

Abstract
Leydig cell transplantation is a better alternative in the treatment of androgen‐deficient males. The main purpose of this study was to investigate the effects of induced pluripotent stem cell‐derived conditioned medium (iPS‐CM) on the anti‐apoptosis, proliferation and function of immature Leydig cells (ILCs), and illuminate the underlying mechanisms. ILCs were exposed to 200 μmol/L hydrogen peroxide (H2O2) for 24 hours with or without iPS‐CM treatments. Cell apoptosis was detected by flow cytometric analysis. Cell proliferation was assessed using cell cycle assays and EdU staining. The steroidogenic enzyme expressions were quantified with Western blotting. The results showed that iPS‐CM significantly reduced H2O2‐induced ILC apoptosis through down‐regulation of autophagic and apoptotic proteins LC3‐I/II, Beclin‐1, P62, P53 and BAX as well as up‐regulation of BCL‐2, which could be inhibited by LY294002 (25 μmol/L). iPS‐CM could also promote ILC proliferation through up‐regulation of β‐catenin and its target proteins cyclin D1, c‐Myc and survivin, but was inhibited by XAV939 (10 μmol/L). The level of bFGF in iPS‐CM was higher than that of DMEM‐LG. Exogenous bFGF (20 ng/mL) or Wnt signalling agonist lithium chloride (LiCl) (20 mmol/L) added into DMEM‐LG could achieve the similar effects of iPS‐CM. Meanwhile, iPS‐CM could improve the medium testosterone levels and up‐regulation of LHCGR, SCARB1, STAR, CYP11A1, HSD3B1, CYP17A1, HSD17B3 and SF‐1 in H2O2‐induced ILCs. In conclusion, iPS‐CM could reduce H2O2‐induced ILC apoptosis through the activation of autophagy, promote proliferation through up‐regulation of Wnt/β‐catenin pathway and enhance testosterone production through increasing steroidogenic enzyme expressions, which might be used in regenerative medicine for future.
Keywords: apoptosis, immature Leydig cells, induced pluripotent stem cell‐derived conditional medium, pathway, proliferation
1. INTRODUCTION
Leydig cells, distributing in clusters between the seminiferous tubules in the testis, are responsible for androgen production in the male.1, 2 Testosterone, referred to as the male hormone, plays a critical role in maintaining sexual function, muscle bulk and bone health.3 Testosterone synthesis in Leydig cells depends on the luteinizing hormone (LH) secreted by the pituitary grand.4 LH binds LH receptors (LHCGR, encoded by Lhcgr) on the Leydig cell plasma membrane and then leads to intracellular cyclic adenosine monophosphate (cAMP) cascade,5 which further results in the rapid transport of cholesterol from the outer to the inner mitochondrial membrane, mediated by lipoprotein receptor (SCARB1, encoded Scarb1) and steroidogenic acute regulatory protein (STAR, encoded by Star). Subsequently, testosterone is synthesized through a series of steroidogenic enzymes: the cytochrome P450 cholesterol side‐chain cleavage enzyme (CYP11A1, encoded by Cyp11a1), 3β‐hydroxysteroid dehydrogenase (HSD3B1, encoded by Hsd3b1), cytochrome P450 17α‐hydroxylase (CYP17A1, encoded by Cyp17a1) and 17β‐hydroxysteroid dehydrogenase 3 (HSD17B3, encoded by Hsd17b3),6 while another critical factor for Leydig cells is the steroidogenic factor 1 (SF‐1, or named NR5A1), which is essential for the commitment of stem Leydig cells to testosterone‐producing Leydig cells.7 It is well known that SF‐1 is an essential factor for Leydig cell development and survival.8 Overexpression of SF‐1 has been shown to be capable of promoting ESCs into Leydig‐like cells.9
Induced pluripotent stem cells (iPSCs) are a type of pluripotent stem cells that can be generated directly from adult cells.10, 11, 12 iPSCs share the features of ESCs that are capable of self‐renewal and differentiation into three germ layers.13 iPSCs can address immune rejection and ethical issues of the autologous cell transplantation. They also offer an attractive approach to disease model, pharmaceutical screening, toxicology research and so on.14, 15 Conditioned medium can affect cell functions via biologically active components. It was reported that cytokines such as basic fibroblast growth factor (bFGF), nerve growth factor (NGF), stem cell factor (SCF), hepatocyte growth factors (HGF), vascular endothelial growth factor (VEGF), insulin‐like growth factor (IGF‐1) and brain derived neurotrophic factor (BDNF), which were secreted in the medium by cultured stem cells, could encourage the growth of cells.16, 17, 18 Li et al11 reported that induced pluripotent stem cell‐derived conditioned medium (iPS‐CM) potentially restored the bronchial microstructure in acute lung injury (ALI). Zhang et al19 mentioned that iPS‐CM contributed to recovery from the effect of endotoxin‐induced ALI in mice. The stimulating proliferation and anti‐apoptosis from iPS‐CM were the result of cytokines secretion, which were generally safe and would not produce tumours.19, 20, 21
The Wnt/β‐catenin signalling pathway is the classic pathway involved in cell proliferation.22 Basic fibroblast growth factor (bFGF) could promote the proliferation and migration of fibroblasts through the activation of Wnt/β‐catenin signalling pathway.23 Autophagy has been known as a conserved catabolism to sustain cellular homeostasis.24, 25 It plays critical roles in eliminating dysfunctional or surplus proteins and damaged intracellular organelles.26 A major event in autophagosome formation is to generate LC3‐I, which is then conjugated to phosphatidylethanolamine, generating membrane‐bound LC3‐II.27, 28 Beclin‐1 is a key autophagic protein regulating autophagosome formation.29, 30 Meanwhile, P62 is a specific autophagic substrate protein and the hallmark representing autophagic flu.30 It is known that the activation of autophagy can protect cells against apoptosis and inflammation.31, 32
In this research, we would investigate the effects of iPS‐CM on the proliferation and the H2O2‐induced apoptosis of immature Leydig cells (ILCs), and evaluate the influences of iPS‐CM on testosterone production of ILCs. Moreover, the potential mechanisms underlying the effects of iPS‐CM would be explored. This study was to develop a new way to improve the activities and function of ILCs used for future clinical cell transplantation.
2. MATERIALS AND METHODS
2.1. Chemicals and kits
Etiocholanolone, nicotinamide adenine dinucleotide (NAD+), collagenase, DNase, Percoll, hydrogen peroxide (H2O2), Y‐27632, Matrigel, M‐199 buffer, Triton X‐100, DAPI, XAV939, LY294002 and lithium chloride (LiCl) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Low‐glucose Dulbecco's modified Eagle's medium, fatal bovine serum (FBS), 1% penicillin and streptomycin, and 0.05% trypsin‐EDTA were purchase from Gibco Company (NY, USA). Annexin V‐FITC/PI apoptosis detection kit, 5, 59, 6, 69‐tetrachloro‐1, 19, 3, 39‐tetraethylbenzimi‐dazolylcarbocyanine iodide (JC‐1) assay kit was purchased from Nanjing KeyGEN Biotech (Nanjing, China). 2′7′‐Dichlorofluorescin diacetate (DCFH‐DA) assay kit was purchased from Qcbio Science & Technologies Co., Ltd. (Shanghai, China).
2.2. Animals
The Sprague‐Dawley rats were provided by Laboratory Animal Center, Wenzhou Medical University, Wenzhou, China. They were raised in a 12‐hour dark/light cycle temperature at 23 ± 2°C, and relative humidity of 45% to 55%. Water and food were accessed ad libitum. This study was approved by the Wenzhou Medical University's Animal Care and Use Committee, and was performed in accordance with the Guide for the Care and Use of Laboratory Animals.
2.3. Immature Leydig cells isolation
Eighteen 35‐day‐old male Sprague‐Dawley rats were killed in CO2 tank for the isolation of immature Leydig cells (ILCs). ILCs express all androgen biosynthetic enzymes33 and are capable of proliferation.34 The isolation of rat ILCs was performed as previously described.33 Briefly, the testes were removed, perfused with collagenase via the testicular artery and digested with M‐199 buffer containing collagenase (0.25 mg/mL) and DNase (0.25 mg/mL) for 15 minutes. Then, the cell suspension was filtered through 100‐μm nylon mesh (BD, CA, USA) and the cells were separated by the Percoll gradient. The cells with the density of 1.07‐1.088 g/mL were collected. The purity of ILCs was evaluated by immunohistochemical staining HSD3B1, the biomarker of ILCs, as previously described.35 The HSD3B1 staining solution contained with 0.4 mmol/L etiocholanolone as the steroid substrate and NAD+ as a cofactor.35 The purity of ILCs was more than 95%.
2.4. Culture of ILCs
The isolated ILCs were directly seeded into wells in the 24‐well culture plates with the density of 2 × 104 cells/well and incubated at a 37°C, 5% CO2 incubator. The culture medium (DMEM‐LG) contains low‐glucose Dulbecco's modified Eagle's medium, 10% fatal bovine serum (FBS), and 1% penicillin and streptomycin as control.
2.5. The culture of iPSCs and preparation of iPSC supernatant
iPSCs were cultured as previously described.36 In brief, culture plates were coated with 1% (v/v) Matrigel for 0.5 hours in advance, and then, iPSCs were cultured in mTeSR1 medium (StemCell Technologies Inc., Canada) at a 37°C, 5% CO2 incubator. These cells would be passaged every 6 days using 0.05% trypsin‐EDTA. Y‐27632 (10 mmol/L) as ROCK inhibitor was added into the plates on the first day after passage. The supernatant of iPSC was derived from the mTeSR1 medium cultured with iPSCs for 24 hours. The supernatant was filtered (0.22 μm) to remove dead cells and then stored at −80°C for at least 2 weeks. iPS‐CM was DMEM‐LG mixed with iPSC supernatant at the ratio of 2:1.
2.6. CCK‐8 assay
CCK‐8 was used to detect the cell viability of ILCs with different treatments. Briefly, isolated ILCs were seeded in the 96‐well plates at the density of 1 × 104 cells/well. Then, cells were cultured in different mediums and incubated at a 37°C, 5% CO2 incubator for 48 hours. Subsequently, the each well was supplemented with 10 μL CCK‐8 (BestBio, Shanghai, China) solution and incubated at 37°C for 4 hours. At last, the absorbance at 450 nm was measured with a microplate reader (Thermo, MA, USA).
2.7. Cell cycle assay
Freshly isolated ILCs were seeded in the 12‐well plates at the density of 5 × 105 cells/well and incubated for 24 hours. Then, the medium was changed into DMEM‐LG or iPS‐CM with 200 μmol/L H2O2. Cells were cultured for 48 hours. Then, cells were harvested and fixed with 75% cold ethanol overnight at 4°C. The fixed cells were washed once with phosphate‐buffered saline (PBS) and stained darkly with propidium iodide (PI) for 30 minutes at room temperature. The stained cells were analysed by flow cytometer (BD FACSAria, San Diego, CA, USA).
2.8. Annexin V and PI assay
ILCs were planted into the 12‐well plates with the density of 5 × 105 cells/well and incubated for 24 hours. Then, the medium was changed into DMEM‐LG or iPS‐CM with 200 μmol/L H2O2. Cells were cultured for 48 hours. To evaluate early and lately apoptotic activity, an Annexin V‐FITC/PI apoptosis detection kit was used as the manufacturer's instructions. Cells were harvested and washed with cold PBS and then were resuspended in 200 μL the Annexin V‐binding buffer. After cells were stained with 5 μL of FITC‐labelled Annexin V and 5 μL of PI, they were instantly measured using flow cytometer.
2.9. Measurement of mitochondrial membrane potential (▵Ψm)
▵Ψm was evaluated with JC‐1 assay kit. Approximately 5 × 105 cells/well‐isolated ILCs were plated into the 12‐well plates and incubated for 24 hours. Then, the medium was changed into DMEM‐LG or iPS‐CM with 200 μmol/L H2O2. Cells were cultured for 48 hours. Subsequently, culture medium was removed and cells were washed in cold PBS twice. Cells were incubated for 30 minutes at 37°C with 500 μL JC‐1 (5 mmol/L). Cells were collected by centrifugation at 500 g for 5 minutes and washed twice again with 1× incubation buffer. Cells were then resuspended with 500 μL 1× incubation buffer. Red and green fluorescence emissions were analysed by flow cytometer using an excitation wavelength of 488 nm and emission wavelengths of 530 nm (green fluorescence)/585 nm (red fluorescence). Mitochondrial depolarization was assessed by a decrease in the intensity ratio of the red/green fluorescence.
2.10. Measurement of cellular H2O2‐induced reactive oxygen species
Reactive oxygen species (ROS) production was measured with the fluorescence dye DCFH‐DA assay kit. Briefly, 5 × 105 cells/well‐isolated ILCs were plated into the 12‐well plates and incubated for 24 hours. Then, the medium was changed into DMEM‐LG or iPS‐CM with 200 μmol/L H2O2. Cells were cultured for 48 hours. Thereafter, cells were harvested and suspended with 200 μL DCFH‐DA for 20 minutes at 37°C in the dark. Cells were washed twice with PBS, and fluorescence intensity determined by flow cytometer was used to measure ROS.
2.11. Cell treatments
For exploration of the underlying mechanisms, 1 × 106 cells/well‐isolated ILCs were plated into the 6‐well plates and incubated for 24 hours. For exploration the apoptosis mechanism, the mediums were changed into DMEM‐LG (control) or iPS‐CM with 200 μmol/L H2O2 to establish control+H2O2 group or iPS‐CM+H2O2 group. Exogenous bFGF (20 ng/mL) was added into DMEM‐LG+H2O2 to obtain the control+H2O2+bFGF group. LY294002 (LY, 25 μmol/L) were added into iPS‐CM+H2O2 to get iPS‐CM+H2O2+LY group. Then, cells were cultured for another 48 hours and harvested for Western blotting. For exploration of the proliferation mechanism, the mediums were changed into DMEM‐LG (control) or iPS‐CM to establish control group or iPS‐CM group. XAV939 (XAV, 10 μmol/L) was added into iPS‐CM to get iPS‐CM+XAV group. Exogenous bFGF (20 ng/mL) was added into DMEM‐LG to get control+bFGF group. LiCl (20 mmol/L) was added into DMEM‐LG to get control+LiCl group. Then, cells were cultured for another 48 hours and harvested for Western blotting.
2.12. Western blotting
Cells were washed twice with cold PBS and then were lysed in the radio immunoprecipitation assay buffer (Bocai Biotechnology, Shanghai, China) supplemented with a protease inhibitor (Amyjet Scientific Inc, Wuhan, China). Lysate was centrifuged at 12 000 g for 15 minutes at 4°C. The protein concentrations in the supernatants were measured using the BCA assay kit (Takara, Japan) as the manufacturer's instructions. Sample proteins (50 μg) were subjected to 10% polyacrylamide gel containing sodium dodecyl and then transferred into the polyvinylidene fluoride membrane. After being blocked with 5% free‐fat milk in Tween 20‐containing Tris‐buffered saline for 2 hours at 4°C, the membranes were incubated with primary antibodies over night at 4°C (listed in Table 1). Then, membranes were washed with Tween 20‐containing Tris‐buffered saline for five times and incubated with horseradish peroxidase‐conjugated secondary antibody (1:5000, Bioword, MN, USA) for 1 hour at room temperature and then were washed with the buffer for three times again. The protein bands were visualized with enhanced chemiluminescence (Pierce Chemical Co, IL, USA). The intensities of proteins were quantified using Image J software.
Table 1.
Antibodies
| Antibody | Species | Vendor (city, state, catalogue) | WB |
|---|---|---|---|
| β‐Actin | Rabbit | Cell Signaling Technology (Danvers, MA, 12620S) | 1:1000 |
| LHCGR | Goat | Santa Cruz (Santa Cruz, CA, sc‐26343) | 1:1000 |
| SCARB1 | Rabbit | Abcam (San Francisco, CA, ab217318) | 1:1000 |
| STAR | Rabbit | Abcam (San Francisco, CA, ab133657) | 1:1000 |
| CYP11A1 | Rabbit | Santa Cruz (Santa Cruz, CA, sc‐18043) | 1:1000 |
| HSD3B1 | Rabbit | Abcam (San Francisco, CA, ab65156) | 1:2000 |
| CYP17A1 | Rabbit | Santa Cruz (Santa Cruz, CA, sc‐66850) | 1:1000 |
| HSD17B3 | Rabbit | Abcam (San Francisco, CA, Ab70088) | 1:2000 |
| SF‐1 | Rabbit | Santa Cruz (Santa Cruz, CA, sc‐10976) | 1:1000 |
| BAX | Rabbit | Cell Signaling technology (Danvers, MA, 2774S) | 1:1000 |
| BCL2 | Rabbit | Cell Signaling technology (Danvers, MA, 4223S) | 1:1000 |
| β‐catenin | Rabbit | Cell Signaling technology (Danvers, MA, 8480) | 1:1000 |
| Cyclin D1 | Mouse | Santa Cruz (Santa Cruz, CA, sc‐8396) | 1:2000 |
| c‐Myc | Mouse | Santa Cruz (Santa Cruz, CA, sc‐40) | 1:2000 |
| Survivin | Mouse | Santa Cruz (Santa Cruz, CA, sc‐73082) | 1:2000 |
| LC3‐I/II | Rabbit | Cell Signaling technology (Danvers, MA, 12741S) | 1:1000 |
| Beclin‐1 | Rabbit | Cell Signaling technology (Danvers, MA, 3495S) | 1:1000 |
| P62 | Rabbit | Cell Signaling technology (Danvers, MA, 39749S) | 1:1000 |
| P53 | Mouse | Cell Signaling technology (Danvers, MA, 2524S) | 1:1000 |
2.13. Testosterone measurement by radioimmunoassay
Medium testosterone concentrations were measured with a tritium‐based radioimmunoassay using antitestosterone antibody as previously described.37 Standards ranging between 10 and 2000 pg/mL testosterone were prepared in triplicate. Standards and samples were incubated with tracer, and charcoal‐dextran suspension was used to separate the bound and free steroids. The bound steroids were mixed with a scintillation buffer and counted in a β scintillation counter (PE, CA, USA). The minimum detectable concentration for testosterone was 5 pg/mL. Quality control samples contain 100 pg/mL testosterone. The intra‐assay and interassay coefficients of variation were within 10%.
2.14. EdU staining assays
EdU staining was conducted with a Click‐iT® EdU imaging kit (Invitrogen, CA, USA) as per manufacturer's instruction. Briefly, isolated ILCs were seeded in the 12‐well plates at the density of 5 × 105 cells/well and incubated for 24 hours. Then, cells were incubated with EdU at a final concentration of 20 μmol/L at 37°C incubator for 2 hours. Cells were harvested by digestion and centrifugation, and were fixed with 4% paraformaldehyde for 15 minutes and permeabilized using Triton X‐100 solution for 30 minutes at room temperature darkly. Then, cells were centrifuged and resuspended in 0.5 mL of PBS. Fixed cells were stained with the Click‐iT™ reaction mixture. Then, cell nucleus was stained with 1 μg/mL DAPI at room temperature for 30 minutes darkly. Lastly, the stained cells were observed at an inverted fluorescence microscopy and were analysed using flow cytometry.
2.15. ELISA
The samples were treated with enzyme‐linked immunosorbent assay (ELISA) kit according to the manufacturer's instruction (Chemicon, CA, USA). Briefly, 50 μL assay diluent and 200 μL samples were added to pre‐coated wells of 96‐well plates. The plates were incubated at room temperature for 2 hours and washed five times with washing buffer. 100 μL peroxidase‐conjugated IgG anti‐bFGF of solution was added to each well at room temperature for 2 hours. Then, plates were washed five times with washing buffer. Then 100 μL substrate buffers were added to each well and incubated in dark at room temperature for 30 minutes. The enzyme reaction was stopped by 50 μL stop solution. Optical densities were obtained for the quantification of bFGF levels by a microplate reader at 550 nm with correction wavelength at 450 nm. Data were conducted by GraphPad Prism 5 software.
2.16. Statistical analysis
All data are presented as the mean ± standard errors (SE). Statistical significance was analysed using one‐way ANOVA followed by ad hoc Turkey multiple comparisons to the control. Statistical analyses were performed using GraphPad Prism (version 6, GraphPad Software Inc., San Diego, CA, USA). *P < .05, **P < .01 or ***P < .001 were considered statistically significant.
3. RESULTS
3.1. The optimal iPS‐CM ratio and apoptosis model of H2O2‐induced immature Leydig cells
The proliferation viability of ILCs was analysed by CCk‐8 to assess the optimal proportion of DMEM‐LG/iPS‐supernatant. ILCs were treated with different proportions of DMEM‐LG/iPS‐supernatant (1:0, 1:1, 2:1, 1:2, 0:1) for 48 hours. The result showed that the proliferation viability of ILCs was the most obvious at the ratio of 2:1 compared with other proportions (Figure 1A). Base on these data, we considered this proportion as iPS‐CM to treat ILCs in following studies.
Figure 1.
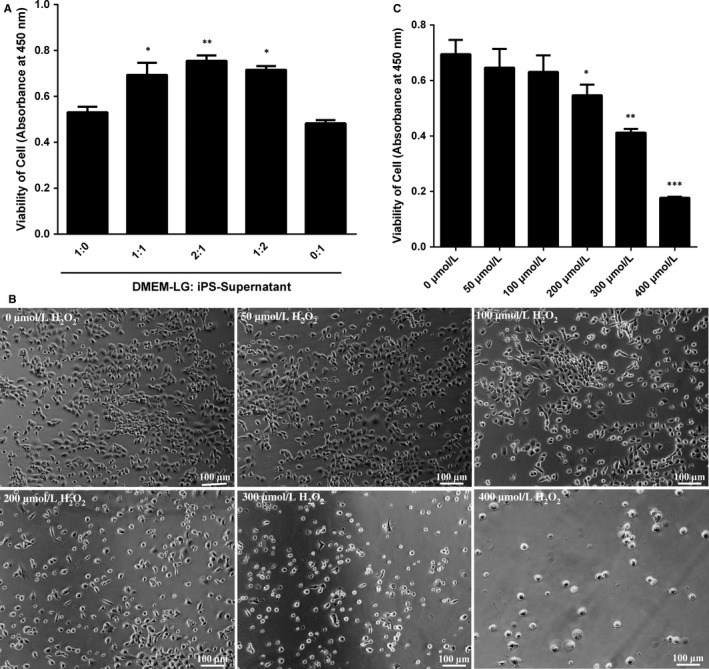
Exploration the optimal proportion of DMEM‐LG/iPS‐supernatant and apoptosis model of H2O2‐induced immature Leydig cells (ILCs). (A) The proliferation viability of ILCs in different proportion of DMEM‐LG/iPS‐supernatant was analysed by CCK‐8. (B) The bright field of ILCs exposed to various concentrations of H2O2 under an inverted microscope. (C) The viability effect of iPS‐CM on ILCs exposed to H2O2 at various doses by CCK‐8 assay. Mean ± SE, n = 5. *P < .05, **P < .01, ***P < .001 designate significant differences when compared to control (1:0 or 0 μmol/L)
To establish the optimal apoptosis model, ILCs were exposed to different concentrations of H2O2 at 0, 50, 100, 200, 300 μmol/L and then were cultured for 24 hours. The death cells were washed away with PBS. The attached cells were imaged using an inverted phase light microscope (Figure 1B). The morphological features of H2O2‐induced ILCs were examined. The hallmark of apoptosis cells such as cell detachment and cytoplasmic condensation would worsen due to increasing dosage of H2O2 exposure beginning from the dosage of 200 μmol/L. The result of CCK‐8 assay was consistent with above performance (Figure 1C). These data indicated that H2O2 at the dose of 200 μmol/L was optimal for the apoptosis model.
3.2. Effects of iPS‐CM on the anti‐apoptosis of H2O2‐induced immature Leydig cells
Annexin V and PI assay was conducted to assess the effects of iPS‐CM on the apoptosis of H2O2‐induced ILCs. In the early stage of apoptosis, cells have the intact cell membranes that cannot be stained by PI. However, externalization of phosphatidylserine can be stained by Annexin V. In the late stage of apoptosis, the damaged cell membranes allow Annexin V and PI to enter into cells. Hence, ILCs in early apoptosis were Annexin V positive and PI negative but in late apoptosis were both Annexin V and PI positive. The percentage of apoptotic cells was calculated from the Q1‐LR (early stage of apoptosis) and Q1‐UR (late stage of apoptosis). Annexin V and PI assays showed that iPS‐CM could decrease the apoptosis ratio of H2O2‐induced ILCs (Figure 2A). The control group (ILCs in DMEM‐LG) almost had no apoptotic cells, with only 1.98 ± 0.15%. The control+H2O2 group had 25.59 ± 0.37% apoptotic cells, which is much higher than that of control (**P < .01). The apoptotic ratio in iPS‐CM+H2O2 group was 7.97 ± 0.21%, which was higher than that of control group (*P < .05) but lower than that of control+H2O2 group (**P < .01) (Figure 2B). This result demonstrated that iPS‐CM could inhibit H2O2‐induced ILC apoptosis.
Figure 2.
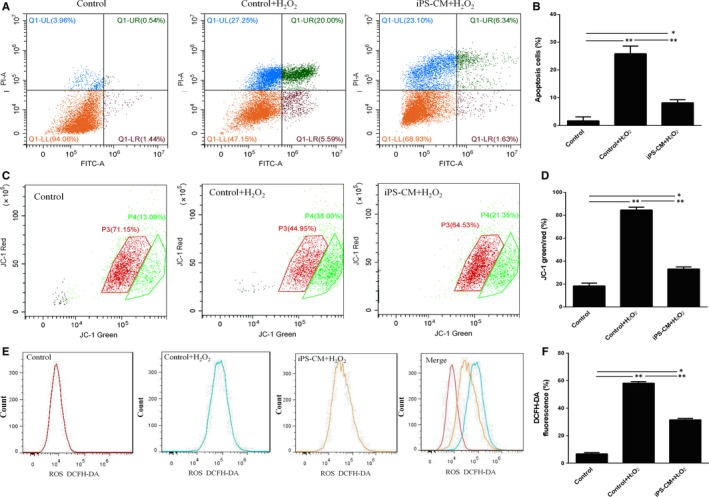
iPS‐CM inhibited the apoptosis of H2O2‐induced immature Leydig cells (ILCs). (A) Annexin V and propidium iodide (PI) assay was used to analyse the cell apoptosis in different groups. (B) Quantification of the Annexin V and PI assay. (C) Mitochondrial membrane potential (▵Ψm) assay was used to analyse the loss of ▵Ψm of ILCs in different groups. (D) Quantification of ▵Ψm. (E) Induced reactive oxygen species (ROS) assay was used to analyse the generation of ROS in different groups. (F) Quantification of intracellular ROS. Mean ± SE, n = 3. *P < .05 (iPS‐CM+H2O2 vs control), **P < .01 (iPS‐CM+H2O2 or control vs control+H2O2) designate significant differences
As the loss of ▵Ψm was associated with early apoptosis, JC‐1 probe was used to analyse the effects of iPS‐CM on the loss of ▵Ψm in H2O2‐induced ILCs. The ▵Ψm of living cells is higher than that of apoptotic cells. JC‐1 can specially enter the mitochondria. When the ▵Ψm is high, JC‐1 will form polymer and glow red fluorescent. When the ▵Ψm is low, it will form the monomer and glow green fluorescence. The ratio of JC‐1‐green/red can indirectly reflect the result of apoptosis. Low ratio presents less apoptosis than high ratio. The loss of ▵Ψm in ILCs after H2O2 treatment increased obviously compared with that of control group (ILCs in DMEM‐LG), and was prevented by iPS‐CM (Figure 2C). The data showed that the ratio of JC‐1‐green/red in control group was 18.39 ± 2.31%, which was less than that of control+H2O2 group (84.5 ± 4.54%) (**P < .01) and iPS‐CM+H2O2 group (33.08 ± 2.24%) (*P < .05). The ratio of JC‐1‐green/red in iPS‐CM+H2O2 group was also less than that of control+H2O2 group (**P < .01) (Figure 2D). This result showed that iPS‐CM could inhibit the loss of ▵Ψm in H2O2‐induced ILCs.
DCFH‐DA was used to detect the effects of iPS‐CM on the generation of ROS in H2O2‐induced ILCs. The generation of ROS in ILCs after H2O2 treatment increased remarkably compared with that of control group (ILCs in DMEM‐LG), and then, iPS‐CM decreased the ROS generation of H2O2‐induced ILCs again (Figure 2E). The result showed that the value of DCFH‐DA fluorescence in control group was 7.8 ± 0.48%, which was less than that of control+H2O2 group (58.2 ± 3.58%) (**P < .01) and iPS‐CM+H2O2 group (31.5 ± 1.48%) (*P < .05). Moreover, the value of DCFH‐DA fluorescence in iPS‐CM+H2O2 group was less than control+H2O2 group, having approximately a onefold decrease (**P < .01) (Figure 2F). The value of DCFH‐DA fluorescence could reflect the generation of ROS. This result documented that iPS‐CM could inhibit the generation of ROS in H2O2‐induced ILCs.
All the above data showed that iPS‐CM could inhibit the apoptosis, the loss of ▵Ψm and the generation of ROS in H2O2‐induced ILCs.
3.3. Effects of iPS‐CM on the BAX and BCL‐2 expressions of H2O2‐induced immature Leydig cells
To explore the potential molecular mechanisms involved in the anti‐apoptosis of iPS‐CM on H2O2‐induced ILCs, the expression levels of apoptotic signalling molecules in the mitochondria including BAX and BCL‐2 were detected by Western blotting in different groups. The result showed that H2O2 could apparently up‐regulate the expression of BAX and down‐regulate the expression of BCL‐2. Compared with control group (ILCs in DMEM‐LG), the expression level of BAX in iPS‐CM was less (*P < .05). When cells treated with H2O2, BAX expressions were up‐regulated in control+H2O2 and iPS‐CM+H2O2 groups, and the level in iPS‐CM+H2O2 group was less than that of control+H2O2 group (*P < .05) (Figure 3A). The tendency of BCL‐2 expression was almost opposite to the BAX expression. The BCL‐2 expression level in iPS‐CM group was more than control group (*P < .05), and H2O2 could significantly inhibit the expression of BCL‐2 in control+H2O2 and iPS‐CM+H2O2 groups. In addition, the level of BCL‐2 in iPS‐CM+H2O2 group was higher than that of control+H2O2 group (*P < .05) (Figure 3B). These results showed that iPS‐CM could suppress the up‐regulation of BAX and down‐regulation of BCl‐2 in H2O2‐induced ILCs.
Figure 3.
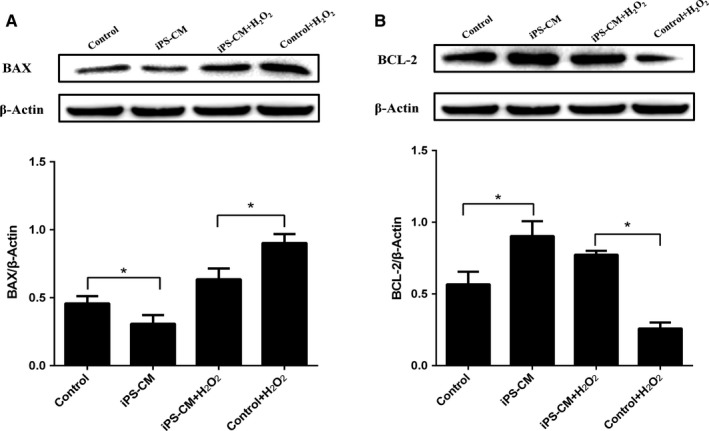
iPS‐CM suppressed BAX up‐regulation and BCl‐2 down‐regulation of H2O2‐induced immature Leydig cells (ILCs). (A) The protein expression of BAX in different groups. (B) The protein expression of BCL‐2 in different groups. Mean ± SE, n = 3. *P < .05 designates significant differences
3.4. Effects of iPS‐CM on the proliferation of H2O2‐induced immature Leydig cells
The cell cycle assay was used to assess the effects of iPS‐CM on the proliferation of H2O2‐induced ILCs. Compared with control (ILCs in DMEM‐LG), H2O2 could inhibit cell proliferation, but iPS‐CM partly rescued the proliferation of H2O2‐induced ILCs (Figure 4A). The percentage of cell entering the S and G2 phases in control group was 50.43 ± 1.37%, which was less than that of iPS‐CM group (57.72 ± 1.45%) (**P < .01). Meanwhile, the percentage of cell entering the S and G2 phases in iPS‐CM+H2O2 group (50.56 ± 1.31%) was also more than that of control+H2O2 group (40.55 ± 1.12%) (**P < .01) (Figure 4B). In addition, EdU staining showed that the EdU‐positive cells in control group was less than that of iPS‐CM group, and iPS‐CM+H2O2 group was more than control+H2O2 group (Figure 4C). Meanwhile, the flow cytometry assays also demonstrated that the fluorescent density of EdU‐positive cells in control group was less than iPS‐CM group (**P < .01), and iPS‐CM+H2O2 group was more than that of control+H2O2 group (**P < .01) (Figure 4D,E). These results suggested that iPS‐CM could promote the proliferation of ILCs.
Figure 4.
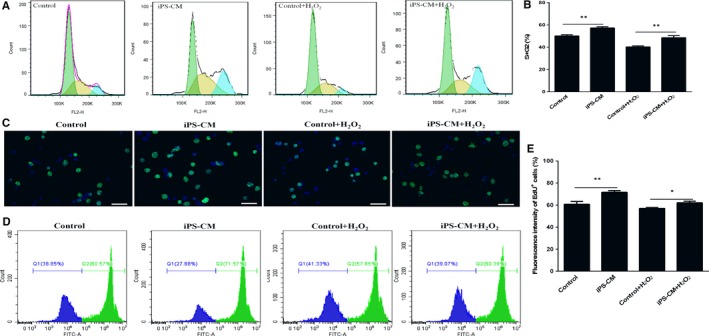
iPS‐CM promoted the proliferation viability of H2O2‐induced immature Leydig cells (ILCs). (A) The cell cycle assay was conducted to analyse the proliferation viability in different groups. (B) Quantification of the cell cycle distribution. (C) EdU staining images at an inverted fluorescence microscope. (D) Fluorescent density of EdU‐positive cells was detected by flow cytometry assays. (E) Quantification of the EdU‐positive cells. Mean ± SE, n = 3. *P < .05, **P < .01 designate significant differences. Scale bars 100 μm
3.5. Effects of iPS‐CM on medium testosterone (T) levels and steroidogenic enzyme expressions of immature Leydig cells
To explore the effects of iPS‐CM administration on the testosterone synthesis of ILCs, the medium testosterone levels in different groups were detected by radioimmunoassay. The result showed that iPS‐CM could improve the medium testosterone levels compared with control (ILCs in DMEM‐LG) (**P < .01) and could also inhibit the decrease in the medium testosterone levels in H2O2‐induced ILCs (***P < .001) (Figure 5A). These data suggested that iPS‐CM could promote the testosterone synthesis of ILCs.
Figure 5.
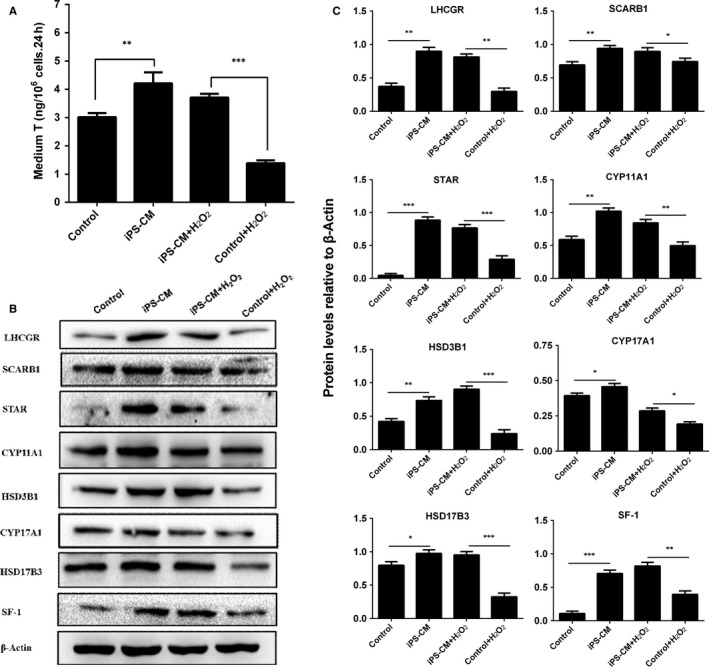
Medium testosterone (T) and protein expression levels of H2O2‐induced immature Leydig cells (ILCs) with or without iPS‐CM treatments. (A) Medium testosterone (T) levels in different groups. Leydig cell proteins: (B) Western blotting band in different groups, (C) quantification of protein levels. Mean ± SE, n = 3. *P < .05, **P < .01, ***P < .01 designate significant differences
Western blotting was conducted to detect the steroidogenic enzyme expression levels of LHCGR, SCARB1, STAR, CYP11A1, HSD3B1, CYP17A1, HSD17B3 and SF‐1 in ILCs with different treatments (Figure 5B). Statistically, we found that iPS‐CM could significantly up‐regulate the expression levels of LHCGR, SCARB1, STAR, CYP11A1, HSD3B1, CYP17A1, HSD17B3 and SF‐1 compared with control. Meanwhile, iPS‐CM could also inhibit the down‐regulation of LHCGR, SCARB1, STAR, CYP11A1, HSD3B1, CYP17A1, HSD17B3 and SF‐1 in H2O2‐induced ILCs (Figure 5C). This result confirmed that iPS‐CM could promote the steroidogenic enzyme expressions of ILCs.
3.6. The potential mechanisms underlying the promotion anti‐apoptosis and proliferation of immature Leydig cell by iPS‐CM
To understand the potential mechanisms underlying the promotion anti‐apoptosis and proliferation of ILCs by iPS‐CM, we firstly compared the levels of several special growth factors such as bFGF, IGF‐1 and VEGF in the medium of iPS‐CM group with those of control group (ILCs in DMEM‐LG) at the H2O2 induction or not by ELISA. There was just bFGF but not IGF‐1 and VEGF (data not shown) having significant difference. The level of bFGF in iPS‐CM was higher than that of DMEM‐LG with or without H2O2 treatment (***P < .001) (Figure 6A). Based on this result, exogenous bFGF (20 ng/mL) was added into the medium of control (DMEM‐LG) to establish a new group for mimicking the iPS‐CM group, and the underlying signalling pathways were further explored.
Figure 6.
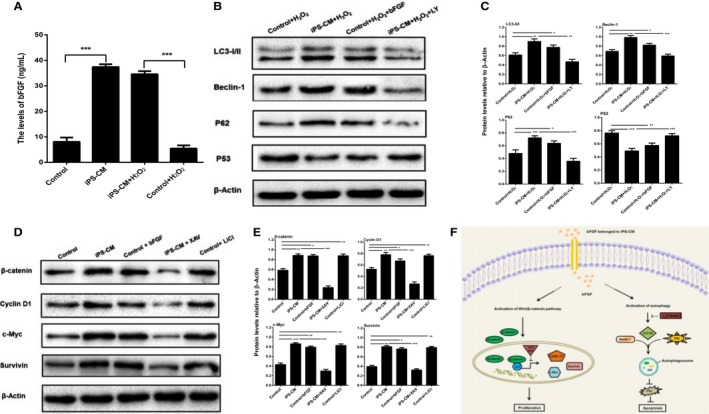
The potential mechanisms underlying the promotion of immature Leydig cell (ILC) anti‐apoptosis and proliferation by iPS‐CM. (A) Quantification of the bFGF levels of medium in different groups using ELISA. (B) Assessment of protein expression levels of LC3 I/II, Beclin‐1, P62 and P53 in different groups using Western blotting assays. (C) Quantification of Western blotting assays. (D) Assessment of protein expression levels of β‐catenin, cyclin D1, c‐Myc and survivin in different groups using Western blotting assays. (E) Quantification of Western blotting assays. (F) Diagram of the potential mechanisms underlying the promotion of ILC anti‐apoptosis and proliferation by iPS‐CM. Mean ± SE, n = 3. *P < .05, **P < .01, ***P < .01 designate significant differences
To elucidate the role of iPS‐CM in the activation of autophagy to suppress apoptosis of H2O2‐induced ILCs, the expression levels of autophagic proteins such as LC3‐I/II, Beclin‐1, P62, and apoptotic protein P53 among different groups were assessed by Western blotting (Figure 6B). The ratio of LC3 II/I and the levels of Beclin‐1 and P62 were significantly up‐regulated, but the expression of P53 was inhibited by iPS‐CM treatment in H2O2‐induced ILCs compared with the control. Additionally, exogenous bFGF added into DMEM‐LG could achieve the similar effects of iPS‐CM. However, the up‐regulated autophagic proteins and the down‐regulated apoptotic protein were also inhibited in iPS‐CM+H2O2 by LY294002 (LY, 25 μmol/L) (Figure 6C), an inhibitor of autophagy.38 These results suggested that iPS‐CM might markedly depend on inherent bFGF to suppress the apoptosis of ILCs through activation of autophagy pathway.
The Wnt/β‐catenin signalling pathway is the classic pathway controlling cell proliferation.22 The results of Western blotting analysis showed that the levels of β‐catenin and its target proteins cyclin D1, c‐Myc and survivin in iPS‐CM group were higher than those in control group (ILCs in DMEM‐LG). At the same time, all proteins could be up‐regulated by exogenous bFGF added into DMEM‐LG. Furthermore, those proteins could also be up‐regulated by LiCl (20 mmol/L) added into DMEM‐LG, which is a widely used in the activation of Wnt/β‐catenin signalling.39, 40 However, those proteins were again down‐regulated in iPS‐CM by XAV939 (XAV, 10 μmol/L) (Figure 6D,E), an inhibitor of the Wnt/β‐catenin signalling pathway.41 These results indicated that iPS‐CM might be mainly relied on inherent bFGF to promote the proliferation of ILCs through Wnt/β‐catenin signalling pathway. And the diagram of the mechanisms underlying the promotion anti‐apoptosis and proliferation of ILCs by iPS‐CM is shown in Figure 6F.
4. DISCUSSION
Male hypogonadism is characterized by the low production of testosterone, which is associated with typical symptoms including mood disturbance, sexual dysfunction, decreased muscle mass and strength, and decreased bone mineral density.42 Currently, testosterone replacement therapy is applied to treat androgen‐deficient males with primary Leydig cell failure and can achieve some alleviation of symptoms. However, the therapy is limited by the risks of side effects, such as reducing the rate of spermatogenesis, increasing cardiovascular and prostate complications.43 Leydig cell transplantation may be a better alternative in providing physiological patterns of hormone for a longer period of time.44 In this study, we systematically investigated iPS‐CM could promote viabilities including proliferation and anti‐apoptosis, and testosterone synthesis of immature Leydig cell (ILCs), which would be better used for future clinical cell transplantation.
iPSCs cultured in vitro could secrete many cytokines, chemokines, growth factors, metabolites and bioactive lipids into the medium. A wide range of products from iPSC secretion could reduce apoptosis, oxidative stress and fibrosis, as well as improve cardiac function in diabetic model of rats.45 Apoptosis, as a basic character of cells, is a physiological process of cell death that plays a key role in a variety of biologic systems.46, 47 H2O2 is very ideal inductor for establishment apoptosis model. In this study, ILCs were exposed to different concentrations of H2O2, and we scan out the optimal dose of 200 μmol/L for the apoptosis model. The mechanisms for apoptosis contain direct damage to the mitochondria by ROS and indirect mitochondrial depolarization by apoptotic‐related BCL‐2 family proteins.48 BAX, a pro‐apoptotic and pore‐forming cytoplasmic protein in the BCL‐2 family, is translocated to the outer mitochondrial membrane and affects permeability from the intermembrane space into the cytosol, which later causes cell death.49 Conversely, BCL‐2, an anti‐apoptotic protein in the BCL‐2 family, lies in the cytoplasm of the outer mitochondrial membrane, nuclear envelope and endoplasmic reticulum.50, 51 It was demonstrated that BCL‐2 in fibroblasts was closely related to mitochondrial homeostasis and cell viability.52 In this study, H2O2‐treated ILCs showed an increase in intracellular ROS, a loss of ▵Ψm, up‐regulation in the expression of BAX and down‐regulation in the expression of BCL‐2. However, the generation of ROS was inhibited by treatment with iPS‐CM. We demonstrated that iPS‐CM protected ILCs against H2O2‐induced apoptosis by stabilizing ▵Ψm and decreasing the expression of BAX, and increasing the expression of BCL‐2. These results indicated that iPS‐CM had protective effects of H2O2‐induced ILCs. Neel and Singla also found that iPS‐CM could decrease the number of cardiac apoptosis in the streptozotocin of diabetic cardiomyopathy (SIDC) rat model.21
Zhang et al20 reported that iPS‐CM could not only suppress apoptosis by inhibiting p53/p21 and p16/pRb pathways, but also promote proliferation by attenuating G1 phase arrest of cell cycle in H9C2 cells. In our study, we also found that iPS‐CM could increase the percentage of ILCs entering the S and G2 phases and EdU staining‐positive ILCs with or without H2O2 treatment, which indicated that iPS‐CM could significantly promote the proliferation of ILCs. iPS‐CM could enhance alveolar epithelial regeneration in vivo partially due to containing hepatocyte growth factor.53 iPS‐CM could also promote the growth of other cells.54, 55
In addition, we also found that iPS‐CM could significantly increase the medium testosterone levels of ILCs with or without H2O2 treatments. The testosterone homeostasis in rat ILCs mainly depends on the cholesterol membrane receptor and transporters such as LHCGR, SCARB1 and STAR, and the testosterone biosynthetic enzymes such as CYP11A1, HSD3B1, CYP17A1 and HSD17B3. The results of Western blotting showed that iPS‐CM could significantly increase the expression levels of membrane receptor LHCGR, cholesterol transporters SCARB1 and STAR, and testosterone biosynthetic enzymes CYP11A1, HSD3B1, CYP17A1 and HSD17B3, which could be contributed to the improvement of testosterone levels. Their increased levels were consistent with SF‐1 up‐regulation, suggesting that iPS‐CM up‐regulated these proteins via increasing SF‐1 expression. Indeed, many studies had showed that CYP11A1, HSD3B1, CYP17A1 and HSD17B3 promoters had SF‐1 binding sites.56, 57 Although the exact mechanism is still unclear, the significant increase in SF‐1, a critical transcription factor for the expression of steroidogenic enzymes,7 might be involved in possible inherent mechanism.
As is known to all that the activation of autophagy can protect cells against apoptosis and inflammation,31, 32 in this study, it was discovered that the level of bFGF in iPS‐CM was higher than that of control (DMEM‐LG) with or without H2O2 treatment. iPS‐CM could up‐regulate the expression of autophagic proteins LC3 II/I, Beclin‐1 and P62 but down‐regulate the expression of apoptotic protein P53 in H2O2‐induced ILCs. However, the up‐regulated autophagic proteins and the down‐regulated apoptotic protein could be inhibited by LY294002 (25 μmol/L), an inhibitor of autophagy.38 Additionally, exogenous bFGF (20 ng/mL) added into DMEM‐LG could achieve the similar effects of iPS‐CM. These results suggested that iPS‐CM might act as an agonist of autophagy pathway to inhibit apoptosis of ILCs, which was markedly contributed to the inherent bFGF. The Wnt/β‐catenin signalling pathway is the classic pathway‐mediated cell proliferation.22 A closely related report demonstrated that XAV939 (an inhibitor of the Wnt/β‐catenin signalling pathway) could counteract the proliferation of preterm umbilical cord mesenchymal stem cells (UC‐MSCs) compared to term UC‐MSCs.58 In this study, it was showed that iPS‐CM could significantly promote the expression levels of Wnt/β‐catenin signalling proteins such as β‐catenin, cyclin D1, c‐Myc and survivin, but could be inhibited by XAV939 (10 μmol/L). Exogenous bFGF (20 ng/mL) or LiCl (20 mmol/L) added into DMEM‐LG could obtain almost similar effects of iPS‐CM. LiCl is a widely used GSK‐3β inhibitor that results in the activation of Wnt/β‐catenin signalling pathway.59 These data suggested that iPS‐CM might mainly rely on inherent bFGF to promote the proliferation of ILCs through up‐regulation of Wnt/β‐catenin signalling pathway. Taken as a whole, our study discovered that both the inhibition ILC apoptosis mediated by the activation of autophagy pathway and the promotion ILC proliferation mediated by the Wnt/β‐catenin signalling pathway might be attributed to the inherent bFGF of iPS‐CM. While this study just hints the potential mechanisms underlying the promotion anti‐apoptosis and proliferation of ILCs by iPS‐CM, the convincing molecular mechanisms should need further study in the future.
In conclusion, we reported that iPS‐CM might mainly rely on inherent bFGF to dramatically inhibit H2O2‐induced apoptosis, stimulate proliferation and enhance testosterone production of ILCs. The anti‐apoptotic role of iPS‐CM was correlated with the down‐regulation of BAX and P53 as well as up‐regulation of BCL‐2. The promotion proliferation role of iPS‐CM was involved in the up‐regulation of β‐catenin, cyclin D1, c‐Myc and survivin. Meanwhile, iPS‐CM could promote the testosterone production through up‐regulation of the steroidogenic enzyme expressions, which might be due to the increase of SF‐1 expression. Additionally, the potential mechanisms including activation of autophagy pathway participated in ILC anti‐apoptotic and the Wnt/β‐catenin signalling pathway involved in promotion ILC proliferation after iPS‐CM treatment were illuminated. This study had developed a new way to improve the viabilities and function of ILCs for future clinical cell transplantation.
COMPETING INTERESTS
The authors have declared that no competing interests exist.
ACKNOWLEDGEMENTS
This work was supported by the National Nature Science Foundation of China (81701426, 81730042 and 81771636), the Medical and Health Research Science and Technology Plan Project of Zhejiang Province (2018KY523, 2017KY473 and 2017ZB067), and the Public Welfare Science and Technology Plan Project of Wenzhou City (Y20170151, Y20160069 and ZS2017012).
Guo X, Chen Y, Hong T, et al. Induced pluripotent stem cell‐derived conditional medium promotes Leydig cell anti‐apoptosis and proliferation via autophagy and Wnt/β‐catenin pathway. J Cell Mol Med. 2018;22:3614–3626. https://doi.org/10.1111/jcmm.13641
Xiaoling Guo and Yong Chen contributed equally to this work.
REFERENCES
- 1. Zirkin BR, Chen H. Regulation of Leydig cell steroidogenic function during aging. Biol Reprod. 2000;63:977‐981. [DOI] [PubMed] [Google Scholar]
- 2. Chen H, Ge R‐S, Zirkin BR. Leydig cells: from stem cells to aging. Mol Cell Endocrinol. 2009;306:9‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Midzak AS, Chen H, Papadopoulos V, Zirkin BR. Leydig cell aging and the mechanisms of reduced testosterone synthesis. Mol Cell Endocrinol. 2009;299:23‐31. [DOI] [PubMed] [Google Scholar]
- 4. Chen H, Wang Y, Ge R, Zirkin BR. Leydig cell stem cells: Identification, proliferation and differentiation. Mol Cell Endocrinol. 2017:445:65‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu H‐C, Zhu D, Wang C, et al. Effects of etomidate on the steroidogenesis of rat immature Leydig cells. PLoS ONE. 2015;10:e0139311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ge RS, Hardy MP. Variation in the end products of androgen biosynthesis and metabolism during postnatal differentiation of rat Leydig cells. Endocrinology. 1998; 139: 3787‐3795. [DOI] [PubMed] [Google Scholar]
- 7. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359‐E386. [DOI] [PubMed] [Google Scholar]
- 8. Shima Y, Miyabayashi K, Baba T, et al. Identification of an enhancer in the Ad4BP/SF‐1 gene specific for fetal Leydig cells. Endocrinology. 2012;153:417‐425. [DOI] [PubMed] [Google Scholar]
- 9. Karpova T, Ravichandiran K, Insisienmay L, Rice D, Agbor V, Heckert LL. Steroidogenic factor 1 differentially regulates fetal and adult Leydig cell development in male Mice1. Biol Reprod. 2015;93:83, 1‐15‐83, 1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Firas J, Liu X, Nefzger CM, Polo JM. GM‐CSF and MEF‐conditioned media support feeder‐free reprogramming of mouse granulocytes to iPS cells. Differentiation. 2014;87:193‐199. [DOI] [PubMed] [Google Scholar]
- 11. Li L‐F, Liu Y‐Y, Yang C‐T, et al. Improvement of ventilator‐induced lung injury by IPS cell‐derived conditioned medium via inhibition of PI3K/Akt pathway and IP‐10‐dependent paracrine regulation. Biomaterials. 2013;34:78‐91. [DOI] [PubMed] [Google Scholar]
- 12. Solari C, Losino N, Luzzani C, et al. Induced pluripotent stem cells’ self‐renewal and pluripotency is maintained by a bovine granulosa cell line‐conditioned medium. Biochem Biophys Res Comm. 2011;410:252‐257. [DOI] [PubMed] [Google Scholar]
- 13. Kitazawa A, Shimizu N. Differentiation of mouse induced pluripotent stem cells into neurons using conditioned medium of dorsal root ganglia. New Biotechnol. 2011;28:326‐333. [DOI] [PubMed] [Google Scholar]
- 14. Lin J, Fernandez I, Roy K. Development of feeder‐free culture systems for generation of ckit+sca1 + progenitors from mouse iPS cells. Stem Cell Rev. 2011;7:736‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kasuda S, Tatsumi K, Sakurai Y, et al. Expression of coagulation factors from murine induced pluripotent stem cell‐derived liver cells. Blood Coag Fibrinol. 2011;22:271‐279. [DOI] [PubMed] [Google Scholar]
- 16. Pluchino S, Cossetti C. How stem cells speak with host immune cells in inflammatory brain diseases. Glia. 2013;61:1379‐1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ribeiro CA, Fraga JS, Grãos M, et al. The secretome of stem cells isolated from the adipose tissue and Wharton jelly acts differently on central nervous system derived cell populations. Stem Cell Res Ther. 2012;3:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ranganath SH, Levy O, Inamdar MS, Karp JM. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell. 2012;10:244‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang Y, Deng C, Qian J, Zhang M, Li X. Improvement of radiotherapy‐induced lacrimal gland injury by induced pluripotent stem cell‐derived conditioned medium via MDK and inhibition of the p38/JNK pathway. Int J Mol Sci. 2014;15:18407‐18421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y, Wang D, Cao K, Chen M, Yang X, Tao Y. Rat induced pluripotent stem cells protect H9C2 cells from cellular senescence via a paracrine mechanism. Cardiology. 2014;128:43‐50. [DOI] [PubMed] [Google Scholar]
- 21. Neel S, Singla DK. Induced pluripotent stem (iPS) cells inhibit apoptosis and fibrosis in streptozotocin‐induced diabetic rats. Mol Pharm. 2011;8:2350‐2357. [DOI] [PubMed] [Google Scholar]
- 22. Fan HB, Zhai ZY, Li XG, et al. CDX2 stimulates the proliferation of porcine intestinal epithelial cells by activating the mTORC1 and Wnt/beta‐Catenin signaling pathways. Int J Mol Sci. 2017;18:2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang X, Zhu Y, Sun C, et al. Feedback activation of basic fibroblast growth factor signaling via the Wnt/beta‐Catenin pathway in skin fibroblasts. Front Pharmacol. 2017;8:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cenci S. Autophagy, a new determinant of plasma cell differentiation and antibody responses. Mol Immunol. 2014;62:289‐295. [DOI] [PubMed] [Google Scholar]
- 25. Matsuzawa‐Ishimoto Y, Hwang S, Cadwell K. Autophagy and inflammation. Annu Rev Immunol. 2017;36:55‐83. [DOI] [PubMed] [Google Scholar]
- 26. Vlada CA, Kim JS, Behrns KE. Autophagy: self‐preservation through cannibalism of proteins and organelles. Surgery. 2015;157:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hemelaar J, Lelyveld VS, Kessler BM, Ploegh HL. A single protease, Apg4B, is specific for the autophagy‐related ubiquitin‐like proteins GATE‐16, MAP1‐LC3, GABARAP, and Apg8L. J Biol Chem. 2003;278:51841‐51850. [DOI] [PubMed] [Google Scholar]
- 28. Tanida I, Sou YS, Ezaki J, Minematsu‐Ikeguchi N, Ueno T, Kominami E. HsAtg4B/HsApg4B/autophagin‐1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule‐associated protein light chain 3‐ and GABAA receptor‐associated protein‐phospholipid conjugates. J Biol Chem. 2004;279:36268‐36276. [DOI] [PubMed] [Google Scholar]
- 29. Brady NR, Hamacher‐Brady A, Yuan H, Gottlieb RA. The autophagic response to nutrient deprivation in the hl‐1 cardiac myocyte is modulated by Bcl‐2 and sarco/endoplasmic reticulum calcium stores. FEBS J. 2007;274:3184‐3197. [DOI] [PubMed] [Google Scholar]
- 30. Ma X, Liu H, Foyil SR, et al. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170‐3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Deretic V, Levine B. Autophagy balances inflammation in innate immunity. Autophagy. 2017;17:1‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yin ZY, Yin J, Huo YF, Yu J, Sheng LX, Dong YF. Rapamycin facilitates fracture healing through inducing cell autophagy and suppressing cell apoptosis in bone tissues. Eur Rev Med Pharmacol Sci. 2017;21:4989‐4998. [PubMed] [Google Scholar]
- 33. Ge RS, Hardy MP. Variation in the end products of androgen biosynthesis and metabolism during postnatal differentiation of rat Leydig cells. Endocrinology. 1998;139:3787‐3795. [DOI] [PubMed] [Google Scholar]
- 34. Ge RS, Hardy MP. Decreased cyclin A2 and increased cyclin G1 levels coincide with loss of proliferative capacity in rat Leydig cells during pubertal development. Endocrinology. 1997;138:3719‐3726. [DOI] [PubMed] [Google Scholar]
- 35. Payne AH, Wong K‐L, Vega MM. Differential effects of single and repeated administrations of gonadotropins on luteinizing hormone receptors and testosterone synthesis in two populations of Leydig cells. J Biol Chem. 1980;255:7118‐7122. [PubMed] [Google Scholar]
- 36. Guo X, Lian R, Guo Y, Liu Q, Ji Q, Chen J. bFGF and Activin A function to promote survival and proliferation of single iPS cells in conditioned half‐exchange mTeSR1 medium. Hum Cell. 2015;28:122‐132. [DOI] [PubMed] [Google Scholar]
- 37. Mitchell R, Childs A, Anderson R, et al. Do phthalates affect steroidogenesis by the human fetal testis? Exposure of human fetal testis xenografts to di‐n‐butyl phthalate. J Clin Endocrinol Metab. 2012;97:E341‐E348. [DOI] [PubMed] [Google Scholar]
- 38. Sawai H. Desipramine‐induced lysosomal vacuolization is independent of autophagy. Cell Biol Int. 2017;42:248‐253. [DOI] [PubMed] [Google Scholar]
- 39. Akiva I, Birgul Iyison N. MGAT1 is a novel transcriptional target of Wnt/beta‐catenin signaling pathway. BMC Cancer. 2018;18:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase‐3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664‐1668. [DOI] [PubMed] [Google Scholar]
- 41. Guo X, Yu R, Xu Y, et al. PAC1R agonist maxadilan enhances hADSC viability and neural differentiation potential. J Cell Mol Med. 2016;20:874‐890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Howell SJ, Radford JA, Smets EMA, Shalet SM. Fatigue, sexual function and mood following treatment for haematological malignancy: the impact of mild Leydig cell dysfunction. Br J Cancer. 2000;82:789‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bassil N, Alkaade S, Morley JE. The benefits and risks of testosterone replacement therapy: a review. Ther Clin Risk Manag. 2009;5:427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pfeil E, Dobs AS. Current and future testosterone delivery systems for treatment of the hypogonadal male. Expert Opin Drug Deliv. 2008;5:471‐481. [DOI] [PubMed] [Google Scholar]
- 45. Jin M, Huang Q, Zhao K, Shang P. Biological activities and potential health benefit effects of polysaccharides isolated from Lycium barbarum L. Int J Biol Macromol. 2013;54:16‐23. [DOI] [PubMed] [Google Scholar]
- 46. Yao K, Tan J, Gu W‐z, Ye P‐P, Wang K‐j. Reactive oxygen species mediates the apoptosis induced by transforming growth factor β 2 in human lens epithelial cells. Biochem Biophys Res Comm. 2007;354:278‐283. [DOI] [PubMed] [Google Scholar]
- 47. Nagata S. Apoptosis by death factor. Cell. 1997;88:355‐365. [DOI] [PubMed] [Google Scholar]
- 48. Qi B, Ji Q, Wen Y, et al. Lycium barbarum polysaccharides protect human lens epithelial cells against oxidative stress–induced apoptosis and senescence. PLoS ONE. 2014;9:e110275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu J, Ye J, Liu X, Han Y, Wang C. Protective effect of L‐carnitine against H2O2‐induced neurotoxicity in neuroblastoma (SH‐SY5Y) cells. Neurol Res. 2011;33:708‐716. [DOI] [PubMed] [Google Scholar]
- 50. Zhao Z, Yu R, Yang J, et al. Maxadilan prevents apoptosis in iPS cells and shows no effects on the pluripotent state or karyotype. PLoS ONE. 2012;7:e33953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chiou H‐L, Hsieh Y‐S, Hsieh M‐R, Chen T‐Y. HCV E2 may induce apoptosis of Huh‐7 cells via a mitochondrial‐related caspase pathway. Biochem Biophys Res Comm. 2006;345:453‐458. [DOI] [PubMed] [Google Scholar]
- 52. Sobral LM, Bufalino A, Lopes MA, Graner E, Salo T, Coletta RD. Myofibroblasts in the stroma of oral cancer promote tumorigenesis via secretion of activin A. Oral Oncol. 2011;47:840‐846. [DOI] [PubMed] [Google Scholar]
- 53. Yang M, Gao N, Zhao Y, Liu L‐X, Lu X‐J. Protective effect of Lycium barbarum polysaccharide on retinal ganglion cells in vitro. Int J Ophthalmol. 2011;4:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lian R‐L, Guo X‐L, Chen J‐S, Guo Y‐L, Zheng J‐F, Chen Y‐W. Effects of induced pluripotent stem cells‐derived conditioned medium on the proliferation and anti‐apoptosis of human adipose‐derived stem cells. Mol Cell Biochem. 2016;413:69. [DOI] [PubMed] [Google Scholar]
- 55. Drago D, Cossetti C, Iraci N, et al. The stem cell secretome and its role in brain repair. Biochimie. 2013;95:2271‐2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schimmer BP, Tsao J, Cordova M, Mostafavi S, Morris Q, Scheys JO. Contributions of steroidogenic factor 1 to the transcription landscape of Y1 mouse adrenocortical tumor cells. Mol Cell Endocrinol. 2011;336:85‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hu M‐C, Hsu N‐C, Pai C‐I, Wang C‐KL, Chung B‐c. Functions of the upstream and proximal steroidogenic factor 1 (SF‐1)‐binding sites in the CYP11A1 promoter in basal transcription and hormonal response. Mol Endocrinol. 2001;15:812‐818. [DOI] [PubMed] [Google Scholar]
- 58. Iwatani S, Shono A, Yoshida M, et al. Involvement of WNT signaling in the regulation of gestational age‐dependent umbilical cord‐derived mesenchymal stem cell proliferation. Stem Cells Int. 2017;2017:8749751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kuphal S, Bosserhoff AK. Phosphorylation of beta‐catenin results in lack of beta‐catenin signaling in melanoma. Int J Oncol. 2011;39:235‐243. [DOI] [PubMed] [Google Scholar]