All assays were performed 7 d after the final tamoxifen injection. Tamoxifen (Sigma-Aldrich catalog #10540-29-1) was dissolved to 20 mg/ml in corn oil.
DRG neuron isolation and culturing.
Mice were anesthetized with inhaled isoflurane and decapitated. All lumbar DRGs were removed bilaterally from naïve animals. For spared nerve injury (SNI) animals (described below), only ipsilateral lumbar 4–6 DRG neurons were removed 16 weeks after SNI was performed. Isolated DRGs were incubated in DMEM/Ham's F12 medium (DMEM) containing 10 mg/ml collagenase for 40 min and 0.5% trypsin for 45 min. Then, DRG neurons were mechanically dissociated and plated onto laminin-coated glass coverslips. DRG neurons were incubated overnight in DMEM supplemented with 10% heat-inactivated horse serum, 2 mm l-glutamine, 1% glucose, 100 units/ml penicillin, and 100 μg/ml streptomycin (Zappia et al., 2016). No exogenous growth factors were added to the medium.
Immunofluorescence (IF).
Tissues were removed from CGRP-Arch Cre+ and Cre− mice. Whole DRGs were collected as described above. Additionally, glabrous hindpaw skin was harvested. All tissues were immediately fixed in Zamboni's fixative for 30 min at 4°C and then incubated in 30% sucrose for 24–48 h at 4°C. Tissue was embedded in Optimal Cutting Temperature compound, cryosectioned at 12 μm (DRGs), and mounted onto poly-L-lysine-coated slides; skin was cryosectioned at 50 μm and then stored at 4°C in PBS.
Slides containing tissue sections were dried at room temperature overnight and then washed 3 times with TBS/0.3% Triton-X (TBS/TX) and rinsed with PBS. Sections were blocked in 10% normal donkey serum (NDS, Abcam catalog #ab7475) in TBS/TX. Slides were then incubated at room temperature overnight in 10% NDS in TBS/TX that contained the following primary antibody: rabbit anti-CGRP (1:1000, Sigma-Aldrich catalog #C8198, RRID:AB_259091). After 4 TBS/TX washes followed by 3 PBS washes, slides were incubated in 10% NDS for 30 min at room temperature. The secondary antibody (donkey anti-rabbit 594, 1:200, Thermo Fisher Scientific catalog #R37119 RRID:AB_2556547) was applied for 3 h at room temperature. After the secondary incubation, the slides were washed 3 times with TBS/TX and then PBS and coverslipped.
Floating skin sections were washed 3 times with TBS/TX and then incubated in 10% NDS for 1 h at room temperature. Next, sections were incubated in 10% NDS, TBS/TX solution containing the primary antibodies (rabbit anti-CGRP 1:1000 or rabbit anti-keratin 14, 1:500, BioLegend catalog #905304, RRID:AB_2616896) overnight at room temperature. The following day, sections were washed 4 times with TBS/TX followed by a 30 min incubation in 10% NDS. The secondary mixture (donkey anti-rabbit 594, 1:200 or donkey anti-rabbit 594, 1:200 and DRAQ5, 1:1000, Abcam catalog #ab108410 RRID:AB_2314341) was added to the sections and incubated for 5 h at room temperature. Next, the sections were washed 2 times with TBS/TX followed by 4 washes with PBS. Sections were mounted onto slides, dried overnight, and coverslipped.
Microscopy.
The experimenter was blinded to genotype and treatment throughout imaging and cell counting. Images were taken using a Nikon Eclipse E800 confocal microscope equipped with 488, 561, and 635 nm lasers. Images were collected using an EZ-C1 viewer (Nikon Instruments) and assessed in ImageJ (version 1.51, National Institutes of Health, Bethesda, MD) and Adobe Illustrator software (Adobe Systems). Only global adjustments to brightness and contrast were made.
Spared Nerve Injury (SNI).
SNI surgery was performed on mice as previously described, by sparing the sural nerve (Decosterd and Woolf, 2000; Smith et al., 2013). The SNI model was selected due to its immediate and long-lasting robust mechanical hypersensitivity. Animals were anesthetized with 1.5% isoflurane throughout surgery. The left tibial and common peroneal nerves were ligated just peripheral to where they branch off the sciatic nerve and transected 2–4 mm distal to the ligature. The sural nerve remained intact and untouched. Sham animals were anesthetized and skin and muscle incisions were made without tibial and peroneal nerve ligation and axotomy.
Incisional pain model of inflammatory pain.
Plantar incision through the skin and muscle was performed as previously described (Brennan et al., 1996; Pogatzki and Raja, 2003; Banik et al., 2006; Barabas and Stucky, 2013). This model was chosen as it is one of the most clinically relevant models of inflammatory pain (Ghasemlou et al., 2015); and because ∼20% of patients who have surgery, later suffer persistent postoperative pain after healing (Vadivelu et al., 2010; Gan et al., 2014). Animals were anesthetized with 1.5% isoflurane throughout surgery. A 5 mm cutaneous longitudinal incision was made 2 mm from the proximal edge of the heel toward the toes using a number 11 blade scalpel. The plantaris muscle was elevated and incised longitudinally through the belly of the muscle. The skin was closed with two sutures, which remained in place throughout testing. Animals whose sutures were pulled out before postoperative day 2 were removed from the study. Sham mice were anesthetized, but not incised. Bacitracin cream was applied to the plantar hindpaw over the surgical site for all animals following incision or sham surgery.
Patch-clamp electrophysiology.
The experimenter was blinded to surgery (i.e., naïve vs SNI). Lumbar DRG neurons from naïve or 16 weeks post-SNI CGRP-Arch Cre+ animals were used for patch-clamping after culturing overnight as described above (Weyer et al., 2015). Coverslips were placed in a perfusion chamber above an Eclipse TE200 inverted microscope (Nikon). Cells were continuously superfused with an extracellular HEPES solution containing the following: 140 mm NaCl, 5 mm KCl, 2 mm CaCl2,1 mm MgCl2,10 mm HEPES, and 10 mm glucose, pH 7.4 ± 0.03, and 310 ± 3 mOsm. Borosilicate pipettes with resistances of 2–5 mΩ that were filled with intracellular HEPES solution (135 mm KCl, 10 mm NaCl,1 mm MgCl2,1 mm EGTA, 0.2 mm NaGTP, 2.5 mm ATPNa2, and 10 mm HEPES, pH 7.20 ± 0.03, and 290 ± 3 mOsm) were used to patch-clamp neurons. Compensation was applied for neuronal capacitance and series resistance (maintained <10 mΩ). In current-clamp mode, rheobase current (i.e., the minimum current adequate to induce an action potential during sustained depolarization) was determined using stepwise square pulse current injections. In voltage-clamp mode, mechanically induced currents were elicited using a 1–2 μm tip borosilicate glass pipette driven by a piezo stack actuator (PA25, PiezoSystem) with a velocity of 106.25 μm/ms. Neurons were stimulated with increasing mechanical displacements (1.7 μm/V, 200 ms duration) every 60 s. CGRPα+/Arch+ neurons were targeted by selecting only the GFP+ cells, which tagged Arch. Therefore, only DRGs from Cre+ animals were used to identify and patch only CGRPα+ cells. Treatment (light off or on) was randomized between cells. Different neurons were used for each light treatment due to neurons dying upon the final mechanical stimulation. For the light on condition, neurons were exposed to the microscope light at maximum power and 590 nm LED (5 ± 0.2 mW) for a continuous 60 s before recording and during the recording period. Continuous light stimulation of Arch does not result in desensitization of the chloride pump (Chow et al., 2012b; Daou et al., 2016; El-Gaby et al., 2016; Wiegert et al., 2017). The microscope light was turned off at least 60 s before and during recordings for the light off condition. Recordings were made using an EPC9 amplifier (HEKA Electronics) and Pulse software (HEKA Electronics).
Behavior.
For all assays, animals were tested between 9:00 A.M. and 3:00 P.M. and habituated in individual Plexiglas enclosures for 1 h before testing. The experimenter was blinded to genotype and treatment of the animals throughout data collection and analyses. Mice 8–18 weeks of age were used. For LED treatment, the 590 nm LED (17.5 mW; Thorlabs, # M590L3) or control 490 LED (21.6 mW; Thorlabs, # M490L4) was shone on the plantar surface of the hindpaw through the mesh or glass used to test mechanical thresholds or thermal latencies continuously for 30 s before and throughout testing. Total time of stimulation was ∼2 min. For CGRP8–37 (Tocris Bioscience, #1169) inhibitor administration, 20 μl of 1× PBS or 533.16 μm of CGRP8–37 (Hsieh et al., 2012a; Ishida et al., 2014) was subcutaneously injected into the plantar hindpaw of mice 45 min before testing. The site of stimulus (mechanical or thermal) application to the hindpaw skin varied depending upon the injury model. For SNI, the stimuli were applied to the lateral aspect of the hindpaw because this area is anatomically where the sural nerve innervates and is the most sensitive after spared sural SNI (Swett and Woolf, 1985; Decosterd and Woolf, 2000). For the postoperative pain model, the stimuli were applied to the medioposterior aspect of the hindpaw because this is the most sensitive area after perioperative incision (Brennan et al., 1996). For sham and injury model mice, the stimuli were applied to the ipsilateral hindpaw. Flaring of the toes and lifting, flicking, licking, and biting of the stimulated paw were all considered positive responses.
Mechanical threshold sensory testing (von Frey up-down assay).
Calibrated von Frey monofilaments ranging from 0.09 to 19.6 mN were applied to the plantar hindpaw, and the 50% withdrawal thresholds were determined using the up-down method (Dixon, 1980; Chaplan et al., 1994).
Noxious mechanical sensory testing.
The beveled tip of a spinal needle was applied to the plantar hindpaw surface 10 times with at least 10 s between each stimulation (Hogan et al., 2004; Garrison et al., 2012). The needle was applied with enough force to indent, but not puncture, the skin. The needle indents the skin an average of 0.0725 ± 0.0066 mm (mean ± SD). Responses were categorized into normal (lifting the paw and flaring of the toes), hyperalgesic (biting, licking, flicking the paw), or no response (Garrison et al., 2012; Moehring et al., 2016).
Low threshold punctate mechanical testing.
The frequency of withdrawal to a punctate light force was determined using a 0.68 mN von Frey monofilament (Garrison et al., 2012; Moehring et al., 2016). The monofilament was applied to the plantar hindpaw 10 times with at least 10 s between each stimulation and the number of responses was recorded (Garrison et al., 2012).
Dynamic light mechanical sensory testing.
A cotton swab and a soft bristled paintbrush were used to measure responsiveness to a dynamic light stimulus. Either a puffed out cotton swab (puffed to at least 3 times the normal size) or a soft bristled paintbrush was swept from heel to toe on the plantar hindpaw 10 times with 10 s between each stimulation and the number of responses was recorded (Garrison et al., 2012, 2014; Duan et al., 2014; Moehring et al., 2016).
Hargreaves test.
Heat sensitivity was measured using the Hargreaves assay as previously established (Hargreaves et al., 1988; Jackson et al., 1995; Barabas and Stucky, 2013). Withdrawal latencies to a radiant heat source (IITC, Life Sciences Instruments) were recorded 3 times for each light treatment (590 and 490 nm LEDs) with a cutoff of 20 s. The 3 withdrawal latencies for each light treatment were averaged for each animal.
Cold plantar assay.
Cold sensitivity was measured using the cold plantar assay as previously described (Brenner et al., 2012) with modifications. Powdered dry ice was packed into a 10 ml syringe that had the tip cutoff and that had 3, 18 gauge, holes on two opposing sides. Compressed dry ice was applied to 2.5-mm-thick glass beneath the plantar surface of the hindpaw. Withdrawal latencies were recorded 3 times for each light treatment (590 and 490 nm LEDs) with a cutoff of 20 s. The 3 withdrawal latencies for each light treatment were averaged for each animal.
Analgesic place preference for spontaneous pain.
A Plexiglas two chamber box (32.5 × 21.5 × 30.5 cm) was used to analyze nonevoked behavior. The two chambers were divided by an opaque black Plexiglas wall with a 5.1 × 5.1 cm passageway between the two chambers. Each chamber rested on a Plexiglas floor under which LED light strips (595 nm, 351.8 μW; Environmental Lights catalog amber3528-120-10-reel or 460 nm, 54.3 μW; Environmental Lights catalog blue3528-120-10-reel) were mounted. LED power was controlled by a 60 W 12 VDC adapter with a dimming knob (Environmental Lights catalog Adapter-60-12D-NA). Chambers (e.g., baseline light aversion) were balanced by placing a transparent black Plexiglas sheet on top of the 460 nm LED side. Animals were allowed to freely roam between chambers throughout the 30 min experiment. During the first 5 min, floor lights were off, LEDs were then turned on for the next 5 min (continuous light stimulation); this 5 min on/5 min off pattern was repeated over the 30 min trial (3 off/on trials) for each mouse. The mouse was continuously tracked and behavior was analyzed using Anymaze software (Stoelting). For the incisional pain model, both plantar hindpaws were incised to produce robust behavioral responses since incised animals are not as hypersensitive as SNI animals to stimuli. Mobility of mice 1 and 2 d following incision was not significantly affected compared with mobility of mice following unilateral SNI surgery (data not shown: two-way ANOVA with a Tukey post test; surgery: F(2,112) = 0.1852, p = 0.8312).
Experimental design and statistical analysis.
Because no significant differences between males and females were found for any assay, the data from animals of both sexes were combined. Statistical significance was calculated using Prism 7 Software (GraphPad) and SPSS Statistics (IBM) for all experiments. A biostatistician was consulted for statistical analyses of datasets. All graphs represent mean ± SEM.
IF.
For IF studies, there were 3 animals per genotype (CGRP-Arch Cre− and Cre+). Three were male and 3 were female. Mice were 8–16 weeks old. For quantification of staining, 5–11 representative images were chosen per animal. A two-tailed unpaired t test was used to determine statistical significance.
Patch-clamp electrophysiology.
For patch-clamp studies, neurons were used if they had a leak current <300 pA. Data were analyzed using Pulse or Fitmaster software (HEKA Electronics). The time constants of inactivation (τ) for each mechanically induced inward current were obtained from the decay kinetics. τ was used to categorize neurons into rapidly adapting (RA, τ <10 ms), intermediately adapting (IA, 10 ms ≤ τ ≤30 ms), or slowly adapting (SA, τ >30 ms). Neurons were considered to be mechanically insensitive (MI) if they did not respond to at least 3 focal mechanical probing stimulations with an inward current amplitude of ≥20 pA to any stimulation. Six naïve male mice were used (n = 38–40 cells), and 12 male mice were used for SNI (n = 28 cells). Mice were 20–24 weeks old.
Naïve behavior with optogenetics.
The same cohort of animals were used for all assays (mechanical threshold, Hargreaves, and cold plantar) with at least 1 h of recovery between assays. A total of 9 animals were used per group (Cre− and Cre+). Nine were male and 9 were female. A two-way repeated-measures ANOVA with Tukey post hoc was used to determine statistical significance for each assay.
SNI and incision behavior with optogenetics.
The same animals were used for all behavioral assays (mechanical threshold, Hargreaves, and cold plantar) within a given injury model, with at least 1 h recovery between assays. A total of 9 animals were used per group (CGRP-Arch Cre− and Cre+). Nine were male and 9 were female. A three-way ANOVA with a Tukey or Sidak post hoc was used to determine statistical significance for each assay and injury model. A separate group of animals was used for noxious mechanical, punctate light mechanical, and dynamic light mechanical assays. A total of 10 mice per genotype (CGRP-Arch Cre− and Cre+) and treatment (sham or SNI) were used. Ten were male and 10 were female. A three-way ANOVA with a Tukey or Sidak post hoc was used to determine statistical significance for each assay. To perform the analgesic place preference assay, 9 CGRP-Arch Cre− mice and 10 CGRP-Arch Cre+ mice that all received SNI surgery were used. A two-way repeated-measures ANOVA with a Tukey post hoc was used to determine statistical significance.
SNI and incision behavior with CGRP8–37 inhibitor.
The same animals were used for all assays (mechanical threshold, Hargreaves, and cold plantar) within a given injury model. A total of 8 animals were used per group (sham/surgery and PBS/CGRP8–37). Adult 8-week-old C57BL/6J mice were used at the start of these experiments. For each treatment group, 4 mice were female and 4 mice were male. A three-way ANOVA with a Tukey or Sidak post hoc was used to determine statistical significance for each assay and injury model.
Results
Histological verification of CGRPα-Arch mouse model
To selectively express Arch tagged with GFP in CANs, Ai35D and CGRPαCreER (Song et al., 2012) mouse lines were mated to produce CGRPα Cre+ Arch/Arch (Cre+) and CGRPα Cre− Arch/Arch (Cre−) littermates (Fig. 1A,B). Importantly, CGRPα is expressed only in sensory neurons and CNS neurons (Assas et al., 2014; Russell et al., 2014; Hay et al., 2018); therefore, other peripheral cell types would not be affected by the optogenetic inhibition. Five repeated tamoxifen injections within 1 week were used to induce Cre activation and subsequent Arch expression in CANs (Fig. 1A). Arch activation (peak photocurrent between 550 and 600 nm) results in outward pumping of H+ ions, which ultimately leads to hyperpolarization of the cell membrane (Chow et al., 2010). Thus, using this mouse line, we were able to transiently inhibit CANs with 550–600 nm light while leaving neuronal circuits intact.
Figure 1.
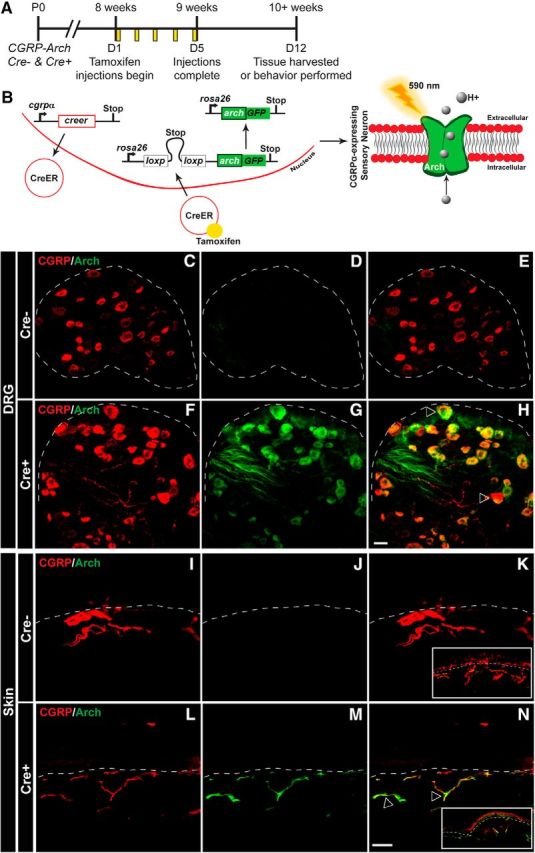
Generation of mouse expressing Arch-GFP selectively in CANs. A, CGRPα-CreER was induced with tamoxifen injections given once a day for 5 consecutive days in adult mice (8+ weeks). All experiments were performed at least 7 d after the last tamoxifen injection. B, Left, Schematic of CGRPα-CreER induction and removal of floxed stop codon, which subsequently drives the expression of Arch-GFP in CANs. Right, Arch is a proton pump that when activated by 590 nm light, pumps H+ ions out of the cell, and thereby hyperpolarizes the cell. C–H, Representative confocal images of lumbar DRG from CGRP-Arch Cre− mice (C–E) and Cre+ mice (F–H) stained with a CGRPα antibody (red) and excited Arch-GFP (green) with a 488 laser. Dotted line indicates border of DRG. C, F, Similar immunoreactivity was observed for CGRPα in both Cre− (C) and Cre+ (F) animals. D, G, Whereas negligible fluorescence was observed for Arch-GFP in Cre− animals (D), Arch+ fibers were observed throughout the DRG in Cre+ animals (G). E, H, Merged images showed no overlap between CGRPα and Arch-GFP in Cre− animals (E), but significant overlap in Cre+ animals (H). I–N, Representative confocal images of glabrous skin from CGRP-Arch Cre− mice (I–K) and Cre+ mice (L–N) stained with a CGRPα antibody (red) and excited Arch-GFP (green). Dotted line indicates epidermal/dermal border. I, L, CGRPα immunoreactivity was observed throughout neurons of the skin in Cre− (I) and Cre+ (L) animals. J, M, No Arch+ fibers were observed in Cre− mice (J), but many Arch+ fibers were observed in the skin of Cre+ animals (M). K, N, Merging showed no overlap between CGRPα and Arch-GFP in Cre− animals (K), but significant overlap in Cre+ animals (N). Inset, Image of skin at 6× lower magnification. Open arrows indicate examples of CGRPα and Arch-GFP overlapping expression. Scale bar, 25 μm. DRG marker quantification is shown in Table 1.
IF was used to confirm that Arch expression was restricted to CANs. Compared with Cre− littermates, Cre+ mice exhibited high Arch expression in the DRG and skin (Fig. 1C–H). Arch was expressed in ∼95% of Cre+ DRG CANs (Fig. 1F–H, quantified in Table 1, unpaired t test, p < 0.0001). As expected, Arch+ fibers and CGRP-IR colocalized in Cre+ skin as well (Fig. 1I–N). Overall, these data indicate that the majority of CANs express Arch in adult Cre+ mice, while the expression is minimal in Cre− mice.
Table 1.
DRG neuron marker quantificationa
| % of CGRP+ (Arch/CGRP) | |
|---|---|
| Cre− | Cre+ |
| 0.000 ± 0.000 (0/372) | 94.520 ± 0.591 * (508/539) |
aArch+ fibers and CGRP antibody staining in DRGs overlap extensively. Counts were from sections of lumbar 1–6 DRG from CGRP-Arch Cre− and Cre+ animals (5–11 representative sections per animal, n = 3 per genotype). The percentage of Arch-GFP+, CGRP+ neurons was calculated for Cre− and Cre+ animals. Cre− animals did not express Arch-GFP, whereas Arch+ fibers extensively overlapped with CGRP antibody staining in Cre+ animals. The exact number of neurons counted for each animal is shown in parentheses below the percentage. Percentages are mean ± SEM.
*p < 0.0001 (unpaired t test).
Inhibition of peripheral CAN terminals alters thermal detection in naïve animals
To determine whether transient inhibition of intact CAN terminals in the hindpaw would affect mechanical and thermal detection in uninjured naïve animals, we stimulated one plantar hindpaw with 590 nm (Arch-selective) (Chow et al., 2010) or 490 nm (control) light during mechanical and thermal behavioral testing. First, we tested the contributions of CAN terminals to mechanosensation using the von Frey up-down method to test mechanical thresholds (Dixon, 1980; Chaplan et al., 1994). Neither 590 nm nor 490 nm light affected paw withdrawal thresholds in Cre+ or Cre− mice (Fig. 2A; two-way repeated-measures ANOVA with a Sidak post test; genotype and light: F(1,16) = 0.3688, p = 0.5522). Next, we tested whether cold sensitivity was altered using the cold plantar assay (Brenner et al., 2012). Optogenetic inhibition of CAN terminals in Cre+ mice decreased withdrawal latencies (i.e., increased the baseline cold sensitivity) by twofold; no change in cold sensitivity was noted in Cre− mice with 590 nm light or in either genotype during 490 nm light exposure (Fig. 2B; two-way repeated-measures ANOVA with a Sidak post test; genotype and light: F(1,16) = 64.46, p < 0.0001). Finally, we tested heat sensitivity using the Hargreaves assay. Optogenetic inhibition of CAN terminals in Cre+ mice increased the heat paw withdrawal latency by twofold (i.e., decreased the baseline heat sensitivity); similar exposure to 590 nm light had no effect in Cre− mice. The 490 nm control light had no effect on Cre+ or Cre− animals (Fig. 2C; two-way repeated-measures ANOVA with a Sidak post test; genotype and light: F(1,16) = 213.9, p < 0.0001). Together, these data indicate that CANs are essential for peripheral heat detection and are also critical for tonically suppressing the perception of peripheral cold stimuli at the level of the spinal cord (McCoy et al., 2013), but CANs do not mediate the transmission of mechanical stimuli in the naïve uninjured state.
Figure 2.
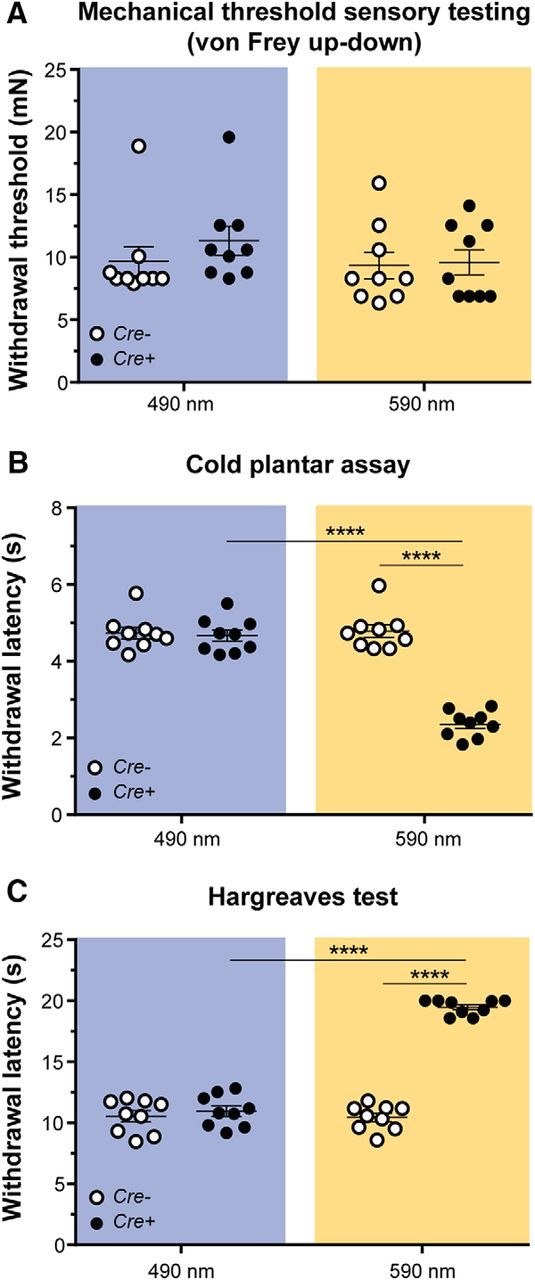
Effects of transient inhibition of CANs in naïve animals. A, Mechanical threshold sensory testing was done using the von Frey up-down method. There was no change in mechanical threshold with either control 490 nm (p = 0.5449) or Arch-activating 590 nm (p = 0.9852) light treatment of CGRP-Arch Cre− and Cre+ animals. B, Cold plantar testing was done using the cold plantar assay to determine withdrawal latencies to noxious cold. Inhibition of CANs with 590 nm light increased cold sensitivity only in Cre+ animals (****p < 0.0001), whereas 490 nm light had no effect on cold sensation (p = 0.9385). C, Hargreaves paw withdrawal testing was done using a radiant heat source shone on the plantar hindpaw. Inhibition of CANs with 590 nm light decreased heat sensitivity only in Cre+ animals (****p < 0.0001), whereas 490 nm light had no effect on heat sensitivity (p = 0.5686). Blue background represents 490 nm light treatment. Amber background represents 590 nm light treatment. Data are mean ± SEM and analyzed using two-way repeated-measures ANOVA; n = 9 animals/genotype.
Inhibition of peripheral CAN terminals alleviates evoked and spontaneous neuropathic pain
After determining the role of CANs in the noninjured setting, we aimed to uncover their roles after nerve injury. Because cutaneous CANs first degenerate peripherally and then sprout abnormally following SNI (Duraku et al., 2012; Nascimento et al., 2015), it is possible that the anatomical presence of these fibers may be important in driving SNI-induced hypersensitivity. SNI typically causes hypersensitivity to evoked mechanical, cold, and heat cutaneous stimulation (Decosterd and Woolf, 2000; Decosterd et al., 2004; Duraku et al., 2012; Smith et al., 2013). Therefore, we assessed mechanical and thermal sensation before and after SNI surgery in Cre+ and Cre− mice in the presence of 590 or 490 nm light.
SNI produced a rapid decrease in mechanical threshold by 6 and 24 h that lasted at least 8 weeks. The early hypersensitivity observed at 6 and 24 h was not reversed by 590 nm light. Interestingly, by 1 week after SNI, 590 nm light exposure of Cre+ animals caused a partial reversal of the mechanical hypersensitivity, and the hypersensitivity continued to be transiently reversed for at least 8 weeks following SNI. This phenomenon was not observed in Cre− control animals (Fig. 3A; three-way ANOVA with a Tukey post test; time: F(6,252) = 121.7, p < 0.0001; genotype and light: F(1,252) = 37.89, p < 0.0001). Further, SNI also induced chronic allodynia to punctate light touch (measured at 10 weeks after SNI), and this sensitization was fully reversed by treatment with 590 nm light in Cre+ mice (Fig. 3B; three-way ANOVA with a Tukey post test; surgery: F(1,72) = 315.8, p < 0.0001; genotype and light: F(1,72) = 29.64, p < 0.0001). Moreover, SNI induced chronic allodynia to dynamic light mechanical stimulation (measured at 3 and 10 weeks after SNI). However, unlike punctate allodynia, the dynamic mechanical allodynia was not affected by inhibition of CAN terminals (Fig. 3C–E; three-way ANOVA with a Tukey post test; Fig. 3C: surgery: F(1,56) = 140.1, p < 0.0001; genotype and light: F(1,56) = 0.09211, p = 0.7626; Fig. 3D: surgery: F(1,56) = 60.15, p < 0.0001; genotype and light: F(1,56) = 0.3559, p = 0.5532; Fig. 3E: surgery: F(1,72) = 20.61, p < 0.0001; genotype and light: F(1,72) = 1.288, p = 0.2601). Next, we tested mechanical stimuli that we are confident are noxious by probing the hindpaw with the tip of a spinal needle. SNI increased the frequency/percentage of hyperalgesic responses to punctate probing with a needle tip (measured at 10 weeks after SNI). Treatment with 590 nm light produced a striking shift of hyperalgesic responses to normal responses in Cre+ mice only (Fig. 3F; Cre+ three-way ANOVA with a Tukey post test; surgery and response: F(2,108) = 63.12, p < 0.0001; light and response: F(2,108) = 27.79, p < 0.0001. Cre− three-way ANOVA with a Tukey post test; surgery and response: F(2,108) = 137.9, p < 0.0001; light and response: F(2,108) = 2.282, p = 0.1069). Therefore, inhibition of CANs after nerve injury results in decreased chronic mechanical hypersensitivity in response to all intensities and types of punctate force but does not affect dynamic mechanical stimulation.
Figure 3.
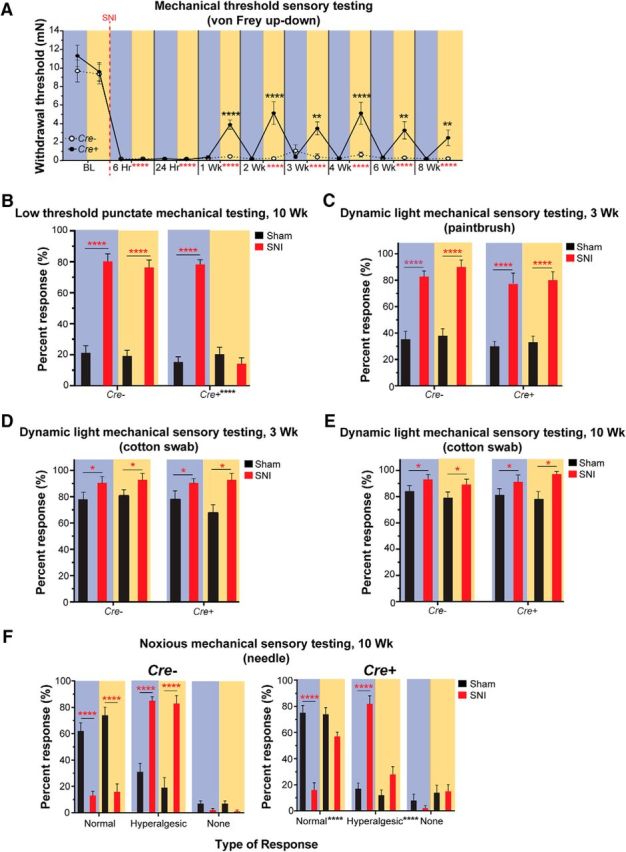
Effects of CAN inhibition on mechanical sensitivity following nerve injury. A, Longitudinal measurements of von Frey thresholds were recorded at baseline (BL, uninjured) and 6 h through 8 weeks after SNI. Animals became hypersensitive to von Frey testing following SNI starting at 6 h and lasting at least 8 weeks. Treatment with 590 nm light partially reversed the mechanical hypersensitivity beginning 1 week after SNI and lasted 8 weeks. Vertical red dashed line indicates when SNI surgery was performed. B, Light punctate mechanical sensory testing was tested 10 weeks following SNI using repeated application of a 0.68 mN von Frey filament. SNI animals were hypersensitive to the 0.68 mN von Frey stimulus compared with sham animals. The 590 nm light treatment completely reversed the hypersensitivity in Cre+, but not Cre−, animals. C, Dynamic light mechanical sensory testing was performed 3 weeks after SNI using a paintbrush. Responses to repeated paintbrush swiping are expressed as percentage of total number of responses over the number of times tested. SNI animals were hypersensitive to the dynamic paintbrush stimulus compared with sham animals. Treatment with 590 nm light had no effect on Cre− or Cre+ animals. D, E, Dynamic light mechanical sensory testing was tested 3 weeks (D) and 10 weeks (E) after SNI using the cotton swab assay. Responses to repeated cotton swab swiping are expressed as percentage of total number of responses over the number of times tested. SNI animals were hypersensitive to the dynamic stimulus compared with sham animals. Treatment with 590 nm light had no effect on Cre+ or Cre− animals. F, Noxious mechanical sensory testing was performed 10 weeks after SNI by applying a spinal needle to the lateral plantar hindpaw. Normal, hyperalgesic, and no responses are expressed as percentage of total number of responses over the number of times tested. Left, Cre− animals, where SNI animals displayed more hyperalgesic responses to the needle compared with sham animals and light treatment had no effect on their responses. Right, Cre+ animals, where SNI animals displayed more hyperalgesic responses to the needle compared with sham animals, similar to Cre− animals. Unlike Cre− animals, 590 nm light treatment of SNI Cre+ animals exhibited responses similar to sham levels. A–F, Black asterisks indicate significant differences between 490 and 590 nm light treatment in Cre+ animals. Red asterisks indicate significant differences between BL/sham and post-SNI hypersensitivity. Blue background bars represent 490 nm light treatment. Amber background bars represent 590 nm light treatment. Hr, hours; Wk, weeks. n = 10/genotype and surgery. Data are mean ± SEM and analyzed using three-way ANOVA. ****p < 0.0001, **p < 0.005, *p < 0.05. ns, Not significant (p > 0.05).
We next investigated responses to cold because individuals with neuropathic pain report higher cold-provoked pain scores (Rasmussen et al., 2004) and animals with nerve injury display increased responses to cold stimulation (Decosterd and Woolf, 2000; Nascimento et al., 2015). Unlike the immediate mechanical hypersensitivity, SNI produced a delayed cold hypersensitivity that developed by 2 weeks and lasted at least 8 weeks after SNI. In complete contrast to baseline (naïve), where Arch activation (inhibition) of CANs induced cold hypersensitivity, at 6 and 24 h after SNI, exposure of Cre+ animals to 590 nm light induced pronounced cold hyposensitivity. By 3 weeks following SNI, 590 nm light reversed the hypersensitivity to the cold stimulus, and the reversal lasted at least 8 weeks in Cre+ animals (Fig. 4A; three-way ANOVA with a Tukey post test; F(9,360) = 40.09, p < 0.0001; genotype and light: F(1,360) = 45.23, p < 0.0001). Therefore, inhibition of CANs following nerve injury results in specific mechanical and cold phenotypes that differ markedly from the phenotypes observed following CAN inhibition in noninjured, naïve animals.
Figure 4.
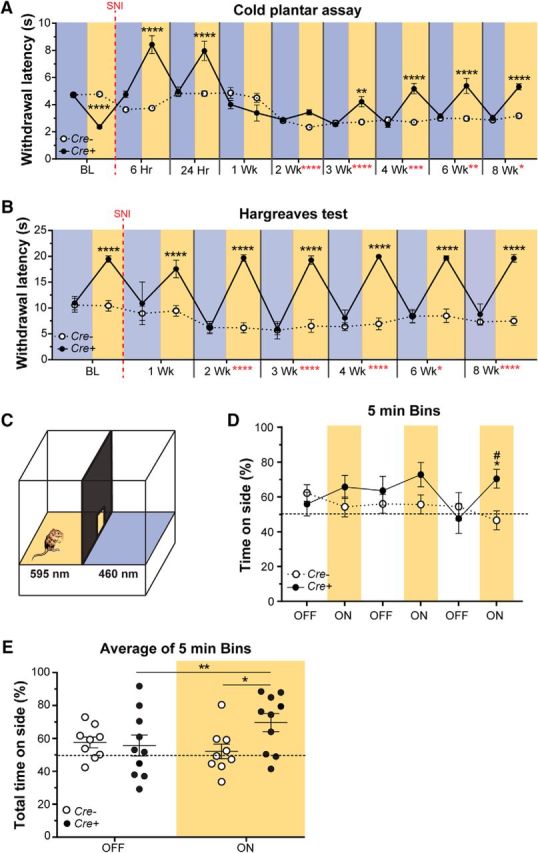
Effects of CAN inhibition on thermal sensitivity and spontaneous pain following nerve injury. A, Longitudinal measurement of cold sensitivity before and after SNI using the cold plantar assay. Hypersensitivity to the cold stimulus occurred 2 weeks after SNI and lasted 8 weeks. Treatment of Cre+ animals with 590 nm light-induced cold hyposensitivity at 6 and 24 h after SNI, and partially reversed cold hypersensitivity beginning by 3 weeks after SNI and lasting 8 weeks. B, Longitudinal measurement of sensitivity to a radiant heat source before and after SNI using the Hargreaves paw withdrawal assay. Hypersensitivity to the heat stimulus occurred 2 weeks after SNI and lasted 8 weeks. Treatment of Cre+ animals with 590 nm light-induced heat hyposensitivity immediately at BL and lasted 8 weeks following SNI. A, B, Vertical red dashed line indicates the time point that SNI surgery was performed; n = 10 animals/genotype and surgery. Black asterisks indicate significant differences between 490 and 590 nm light treatment in Cre+ animals. Red asterisks indicate significant differences between BL and post-SNI hypersensitivity. Blue background bars represent 490 nm light treatment. Amber background bars represent 590 nm light treatment. Hr, hours; Wk, weeks. Data are mean ± SEM and analyzed using three-way ANOVA. ****p < 0.0001, ***p < 0.0005, **p < 0.005, *p < 0.05. ns, Not significant (p > 0.05). C, Diagram represents the real-time place preference setup, where at 10 weeks after SNI, Cre+ and Cre− animals were allowed to choose to spend time in a chamber with a floor lit with either 595 and 460 nm light. The lights were off for 5 min and then turned on for 5 min; this sequence was repeated 3 times for a total of 30 min. D, Percentage time spent on the 595 nm side of the box during each 5 min bin. By the last 5 min ON bin, the Cre+ animals significantly preferred the 595 nm side of the box compared with light OFF (*p = 0.0205) and opposed to Cre− animals (#p = 0.0471). E, The time spent on the 595 nm side of the box is represented as percentage of total time (15 min) during the light OFF and light ON conditions. Cre+ animals preferred the 595 nm side of the box when the lights were on (**p = 0.0047). Cre+ animals had a higher preference for the 595 nm side of the box when the lights were on compared with the Cre− animals (*p = 0.0439). D, E, Amber bars represent 595 nm light treatment Data are mean ± SEM and analyzed using two-way repeated-measures ANOVA; n = 9 for Cre− animals and n = 10 for Cre+ animals.
Because individuals with neuropathies also report greater heat-evoked pain (Koltzenburg et al., 1994; Rowbotham and Fields, 1996; Colleoni and Sacerdote, 2010), we next tested heat sensitivity in our SNI mice. Previous studies have shown that animals are hypersensitive to heat stimuli by 2 weeks after SNI (Decosterd and Woolf, 2000; Baliki et al., 2005; Duraku et al., 2014). Here, animals displayed delayed heat hypersensitivity in the form of decreased withdrawal latencies to a radiant heat source by 2 weeks following SNI, and the hypersensitivity lasted at least 8 weeks after SNI. Treatment with 590 nm light resulted in immediate heat hyposensitivity, which included elimination of all SNI-induced heat hypersensitivity at each time point (2–8 weeks after SNI) in Cre+ animals (Fig. 4B; three-way ANOVA with a Tukey post test; F(6,252) = 35.02, p < 0.0001; genotype and light: F(1,252) = 1128, p < 0.0001). Importantly, the effects of 590 nm light were not observed in Cre− control animals or in either genotype during the control 490 nm light exposure for any of the behavior assays tested (Figs. 3A–F, 4A,B). These data are consistent with those observed in uninjured naïve animals and indicate that CANs are essential for all heat detection under both naïve and nerve injury conditions.
Because individuals with neuropathic pain report greater superficial, cutaneous ongoing spontaneous pain scores than individuals without neuropathic injuries (Rasmussen et al., 2004; Truini et al., 2013; Lax et al., 2014), we assessed nonevoked spontaneous pain in freely moving Cre+ and Cre− mice. We developed a real-time place preference assay in which CGRP-Arch animals underwent SNI and 10 weeks later, were given the choice of spending time on a 595 nm or a 460 nm LED floor (Fig. 4C). Here we used an alternating off/on paradigm, where the LED floor was turned off for 5 min then turned on for 5 min (1 bin); this sequence was repeated for a total of 30 min (3 bins overall). The animals had no preference for a side when the lights were off. When the lights were on, during the first 2 bins, Cre+ animals trended to prefer the 595 nm side of the box and by the third bin, a significant preference for the 595 nm floor developed (Fig. 4D; two-way repeated-measures ANOVA with a Tukey post test; genotype and light: F(5,85) = 2.957, p = 0.0165). When data from all bins were averaged together, Cre+ animals spent more time in the 595 nm floor-paired chamber when the light was on compared with when the light was off. Additionally, Cre+ mice also spent more time in the 595 nm chamber than Cre− animals during the light on bins (Fig. 4E; two-way repeated-measures ANOVA with a Sidak post test; genotype and light: F(1,17) = 11.59, p = 0.0034). These findings suggest that sensitization of peripheral fibers is necessary for ongoing spontaneous pain and that CANs play a significant role in maintaining SNI-induced spontaneous pain behavior.
SNI increases mechanically evoked currents in isolated CANs, and optogenetic inhibition of these neurons reverses the mechanical sensitization
There is a paucity of studies that have investigated whether primary afferent neurons are sensitized to mechanical stimuli after peripheral nerve injury. A previous study from our laboratory showed that peripheral terminals of C and Aδ nociceptors are sensitized to mechanical stimuli (Smith et al., 2013). To our knowledge, no studies have assessed the mechanical response properties of the somata of sensory neurons in any nerve injury paradigm. To address this, we used whole-cell patch-clamp electrophysiology together with focal mechanical probing of the soma membrane. DRG neurons from only Cre+ animals were used to identify and patch CGRPα+ cells because Cre+ animals express Arch-GFP in CANs. First, the electrophysiological properties of lumbar DRG CANs that projected to the sciatic nerve from uninjured naïve animals and from animals 16 weeks after SNI were determined with and without light treatment. No differences in the resting membrane potential of CAN somata were found after SNI compared with naïve DRG neurons with the light off. As expected, because Arch is an outward-rectifying proton pump, the application of 590 nm light significantly decreased the resting membrane voltage of both naïve and SNI Arch+ neurons on average by 5.5 mV compared with light off conditions; these data indicate that Arch is functional in CAN somata (Fig. 5A; two-way ANOVA with a Sidak post test; surgery: F(1,130) = 3.09, p = 0.0811 light: F(1,130) = 22.28, p < 0.0001). In addition to resting membrane potential, the rheobase current, a measure of action potential threshold, was recorded. No differences in rheobase were found between naïve and SNI neurons with the light off. However, exposure of naïve and SNI Arch-expressing CANs to 590 nm light increased their rheobase by more than twofold (Fig. 5B; two-way ANOVA with a Sidak post test; light: F(1,131) = 0.6471, p < 0.4226; surgery: F(1,131) = 29.59, p < 0.0001). These data confirm that the 590 nm light functionally activates Arch in the somata of CANs and significantly decreases their electrical excitability.
Figure 5.
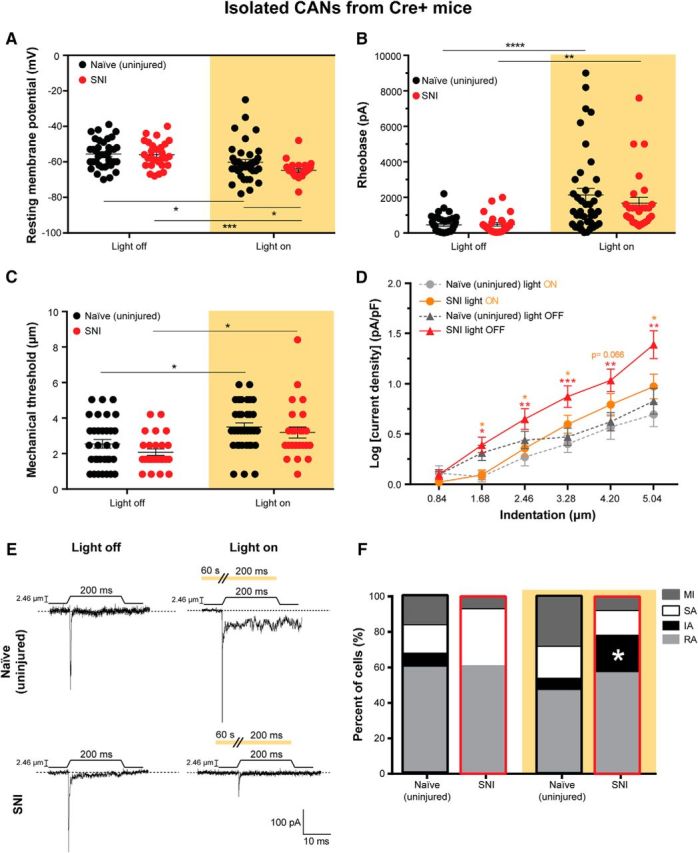
SNI increases mechanical responsiveness of CAN somata, which is reversed by Arch activation. Neurons from only the Cre+ CGRP-Arch mice were used for these experiments to be able to specifically identify CANs via their GFP expression. A, Resting membrane potential of naïve (uninjured) and SNI DRG neurons with (right) and without (left) Arch activation. Treatment of naïve (*p = 0.0254) and SNI (***p = 0.0002) neurons with 590 nm light decreased their resting membrane potentials compared with no light treatment. The 590 nm light decreased the resting membrane potential of the SNI neurons significantly more than naïve neurons (*p = 0.0474), two-way ANOVA. B, Rheobase (amount of current required to induce an action potential) in naïve and SNI neurons with (right) and without (left) Arch-activating light treatment. Treatment of naïve (****p < 0.0001) and SNI (**p = 0.0074) neurons with 590 nm light increased their rheobases compared with no light treatment, two-way ANOVA. C, Level of mechanical indentation required to elicit an inward current (mechanical threshold) in naïve and SNI neurons with (right) and without (left) light treatment. Treatment of naïve (*p = 0.0237) and SNI (*p = 0.0184) neurons with 590 nm light increased their mechanical threshold compared with no light treatment, two-way ANOVA. D, Log of mechanically gated current densities in response to a series of 6 increasing indentations of the DRG membrane of naïve neurons during light off (dark gray) and light on (light gray) and SNI neurons during light off (red) and light on (orange). Treatment of naïve neurons with 590 nm had no effect. SNI neurons had a greater current density in response to mechanical probing than naïve neurons. This increase in current density in response to mechanical probing in SNI neurons was reversed by 590 nm light treatment. Red asterisks indicate significant differences between naïve and SNI light off. Orange asterisks indicate significant differences between SNI light off and light on conditions. ***p < 0.0005 (mixed-model analysis). **p < 0.005 (mixed-model analysis). *p < 0.05 (mixed-model analysis). ns, Not significant (p > 0.05). E, Representative current traces from 2.46 μm indentation of naïve and SNI neurons during light off (left) and light on (right) conditions indicated by amber background. Amber bar above current traces during light on conditions (right) represents a continuous duration of light stimulation (60 s before the mechanical stimulus and throughout the 200 ms mechanical stimulus). F, Current kinetics in response to mechanical probing of naïve and SNI neurons during light off (left) and on (right) conditions. SNI neurons with no light treatment did not display IA currents, but SNI neurons treated with 590 nm light displayed a return of IA currents similar to naïve controls (*p = 0.0232). Black outline indicates naïve neurons. Red outline designates SNI neurons. Analysis was done using χ2 and Fisher's exact test. Amber background bars represent Arch-activating light treatment. Data are mean ± SEM. Naïve light off, n = 38 cells and n = 3 animals. Naïve light on, n = 40 cells and n = 3 animals. SNI light off, n = 28 cells and n = 6 animals. SNI light on, n = 28 cells and n = 6 animals.
Because we found that, on the behavioral level, the mechanical sensitization in SNI animals could be reversed by 590 nm light, we next asked whether the mechanical responsiveness of Arch-positive lumbar CAN DRG from naïve and SNI mice could also be altered by 590 nm light stimulation. No differences were found in the mechanical thresholds between naïve uninjured and SNI CAN somata with the light off. However, optogenetic activation of Arch increased the mechanical indentation thresholds of both the naïve and SNI CAN somata (Fig. 5C; two-way ANOVA with a Sidak post test; surgery: F(1,115) = 2.512, p = 0.1157; light: F(1,115) = 17.72, p < 0.0001), thereby decreasing mechanical sensitivity of the soma membrane. Furthermore, the current densities in response to incremental mechanical probing were significantly larger in the SNI CAN somata than the naïve CAN somata (without light treatment), a finding that indicates that the SNI model in vivo sensitized the primary afferent neurons to mechanical stimuli at the level of the soma membrane (Fig. 5D, red line; mixed-model analysis with a Sidak post test; stimulus, surgery, and light: F(21, 425.477) = 3.015, p < 0.0001). Of note, this sensitization was long lasting and a phenotype of the neuron. The cellular mechanical sensitization was present 16 weeks following nerve injury and was present in somata that had been removed from the animal for 18–24 h as well as dissociated from their peripheral processes. Consistent with our behavioral data, which showed no effect of Arch activation on behavioral mechanical thresholds in naïve animals, Arch activation in isolated somata did not affect the naïve CAN somata current density in response to incremental mechanical probing (Fig. 5D, light gray line). However, Arch activation completely reversed the amplified mechanical current densities in SNI CAN somata back to naïve, noninjury levels (Fig. 5D, orange line). Example traces for currents in response to mechanical stimulation during light off and on conditions are shown in Figure 5E. These results show that the somata of lumbar CAN DRG are sensitized to mechanical stimuli as long as 16 weeks after SNI, that this sensitization persists in neurons isolated from the animal, and that this mechanical sensitization is fully reversed by Arch activation. Therefore, these data support our behavioral data in Figure 2 and our hypothesis that sensitization of peripheral fibers is required for chronic SNI-induced hypersensitivity.
To further characterize the functional changes that occur in CAN somata after SNI (16 weeks), we studied the current kinetics of both naïve and SNI CAN DRG neurons in the absence and presence of 590 nm light. SNI also altered the type of current kinetics displayed in response to focal mechanical probing (no light exposure). Unexpectedly, CANs from SNI animals no longer displayed IA currents and had a trend toward decreased MI currents (i.e., more mechanically responsive currents in total), but had no significant change in SA or RA currents compared with naïve neurons (Fig. 5F; Fisher's exact test, IA: p = 0.2559; MI: p = 0.2820; SA: p = 0.1443; RA: p > 0.9999). With Arch stimulation, the current kinetics of naïve CANs were unaltered (Fig. 5F; Fisher's exact test, IA: p > 0.9999; MI: p = 0.4243; SA: p > 0.9999; RA: p = 0.3754). Surprisingly, the current kinetics of CANs from SNI animals were significantly altered by Arch activation; with brief 590 nm light exposure, the IA currents reappeared at levels that were not significantly different from those in naïve CANs with or without 590 nm light exposure. In addition, there was also a trend for decreased SA currents in SNI neurons during Arch activation. However, the proportion of MI and RA currents was unchanged in SNI CANs exposed to 590 nm light (Fig. 5F; Fisher's exact test, SNI light off vs SNI light on: IA: p = 0.0232; MI: p > 0.9999; SA: p = 0.2047; RA: p > 0.9999). Overall, these functional data in primary sensory somata correspond well with our behavioral data in Figures 2A and 3A–F, which demonstrated that CANs convey punctate mechanical sensitivity after chronic nerve injury. Furthermore, we demonstrate, for the first time, that peripheral nerve injury is capable of sensitizing the membrane of primary sensory neurons to mechanical stimuli, and this sensitization persists for at least 24 h when neurons are ex vivo. This finding at the DRG soma membrane level, together with the finding that suprathreshold mechanical stimulation of nociceptor terminals in skin-nerve preparations from mice with SNI results in increased firing compared with sham preparations (Smith et al., 2013), suggests that peripheral sensitization of primary afferent neurons is a potentially significant driver of the chronic mechanical hypersensitivity that follows nerve injury.
Peripheral CAPS is not required to maintain neuropathic pain
We have demonstrated that CANs are critical for SNI-mediated hypersensitivity because ∼30 s of CAN inhibition with 590 nm light repeatedly reversed the chronic SNI-induced mechanical and thermal hypersensitivity over the longitudinal time course of 1–8 weeks (Figs. 3A, 4A,B). In addition, CAN inhibition relieved the SNI-induced ongoing pain (Fig. 4D,E). Of note, the brief inhibition of CANs we used would not likely be long enough to inhibit, degrade, or affect the levels of CGRPα peptide that these CANs might have previously released from their peripheral terminals. Therefore, at this point, we could not rule out the involvement of peripheral CAPS in neuropathic pain.
Peripheral CGRPα peptide levels are elevated after SNI (W. Ma and Quirion, 2006) and CGRP− fibers and non-neuronal cells (keratinocytes, blood vessels, mast cells, etc.) express the CGRPα receptor, CGRP1 (Russell et al., 2014; Iyengar et al., 2017; Hay et al., 2018). Therefore, it follows that secreted CGRPα could act on nearby CGRP− neuronal and non-neuronal cells in the skin to induce hypersensitivity following SNI. To test this, we locally injected the CGRP1 receptor antagonist CGRP8–37 at a concentration known to be effective in other pain models (Hsieh et al., 2012b; Ishida et al., 2014) or PBS into the plantar hindpaw of SNI and sham animals weekly 45 min before testing and measured their responses to mechanical and thermal stimuli at 1, 2, and 3 weeks after SNI. SNI produced prominent mechanical hypersensitivity by 1 week that lasted throughout the 3 weeks of testing. Peripheral CGRP8–37 had no effect on mechanical hypersensitivity compared with PBS (Fig. 6A; three-way ANOVA with a Tukey post test; surgery: F(1,112) = 244.7, p < 0.0001; drug and surgery: F(1,112) = 0.02969, p = 0.8635). SNI resulted in cold hypersensitivity by 1 week that lasted at least 3 weeks. Peripheral CGRP8–37 administration also had no effect on cold hypersensitivity compared with PBS (Fig. 6B; three-way ANOVA with a Tukey post test; surgery: F(1,112) = 274.3, p < 0.0001; surgery and drug: F(1,112) = 0.6313, p = 0.4286). Further, heat hypersensitivity developed 2 weeks following SNI that lasted at least 3 weeks. This heat hypersensitivity was also not affected by peripheral CGRP8–37 or PBS administration (Fig. 6C; three-way ANOVA with a Tukey post test; surgery: F(1,112) = 55.33, p < 0.0001; surgery and drug: F(1,112) = 0.3206, p = 0.5724). Therefore, these data indicate that peripheral CAPS is not required for SNI-induced hypersensitivity.
Figure 6.
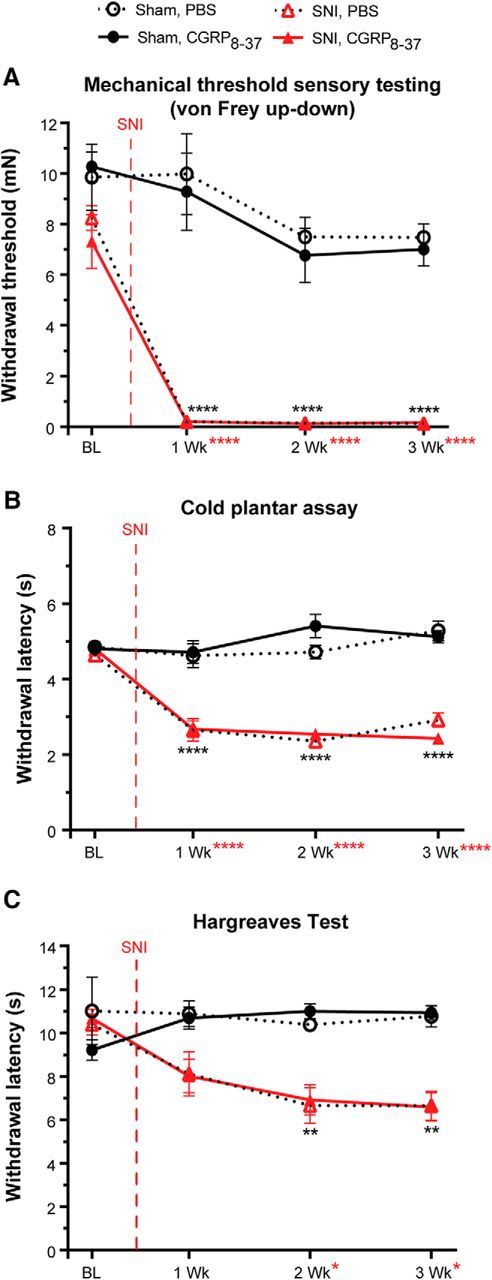
Peripheral CAPS is not required for SNI mechanical and thermal hypersensitivity. A–C, Mice were administered intraplantar CGRP8–37 (533 μm) or PBS each test day 45 min before testing. A, Measurement of paw withdrawal threshold using the von Frey up-down method. CGRP8–37 had no effect on mechanical hypersensitivity in SNI animals or sham controls. B, Measurement of cold sensitivity using the cold plantar assay. CGRP8–37 had no effect on the SNI-induced cold hypersensitivity in SNI or sham animals. C, Measurement of sensitivity to a radiant heat source using the Hargreaves paw withdrawal assay. CGRP8–37 had no effect on heat hypersensitivity in SNI or sham animals. Vertical red dashed line indicates when SNI surgery was performed. Red asterisks indicate significant differences between naïve and SNI surgery. Black asterisks indicate significant differences between naïve and SNI animals treated with CGRP8–37. Data are mean ± SEM and analyzed using three-way ANOVA; n = 8 animals for each treatment group. ****p < 0.0001, **p < 0.005, *p < 0.05. ns, Not significant (p > 0.05).
Inhibition of CANs has no effect on incisional pain
In the periphery, stimulation of sensory nerve endings can lead to release of CGRPα, inducing neurogenic inflammation and pain (Iyengar et al., 2017). The incisional pain model (Brennan et al., 1996) is widely used to study inflammatory pain in rodents as it is a model that closely mimics peripheral inflammatory injuries in humans (Ghasemlou et al., 2015). Therefore, we aimed to investigate whether CANs drive incision-induced mechanical and heat hypersensitivity.
The model of incisional pain is known to cause mechanical and heat hyperalgesia, but not cold hypersensitivity (Brennan et al., 1996; Pogatzki-Zahn et al., 2005; Zhang et al., 2013). Thus, mechanical and heat sensitivities were assessed before and after incision in Cre+ and Cre− mice exposed to either 590 or 490 nm light during testing over a 7 d period. Mechanical hypersensitivity following incision occurred immediately at 2 h and lasted 5 d after incision. Inhibition of CANs did not alter the mechanical sensitivity (Fig. 7A; three-way ANOVA with a Tukey post test; time: F(8,288) = 98.64, p < 0.0001; light and genotype: F(1,288) = 0.7719, p = 0.3803). Incisional heat hypersensitivity occurred immediately at 2 h and lasted 6 d after incision. Again, inhibition of CANs had no effect on heat hypersensitivity. However, 7 d after incision, when heat sensitivity had recovered to baseline levels, inhibition of CANs resulted in heat hyposensitivity similar to that observed at baseline and in uninjured naïve Cre+ animals in Figure 2A (Fig. 7B; three-way ANOVA with a Tukey post test; time: F(8,288) = 186.9, p < 0.0001; light and genotype: F(1,288) = 30.09, p < 0.0001). The Arch-induced heat hyposensitivity by 7 d is most likely a result of the incision being healed and the hypersensitivity being resolved back to baseline by 7 d (Brennan, 2011; Barabas and Stucky, 2013; Ghasemlou et al., 2015).
Figure 7.
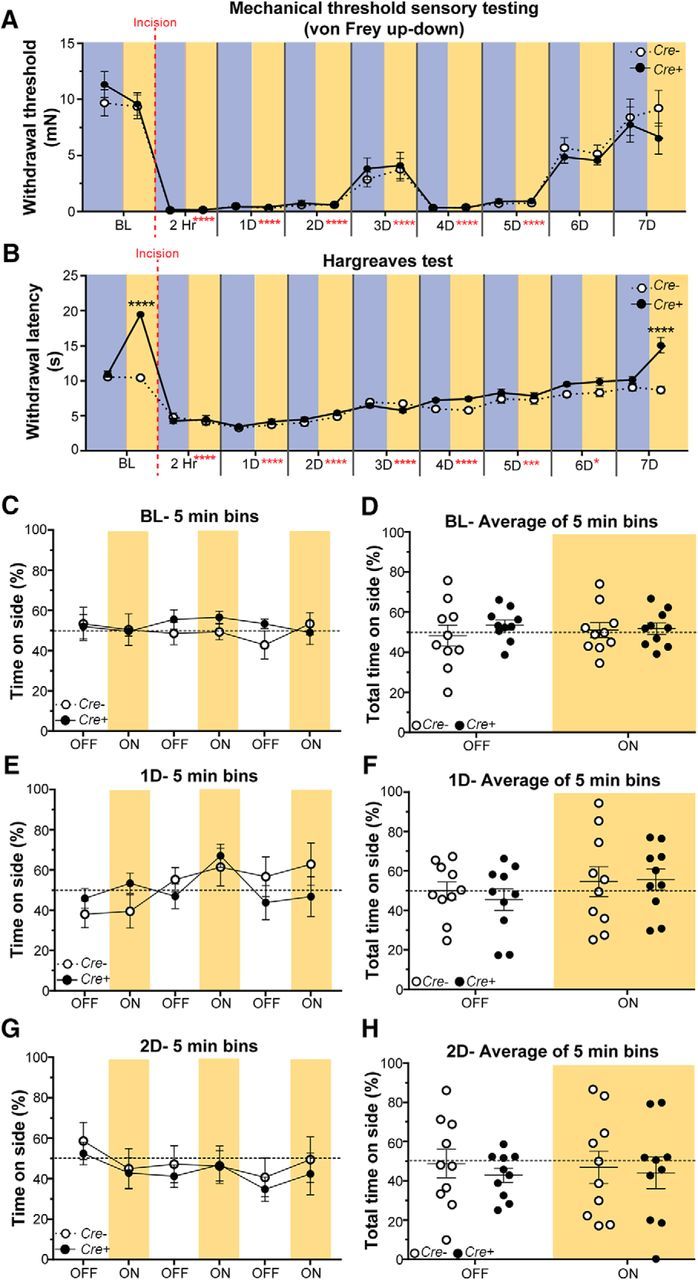
Brief optogenetic inhibition of CANs has no effect on incisional pain. A, Measurement of paw withdrawal threshold using the von Frey up-down method. The 590 nm light had no effect on mechanical sensitivity. B, Measurement of sensitivity to a radiant heat source using the Hargreaves assay. The 590 nm light-induced heat hyposensitivity before incision, and also when the incision-induced hypersensitivity was gone at 7 d after incision; however, 590 nm light had no effect on incision-induced heat hypersensitivity. Blue background represents 490 nm light treatment. Amber background represents 590 nm light treatment. Vertical red dashed line indicates when the incision surgery was performed. Red asterisks indicate significant differences between baseline and postincision surgery. Black asterisks indicate significant differences between Cre+ and Cre− animals. Data are mean ± SEM and analyzed using three-way ANOVA; n = 9 animals for each treatment group. ****p < 0.0001, ***p < 0.0005, *p < 0.05. ns, Not significant (p > 0.05). C–H, Real-time place preference where incision Cre− and Cre+ animals were allowed to choose between a 595 and 460 nm lit floor. The lights were off for 5 min and then turned on for 5 min; this was repeated 3 times for a total of 30 min. C, E, G, Percentage time spent on the 595 nm side of the box during each 5 min bin before incision (BL) (C), 1 d (1D) following incision (E), and 2 d (2D) following incision (G). Neither Cre+ nor Cre− exhibited preference (C: p = 0.6884; E: p = 0.1082; G: p = 0.1237). D, F, H, The time spent on the 595 nm side of the box is represented as percentage of total time (15 min) during the light OFF and light ON conditions before incision (BL) (D), 1 d (1D) following incision (F), and 2 d (2D) following incision (H). Neither Cre+ nor Cre− animals exhibited a preference (D: p = 0.5428; F: p = 0.5498; H: p = 0.6557). C–H, Amber bars represent 595 nm light treatment. Data are mean ± SEM and analyzed using two-way repeated-measures ANOVA; n = 9 for Cre− animals and n = 10 for Cre+ animals.
Because mice exhibit spontaneous pain behaviors up to 48 h following incision (Pogatzki and Raja, 2003; Sahbaie et al., 2014), we assessed nonevoked spontaneous pain in freely moving Cre+ and Cre− mice before incision (BL) and 1 and 2 d following incision. We used the same real-time place preference assay as Figure 4C, where mice were given the choice of spending time on a 595 nm or a 460 nm LED floor. The same alternating off/on paradigm as in Figure 4D, E was used, where the LED floor was turned off for 5 min then turned on for 5 min (1 bin); this sequence was repeated for a total of 30 min (3 bins overall). The animals had no preference for a side regardless of the light treatment (Fig. 7C,E,G; two-way repeated-measures ANOVA with a Tukey post test; Fig. 7C: genotype and light: F(5,90) = 0.6153, p = 0.6884; Fig. 7E: genotype and light: F(5,90) = 1.866, p = 0.1082; Fig. 7G: genotype and light: F(5,90) = 0.1237, p = 0.9868). When data from all bins were averaged together, neither Cre+ nor Cre− animals exhibited a preference for either the 595 or 460 nm floor (Fig. 7D,F,H; two-way repeated-measures ANOVA with a Tukey post test; Fig. 7D: genotype and light: F(1,18) = 0.3849, p = 0.5428; Fig. 7F: genotype and light: F(1,18) = 0.5498, p = 0.4679; Fig. 7H: genotype and light: F(1,18) = 0.2055, p = 0.6557). Overall, these results indicate that CANs do not convey the mechanical or heat hypersensitivity, or spontaneous pain associated with incision.
Peripheral blockade of CAPS alleviates incisional pain
Because brief inhibition of CANs did not alleviate the mechanical and heat hypersensitivity observed after incision and brief inhibition would not allow sufficient time for the degradation of the already present CGRPα peptide released after incisional injury, we next asked whether peripheral CAPS mediates these pain phenotypes. Because CAPS is known to drive inflammation (Iyengar et al., 2017), we hypothesized that blockade of peripheral CAPS would decrease the mechanical and heat hypersensitivity displayed after incision. To test this, the same concentration of CGRP8–37 that was used in SNI experiments and that has been shown to be effective in other pain models (Hsieh et al., 2012a; Ishida et al., 2014) or PBS was locally injected into the plantar hindpaw of incision and sham animals daily 45 min before testing responses to mechanical and heat stimuli. Neither CGRP8–37 nor PBS had an effect at baseline. Incisional mechanical hypersensitivity was present 1 d after surgery and lasted 5 d, similar to data in Figure 6. However, administration of CGRP8–37 reduced the mechanical hypersensitivity in incised animals after 3 d of treatment (Fig. 8A; three-way ANOVA with a Tukey post test; surgery: F(1,224) = 155.6, p < 0.0001; surgery and drug: F(1,224) = 8.72, p = 0.0035). Incision-induced heat hypersensitivity was present 1 d after incision and lasted 5 d. The heat hypersensitivity was immediately reversed with CGRP8–37 administration (Fig. 8B; three-way ANOVA with a Tukey post test; surgery: F(1,224) = 95.74, p < 0.0001; surgery and drug: F(1,224) = 41.67, p < 0.0001). These data indicate that peripheral CAPS is involved in mediating both mechanical and heat hypersensitivity following perioperative incision.
Figure 8.

Peripheral antagonism of CAPS alleviates incisional pain. A, B, Mice were administered intraplantar CGRP8–37 (533 μm) or PBS each test day 45 min before testing. A, Measurement of paw withdrawal threshold using the von Frey up-down method. CGRP8–37 partially reversed the mechanical hypersensitivity of incision animals by 3 d and had no effect on sham animals. B, Measurement of sensitivity to a radiant heat source using the Hargreaves assay. CGRP8–37 immediately reversed the heat hypersensitivity following incision but had no effect on sham animals. Vertical red dashed line indicates when the incision surgery occurred. Red asterisks indicate significant differences between incision CGRP8–37 and incision PBS animals. Black asterisks indicate significant differences between sham CGRP8–37 and incision CGRP8–37. Data are mean ± SEM and analyzed using three-way ANOVA; n = 8 animals for each treatment group. ****p < 0.0001, ***p < 0.0005, **p < 0.005, *p < 0.05. ns, Not significant (p > 0.05).
Injury does not induce ectopic Arch expression
It has been demonstrated that CGRP IF is significantly increased in keratinocytes following spinal nerve ligation, sciatic nerve chronic constriction injury, and Complete Freud's Adjuvant in rats (Hou et al., 2011). However, the CGRP isoform (α or β) was not distinguished because rat anti-CGRP antibodies cannot distinguish between the isoforms. IF was used to confirm that Arch expression was restricted to CANs and to ensure that the behavioral effects observed from Arch activation were specific to CANs. Compared with sham Cre+ mice, SNI and incision Cre+ mice did not exhibit Arch-GFP expression in keratinocytes (Fig. 9A–L). Additionally, as expected, Arch+ fibers were significantly reduced in skin 3 weeks following SNI (Fig. 9F). Overall, these data indicate that Arch expression is restricted to CANs after injury similar to sham animals and the behavioral effects observed are due to inhibition of CANs.
Figure 9.

Injury does not induce ectopic expression of Arch. A–D, Representative confocal images of glabrous skin from sham CGRP-Arch Cre+ mice stained with a keratin 14 antibody (red) and DRAQ5 nuclear stain (blue) and excited Arch-GFP (green) with a 488 laser. D, Merged image showed no overlap between Arch-GFP and keratin 14. E–H, Representative confocal images of glabrous skin from CGRP-Arch Cre+ mice 3 weeks following SNI stained with a keratin 14 antibody (red) and DRAQ5 nuclear stain (blue) and excited Arch-GFP (green) with a 488 laser. F, Arch-GFP CANs are significantly decreased compared with sham. H, Merged image shows no overlap between Arch-GFP and keratin 14. I–L, Representative confocal images of glabrous skin from CGRP-Arch Cre+ mice 1 d following incision stained with a keratin 14 antibody (red) and DRAQ5 nuclear stain (blue) and excited Arch-GFP (green) with a 488 laser. J, Arch-GFP CANs are not changed compared with sham. L, Merged image shows that there is no overlap between Arch-GFP and keratin 14. Scale bar, 25 μm.
Together, these data suggest that CANs have distinct roles in neuropathic and incisional pain. Our data indicate that CANs drive nerve injury-induced evoked mechanical, cold, and heat hypersensitivity, ongoing spontaneous pain, and are directly sensitized at the membrane level to mechanical stimuli following nerve injury. CANs do not play a direct role in transmitting incision-induced mechanical and heat hypersensitivity to the CNS. In contrast to neuropathic pain, peripheral CAPS plays a major role in mediating surgical incision-induced inflammatory pain.
Discussion
Our study demonstrates that intact peripheral CANs are sensitized and drive transmission of SNI-induced persistent mechanical, cold, and heat hypersensitivity, and spontaneous pain to the spinal cord. In contrast, CANs do not drive the transmission of incisional pain but instead amplify incisional pain via their peripheral release of CGRPα and peripheral CAPS (Table 2).
Table 2.
Sensory modalities affected by CAN inhibition or blockade of CAPSa
| Sensitivity | Naïve (uninjured) |
SNI |
Incision |
|||
|---|---|---|---|---|---|---|
| 590 nm | CGRP8–37 | 590 nm | CGRP8–37 | 590 nm | CGRP8–37 | |
| Mechanical | ↔ | ↔ | ↓ | ↔ | ↔ | ↓ |
| Cold | ↑ | ↔ | ↓ | ↔ | — | — |
| Heat | ↓ | ↔ | ↓ | ↔ | ↔ | ↓ |
aIn naïve (uninjured) animals, brief CAN inhibition with 590 nm light did not affect mechanical sensitivity; however, it increased cold sensitivity and decreased heat sensitivity. CGRP8–37 had no effect on any modality in naïve animals. After SNI, once mechanical, cold, and heat hypersensitivity developed, brief inhibition of CANs with 590 nm light reduced the observed hypersensitivity for each modality. CGRP8–37 had no effect on these modalities following SNI. After incision, mechanical and heat hypersensitivity occurred, but cold sensitivity was unaltered. Brief CAN inhibition with 590 nm light had no effect on mechanical or heat hypersensitivity after incision. However, blockade of peripheral CAPS with CGRP8–37 reversed the mechanical and heat hypersensitivity induced by surgical incision.
CANs are required for maintenance of chronic pain after nerve injury
SNI induced pronounced mechanical hypersensitivity by 6 h that persisted for at least 8 weeks. Optogenetic inhibition of CANs had no effect at 6 and 24 h after SNI, but by 1 week, partially and consistently reversed the mechanical hypersensitivity for 8 weeks. SNI is known to cause denervation of skin and muscle where CGRPα staining is nearly gone within the first 2 weeks (Duraku et al., 2012; Nascimento et al., 2015). The lack of effect at early time points may be due to severely reduced CAN innervation where the mechanical hypersensitivity is likely mediated by CGRP-negative fibers that are sensitized by factors released from neighboring injured fibers or support cells (Berta et al., 2014, 2017; Weaver et al., 2017). The most parsimonious explanation for the inhibition that begins at 1 week is that spared collateral CANs reinnervate the hindpaw (Duraku et al., 2012; Nascimento et al., 2015) and continue to maintain transmission of mechanical neuropathic pain chronically. Our finding, that chronic (10 weeks after SNI) hypersensitivity to punctate innocuous and noxious touch was reversed by CAN inhibition but not dynamic stimuli, suggests that CANs may mediate only punctate chronic neuropathic allodynia (Koltzenburg et al., 1992; La and Chung, 2017).
CANs mediate chronic thermal hypersensitivity following nerve injury
SNI-induced thermal hypersensitivity developed later than mechanical hypersensitivity. Both cold and heat hypersensitivity did not begin until 2 weeks following SNI, a time point at which collateral CANs reinnervate the plantar hindpaw (Nascimento et al., 2015). Optogenetic inhibition of CANs in naïve animals elicited a pronounced cold hypersensitivity that is caused by the unmasking of CAN-mediated tonic inhibition of spinal cord neurons that convey cold information to the brainstem and cortex (McCoy et al., 2013). Unexpectedly, SNI rapidly reversed the cold function of CANs compared with the naïve state in that by at 6 and 24 h, CAN inhibition elicited pronounced cold hyposensitivity and reversed the cold hypersensitivity that began at 3 weeks. This reversed cold function may be due either to altered peripheral CAN transmission to the spinal cord that results from the acute loss of damaged afferents after SNI (Nascimento et al., 2015) or to changes in central terminals of cold-sensing neurons (Ran et al., 2016). Alternatively, silent thermosensors may play a role in this rapid change in function following injury. Silent thermosensors are unresponsive to thermal stimuli before injury but respond robustly to cold stimuli after injury (Yarmolinsky et al., 2016). This population is a subset of TRPM8+ neurons that express CGRP. Therefore, it is possible that silent thermosensors may be required to sense the onset of cold stimuli after injury and that our optogenetic inhibition paradigm transiently inhibits them.
In contrast to cold, the heat function of CANs was minimally altered by nerve injury as optogenetic inhibition immediately and continually produced heat hyposensitivity in both naïve and SNI animals throughout the acute to chronic time course. The finding that optogenetic inhibition immediately and persistently induces heat hyposensitivity after SNI may indicate that CANs are innately required for heat sensation in both naïve and nerve-injured states. Together, our data show that peripheral CANs are required to drive the transmission of chronic mechanical, cold, and heat hypersensitivity in a time-dependent manner following SNI.
Peripheral CANs are required for maintenance of ongoing nerve injury pain
Superficial spontaneous pain is a symptom most frequently reported by neuropathic pain patients (Rasmussen et al., 2004; Truini et al., 2013), and preclinical models exhibit ongoing pain following nerve injury (Decosterd and Woolf, 2000; Djouhri et al., 2006; Dworkin et al., 2007; King et al., 2009; Lax et al., 2014). Ten weeks after SNI, animals preferred to be in an environment where the peripheral terminals of CANs were inhibited, suggesting that CANs contribute to chronic ongoing neuropathic pain. These assays were performed >8 weeks following SNI, when central sensitization has developed and likely contributes to spontaneous pain (Woolf, 1983; Coderre et al., 1993; Weyer et al., 2016). CAN inhibition-induced place preference indicates that nerve injury-induced chronic spontaneous pain is driven in part by the peripheral nervous system.
Peripheral nerve injury induces sensitization of the primary afferent somata
Mechanical hypersensitivity is the most prominent evoked phenotype following SNI (Decosterd and Woolf, 2000; Duraku et al., 2012; Smith et al., 2013; Nascimento et al., 2015). Studies have examined changes in gene expression, chemical sensitization, and spontaneous activity in DRG neurons following nerve injury, but not mechanical sensitization (Djouhri et al., 2006; Laedermann et al., 2013; Duan et al., 2014; X. L. Ma et al., 2015; Peirs et al., 2015; Daou et al., 2016; Ventéo et al., 2016).
We found that the somata of CANs have amplified mechanically gated currents 16 weeks following SNI. The amplified currents may result from sensitized mechanically sensitive proteins, such as Piezo2 (Coste et al., 2010; Eijkelkamp et al., 2013) or STOML3 (Wetzel et al., 2007, 2017), which underlie the mechanical hypersensitivity in other neuropathies. Alternatively, dysregulated voltage-gated ion channels (Laedermann et al., 2013), such as Nav1.3, may indirectly contribute to the mechanical sensitivity (Hains et al., 2004; Nassar et al., 2005; Laedermann et al., 2013).
The distribution of mechanically gated current subtypes in CANs was also altered as no IA currents were observed after SNI. Because the IA currents reappear with brief Arch activation, the decreased IA currents may be due to SNI-induced functional changes in mechanically and/or voltage-gated ion channels. For example, Piezo2 mediates RA currents (Coste et al., 2010; Ranade et al., 2015), and TRPA1 has been shown to mediate SA currents and the IA current amplitude in small-diameter neurons (Vilceanu and Stucky, 2010; Brierley et al., 2011). Additionally, many (∼17) Piezo2 splice variants exist in mouse and human sensory neurons, which have differences in kinetics, permeability, and calcium regulation (Szczot et al., 2017). It is possible that specific Piezo2 splice variants might underlie IA currents and that SNI may alter their sensitivity to membrane voltage. Alternatively, Arch hyperpolarization may alter the activity of mechanically sensitive voltage-gated potassium and sodium ion channels (Drew, 2011; Ranade et al., 2015). Additional studies are needed to fully explain these changes.
Peripheral CAPS mediates incisional hypersensitivity
At the injury site, CGRPα causes neurogenic inflammation and activates CGRP1 receptors found on endothelial cells, keratinocytes, and CGRP− neuronal (Cottrell et al., 2005; Eftekhari and Edvinsson, 2011; Russell et al., 2014; Hay et al., 2018). CGRP1 receptor antagonism alleviates Complete Freud's Adjuvant-induced mechanical hypersensitivity (Hirsch et al., 2013). Here, peripheral antagonism of CGRP1 via CGRP8–37 alleviates incisional pain, whereas brief inhibition of CANs has no effect (Table 2). It is possible that optogenetic inhibition of CANs does not prevent CGRP peptide release. Prolonged optogenetic activation of Nav1.8+ neurons, some of which express CGRP, did not elicit neurogenic inflammation (Daou et al., 2013). Thus, optogenetic channel activation may not stimulate the same intracellular signaling cascades that lead to CGRP release or prevention of release. Alternatively, optogenetic inhibition may have no effect on incisional pain because it may be mediated by CGRP− neuronal. Isolectin-B4-binding sensory neurons express CGRP1 and are sensitized following incision (Barabas and Stucky, 2013). Together, these findings suggest that incision likely induces local CGRPα peptide release from peptidergic afferents that acts on CGRP1 receptors expressed by CGRP− sensory neurons and non-neuronal cells.
Optogenetic inhibition and neuronal ablation produce different results following injury
Optogenetic approaches leave neuronal anatomy, circuits, and function intact, whereas neuronal ablation eliminates a population of neurons (Chow et al., 2012a; Maksimovic et al., 2014; Baumbauer et al., 2015; Park et al., 2015; Copits et al., 2016; Daou et al., 2016). In naïve animals, transient inhibition of CANs produced results (Table 2) similar to diphtheria toxin-driven ablation of CANs (McCoy et al., 2013). Neither approach affected mechanical sensitivity, and both approaches increased cold sensitivity and decreased heat sensitivity. However, after injury, optogenetic inhibition (Table 2) and neuronal ablation of CANs (performed before injury) (McCoy et al., 2013) produced different outcomes. Neuronal ablation did not affect SNI-induced mechanical hypersensitivity, whereas CAN inhibition partially and consistently reversed the SNI-induced mechanical hypersensitivity. Additionally, CAN ablation prevented complete Freund's adjuvant-induced inflammatory heat hypersensitivity (McCoy et al., 2013), but transient CAN inhibition had no effect on incisional heat hypersensitivity (Table 2).
Divergent results were also found for optogenetic inhibition and ablation of Nav1.8+ neurons. When ablation was performed (before injury), Nav1.8+ neurons were found to not be involved in neuropathic pain (Abrahamsen et al., 2008), whereas optogenetic inhibition of Nav1.8+ neurons alleviated the neuropathic-induced mechanical hypersensitivity (Daou et al., 2016). These discrepancies between studies may be attributed to injury-induced changes to intact cells and circuits. Also, both central and peripheral sprouting of primary afferent fibers alters dorsal horn synaptic transmission and behavioral phenotypes after SNI (Kohno et al., 2003; Duraku et al., 2012). Because ablation eliminates a neuronal population whereas optogenetics does not, the alterations due to sprouting may differ.
Although both the mouse line used here and that used by McCoy and colleagues involves insertion of Arch or the human diphtheria toxin receptor into the Calca locus (Song et al., 2012; McCoy et al., 2013), respectively, these mouse lines may not affect exactly the same neurons. McCoy et al. (2013) used heterozygous mice leaving one functional Calca allele. In our study, homozygous mice were used that express Arch in all CANs. Furthermore, McCoy et al. (2013) used intraperitoneal injections of diphtheria toxin, which ablates CANs systemically, whereas our optogenetic approach inhibited the peripheral input from plantar-projecting CANs. Thus, differences between mouse models may also account for the discrepant results.
The value of using optogenetic approaches is that neuronal populations anatomically remain intact before and after injury and can be transiently and repeatedly inhibited. Therefore, we chose to use optogenetic approaches following the pioneering work of Gereau and Rogers who established optogenetics for the somatosensory field (Park et al., 2015; Copits et al., 2016). Increased use of optogenetics in animal models may result in more accurate identification of mechanisms driving chronic pain and more faithfully predict clinically relevant therapies.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grants NS040538 and NS070711 to C.L.S. and Grant F31GM123778 to A.M.C. This work was supported in part by Research and Education Component of the Advancing a Healthier Wisconsin Endowment at the Medical College of Wisconsin. We thank Dr. Aniko Szabo (Medical College of Wisconsin Biostatistics Consulting Service) for advice and assistance with statistical analysis throughout the manuscript; Dr. Quinn Hogan, Dr. Katelyn Sadler, Anthony Menzel, and Sarah Langer for providing advice on data analysis, organization, and direction of the manuscript; Dr. Pao-Tien Chuang for the gift of the CGRPαCreER mice; Dr. Mark J. Zylka and Bonnie Taylor-Blake for insightful discussions on this research and aid with immunohistochemical protocols; and Dr. Smriti Iyengar for review of the manuscript.
The authors declare no competing financial interests.
References
- Abrahamsen B, Zhao J, Asante C, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN (2008) The cell and molecular basis of mechanical, cold, and inflammatory pain. Science 321:702–705. 10.1126/science.1156916 [DOI] [PubMed] [Google Scholar]
- Assas BM, Pennock JI, Miyan JA (2014) Calcitonin gene-related peptide is a key neurotransmitter in the neuro-immune axis. Front Neurosci 8:1–9. 10.3389/fnins.2014.00023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliki M, Calvo O, Chialvo DR, Apkarian AV (2005) Spared nerve injury rats exhibit thermal hyperalgesia on an automated operant dynamic thermal escape task. Mol Pain 1:18. 10.1186/1744-8069-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banik RK, Woo YC, Park SS, Brennan TJ (2006) Strain and sex influence on pain sensitivity after plantar incision in the mouse. Anesthesiology 105:1246–1253. 10.1097/00000542-200612000-00025 [DOI] [PubMed] [Google Scholar]
- Barabas ME, Stucky CL (2013) TRPV1, but not TRPA1, in primary sensory neurons contributes to cutaneous incision-mediated hypersensitivity. Mol Pain 9:1–14. 10.1186/1744-8069-9-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D (2009) Cellular and molecular mechanisms of pain. Cell 139:267–284. 10.1016/j.cell.2009.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbauer KM, Deberry JJ, Adelman PC, Miller RH, Hachisuka J, Lee KH, Ross SE, Koerber HR, Davis BM, Albers KM (2015) Keratinocytes can modulate and directly initiate nociceptive responses. Elife 4:1–14. 10.7554/eLife.09674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berta T, Park CK, Xu ZZ, Xie RG, Liu T, Lü N, Liu YC, Ji RR (2014) Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-α secretion. J Clin Invest 124:1173–1186. 10.1172/JCI72230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berta T, Perrin FE, Pertin M, Tonello R, Liu YC, Chamessian A, Kato AC, Ji RR, Decosterd I (2017) Gene Expression Profiling of Cutaneous Injured and Non-Injured Nociceptors in SNI Animal Model of Neuropathic Pain. Scientific Reports 7:9367. 10.1038/s41598-017-08865-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boada MD, Martin TJ, Peters CM, Hayashida K, Harris MH, Houle TT, Boyden ES, Eisenach JC, Ririe DG (2014) Fast-conducting mechanoreceptors contribute to withdrawal behavior in normal and nerve injured rats. Pain 155:2646–2655. 10.1016/j.pain.2014.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan TJ. (2011) Pathophysiology of postoperative pain. Pain 152:S33–S40. 10.1016/j.pain.2010.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan TJ, Vandermeulen EP, Gebhart GF (1996) Characterization of a rat model of incisional pain. Pain 64:493–501. 10.1016/0304-3959(95)01441-1 [DOI] [PubMed] [Google Scholar]
- Brenner DS, Golden JP, Gereau RW IV (2012) A novel behavioral assay for measuring cold sensation in mice. PLoS One 7:1–8. 10.1371/journal.pone.0039765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brierley SM, Castro J, Harrington AM, Hughes PA, Page AJ, Rychkov GY, Blackshaw LA (2011) TRPA1 contributes to specific mechanically activated currents and sensory neuron mechanical hypersensitivity. J Physiol 14:3575–3593. 10.1113/jphysiol.2011.206789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh DJ, Lee H, Lo L, Shields SD, Zylka MJ, Basbaum AI, Anderson DJ (2009) Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc Natl Acad Sci U S A 106:9075–9080. 10.1073/pnas.0901507106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53:55–63. 10.1016/0165-0270(94)90144-9 [DOI] [PubMed] [Google Scholar]
- Chow BY, Han X, Dobry AS, Qian X, Chuong AS, Li M, Henninger MA, Belfort GM, Lin Y, Monahan PE, Boyden ES (2010) High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature 463:98–102. 10.1038/nature08652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow BY, Han X, Bernstein JG, Monahan PE, Boyden ES (2012a) Light-activated ion pumps and channels for temporally precise optical control of activity in genetically targeted neurons. Neuromethods 67:305–338. [Google Scholar]
- Chow BY, Han X, Boyden ES (2012b) Genetically encoded molecular tools for light-driven silencing of targeted neurons. Prog Brain Res 196:49–61. 10.1016/B978-0-444-59426-6.00003-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coderre TJ, Katz J, Vaccarino AL, Melzack R (1993) Contribution of central neuroplasticity to pathological pain: review of clinical and experimental evidence. Pain 52:259–285. 10.1016/0304-3959(93)90161-H [DOI] [PubMed] [Google Scholar]
- Colleoni M, Sacerdote P (2010) Murine models of human neuropathic pain. Biochim Biophys Acta 1802:924–933. 10.1016/j.bbadis.2009.10.012 [DOI] [PubMed] [Google Scholar]
- Copits BA, Pullen MY, Gereau RW 4th (2016) Spotlight on pain: optogenetic approaches for interrogating somatosensory circuits. Pain 157:2424–2433. 10.1097/j.pain.0000000000000620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A (2010) Piezo1 and Piezo2 are essential components of distinct mechanically-activated cation channels. Science 330:55–60. 10.1126/science.1193270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell GS, Roosterman D, Marvizon JC, Song B, Wick E, Pikios S, Wong H, Berthelier C, Tang Y, Sternini C, Bunnett NW, Grady EF (2005) Localization of calcitonin receptor-like receptor and receptor activity modifying protein 1 in enteric neurons, dorsal root ganglia, and the spinal cord of the rat. J Comp Neurol 490:239–255. 10.1002/cne.20669 [DOI] [PubMed] [Google Scholar]
- Daou I, Tuttle AH, Longo G, Wieskopf JS, Bonin RP, Ase AR, Wood JN, De Koninck Y, Ribeiro-da-Silva A, Mogil JS, Séguéla P (2013) Remote optogenetic activation and sensitization of pain pathways in freely moving mice. J Neurosci 33:18631–18640. 10.1523/JNEUROSCI.2424-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daou I, Beaudry H, Ase AR, Wieskopf JS, Ribeiro-da-Silva A, Mogil JS, Séguéla P (2016) Optogenetic silencing of Nav1.8-positive afferents alleviates inflammatory and neuropathic pain. eNeuro 3:1–12. 10.1523/ENEURO.0140-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ (2000) Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 87:149–158. 10.1016/S0304-3959(00)00276-1 [DOI] [PubMed] [Google Scholar]
- Decosterd I, Allchorne A, Woolf CJ (2004) Differential analgesic sensitivity of two distinct neuropathic pain models. Anesth Analg 99:457–463. 10.1213/01.ANE.0000131967.69309.4F [DOI] [PubMed] [Google Scholar]
- Deen M, Correnti E, Kamm K, Kelderman T, Papetti L, Rubio-Beltrán E, Vigneri S, Edvinsson L, Maassen Van Den Brink A (2017) Blocking CGRP in migraine patients: a review of pros and cons. J Headache Pain 18:96. 10.1186/s10194-017-0807-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon WJ. (1980) Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 20:441–462. 10.1146/annurev.pa.20.040180.002301 [DOI] [PubMed] [Google Scholar]
- Djouhri L, Koutsikou S, Fang X, McMullan S, Lawson SN (2006) Spontaneous pain, both neuropathic and inflammatory, is related to frequency of spontaneous firing in intact C-fiber nociceptors. J Neurosci 26:1281–1292. 10.1523/JNEUROSCI.3388-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew LJ. (2011) Sodium channel mechanosensitivity: pay a-tension to voltage sensor movement. J Physiol 5:1003–1004. 10.1113/jphysiol.2011.205880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan B, Cheng L, Bourane S, Britz O, Padilla C, Garcia-Campmany L, Krashes M, Knowlton W, Velasquez T, Ren X, Ross S, Lowell BB, Wang Y, Goulding M, Ma Q (2014) Identification of spinal circuits transmitting and gating mechanical pain. Cell 159:1417–1432. 10.1016/j.cell.2014.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraku LS, Hossaini M, Hoendervangers S, Falke LL, Kambiz S, Mudera VC, Holstege JC, Walbeehm ET, Ruigrok TJ (2012) Spatiotemporal dynamics of reinnervation and hyperinnervation patterns by uninjured CGRP fibers in the rat foot sole epidermis after nerve injury. Mol Pain 8:61. 10.1186/1744-8069-8-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraku LS, Niehof SP, Misirli Y, Everaers M, Hoendervangers S, Holstege J, Boele HJ, Koekkoek SK, Smits ES, Selles RW, Walbeehm ET (2014) Rotterdam advanced multiple plate: a novel method to measure cold hyperalgesia and allodynia in freely behaving rodents. J Neurosci Methods 224:1–12. 10.1016/j.jneumeth.2013.12.006 [DOI] [PubMed] [Google Scholar]
- Dworkin RH, O'Connor AB, Backonja M, Farrar JT, Finnerup NB, Jensen TS, Kalso EA, Loeser JD, Miaskowski C, Nurmikko TJ, Portenoy RK, Rice AS, Stacey BR, Treede RD, Turk DC, Wallace MS (2007) Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain 132:237–251. 10.1016/j.pain.2007.08.033 [DOI] [PubMed] [Google Scholar]
- Eftekhari S, Edvinsson L (2010) Possible sites of action of the new calcitonin gene-related peptide receptor antagonists. Ther Adv Neurol Disord 3:369–378. 10.1177/1756285610388343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftekhari S, Edvinsson L (2011) Calcitonin gene-related peptide (CGRP) and its receptor components in human and rat spinal trigeminal nucleus and spinal cord at C1-level. BMC Neurosci 12:112–129. 10.1186/1471-2202-12-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelkamp N, Linley JE, Torres JM, Bee L, Dickenson AH, Gringhuis M, Minett MS, Hong GS, Lee E, Oh U, Ishikawa Y, Zwartkuis FJ, Cox JJ, Wood JN (2013) A role for Piezo2 in EPAC1-dependent mechanical allodynia. Nat Commun 4:1–13. 10.1038/ncomms2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisener-Dorman AF, Lawrence DA, Bolivar VJ (2009) Cautionary insights on knockout mouse studies: the gene or not the gene. Brain Behav Immun 23:318–324. 10.1016/j.bbi.2008.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gaby M, Zhang Y, Wolf K, Schwiening CJ, Paulsen O, Shipton OA (2016) Archaerhodopsin selectively and reversibly silences synaptic transmission through altered pH. Cell Rep 16:2259–2268. 10.1016/j.celrep.2016.07.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan TJ, Habib AS, Miller TE, White W, Apfelbaum JL (2014) Incidence, patient satisfaction, and perceptions of post-surgical pain: results from a US national survey. Curr Med Res Opin 30:149–160. 10.1185/03007995.2013.860019 [DOI] [PubMed] [Google Scholar]
- Garrison SR, Dietrich A, Stucky CL (2012) TRPC1 contributes to light-touch sensation and mechanical responses in low-threshold cutaneous sensory neurons. J Neurophysiol 107:913–922. 10.1152/jn.00658.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison SR, Weyer AD, Barabas ME, Beutler BA, Stucky CL (2014) A gain-of-function voltage-gated sodium channel 1.8 mutation drives intense hyperexcitability of A- and C-fiber neurons. Pain 155:896–905. 10.1016/j.pain.2014.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemlou N, Chiu IM, Julien JP, Woolf CJ (2015) CD11b+ Ly6G− myeloid cells mediate mechanical inflammatory pain hypersensitivity. Proc Natl Acad Sci U S A 112:1–10. 10.1073/iti0115112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghitani N, Barik A, Szczot M, Thompson JH, Li C, Le Pichon CE, Krashes MJ, Chesler AT (2017) Specialized mechanosensory nociceptors mediating rapid responses to hair pull. Neuron 95:944–954.e4. 10.1016/j.neuron.2017.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goadsby PJ, Edvinsson L (1993) The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol 33:48–56. 10.1002/ana.410330109 [DOI] [PubMed] [Google Scholar]
- Hagner S, Haberberger RV, Overkamp D, Hoffmann R, Voigt KH, McGregor GP (2002) Expression and distribution of calcitonin receptor-like receptor in human hairy skin. Peptides 23:109–116. 10.1016/S0196-9781(01)00586-1 [DOI] [PubMed] [Google Scholar]
- Hains BC, Saab CY, Klein JP, Craner MJ, Waxman SG (2004) Altered sodium channel expression in second-order spinal sensory neurons contributes to pain after peripheral nerve injury. J Neurosci 24:4832–4839. 10.1523/JNEUROSCI.0300-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32:11. 10.1016/0304-3959(88)90026-7 [DOI] [PubMed] [Google Scholar]
- Hay DL, Garelja ML, Poyner DR, Walker CS (2018) Update on the pharmacology of calcitonin/CGRP family of peptides: IUPHAR Review 25. Br J Pharmacol 175:3–17. 10.1111/bph.14075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch S, Corradini L, Just S, Arndt K, Doods H (2013) The CGRP receptor antagonist BIBN4096BS peripherally alleviates inflammatory pain in rats. Pain 154:700–707. 10.1016/j.pain.2013.01.002 [DOI] [PubMed] [Google Scholar]
- Hogan Q, Sapunar D, Modric-Jednacak K, McCallum JB (2004) Detection of neuropathic pain in a rat model of peripheral nerve injury. Anesthesiology 101:476–487. 10.1097/00000542-200408000-00030 [DOI] [PubMed] [Google Scholar]
- Hou Q, Barr T, Gee L, Vickers J, Wymer J, Borsani E, Rodella L, Getsios S, Burdo T, Eisenberg E, Guha U, Lavker R, Kessler J, Chittur S, Fiorino D, Rice F, Albrecht P (2011) Keratinocyte expression of calcitonin gene-related peptide β: implications for neuropathic and inflammatory pain mechanisms. Pain 152:2036–2051. 10.1016/j.pain.2011.04.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YL, Lin CL, Chiang H, Fu YS, Lue JH, Hsieh ST (2012a) Role of peptidergic nerve terminals in the skin: reversal of thermal sensation by calcitonin gene-related peptide in TRPV1-depleted neuropathy. PLoS One 7:1–15. 10.1371/journal.pone.0050805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YL, Lin CL, Chiang H, Fu YS, Lue JH, Hsieh ST (2012b) Role of peptidergic nerve terminals in the skin: reversal of thermal sensation by calcitonin gene-related peptide in TRPV1-depleted neuropathy. PLoS One 7:e50805 10.1371/journal.pone.0050805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida K, Kawamata T, Tanaka S, Shindo T, Kawamata M (2014) Calcitonin gene-related peptide is involved in inflammatory pain but not in postoperative pain. Anesthesiology 121:1068–1079. 10.1097/ALN.0000000000000364 [DOI] [PubMed] [Google Scholar]
- Iyengar S, Ossipov MH, Johnson KW (2017) The role of calcitonin gene-related peptide in peripheral and central pain mechanisms including migraine. Pain 158:543–559. 10.1097/j.pain.0000000000000831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SM, Montgomery KL, Towne C, Lee SY, Ramakrishnan C, Deisseroth K, Delp SL (2014) Virally mediated optogenetic excitation and inhibition of pain in freely moving nontransgenic mice. Nat Biotechnol 32:274–278. 10.1038/nbt.2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DL, Graff CB, Richardson JD, Hargreaves KM (1995) Glutamate participates in the peripheral modulation of thermal hyperalgesia in rats. Eur J Pharmacol 284:321–325. 10.1016/0014-2999(95)00449-U [DOI] [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, Porreca F (2009) Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci 12:1364–1366. 10.1038/nn.2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Moore KA, Baba H, Woolf CJ (2003) Peripheral nerve injury alters excitatory synaptic transmission in lamina II of the rat dorsal horn. J Physiol 548:131–138. 10.1113/jphysiol.2002.036186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltzenburg M, Lundberg LE, Torebjörk HE (1992) Dynamic and static components of mechanical hyperalgesia in human hairy skin. Pain 51:207–219. 10.1016/0304-3959(92)90262-A [DOI] [PubMed] [Google Scholar]
- Koltzenburg M, Torebjörk HE, Wahren LK (1994) Nociceptor modulated central sensitization causes mechanical hyperalgesia in acute chemogenic and chronic neuropathic pain. Brain 117:579–591. 10.1093/brain/117.3.579 [DOI] [PubMed] [Google Scholar]
- La JH, Chung JM (2017) Peripheral afferents and spinal inhibitory system in dynamic and static mechanical allodynia. Pain 158:2285–2289. 10.1097/j.pain.0000000000001055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laedermann CJ, Cachemaille M, Kirschmann G, Pertin M, Gosselin RD, Chang I, Albesa M, Towne C, Schneider BL, Kellenberger S, Abriel H, Decosterd I (2013) Dysregulation of voltage-gated sodium channels by ubiquitin ligase NEDD4–2 in neuropathic pain. J Clin Invest 123:3002–3013. 10.1172/JCI68996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lax NC, George DC, Ignatz C, Kolber BJ (2014) The mGluR5 antagonist fenobam induces analgesic conditioned place preference in mice with spared nerve injury. PLoS One 9:1–11. 10.1371/journal.pone.0103524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CL, Li KC, Wu D, Chen Y, Luo H, Zhao JR, Wang SS, Sun MM, Lu YJ, Zhong YQ, Hu XY, Hou R, Zhou BB, Bao L, Xiao HS, Zhang X (2016) Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res 26:83–102. 10.1038/cr.2015.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Quirion R (2006) Increased calcitonin gene-related peptide in neuroma and invading macrophages is involved in the up-regulation of interleukin-6 and thermal hyperalgesia in a rat model of mononeuropathy. J Neurochem 98:180–192. 10.1111/j.1471-4159.2006.03856.x [DOI] [PubMed] [Google Scholar]
- Ma XL, Zhang FX, Dong F, Bao L, Zhang X (2015) Experimental evidence for alleviating nociceptive hypersensitivity by single application of capsaicin. Mol Pain 11:1–10. 10.1186/s12990-015-0019-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksimovic S, Nakatani M, Baba Y, Nelson AM, Marshall KL, Wellnitz SA, Firozi P, Woo SH, Ranade S, Patapoutian A, Lumpkin EA (2014) Epidermal Merkel cells are mechanosensory cells that tune mammalian touch receptors. Nature 509:617–621. 10.1038/nature13250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy ES, Taylor-Blake B, Zylka MJ (2012) CGRPα-expressing sensory neurons respond to stimuli that evoke sensations of pain and itch. PLoS One 7:1–11. 10.1371/journal.pone.0036355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy ES, Taylor-Blake B, Street SE, Pribisko AL, Zheng J, Zylka MJ (2013) Peptidergic CGRPa primary sensory neurons encode heat and itch and tonically suppress sensitivity to cold. Neuron 29:997–1003. 10.1016/j.neuron.2013.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehring F, O'Hara CL, Stucky CL (2016) Bedding material affects mechanical thresholds, heat thresholds, and texture preference. J Pain 17:50–64. 10.1016/j.jpain.2015.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento FP, Magnussen C, Yousefpour N, Ribeiro-da-Silva A (2015) Sympathetic fibre sprouting in the skin contributes to pain-related behaviour in spared nerve injury and cuff models of neuropathic pain. Mol Pain 11:1–14. 10.1186/s12990-015-0062-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar MA, Levato A, Stirling LC, Wood JN (2005) Neuropathic pain develops normally in mice lacking both Na(v)1.7 and Na(v)1.8. Mol Pain 1:1–9. 10.1186/1744-8069-1-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MQ, Wu Y, Bonilla LS, von Buchholtz LJ, Ryba NJP (2017) Diversity amongst trigeminal neurons revealed by high throughput single cell sequencing. PLoS One 12:1–22. 10.1371/journal.pone.0185543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, Pollentier S, Lesko LM (2004) Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med 350:1104–1110. 10.1056/NEJMoa030505 [DOI] [PubMed] [Google Scholar]
- Park SI, Brenner DS, Shin G, Morgan CD, Copits BA, Chung HU, Pullen MY, Noh KN, Davidson S, Oh SJ, Yoon J, Jang KI, Samineni VK, Norman M, Grajales-Reyes JG, Vogt SK, Sundaram SS, Wilson KM, Ha JS, Xu R, et al. (2015) Soft, stretchable, fully implantable miniaturized optoelectronic systems for wireless optogenetics. Nat Biotechnol 33:1280–1286. 10.1038/nbt.3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirs C, Williams SP, Zhao X, Walsh CE, Gedeon JY, Cagle NE, Goldring AC, Hioki H, Liu Z, Marell PS, Seal RP (2015) Dorsal horn circuits for persistent mechanical pain. Neuron 87:797–812. 10.1016/j.neuron.2015.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogatzki EM, Raja SN (2003) A mouse model of incisional pain. Anesthesiology 99:1023–1027. 10.1097/00000542-200310000-00041 [DOI] [PubMed] [Google Scholar]
- Pogatzki-Zahn EM, Shimizu I, Caterina M, Raja SN (2005) Heat hyperalgesia after incision requires TRPV1 and is distinct from pure inflammatory pain. Pain 115:296–307. 10.1016/j.pain.2005.03.010 [DOI] [PubMed] [Google Scholar]
- Ran C, Hoon MA, Chen X (2016) The coding of cutaneous temperature in the spinal cord. Nat Neurosci 19:1201–1209. 10.1038/nn.4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranade SS, Syeda R, Patapoutian A (2015) Mechanically activated ion channels 87:1162–1179. 10.1016/j.neuron.2015.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen PV, Sindrup SH, Jensen TS, Bach FW (2004) Symptoms and signs in patients with suspected neuropathic pain. Pain 110:461–469. 10.1016/j.pain.2004.04.034 [DOI] [PubMed] [Google Scholar]
- Rowbotham MC, Fields HL (1996) The relationship of pain, allodynia and thermal sensation in post-herpetic neuralgia. Brain 119:347–354. 10.1093/brain/119.2.347 [DOI] [PubMed] [Google Scholar]
- Russell FA, King R, Smillie SJ, Kodji X, Brain SD (2014) Calcitonin gene-related peptide: physiology and pathophysiology. Physiol Rev 94:1099–1142. 10.1152/physrev.00034.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahbaie P, Sun Y, Liang DY, Shi XY, Clark JD (2014) Curcumin treatment attenuates pain and enhances functional recovery after incision. Anesth Analg 118:1336–1344. 10.1213/ANE.0000000000000189 [DOI] [PubMed] [Google Scholar]
- Smith AK, O'Hara CL, Stucky CL (2013) Mechanical sensitization of cutaneous sensory fibers in the spared nerve injury mouse model. Mol Pain 9:1–6. 10.1186/1744-8069-9-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Yao E, Lin C, Gacayan R, Chen MH, Chuang PT (2012) Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc Natl Acad Sci U S A 109:17531–17536. 10.1073/pnas.1207238109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St John Smith E. (2018) Advances in understanding nociception and neuropathic pain. J Neurol 265:231–238. 10.1007/s00415-017-8641-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swett JE, Woolf CJ (1985) The somatotopic organization of primary afferent terminals in the superficial laminae of the dorsal horn of the rat spinal cord. J Comp Neurol 231:66–77. 10.1002/cne.902310106 [DOI] [PubMed] [Google Scholar]
- Szczot M, Pogorzala LA, Solinski HJ, Young L, Yee P, Le Pichon CE, Chesler AT, Hoon MA (2017) Cell-type-specific splicing of Piezo2 regulates mechanotransduction. Cell Rep 21:2760–2771. 10.1016/j.celrep.2017.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truini A, Garcia-Larrea L, Cruccu G (2013) Reappraising neuropathic pain in humans-how symptoms help disclose mechanisms. Nat Rev Neurol 9:572–582. 10.1038/nrneurol.2013.180 [DOI] [PubMed] [Google Scholar]
- Tso AR, Goadsby PJ (2017) Anti-CGRP monoclonal antibodies: the next era of migraine prevention? Curr Treat Options Neurol 19:1–11. 10.1007/s11940-017-0463-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, Linnarsson S, Ernfors P (2015) Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci 18:145–153. 10.1038/nn.3881 [DOI] [PubMed] [Google Scholar]
- Vadivelu N, Mitra S, Narayan D (2010) Recent advances in postoperative pain management. Yale J Biol Med 83:11–25. [PMC free article] [PubMed] [Google Scholar]
- Ventéo S, Laffray S, Wetzel C, Rivat C, Scamps F, Méchaly I, Bauchet L, Raoul C, Bourinet E, Lewin GR, Carroll P, Pattyn A (2016) Fxyd2 regulates Aδ- and C-fiber mechanosensitivity and is required for the maintenance of neuropathic pain. Sci Rep 6:1–12. 10.1038/srep36407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilceanu D, Stucky CL (2010) TRPA1 mediates mechanical currents in the plasma membrane of mouse sensory neurons. PLoS One 5:1–10. 10.1371/journal.pone.0012177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver JL, Arandjelovic S, Brown G, Mendu SK, Schappe MS, Buckley MW, Chiu YH, Shu S, Kim JK, Chung J, Krupa J, Jevtovic-todorovic V, Desai BN, Ravichand KS, Bayliss DA (2017) Hematopoietic pannexin 1 function is critical for neuropathic pain. Sci Rep 7:42550 10.1038/srep42550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel C, Hu J, Riethmacher D, Benckendorff A, Harder L, Eilers A, Moshourab R, Kozlenkov A, Labuz D, Caspani O, Erdmann B, Machelska H, Heppenstall PA, Lewin GR (2007) A stomatin-domain protein essential for touch sensation in the mouse. Nature 445:206–209. 10.1038/nature05394 [DOI] [PubMed] [Google Scholar]
- Wetzel C, Pifferi S, Picci C, Gök C, Hoffmann D, Bali KK, Lampe A, Lapatsina L, Fleischer R, Smith ES, Bégay V, Moroni M, Estebanez L, Kühnemund J, Walcher J, Specker E, Neuenschwander M, von Kries JP, Haucke V, Kuner R, et al. (2017) Small-molecule inhibition of STOML3 oligomerization reverses pathological mechanical hypersensitivity. Nat Neurosci 20:209–218. 10.1038/nn.4454 [DOI] [PubMed] [Google Scholar]
- Weyer AD, O'Hara CL, Stucky CL (2015) Amplified mechanically gated currents in distinct subsets of myelinated sensory neurons following in vivo inflammation of skin and muscle. J Neurosci 35:9456–9462. 10.1523/JNEUROSCI.0549-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer AD, Zappia KJ, Garrison SR, O'Hara CL, Dodge AK, Stucky CL (2016) Nociceptor sensitization depends on age and pain chronicity. eNeuro 3:1–26. 10.1523/ENEURO.0115-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegert JS, Mahn M, Prigge M, Printz Y, Yizhar O (2017) Silencing neurons: tools, applications, and experimental constraints. Neuron 95:504–529. 10.1016/j.neuron.2017.06.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ. (1983) Evidence for a central component of post-injury pain hypersensitivity. Nature 306:686–688. 10.1038/306686a0 [DOI] [PubMed] [Google Scholar]
- Yarmolinsky DA, Peng Y, Pogorzala LA, Rutlin M, Hoon MA, Zuker CS (2016) Coding and plasticity in the mammalian thermosensory system. Neuron 92:1079–1092. 10.1016/j.neuron.2016.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappia KJ, Garrison SR, Palygin O, Weyer AD, Barabas ME, Lawlor MW, Staruschenko A, Stucky CL (2016) Mechanosensory and ATP release deficits following keratin14-cre-mediated TRPA1 deletion despite absence of TRPA1 in murine keratinocytes. PLoS One 11:1–30. 10.1371/journal.pone.0151602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Liu X, Lu S, Yu A, Fu Z (2013) Increased pain in response to mechanical or thermal stimulation in a rat model of incision-induced pain with nicotine dependence and withdrawal. Exp Ther Med 5:1063–1066. 10.3892/etm.2013.963 [DOI] [PMC free article] [PubMed] [Google Scholar]