

Abstract
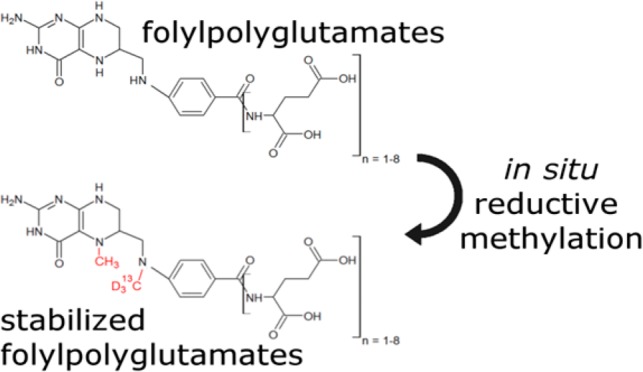
Folate cofactors play a key role in one-carbon metabolism. Analysis of individual folate species is hampered by the low chemical stability and high interconvertibility of folates, which can lead to severe experimental bias. Here, we present a complete workflow that employs simultaneous extraction and stabilization of folates by derivatization. We perform reductive methylation employing stable isotope labeled reagents to retain information on the position and redox state of one-carbon units as well as the redox state of the pteridine ring. The derivatives are analyzed by a targeted LC(HILIC)-MS/MS method without the need for deconjugation, thereby also preserving the glutamation state of folates. The presented method does not only improve analyte coverage and sensitivity as compared to other published methods, it also greatly simplifies sample handling and storage. Finally, we report differences in the response of bacterial and mammalian systems to pharmacological inhibition of dihydrofolate reductase.
One-carbon (C1) metabolism is a central metabolic pathway which distributes C1 units derived from C1 donors to several crucial cellular pathways, namely, purine synthesis,1 thymidine synthesis,2 and the S-adenosyl methionine cycle.3 Alterations in C1 metabolism have been reported in numerous diseases, including neural tube defects,4 cardiovascular disease,5 and cancer.6 In mammals, potential C1 donors include the nonessential amino acids serine and glycine, the essential amino acids histidine and tryptophan, as well as the degradation products of choline, betaine, dimethylglycine, and sarcosine. Folates are essential cofactors of C1 metabolism. They act as carriers which can temporarily bind C1 groups in different oxidation states. Folate species are thought to exist in large part as a stabilized, protein bound form in cells,7 and some are unstable in solution.8 However, quantifying intracellular folate pools requires quenching of enzymatic activity and extraction of folates from their native protein environment, which can result in a loss of analytes. Several LC-MS methods have been published attempting to quantify folate pools individually, most of them focusing on the more stable isoforms or employing isotope dilution approaches to account for losses during sample preparation and analysis9−11 (also see Supporting Information Table S1). Unfortunately, the isotope dilution approach suffers from the limited availability of isotopically labeled standards as well as a loss of sensitivity for less stable folates which undergo interconversion or degradation. Recently, Chen et al.12 employed sodium cyanoborodeuteride to reduce 5,10-methylene THF to isotopically labeled 5-methyl THF, thereby preventing spontaneous decomposition of 5,10-methylene THF to THF and formaldehyde. Using enzymatic deconjugation of polyglutamtes and combined with a second workflow, which includes a heat inactivation step to halt residual enzymatic activity, they were able to quantify five pools of one-carbon-carrying folates. However, the requirement of two extracts for the two separate workflows and residual pH dependent interconversion of 5-formyl THF to 10-formyl THF via 5,10-methenyl THF (Figure 1A) are potential problems of this method.
Figure 1.

Unstable folate species and stabilized derivatives. (A) pH dependent equilibrium of folates carrying one-carbon units at the formic acid oxidation level. (B) General structure of the folate backbone. (C) Dimethylated folate without isotopic labels can be employed as an internal standard. (D–K) Derivatives of 5-formyl THF, 10-formyl THF, 5-methyl THF, 5,10-methylene THF, 5,10-methenyl THF, THF, DHF, and folic acid.
In this study, we show how all naturally occurring folate species can be stabilized by specific derivatization of folates directly in the quenching solution and how the information on the oxidation state of both, the folate ring and C1 unit, can be encoded as isotopologue derivatives. The stable derivatives can be readily analyzed by LC-MS with the added benefit of improved sensitivity compared to nonderivatized folates. Moreover, the derivatization chemistry is compatible with metabolites up- and downstream of C1 metabolism, allowing a complete depiction of C1 metabolism in a single analysis.
Experimental Section
Derivatization of Folate Standards
Standards of 5-methyl THF, 5,10-methylene THF, 5,10-methenyl THF, THF, DHF, folic acid, and 5-formyl THF were purchased from Schircks Laboratories, Bauma, Switzerland. 10-Formyl THF was synthesized from 5-formyl THF as published by Stover et al.13
Folate standards were dissolved in ice cold 80% methanol, containing 30 mM NaCNBD3, 0.2% formaldehyde-13C, d2 (∼67 mM), and 0.1% acetic acid at a concentration of 0.1 mg/mL. Samples were incubated for 60 min on ice and stored at −20 °C until measured.
Cultivation of E. coli K12
A total of 50 mL of LB-Miller medium was inoculated from an overnight culture of E. coli K12 to an OD600 of 0.1 and grown at 37 °C in a 500 mL flask, shaking at 300 rpm. OD was measured at 0, 60, and 90 min and immediately before harvesting. After reaching an OD of 0.4, 10 μM (0.15 mL of a 1 mg/mL stock solution in methanol) trimethoprim was added to the treatment group (5 biological replicates), and the same volume of methanol was added to the control group (5 biological replicates). Thirty minutes after adding Trimethoprim, OD600 was measured (mean OD600 0.71 for control group and 0.63 for treated group), and cells were centrifuged at 4500g for 3 min. The supernatant was removed, and cells were immediately resuspended in 0.5 mL of ice cold 80% methanol, containing 30 mM NaCNBD3, 0.2% formaldehyde-13C, d2, and 0.1% acetic acid by pipetting. Samples were incubated on ice for 60 min, transferred to microcentrifuge tubes, and centrifuged at 16 000g for 1 min. Supernatants were transferred to a new tube, and the pellet was resuspended in 0.5 mL of ice cold 80% methanol and 0.1% acetic acid and incubated in a sonic bath for 15 min. After sonication, the suspension was centrifuged at 16,000 g for 3 min and the supernatant was combined with the previous supernatant. An aliquot of 50 μL was subjected to analysis (pre-concentrated sample). The remaining 950 μL was dried in a rotary evaporator and resuspended by sonication in 50 μL of 80% methanol and 0.1% acetic acid for 15 min. Samples were centrifuged at 16 000g for 3 min, and the supernatant was analyzed (post concentrated sample).
Cultivation of Mammalian Cells
HepG2 cells were acquired from ATCC (ATCC HB-8065) and grown in T75 flasks in RPMI1640 (Gibco, Thermo Fisher Scientific, Switzerland) containing 2 mM glutamine, 2 g/L glucose, and 10% FBS. One milligram of methotrexate hydrate (Sigma-Aldrich, Buchs SG, Switzerland) was dissolved in 10 μL of 1 M sodium hydroxide and diluted with phosphate buffered saline to 1 mL and sterile filtered (2.2 mM Stock solution). At 80% cell confluence, 20 μM methotrexate was added to the treatment group (180 μL stock solution per 20 mL medium, three biological replicates). A total of 180 μL of phosphate buffered saline containing 1% (v/v) 1 M sodium hydroxide was added to the control group. Sixteen hours post treatment, cells were harvested by trypsinization and centrifuged at 4 °C and 400g for 5 min. The supernatant was removed; the cell pellet was briefly washed with 1 mL of PBS and then resuspended in 0.5 mL of ice cold 80% methanol, containing 30 mM NaCNBD3, 0.2% formaldehyde-13C, d2, and 0.1% acetic acid by pipetting. Subsequent steps were carried out as described for E. coli.
LC-MS/MS Method
On the basis of the highly hydrophilic character of both polyglutamated folates and the derivatives, either ion pairing chromatography or hydrophilic interaction liquid chromatography (HILIC) are potential methods for separation of folylpolyglutamates before MS analysis. Since superior sensitivity of folate anaylsis in positive mode was reported before,14 we tested two HILIC phases, namely, the aminopropyl modified silica employed by Lu et al.15 (Luna-NH2, Phenomenex) and a polymer-based zwitterionic stationary phase (ZIC-pHILIC, Merck). The latter was chosen because of the lower noise levels and accompanying better signal-to-noise ratios achieved. As a total of 72 folate species plus several up- and downstream metabolites were measured, a scheduled MRM method was used. Liquid chromatography was carried out on an Agilent Technologies Infinity 1290 UHPLC system. The column was a SeQuant ZIC-pHILIC 150 × 2.1 mm plus guard column 20 × 2.1 mm (Merck, Darmstadt, Germany). The solvents were acetonitrile without any further additives (solvent A) and 20 mM (NH4)2CO3 in H2O, adjusted to pH = 9.2 with ammonium hydroxide solution (solvent B). The following gradient was run at a constant flow rate of 100 μL/min: 0 min, 30% B; 2 min, 30% B; 18 min, 70% B; 20 min, 95% B; 23 min, 95% B; followed by re-equilibration at 30% B for 7 min. The column compartment temperature was 30 °C. Mass spectrometry was performed on an ABSciex 5500 QTrap mass spectrometer in positive, scheduled MRM mode. The detection window was set to 60 s, and the target scan time was 1.5 s. Source parameters were curtain gas, 20 psi; collision gas, medium; ion spray voltage, 5500 V; temperature, 700 °C; ion source gas 1, 40 psi; ion source gas 2, 50 psi. The complete list of transitions and parameters can be found in Supporting Information Table S2. The employed software was Analyst Version 1.6.2 (ABSciex).
Data Analysis
Statistical analysis was performed in Microsoft Excel 2010 and R Studio 1.1.416. If not stated otherwise in the text, a homoscedastic two-tailed student’s t test with a significance threshold of p < 0.01 was used.
Results and Discussion
Preserving the C1 Oxidation State Information during Chemical Stabilization of Folates by Derivatization
The pteridine ring of C1 carrying folates is typically fully reduced to tetrahydrofolate (THF, Figure 1B), and C1 units are either attached to nitrogen 5 (N5), nitrogen 10 (N10), or both forming a bridge between the nitrogen moieties. Unsurprisingly, oxidation of the pteridine ring is one commonly observed degradation reaction of folates, especially when N5 is unsubstituted. Oxidation can be minimized by adding various antioxidants during extraction. More severely, migration or loss of the C1 group can occur in a pH dependent manner. Folates carrying C1 units at the oxidation level of formic acid exist in a pH dependent equilibrium in protein free form.16 Low pH (<4.5) strongly favors 5,10-methenyl THF, which in turn is unstable at higher pH and converts to either 5-formyl THF or 10-formyl THF (Figure 1A). To prevent interconversion of the three species, the free C1 binding site has to be chemically blocked. Both N5 and N10 are secondary amines, and the choice of potential derivatization reactions is limited by the aqueous environment. We have found that unoccupied N5 and N10 are rapidly and quantitatively derivatized by reductive methylation (Figure 1C), a method also used for isotopic labeling17 and stabilization of proteins.18 During derivatization, 5-formyl THF gets methylated at N10, yielding the 5-formyl, 10-methyl THF (Figure 1D), and 10-formyl THF yields the N5-methyl, N10-formyl derivative (Figure 1E). These two structural isomers can be separated by chromatography and can additionally be discriminated by MS2 (Figure S1). Employing heavy isotope labeled reagents allows discrimination between native and synthetic C1 groups of dimethylated species, so derivatized 5-methyl THF (Figure 1F) can be distinguished from derivatized THF (Figure 1G) by mass spectrometry. Furthermore, since bridged species are reduced by sodium cyanoborodeuteride, the initial oxidation state of the native C1 unit is encoded in the isotopic composition of the derivative, with lower oxidation states incorporating more deuterium (Figure 1H and I). Finally, the pteridine ring of folic acid is also readily reduced by sodium cyanoborodeuteride and subsequently methylated at N5. Dependent on the initial oxidation state of the pteridine ring, either one (Figure 1J) or two deuteriums (Figure 1K) are introduced in the pteridine ring.
Taken together, derivatization results in three structurally different and chemically stable analytes, which greatly simplifies analysis and sample handling. Six of the initial folate species are encoded as isotopologues (Figure S2), and a seventh isotopologue can easily be synthesized with unlabeled reagents, providing an isotopic standard (Figure 1C) for derivatives of 5-methyl THF, 5,10-methylene THF, and 5,10-methenyl THF, DHF, THF, and folic acid.
Completeness, Selectivity, and Specificity
Completeness of derivatization was assessed by running LC-MS analysis in product ion scan mode (including m/z range of unfragmented parent ions) for underivatized, single methylated, doubly methylated, and triple (i.e., over-) methylated compounds. The sum of signals for underivatized folates and side products was below 4% of the expected product for all compounds. Among the side products, triple methylated species were the most abundant (Figure S3).
Due to the limited resolution of quadrupole technology, the minimal mass difference of 1 Da between some parent and product masses is a potential source of experimental error. We therefore carefully examined potential cross-talk between channels at different quadrupole resolution settings using separate standards. At unit quadrupole resolution and better, no cross-talk between adjacent channels exceeding the expected effect from natural isotope abundances was observed (Figure 2A). This experiment also shows the high specificity of the derivatization reaction, as only the expected derivatization product was observed for each folate. Since the enrichment of heavy isotopes in labeling reagents is not complete (96–98% according to vendor), also M-1 and M-2 species were considered. The true enrichment of heavy isotopes is slightly higher as specified, as the measured cross-talk was slightly lower for M-1 and M-2 species than the calculated threshold. Accordingly, experimentally determined values were used for subsequent deconvolution of signals. Deconvolution was performed by solving linear equation systems using the solve function of the R software suite.
Figure 2.

Method performance overview. (A) Concentrations of individual isotopologues are calculated by deconvolution. Crosstalk between adjacent channels caused by natural 13C isotopic abundances and imperfect labeling of reagents. Blue: Calculated values from theoretic isotopic distribution and specified labeling extent of reagents. Green: Experimental values from pure standards. (B) Stability of derivatized analytes over time at 4 °C. (C) Stability of derivatized folates during vacuum concentration. (D) Matrix effects of E. coli lysate (purple) and HepG2 lysate (green) at two different spiked in standard concentrations (conc) compared to buffer (blue). *p < 0.01.
Stability of Derivatives
After ensuring complete derivatization, we next assessed the stability of the derivatized analytes and compared it to native folates. Underivatized folates were dissolved in 50:50 methanol/water, 0.1% ascorbic acid, and 20 mM ammonium acetate, at pH = 6.2 (as published by Lu et al.).15 After mixing, folates were incubated at 4 °C in the autosampler and injected repeatedly over a time span of 96 h. As expected, the addition of ascorbic acid effectively slowed oxidation of THF and to some extent also DHF. However, at the near neutral pH, 5,10-methenyl THF degraded with a t1/2 of <3 h at 4 °C to either 5- or 10-formyl THF (undistinguishable by the method of Lu et al.). Moreover, it was highly unstable under the employed chromatographic conditions (pH 9.2, 30 °C), and the signal at the retention time of 10 min was only 11% (AUC) of that obtained by flow injection. Another unstable folate is the 5,10-methylene folate, which was reported to dissociate rapidly to formaldehyde and THF in the absence of excess formaldehyde at nonalkaline pH.12,19 The t1/2 of this compound was below 30 min at 4 °C.
In contrast, the derivatized folates showed excellent stability under both storage (pH = 4.5) and chromatographic conditions (pH = 9.2), as can be seen in Figure 2B. To avoid any effects caused by further sample preparation, no steps to remove excess sodium cyanoborodeuteride were carried out. The resulting hydrogen bubble formation in the sample vial is the reason for the fluctuations at the beginning of the stability experiment. The stability experiment was carried out at 4 °C to prevent evaporation of solvent after multiple injections from the same sample vial. However, the derivatives also show good stability at room temperature. This also enables preconcentration of extracts, e.g., by using a rotary vacuum concentrator while maintaining the ratios of individual folate species. An example of folate species in an E. coli extract pre- and postconcentration is shown in Figure 2C.
Sensitivity of the Method and Matrix Effects
Compared to the methods by Lu et al.15 and Haandel et al.,20 the sensitivity of the method was improved for all monoglutamated folates, with the limit of detection (LOD, signal-to-noise > 5) being <250 fg on column for all species. Linearity of quantification was excellent for almost 4 orders of magnitude with R2 ≥ 0.99 for all analytes in the calibration range (250 fg to 1 μg) for both unweighted and double logarithmically weighted regression (Supporting Information Figure S4).
As matrix effects can play an important role in MS analysis, we tested for different ionization efficiencies in different backgrounds by spiking two concentrations of the light labeled dimethyl THF (Figure 1C) into extraction buffer, E. coli extract, and vacuum concentrated HepG2 cell extract, both derivatized with heavy isotope labels. The observed area under the curve is compared in Figure 2D. No significant matrix effect was observed in the vacuum concentrated HepG2 cell extract. However, a strong matrix effect was observed in unconcentrated E. coli lysate, with signal intensities amounting to only 45% of signal intensities in the spiked extraction buffer.
5-Formimino THF
A potential source of C1 units not covered so far is the histidine degradation product 5-formimino THF. Unfortunately, no standard for 5-formimino THF is commercially available. We therefore screened for the predicted derivatization product in E. coli extracts using a product ion scan method and were able to identify two compounds corresponding to the triple and 4-fold glutamated species. The retention times were consistent with other triple and 4-fold glutamated folates, and the major MS2 fragment fits the predicted heavy labeled, deglutamated 5-formimino THF. On the basis of these data, we included transitions for all 5-formimino THF glutamate species in the method.
Compatibility with Metabolites Up- and Downstream of C1 Pathway
Primary and secondary amines undergo reductive methylation under the employed derivatization conditions. Hence, amino acids such as glycine, serine, and methionine as well as their derivatives, e.g., S -adenosylmethionine (SAM), are dimethylated when stabilizing folate species. However, this does not prevent their quantification, albeit so far only qualitative measurements for these compounds have been conducted. No derivatization was observed for nucleotides and nucleotide building blocks included in the method. An overview of metabolites covered by the method is shown in Figure 3, and the complete set of transitions is listed in Supporting Information Table S2.
Figure 3.

Schematic of one-carbon cycle showing the connection between folate metabolism and methionine cycle (Scheme adapted from ref (21). Copyright Elsevier 2013). Color coding: purple, C1 donors; green, folates; blue, products of C1 metabolism and downstream pathways; white, not covered by method. Abbreviations: CTH, cystathionine γ-lyase; CBS, cystathionine β-synthase; DHF, dihydrofolate; FA, folic acid; Hcy, homocysteine; MAT, methionine adenosyltransferase; MS, methionine synthase; MTHFR, methylenetetrahydrofolate reductase; 5-MTHF, 5-methyl-tetrahydrofolate; MT, methyltransferase; SAH, S-adenosylhomocysteine; SAHH, SAH hydrolase; SAM, S-adenosylmethionine; SHMT, serine hydroxymethyltransferase; THF, tetrahydrofolate; B6, vitamin B6; B12, vitamin B12.
Application to Biological Samples
We first reproduced the trimethoprim inhibition experiment carried out by Lu et al.15 with the major difference that E. coli K12 was grown on complex (LB-Miller) instead of minimal medium. We were able to reproduce the distribution of folylpolyglutamates with n = 3 being most abundant in control cells, followed by n = 4 and n = 5 (Figures 4 and 5A). For this purpose, all MRM peak areas of folate species with a given polyglutamation number but regardless of their one carbon load were summed up and normalized. It has to be noted that standards with high polyglutamation numbers are not commercially available, and the transitions for n = 7 and n = 8 are based on predicted retention times and calculated m/z for doubly charged parents. The folate profile of the control group grown on complex medium differed markedly from the profile reported for minimal medium. While the fully reduced 5-methyl THF was reported to be the dominant folate species on minimal medium, 5,10-methenyl THF was the most abundant folate species in E. coli grown in complex medium, followed by 10-formyl THF. With the capability of our method to distinguish between 5- and 10-formyl THF, we can also conclude that polyglutamated 5-formyl THF is a minor species under the employed conditions, with only polyglutamated folic acid being lower (supplementary Figure S5).
Figure 4.

MRM Chromatogram of derivatized folate extract from E. coli K12 grown on complex medium.
Figure 5.

Effect of DHFR inhibition in bacterial and mammalian systems. (A) Distribution of polyglutamation in control and trimethoprim treated E. coli K12. Peak areas of all folate species with a given polyglutamation number were summed and normalized. (B) Relative changes (log 10-fold) of reduced folates upon trimethoprim treatment of E. coli K12. (C) Selected C1 targets in E. coli. (D) Distribution of polyglutamation in control and methotrexate treated HepG2 cells. (E) Relative changes (log 10-fold) of reduced folates upon methotrexate treatment of HepG2 cells. (F) Selected C1 targets in HepG2 cells. *p < 0.01.
Trimethoprim is an inhibitor of dihydrofolate reductase (DHFR), the enzyme that reduces both folic acid (via DHF) and DHF to THF. DHFR activity is not only necessary to recycle DHF, which is produced from 5,10-methylene THF during thymidine synthesis, but also for reduction of folates de novo synthesized from dihydropteroate. Accordingly, upon treatment of E. coli K12 with trimethoprim, the growth of the treated group slowed, and most folate species carrying one-carbon units decreased significantly. However, also the overall distribution of folate polyglutamation levels changed severely, with an increase of folates with both a low (n = 1–2) and high glutamation (n = 4–6) number, while folylpolyglutamate with n = 3 was strongly reduced (Figure 4A). This is likely an effect of the inhibition of folylpolygamma glutamate synthase by accumulating DHF, as described by Kwon et al. and termed the “domino effect.”22 In accordance with the results of Kwon et al., we observed an increase of reduced folates with a low glutamation number (n = 1–2) after prolonged treatment (30 min) with trimethoprim. Interestingly, only the THF species with three glutamates was reduced significantly in our hands, while THF species with high glutamation numbers (n = 5–6) were increased in trimethoprim treated samples. As our method also allows quantification of 5,10-methenyl THF, 5-formyl THF, and 10-formyl THF individually, we are able to report that 5,10-methenyl THF species with glutamation numbers n = 1–2 are upregulated, while those with glutamation numbers n = 3–5 are significantly decreased (Figure 4B) upon trimethoprim treatment. The most severe decrease in both absolute and relative terms was observed for 10-formyl THF, which was found to be significantly downregulated with glutamation numbers n = 3–6.
The massive changes in folate distribution caused by DHFR inhibition are also reflected in substrates and products of C1 transfer reactions. Thymidine (TMP) was significantly downregulated in treated cells, while the purine base biosynthetic intermediate AICAR was significantly elevated (Figure 4C, p < 0.001). Interestingly, neither SAM nor S-adenosyhomocysteine (SAH) were altered, indicating a precedence of the methylation system over de novo nucleotide synthesis under the employed conditions.
Methotrexate Treatment of HepG2 Hepatoma Cells
While trimethoprim is a potent inhibitor of bacterial DHFR and therefore used as an antibiotic, inhibitors of mammalian DHFR are widely employed in treatment of cancer and autoimmune disorders.23 To investigate differences in the response of bacterial and mammalian systems to DHFR inhibition, we employed the antifolate methotrexate to inhibit DHFR in the human hepatocellular carcinoma cell line HepG2. In contrast to the bacterial system, no significant increase of the oxidized pteridines was observed, with the exception of DHF-Glu2 (data not shown). As expected, most of the reduced folates were significantly lowered (Figure 4D). After 16 h of treatment with methotrexate, especially folates with higher polyglutamation number were significantly decreased (Figure 4E), and growth was completely halted. The level of TMP was not significantly altered despite the depletion of its biosynthetic precursor 5,10-methylen THF, which might be an effect of the observed complete growth arrest. Interestingly, inosine monophosphate (IMP) actually increased significantly alongside AICAR, a tendency also observed in E. coli, albeit not passing the significance threshold. One possible explanation for this is an increase in the nucleotide salvage pathway24 activity. Finally, both SAM (p < 0.02) and SAH were reduced significantly. The SAM/SAH ratio was lower by trend in the methotrexate sample (although not significant, p = 0.075), which is in line with 5-methyl-THF depletion. However, the fall of both SAM and SAH can be either explained by a draining of the entire methionine pool (e.g., toward cysteine and glutathione synthesis) or, more likely, a depletion of the adenosyl pool, as ATP is also required for SAM biosynthesis.
Conclusion
Traditionally, the first step of folate analytics used to be enzymatic deconjugation, i.e., the trimming of the polyglutamate tail to a single glutamate.16 While this approach requires a less sensitive analytical method since it pools several glutamation states into one, it also sacrifices valuable information; for example, the “domino effect” reported by Kwon et al.22 and reproduced here is not observable using this approach. Another problem is the lengthy enzymatic deglutamation step, which promotes interconversion and potential loss of unstable folate species. Especially pteridine ring oxidation and subsequent bond cleavage between the pteridine ring and para-aminobenzoyl glutamate has been reported as one major pathway of folate degradation.25 Finally, the source of folate conjugase, which is in most cases a crude preparation (i.e., charcoal treated) of rat plasma, is a potential source of sample contamination given the low hydrophobicity of even monoglutamated folates. With the availability of highly sensitive LC-MS methods, the compromises made by deconjugation have become unnecessary (Figure S6). The first method published avoiding deconjugation was published already 13 years ago by Garratt et al., who used an ion pairing chromatography setup to separate the highly hydrophilic folylpolyglutamates. In 2007, Lu et al. published the first method analyzing folylpolyglutamates on an HILIC column. However, even methods avoiding deconjugation suffer from the low chemical stability of several folate species, making their analysis a highly challenging task.
In this study we demonstrate how in depth folate analysis is greatly simplified by stabilizing folates by derivatization directly in the quenching solution. In addition to the benefits of easier sample handling, the sensitivity of the method is increased by more than 2 orders of magnitude for the most unstable folates compared to nonderivatized folate analytics. Notably, the sensitivity improvement of folates considered more stable under the respective conditions was lower (factor 4 for 5-methyl THF and 20 for 5-formyl THF, respectively) than for folates highly unstable at elevated pH (5,10-methenyl THF, factor 400 compared to Lu et al.). In contrast, Haandel et al. reported LODs for 5,10-methenyl THF and 5-methyl THF (900 fg on column) for their method using acidic extraction and LC conditions. However, under these conditions, they did not provide any LODs for the acid labile analytes 5- and 10-formyl THF. Indeed, they used TCA acidified samples to quantify the entirety of folates carrying C1 units at the formic acid oxidation state as 5,10-methenyl THF. These prior reports thus underscore the impracticality of assessing all underivatized folates in one method and underpin the benefits of chemically stabilizing folates.
The method described here also allows discrimination of the structural isomers 5-formyl THF and 10-formyl THF. The two analytes are baseline separated by chromatography and can additionally be distinguished by their fragmentation pattern26 (Figure S1). For a complete depiction of C1 metabolism, we also included C1 donors and products of C1 metabolism in the method. While some of the donors and products are also derivatized, this does not hamper their analysis; in one special case, it is even beneficial. The choline degradation products dimethylglycine, sarcosine, and glycine are converted into three isotopologues carrying different numbers of heavy methyl groups allowing a direct quantitative comparison. Another advantage of our method is that folates in the sample will be labeled with heavy isotopes during derivatization. This allows the use of unlabeled standards derivatized with unlabeled reagents as an isotopic standard rather than costly isotope labeled folates.
Conclusively, we have demonstrated how comprehensive folate analysis can be greatly simplified by derivatization using heavy isotope labeled reagents. This approach allows for the first time to analyze all cellular folate species in one method with very high sensitivity. Six out of a total of 9 (including 5-formimino THF) folate species measured are encoded as isotopologues, and the high stability of the derivatives allows prolonged sample storage and sample preparation steps such as vacuum centrifugation. We hope that this method will catalyze a surge of C1 metabolism research, providing a better understanding of how this central biosynthetic pathway is regulated under varying conditions and diseases. Considering that C1 metabolism is still a major target of cancer drugs six decades after the initial steps in chemotherapy,23,27 in depth analysis of folate profiles of varying cancer types and their alterations by drugs is a paramount objective. The greatly simplified sample handling will especially be beneficial in a clinical setting, where strictly time controlled sample preparation is often infeasible. Beyond that, determination of folates in food and feed can also be improved using this approach.
Acknowledgments
This work was supported by the Austrian Science Fund (FWF) Project P 26074 and the FWF Erwin Schrödinger Fellowship J-3983.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b00650.
Figures S1–S7 and Tables S1 and S2 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hartman S. C.; Buchanan J. M. J. Biol. Chem. 1959, 234, 1812–1816. [PubMed] [Google Scholar]
- Carreras C. W.; Santi D. V. Annu. Rev. Biochem. 1995, 64, 721–762. 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- Selhub J. Annu. Rev. Nutr. 1999, 19, 217–246. 10.1146/annurev.nutr.19.1.217. [DOI] [PubMed] [Google Scholar]
- Narisawa A.; Komatsuzaki S.; Kikuchi A.; Niihori T.; Aoki Y.; Fujiwara K.; Tanemura M.; Hata A.; Suzuki Y.; Relton C. L.; Grinham J.; Leung K.-Y.; Partridge D.; Robinson A.; Stone V.; Gustavsson P.; Stanier P.; Copp A. J.; Greene N. D. E.; Tominaga T.; Matsubara Y.; Kure S. Hum. Mol. Genet. 2012, 21, 1496–1503. 10.1093/hmg/ddr585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Huang T.; Zheng Y.; Muka T.; Troup J.; Hu F. B. J. Am. Heart Assoc. 2016, 5, 1–18. 10.1161/JAHA.116.003768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale J. W. Nat. Rev. Cancer 2013, 13, 572–583. 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirch V.; Strong W. B. Arch. Biochem. Biophys. 1989, 269, 371–380. 10.1016/0003-9861(89)90120-3. [DOI] [PubMed] [Google Scholar]
- Stover P. J.; Field M. S. Adv. Nutr. 2011, 2, 325–331. 10.3945/an.111.000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiekens F.; Van Daele J.; Blancquaert D.; Van Der Straeten D.; Lambert W. E.; Stove C. P. J. Chromatogr. A 2015, 1398, 20–28. 10.1016/j.chroma.2015.04.025. [DOI] [PubMed] [Google Scholar]
- Garratt L. C.; Ortori C. A.; Tucker G. A.; Sablitzky F.; Bennett M. J.; Barrett D. A. Rapid Commun. Mass Spectrom. 2005, 19, 2390–2398. 10.1002/rcm.2074. [DOI] [PubMed] [Google Scholar]
- Ringling C.; Rychlik M. Anal. Bioanal. Chem. 2017, 409, 1815–1825. 10.1007/s00216-016-0126-4. [DOI] [PubMed] [Google Scholar]
- Chen L.; Ducker G. S.; Lu W.; Teng X.; Rabinowitz J. D. Anal. Bioanal. Chem. 2017, 409, 5955–5964. 10.1007/s00216-017-0514-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover P.; Schirch V. Anal. Biochem. 1992, 202, 82–88. 10.1016/0003-2697(92)90210-X. [DOI] [PubMed] [Google Scholar]
- Freisleben A.; Schieberle P.; Rychlik M. Anal. Bioanal. Chem. 2003, 376, 149–156. 10.1007/s00216-003-1844-y. [DOI] [PubMed] [Google Scholar]
- Lu W.; Kwon Y. K.; Rabinowitz J. D. J. Am. Soc. Mass Spectrom. 2007, 18, 898–909. 10.1016/j.jasms.2007.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagerstad M.; Jastrebova J. J. Agric. Food Chem. 2013, 61, 9758–9768. 10.1021/jf4028427. [DOI] [PubMed] [Google Scholar]
- Hsu J. L.; Huang S. Y.; Chow N. H.; Chen S. H. Anal. Chem. 2003, 75, 6843–6852. 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- Schittmayer M.; Fritz K.; Liesinger L.; Griss J.; Birner-Gruenberger R. J. Proteome Res. 2016, 15, 1222–1229. 10.1021/acs.jproteome.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strandler H. S.; Patring J.; Jägerstad M.; Jastrebova J. J. Agric. Food Chem. 2015, 63, 2367–2377. 10.1021/jf504987n. [DOI] [PubMed] [Google Scholar]
- Haandel L.; Becker M. L.; Williams T.; Stobaugh J.; Leeder J. S. Rapid Commun. Mass Spectrom. 2012, 26, 1617–1630. 10.1002/rcm.6268. [DOI] [PubMed] [Google Scholar]
- Iacobazzi V.; Castegna A.; Infantino V.; Andria G. Mol. Genet. Metab. 2013, 110, 25–34. 10.1016/j.ymgme.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Kwon Y. K.; Lu W.; Melamud E.; Khanam N.; Bognar A.; Rabinowitz J. D. Nat. Chem. Biol. 2008, 4, 602–608. 10.1038/nchembio.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonen N.; Assaraf Y. G. Drug Resist. Updates 2012, 15, 183–210. 10.1016/j.drup.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Nyhan W. L. Mol. Genet. Metab. 2005, 86, 25–33. 10.1016/j.ymgme.2005.07.027. [DOI] [PubMed] [Google Scholar]
- Reed L. S.; Archer M. C. J. Agric. Food Chem. 1980, 28, 801–805. 10.1021/jf60230a044. [DOI] [Google Scholar]
- Ringling C.; Rychlik M. Eur. Food Res. Technol. 2013, 236, 17–28. 10.1007/s00217-012-1849-x. [DOI] [Google Scholar]
- Vander Heiden M. G.; DeBerardinis R. J. Cell 2017, 168, 657–669. 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.