

Abstract
The ability of a chiral molecule to be able to transform from one structure to another, whilst remembering its original molecular information by means of an appropriate transition state is an abstract notion that may very well play a key role in important synthetic processes, and has thus attracted a considerable amount of attention among the chemistry community. Here, we have highlighted this concept of “memory of chirality” (MOC) and extended it beyond the case of a simple molecule to larger and more complex natural products. Updated approaches that have recently been elucidated to obtain these asymmetric natural products are described, some of which may, until now, have been overcomplicated or overlooked.
Keywords: chiral carbocation, enantioselectivity, memory of chirality, natural products, retention of configuration
In chemistry, we define a structure to be chiral if it cannot be imposed upon its mirror image. As these are the same molecule, just in reverse, their physical properties are identical. The do differ, however, in optical activity by the direction in which they rotate plane‐polarised light. Chemically, however, it is possible for these enantiomers to perform very differently. It is well known that, if an enantiopure sp3‐hybridised centre is trigonalised to sp2, the product will be yielded in an equal/racemic mix of products, in the absence of any external factors (i.e. chiral auxiliaries), as shown in Scheme 1. However, there are situations where this is not the case, and the original stereochemistry of the starting material can be retained to a certain extent in the product through a process known as “memory of chirality” (MOC). Several others have, in the past, reviewed this topic; however, they focused on the concept when applied to precursor molecules or to reaction strategies.1 Here, we extend this notion to strategies concerning molecules that have recently been elucidated, which are naturally occurring or have interest owing to their biological functions.
Scheme 1.
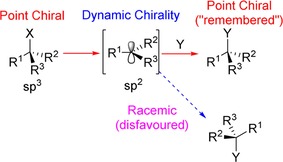
Formation of racemic products from an enantiopure starting material.
There are, however, several requirements that must be met for a system to have any hope of undergoing this process. Firstly, a conformationally chiral intermediate must be formed enantioselectively at the stereogenic centre of a chiral starting material.1b, 2 This chiral intermediate must not readily racemise and must react with high stereospecificity. Depending on the nature of the intermediate, MOC is known to occur through enolates, anions, radicals and cations, the latter of which has been significantly less studied. The first example of cationic memory of chirality was reported by Onomura and co‐workers, who used the electrochemical oxidation of the N‐benzoylated serine derivative A in methanol to give an N, O‐acetal B (Scheme 2).3 Such amino acid derivatives have already seen function in biologically active peptides and pharmaceuticals,4, 5, 6 and thus applying MOC as a synthetic strategy is deemed appropriate. As protecting groups are known to increase reaction selectivity of amino compounds,7 the N‐protecting group has been modified with an extra phenyl group ortho to the amino group in the hope that the steric hindrance would favour the R isomer.8 Batch electrolysis of the compound at −30 °C with methanol and platinum as both electrodes resulted in the methoxide predominantly attacking from the same, less hindered (syn) side as the carboxylate group, allowing for retention of configuration. This is attributed to the stability of the conformation of B and bulkiness of the o‐phenyl group, which would have both unevenly distributed the ease of nucleophilic attack from the anti and syn faces, and its restricted rotation would allow formation of the stabile iminium intermediate, which has the same stable conformation as B.
Scheme 2.
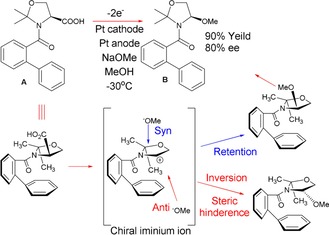
MOC mechanism for the electrochemical oxidation of an N‐benzoylated serine derivative, in which the two possible routes for nucleophilic attack are highlighted.
More recently, a Friedel–Crafts alkylation reaction involving chiral α‐aryl alcohols with a trimethylsilyl group and indole was found to occur under the influence of memory of chirality (Scheme 3).9 The enantioenriched β‐silyl alcohols 1 and 3 were successfully alkylated in good yields with a reasonably high amount of chiral transfer. Conversely, the chiral thiophene compound 5, not containing a silyl group, was alkylated into an almost racemic product (3 % ee). These findings have been attributed to the formation of a conformationally stable carbocation 7, formed from a Lewis acidic iron salt coordinating to the hydroxyl group. This occurs in an antiperiplanar arrangement, which then allows the nucleophilic attack by the indol to occur from the opposite side to the trimethylsilyl group to afford the final product formed with retention of configuration. DFT calculations of the iron intermediate 7 show that the C−Si σ‐bond, located perpendicular to the plane of the cationic carbon, donates into the carbocation (more so than a C−H would, hence β‐silyl effect). It is this hyperconjugation of the C−Si bond that provides stabilisation to the intermediate 7, placing the hydroxyl group and the trimethylsilyl group antiperiplanar to one another. The rotational barrier of 7 is suggested to by much higher than that with a methyl instead of a silyl, allowing the original chirality to be retained.
Scheme 3.
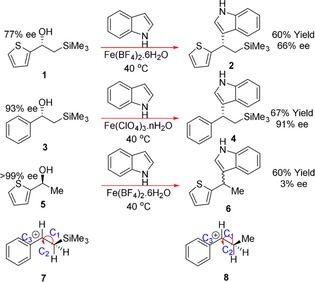
Friedel–Crafts alkylations employing the MOC effect through cationic intermediates.
Continuing with the aforementioned idea of applying MOC to amino acid derivatives, in 2012, Yoshimura et al. reported the asymmetric intermolecular conjugate addition of such compounds proceeding through an axial chiral enolate intermediate with up to 98 % ee retention, which could then be transformed into Manzacidin A, a biologically active bromopyrrole alkaloid (Scheme 4).10
Scheme 4.
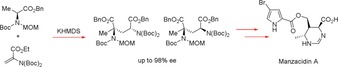
Reaction scheme for an intermolecular conjugation of an amino acid derivative which acts as a precursor for the synthesis of the natural product Manzacidin A.
The same group have more recently expended these concepts to the asymmetric α‐arylation of α‐amino of acid derivatives to afford Benzocyclobutenones (BCBs).11 These have gained popularity for their role as synthetic intermediates12 such as in the total synthesis of natural products.13 Through optimised conditions, the combination of the corresponding axially chiral enolates with in situ generated arynes proceeded via intermolecular asymmetric α‐arylation. This would then be followed by an intramolecular C acylation to afford the BCB products with retention of configuration of up to 99 % ee at the tetrasubstituted carbon (Scheme 5).
Scheme 5.
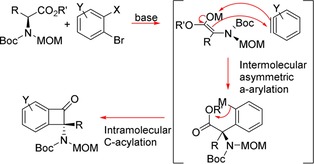
Pathway for the asymmetric synthesis of BCBs.
In 2015, Kim et al. reported the first total synthesis of the natural marine alkaloid penibruguieramine A (PA), using proline as the only chiral source.14 A biomimetic intramolecular aldol reaction operating with MOC was utilised to convert proline ester 9 into the bicyclic product 10. The intramolecular aldol reaction of 9 afforded a single diastereomer of 10 (Scheme 6), irrespective of the nature of the base, although treatment with NaOEt in EtOH gave the highest yield of 77 %. Computational studies revealed that this aldol product 9 is not only the kinetic product, but also the thermodynamically favoured product as well. Total synthesis of PA was completed by reducing the tert‐butyl ester group in 10 to the corresponding alcohol (Scheme 6), resulting in PA being formed with enantiomeric purity greater than 99 %.
Scheme 6.
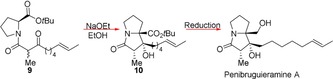
Intramolecular aldol reaction controlled by MOC followed by a proceeding reduction of the tert‐butyl ester group to PA.
As an extension of this work, Hu and co‐workers employed the strategies developed by Kim et al. to synthesise 6 (R)—and 6 (S)‐ fluoropenibruguieramine (Scheme 7), the fluorine analogues of PA.15 A fluorine atom, despite being 19 times the mass, is about the same size as a hydrogen and the highly polarised C−F bond can become involved in several stereoelectronic interactions with neighbouring functional groups such as ammonium, amine and amide,16 thus altering a given molecules conformation when it is incorporated into it. This size similarity means that, as in the above works of Kim et al., MOC will direct the key intramolecular aldol reaction. During the synthesis, the authors followed the synthetic route to the reported by Raines and co‐workers,17 with several major improvements that allowed the precursor 4‐fluoroprolines to be produced with a 38 % increase in yield and 11 a and 11 b to be readily obtained on the gram scale. By using conditions identical to that of Kim et al., the intremolecular aldol reaction proceeded with only a single product being formed. This, therefore, indicates that MOC was controlling the sterochemistry of 12 a and 12 b. To further illistate the effectivness of MOC in asymmetric synthesis during this reaction, it was postulated that the fluorine amide gauche effect may function as a competing reaction. However, this was not the case, as the fluorine gauche effect was completely overpowered by the MOC phenomena.
Scheme 7.
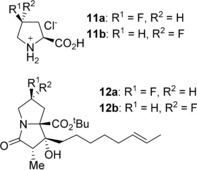
Precursor 4‐fluroprolines (top) and 6 (R)—and 6 (S)‐ fluoropenibruguieramine (bottom).
Kim at al. furthered their work to the asymmetric total synthesis of another marine alkaloid, Lepadiformine C, with d‐proline as the only chiral source.18 Through computational analysis, substrate 13 was designed as the component that would undergo the key synthetic sequence, an intramolecular Michael addition. It was discovered that the cis isomer of 13 is thermodynamically the most favoured; therefore, the reaction had to be carried out under kinetic control for the formation of the trans isomer and, hence, 13 was created from postulation of kinetically relevant transition states. It is, however, possible for 13 to react via other competing reactions such as Dieckmann‐type condensations, owing to the presence of multiple enolisable carbonyl moieties; therefore, screening of reaction conditions was performed, with NaOtBu and DMF as the best performing base and solvent system, respectively (Scheme 8). Control of both reaction time and temperature led the group to choose conditions of 2 h at 0 °C, which gave a good yield of 76 % with a high enantiomeric excess of 92 %. After this, several proceeding steps was carried out to afford the natural product Lepadiformine C.
Scheme 8.
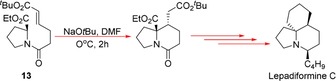
Strategy to the production of Lepadiformine C.
To conclude, MOC is continuously being proven to be a powerful methodology, in which asymmetric synthetic processes can be conducted.19 We have now begun a new era as MOC has evolved into a much more formidable strategy in the generation of valuable natural products from simple chiral compounds, without the need for additional chiral auxiliaries.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
Marie Skłodowska‐Curie Actions COFUND (Grant No 663830) to N.A. is gratefully acknowledged.
T. Hardwick, N. Ahmed, ChemistryOpen 2018, 7, 484.
References
- 1.
- 1a. Kawabata T., Yahiro K., Fuji K., J. Am. Chem. Soc. 1991, 113, 9694–9696; [Google Scholar]
- 1b. Zhao H., Hsu D. C., Carlier P. R., Synthesis 2005, 1, 1–16. [Google Scholar]
- 2. Campolo D., Gastaldi S., Roussel C., Bertrand M. P., Nechab M., Chem. Soc. Rev. 2013, 42, 8434–8466. [DOI] [PubMed] [Google Scholar]
- 3. Matsumura Y., Shirakawa Y., Satoh Y., Umino M., Maki T., Onomura O., Org. Lett. 2000, 2, 1689–1691. [DOI] [PubMed] [Google Scholar]
- 4. Wu X., Zhang D., Zhou S., Gao F., Liu H., Chem. Commun. 2015, 51, 12571–12573. [DOI] [PubMed] [Google Scholar]
- 5. Akif M., Georgiadis D., Mahajan A., Dive V., Sturrock E. D., Isaac R. E., Acharya K. R., J. Mol. Biol. 2010, 400, 502–517. [DOI] [PubMed] [Google Scholar]
- 6. Chang K.-J., Cuatrecasas P., Wei E. T., Chang J.-K., Life Sci. 1982, 30, 1547–1551. [DOI] [PubMed] [Google Scholar]
- 7. Wuts P. G. M., Greene T. W., Greene's Protective Groups in Organic Synthesis, 2nd ed, Wiley, New York, NY, 1991, pp. 41–452. [Google Scholar]
- 8. Ng′ang′a Wanyoik G., Onomura O., Maki T., Matsumura Y., Org. Lett. 2002, 4, 1875–1877. [DOI] [PubMed] [Google Scholar]
- 9. Nokami T., Yamane Y., Oshitani S., Kobayashi J., Matsui S., Nishihara T., Uno H., Hayase S., Itoh T., Org. Lett. 2015, 17, 3182–3185. [DOI] [PubMed] [Google Scholar]
- 10. Yoshimura T., Kinoshita T., Yoshioka H., Kawabata T., Org. Lett. 2013, 15, 864–867. [DOI] [PubMed] [Google Scholar]
- 11. Kasamatsu K., Yoshimura T., Mandi A., Taniguchi T., Monde K., Furuta T., Kawabat T., Org. Lett. 2017, 19, 352–355. [DOI] [PubMed] [Google Scholar]
- 12. Juliá-Hernández F., Ziadi A., A, Nishimura , Martin R., Angew. Chem. Int. Ed. 2015, 54, 9537–9541; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9673–9677. [Google Scholar]
- 13. Xu T., Dong G., Angew. Chem. Int. Ed. 2014, 53, 10733–10736; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10909–10912. [Google Scholar]
- 14. Kim J. H., Lee S., Kim S., Angew. Chem. Int. Ed. Angew.Chem. Int.Ed. 2015, 54, 10875–10878. [DOI] [PubMed] [Google Scholar]
- 15. Liu T., Yan N., Zhao H., Wang Z.-X., Hu X.-G., J. Fluor. Chem. 2018, 207, 18–23. [Google Scholar]
- 16.
- 16a. Hu X. G., Hunter L., Beilstein J. Org. Chem. 2013, 9, 2696–2708; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16b. Hunter L., Beilstein J. Org. Chem. 2010, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chorghade M. S., Mohapatra D. K., Sahoo G., Gurjar M. K., Mandlecha M. V., Bhoite N., Moghe S., Raines R. T., J. Fluor. Chem. 2008, 129, 781–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lee S., Bae M., In J., Kim J. H., Kim S., Org. Lett. 2017, 19, 254–257. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Mahajan P. S., Mhaske S. B., Org. Lett. 2018, 20, 2092–2095; [DOI] [PubMed] [Google Scholar]
- 19b. Veeraswamy V., Goswami G., Mukherjee S., Ghosh K., Saha M. L., Sengupta A., Ghorai M. K., J. Org. Chem. 2018, 83, 1106–1115. [DOI] [PubMed] [Google Scholar]