

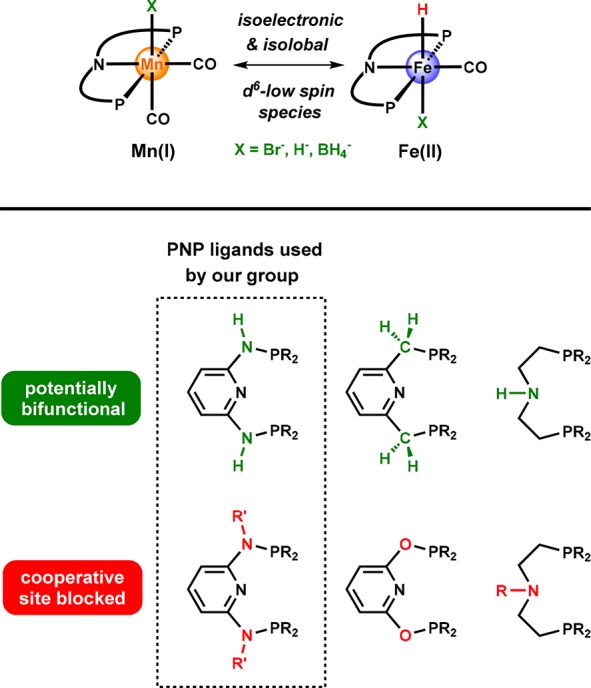
Conspectus

Sustainable processes that utilize nontoxic, readily available, and inexpensive starting materials for organic synthesis constitute a major objective in modern chemical research. In this context, it is highly important to perform reactions under catalytic conditions and to replace precious metal catalysts by earth-abundant nonprecious metal catalysts. In particular, iron and manganese are promising candidates, as these are among the most abundant metals in the earth’s crust, are inexpensive, and exhibit a low environmental impact. As far as chemical processes are concerned, hydrogenations and acceptorless alcohol dehydrogenation (AAD), sometimes in conjunction with hydrogen autotransfer reactions, are becoming important areas of research. While the first is a very important synthetic process representing a highly atom-efficient and clean methodology, AAD is an oxidant-free, environmentally benign reaction where carbonyl compounds together with dihydrogen as a valuable product and/or reactant (autotransfer) and water are formed. Carbonyl compounds, typically generated in situ, can be converted into other useful organic materials such as amines, imines, or heterocycles.
In 2016 several groups, including ours, discovered for the first time the potential of hydride biscarbonyl Mn(I) complexes bearing strongly bound PNP pincer ligands or related tridentate ligands as highly effective and versatile catalysts for hydrogenation, transfer hydrogenation, and dehydrogenation reactions. These complexes are isoelectronic analogues of the respective hydride monocarbonyl Fe(II) PNP compounds and display similar reactivities but also quite divergent behavior depending on the coligands. Moreover, manganese compounds show improved long-term stability and high robustness toward harsh reaction conditions. In light of these recent achievements, this Account contrasts Mn(I) and Fe(II) PNP pincer catalysts, highlighting specific features that are connected to particular structural and electronic properties. It also addresses opportunities and restrictions in their catalytic applications. Apart from classical hydrogenations, it also covers the most recent developments of these catalysts for AAD resulting in the synthesis of complex organic molecules such as heterocycles via multicomponent reactions. The ambivalent hydrogen-based redox chemistry provides access to a variety of synthetically valuable reductive and oxidative coupling reactions. Hence, these catalysts cover a broad scope of catalytic applications and exhibit activities and productivities that are becoming competitive with those of well-established precious metal catalysts. The knowledge about the nature and characteristics of active Mn(I)- and Fe(II)-based systems paves the way for conceptually and mechanistically well-founded research, which might lead to further developments and the discovery of novel catalysts extending the current scope and limitations of reactivity. It underlines that base metal catalysts are beginning to challenge precious metal catalysts and contributes to the further advancement of waste-free sustainable base metal catalysis.
Introduction
The development of sustainable, efficient, and selective syntheses is one of the fundamental research goals in modern chemistry. In this context, it is important to perform reactions under catalytic conditions and to replace precious metal catalysts by earth-abundant nonprecious metal catalysts.1 As far as chemical processes are concerned, hydrogenations and acceptorless alcohol dehydrogenation, sometimes in conjunction with hydrogen autotransfer reactions, are becoming important areas of research.2,3 In particular, iron and manganese constitute promising candidates, as these are among the most abundant metals in the earth’s crust, are inexpensive, and exhibit a low environmental impact. While iron hydrogenation and dehydrogenation catalysts have been the subject of intense investigation over the past decade,4−8 low-valent Mn(I) complexes just recently appeared as new but very powerful players in this field.9−12 Concerning iron, the development of novel catalysts was largely inspired by concepts known from its well-established ruthenium congeners, which are particularly effective for hydrogenative reductions and oxidations of polar substrates. After the first reports on Fe(II)-catalyzed hydrogenations, we even stated “Iron, the New Ruthenium”.13 However, in light of recent achievements accomplished by isoelectronic Mn(I) catalysts, the question arises whether this statement is still valid or has to be reconsidered, since these novel systems appear to show even more similarities to traditional ruthenium than iron chemistry (diagonal relationship Mn–Ru). Nevertheless, the development of base metal catalysts that can even compete with their “noble” analogues remains a challenging task since specific properties of first-row transition metals (e.g., oxidation states, spin states, ionic radii, redox potentials) are fundamentally different and new strategies and concepts have to be developed in order to circumvent unfavorable phenomena in this context. Consequently, the synthesis of isolated and structurally well-defined complexes combined with a fundamental understanding of reaction mechanisms appears to be a primary objective for the rational development of novel catalytic systems.14,15
In this Account, we describe well-defined Fe(II)- and Mn(I)-based catalysts featuring PNP pincer ligands based on 2,6-diaminopyridine that have been developed by our group (Figure 1).16−18 We highlight common and characteristic features that influence the activity and selectivity of these systems and compare them with related PNP pincer systems. While Fe(II) and Mn(I) exhibit similar reactivities in many cases, these isoelectronic metal systems also reveal remarkable divergent behavior that will be emphasized in this contribution.
Figure 1.

Well-defined Fe(II)- and Mn(I)-based catalysts developed by our group (R = iPr).
PNP Pincer Ligands: A Versatile Platform for Iron and Manganese Chemistry
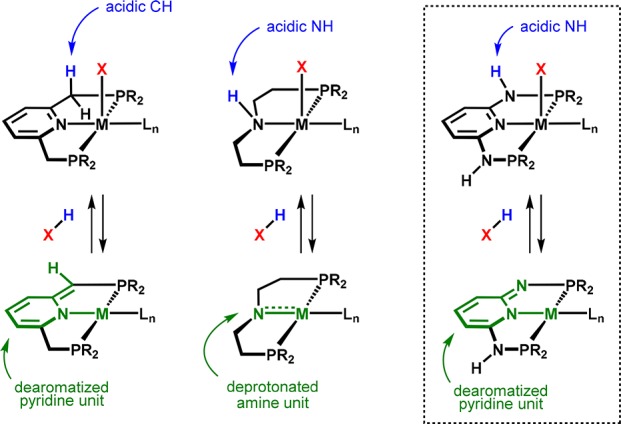
PNP pincer complexes proved to be particularly effective and versatile for hydrogenative reduction and dehydrogenative oxidation reactions of polar substrates,19 where in many cases metal–ligand cooperation plays an important role (Figure 2).20 Pincer complexes with aliphatic ligand backbones possess a secondary amine functionality adjacent to the metal center that is involved through an interplay between the amine/amide forms in H–H and H–heteroatom bond activation and formation steps. Lutidine-derived PNP pincers, on the other hand, are able to undergo reversible deprotonation of a methylene linker, which is accompanied by dearomatization of the heteroaromatic backbone (Figure 3).
Figure 2.
Structural motifs of isoelectronic d6 low-spin Fe(II) and Mn(I) complexes featuring PNP pincer ligands.
Figure 3.
Metal–ligand cooperation in pyridine-based and aliphatic PNP pincer complexes.
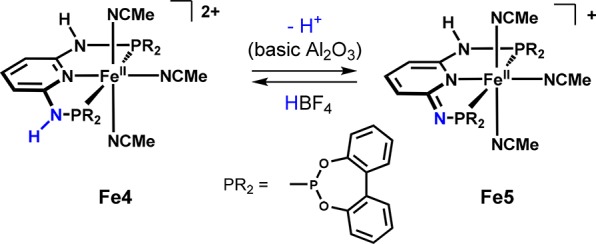
In 2006, we started our research on transition metal PNP pincer complexes by focusing on a specific type of ligand in which the phosphorus donors are connected via NH or NR (R = alkyl, aryl) linkers to a central pyridine unit.21 These ligands, derived from the 2,6-diaminopyridine scaffold or derivatives thereof, are synthesized in a simple and modular fashion that allows access to a broad variety of structural, electronic, and stereochemical modifications. They show excellent chelating ability due to the strong donor groups, forming stable, rigid, and symmetric five-membered metallacycles, which also minimizes the occurrence of different isomers and facilitates spectroscopic characterization. These PNP pincer ligands thus provide an excellent platform to explore base metal chemistry and catalysis, since they constitute an appropriate environment compensating for the intrinsically lower stability of 3d metals. Concerning metal–ligand cooperation, initial investigations revealed that a reversible aromatization/dearomatization reaction is feasible with this type of ligand. For example, the dicationic Fe(II) PNP complex Fe4 affords the deprotonated complex Fe5 in the presence of base (Scheme 1).19,21
Scheme 1. Reversible Deprotonation of an NH Linker in Fe4.
Consequently, it appeared promising to explore the effect of NH and NR linkers in the backbone of pyridine-based PNP ligands in catalytic hydrogenations. However, in contrast to PNP ligands featuring CH2 and O linkers, an advantage of PNP pincers based on 2,6-diaminopyridine is that cooperativity can be either blocked or activated by introducing substituents on the nitrogen linker. Such modifications contribute to the investigation of metal–ligand cooperation but also pave the way for the discovery of new reactivities and concepts. Accordingly, PNP systems based on aminopyridines benefit from both concepts mentioned above.
Hydrogenation of Polar Double Bonds
The first results relevant for the development of our catalysts were obtained by demonstrating that our pincer complexes are capable of activating molecular hydrogen in a bifunctional manner (Scheme 2).22 The biscarbonyl complex trans-[Fe(PNP-iPr)(CO)2Cl]+ (Fe6) reacts with Zn as a reducing agent under a dihydrogen atmosphere to give the Fe(II) hydride complex cis-[Fe(PNP-iPr)(CO)2H]+ (Fe7). The crucial step in this reaction is the reduction of the acidic NH protons of the PNP ligand to afford H2 and the coordinatively unsaturated intermediate [Fe(PNPH-iPr)(CO)2] bearing a dearomatized/deprotonated pyridine moiety. This species is able to bind and heterolytically cleave H2 to give Fe7. However, this hydride complex is catalytically inactive, indicating that bifunctionality is not the only requirement for an active system (vide infra).
Scheme 2. Heterolytic Cleavage of H2.

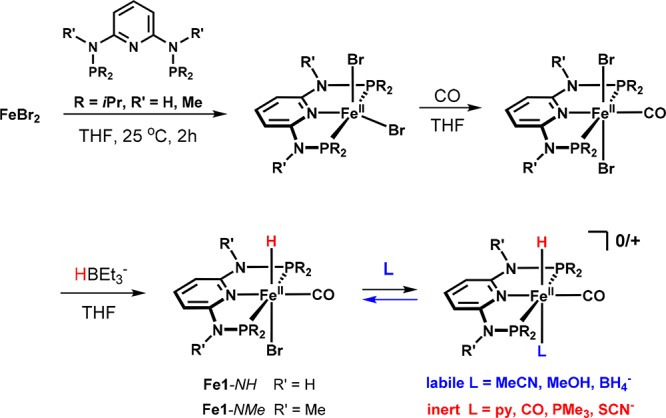
Inspired by the seminal work of Milstein and co-workers,23,24 we focused on [Fe(PNP-iPr)(H)(CO)(Br)] (Fe1-NH), which incorporates a bromide instead of a second carbonyl ligand trans to the hydride. In contrast to the cationic biscarbonyl iron hydride complex tested before, Fe1-NH was an effective hydrogenation catalyst capable of reducing ketones and aldehydes under mild conditions (Scheme 3).25 A structural view of Fe1-NH is depicted in Figure 4.
Scheme 3. Iron-Catalyzed Hydrogenation of Ketones and Aldehydes.

Figure 4.
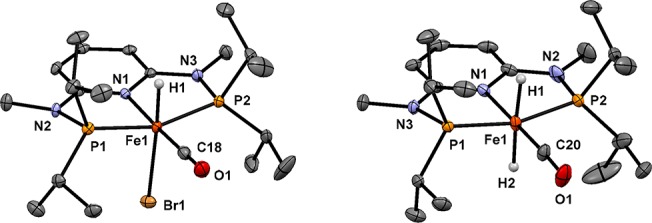
Structural views of Fe1-NH and trans-Fe3-NMe.
Although the catalytic performance was not enhanced in comparison to related catalysts,23,24 this system paved the way for systematic variations that provided valuable information on the structural requirements for obtaining a catalytically active complex. It turned out that the bromide trans to the hydride is substitutionally labile and is readily replaced by anionic or neutral coligands, resulting in a series of different hydride complexes (Scheme 4).
Scheme 4. Preparation of Iron Hydride Complexes Featuring Labile and Inert Coligands.
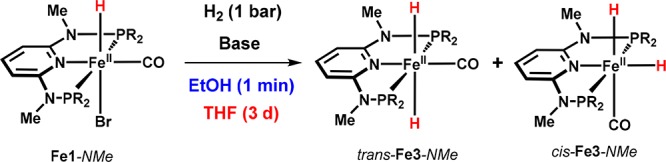
Treatment of Fe1-NMe with H2 in the presence of a strong base readily yields a mixture of trans- and cis-Fe3-NMe (Scheme 5). These complexes could even be structurally characterized by X-ray crystallography (Figure 4). In the case of Fe1-NH featuring acidic NH linkers, the analogous dihydrides are also formed in solution, but they could not be isolated in the solid state.
Scheme 5. Synthesis of cis- and trans-Fe3-NMe.
Initial screening of these complexes in the hydrogenation of acetophenone revealed that only complexes with labile coligands are catalytically active, indicating that the formation of a vacant coordination site is required. Moreover, we investigated the role of metal–ligand cooperation by modifying the substituents on the nitrogen linkers. Accordingly, only Fe1-NH featuring acidic NH groups could hydrogenate ketones, while Fe1-NMe bearing NMe linkers was catalytically inactive.
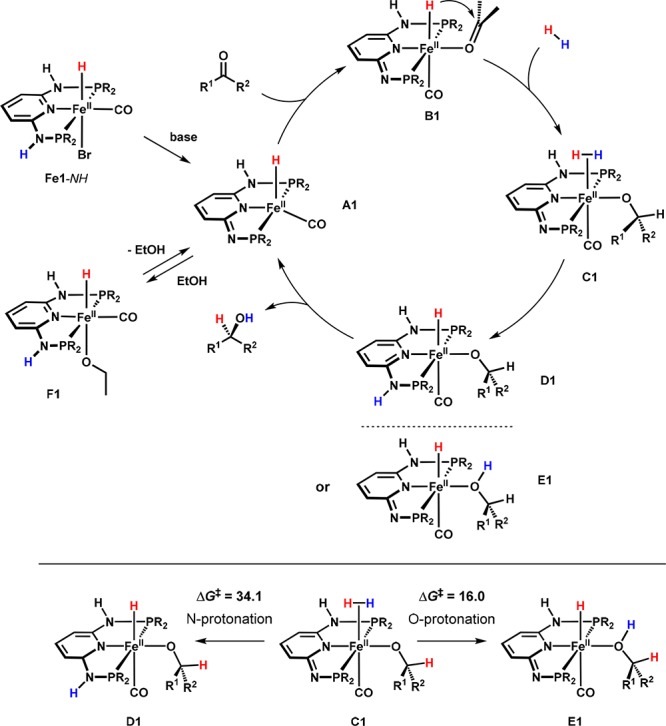
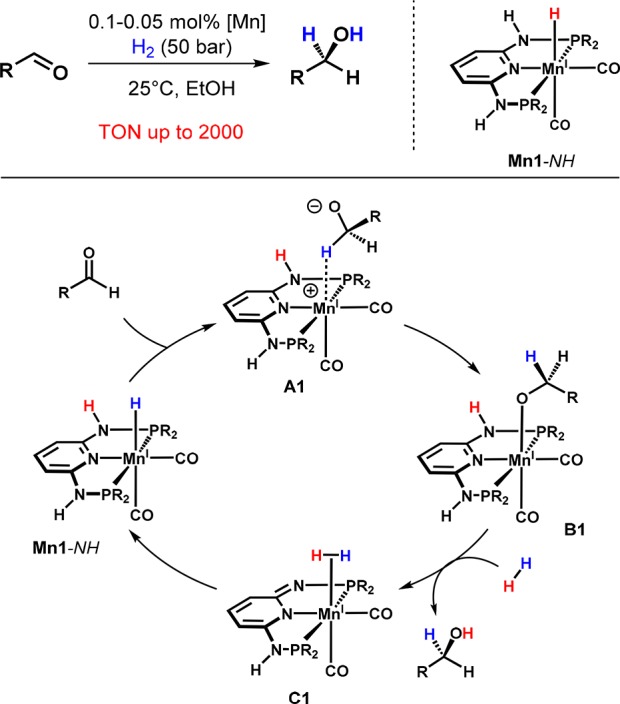
On the basis of these findings, a catalytic cycle is proposed (Scheme 6). The deprotonated intermediate A1 is the catalytically active species and is thought to be stabilized by the reversible addition of the solvent alcohol to give the reprotonated hydride alkoxide complex F1. Since A1 represents a formal 16-electron complex, providing a free coordination site for an incoming substrate, the mechanism is considered to proceed via coordination of the ketone prior to its insertion into the metal hydride bond. In principle, two different pathways for the release of the product alcohol and the regeneration the initial intermediate A1 were considered to be feasible. Either dihydrogen is activated by aromatization of the ligand followed by elimination of the product (D1), which again leads to a dearomatized system, or H2 is directly cleaved between the iron center and the product alkoxide without reprotonation of the nitrogen linker (E1). DFT calculations showed that the latter pathway is energetically more favorable, indicating that deprotonation of the NH linker is not involved in the heterolytic cleavage of H2 but instead is necessary for catalyst activation by generating a vacant coordination site and by increasing the negative charge at the metal center.
Scheme 6. DFT-Calculated Mechanism of the Iron-Catalyzed Hydrogenation of Ketones and Aldehydes.
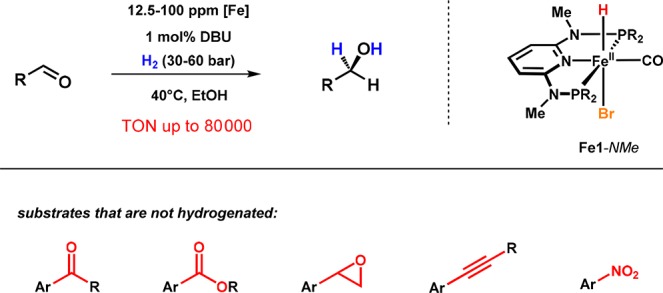
It was thus quite surprising that under the same conditions Fe1-NMe, which is unable to promote the reduction of ketones, was found to catalyze the hydrogenation of aldehydes in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the base (Scheme 7).26 Moreover, this system not only provides complete chemoselectivity for aldehydes over ketones and other reducible functional groups but exhibits extremely high activity and productivity. A variety of different aromatic, heteroaromatic, and aliphatic aldehydes could be quantitatively converted into the corresponding primary alcohols, reaching turnover numbers (TONs) of up to 80 000, which are among the highest reported for an iron-based hydrogenation catalyst. In contrast to the hydrogenation of ketones catalyzed by Fe1-NH, an iron dihydride was identified as key intermediate. A catalytic cycle is depicted in Scheme 8. The precatalyst Fe1-NMe readily forms a mixture of complexes trans- and cis-Fe3-NMe as a result of heterolytic cleavage of dihydrogen promoted by the iron center and the external base. Substrate insertion proceeds through an outer-sphere mechanism in which the nucleophilic dihydride directly attacks the aldehyde’s carbonyl group to give intermediate A3 featuring a H-coordinated alkoxide, which is in equilibrium with B3 where the alkoxide is O-coordinated. Replacement of the alkoxide either by the solvent (ethanol) or dihydrogen leads to the formation of C3 or D3, respectively. The latter features an η2-bound dihydrogen ligand. Subsequent deprotonation of the coordinated H2 finally leads to the regeneration of trans-Fe3-NMe and liberation of the product. Complexes B3 and C3 could be detected by NMR spectroscopy in stoichiometric experiments. DFT calculations revealed that the alkoxide complex B3 represents a resting state outside the catalytic cycle. This reasonably explains why the catalytic reaction proceeds only in protic solvents, which permit catalytic turnovers by solvent-assisted release of the alkoxide. The remarkable substrate selectivity, i.e., aldehydes versus ketones, was recently explained by the relative stability of alkoxide intermediates formed upon aldehyde insertion into an Fe–H bond of trans-Fe3-NMe based on state-of-the-art DFT calculations.27
Scheme 7. Chemoselective Hydrogenation of Aldehydes.
Scheme 8. Hydrogenation of Aldehydes Catalyzed by trans-Fe3-NMe.

In light of the exceptional catalytic performance and the industrial relevance of the chemoselective reduction of aldehydes, we also focused on the immobilization of Fe1-NMe. Since direct immobilization of a sensitive homogeneous catalyst on a solid support often results in partial or complete loss of activity because the active species suffers from a modification of its chemical and physical properties, an alternative and more convenient approach was chosen by preparing a supported ionic-liquid-phase (SILP) catalyst.28
For this purpose, Fe1-NMe was dissolved in an ionic liquid (IL) and impregnated on a porous support (Figure 5). Gratifyingly, the chemical nature of the catalyst was preserved, yet the catalyst was dissolved in a separate phase that was easily separated from the reaction solution. This novel SILP system was successfully applied to the chemoselective hydrogenation of aromatic and aliphatic aldehydes to give alcohols under mild conditions (0.1–0.05 mol % catalyst, 25 °C, 50 bar H2) without leaching of the catalyst. Although immobilization leads to decreased catalytic activity compared with the analogous homogeneous hydrogenation, it was possible to perform recharge experiments by adding additional substrate to the reaction mixture before the end of the reaction, showing that the new SILP system continuously catalyzes the reaction and making it interesting for applications under continuous flow conditions.
Figure 5.
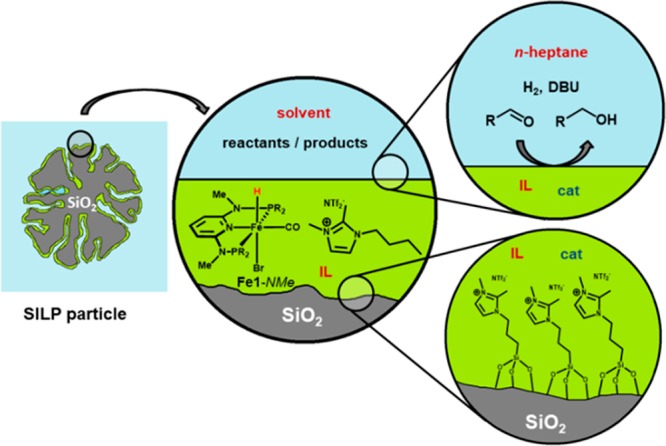
Iron–SILP system as a precatalyst for the chemoselective hydrogenation of aldehydes.
On the basis of the knowledge about the factors that affect the reactivity of our iron complexes, we realized striking similarities when manganese hydrogenation and dehydrogenation catalysts were first reported.29−33 Since complex Fe7 was found to be catalytically inactive, it was surprising to find that the isostructural and isoelectronic complex Mn1 is a highly reactive species.
This difference essentially arises from the higher electron density around manganese in the +I oxidation state, which also has a large influence on the reactivity of the M–H bond. As revealed by DFT calculations, the M–H distances as well as charge distributions in Mn(I) hydride complexes are very similar to those found in the respective Fe(II) dihydride species (Figure 6).34
Figure 6.
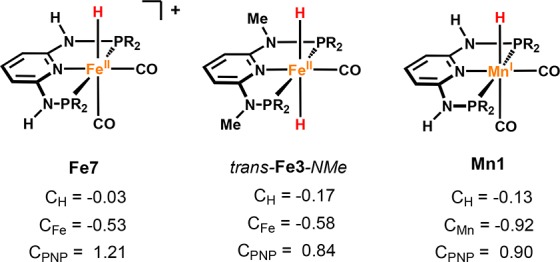
NPA charges of Fe(II) and Mn(I) PNP complexes.
An analysis of the frontier orbitals of the dearomatized species of Fe1-NH and Mn1-NH reveals that Fe(II) hydrido carbonyl and Mn(I) biscarbonyl PNP complexes are isolobal to each other. Both systems are able to promote H–H and H–heteroatom bond-breaking and -making reactions, with the metal center being acidic (LUMO) and the linker (HOMO) being basic (Figure 7). The HOMO corresponds to the ligand π system, with a significant contribution of the lone pair of the deprotonated N atoms (basic site). The LUMO is essentially the metal dz2 orbital pointing toward the empty coordination position (acidic site).
Figure 7.

LUMOs and HOMOs of dearomatized Fe(II) and Mn(I) PNP complexes.
The Mn(I) PNP hydride complexes Mn1-NH and Mn1-NMe were prepared by treating Mn(CO)5Br with 1 equiv of the PNP ligand to yield complexes of the types [Mn(PNP)(CO)2Br)] (Mn11-NH) and/or [Mn(PNP)(CO)3)]Br (Mn12) depending on the amine linker. Both complexes react with [HBEt3]− to afford the hydride complexes Mn1-NH and Mn1-NMe, respectively (Scheme 9).
Scheme 9. Synthesis of Mn1-NH and Mn1-NMe (Structural View of Mn1-NH).

In the same way as the iron dihydride complex trans-Fe3-NMe, the monohydride Mn1-NH was found to be a highly active catalyst for the hydrogenation of aldehydes, while other reducible functionalities including ketones, carboxyl acid derivatives, and C=C double bonds remained unaffected (Scheme 10).35Mn1-NMe was completely inactive, emphasizing the importance of the acidic NH linkers in enabling a bifunctional mechanism. In contrast to Fe1-NMe, which requires the presence of an external base for the activation of H2, Mn1-NH can be used under base-free conditions since hydrogen is cleaved in an intramolecular fashion by cooperation of the metal center and the deprotonated nitrogen linker (bifunctional catalysis). It has to be noted that related Mn(I) PNP pincer complexes were shown to act as catalysts for the hydrogenation of ketones and aldehydes but required higher catalyst loadings (0.1–5.0 mol %), higher temperatures (80–130 °C), and 1–10 mol % base (KOtBu) in toluene as the solvent.36,37
Scheme 10. Chemoselective Manganese-Catalyzed Hydrogenation of Aldehydes.
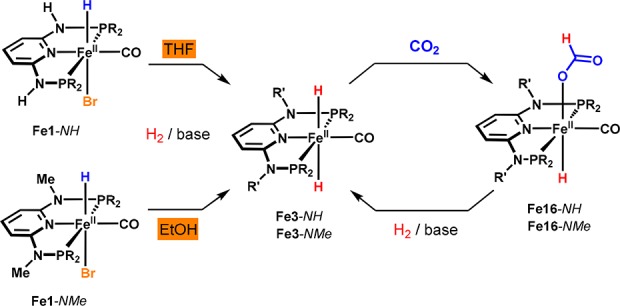
Apart from carbonyl compounds, we investigated the application of these catalysts to the hydrogenation of CO2.38 Both the complexes bearing NH linkers and those containing NMe linkers were found to promote the catalytic hydrogenation of CO2 and NaHCO3 to formates (Scheme 11). NMR and DFT studies highlight the role of dihydride and hydride complexes in the catalytic process. The mechanism strongly resembles the one previously proposed for the selective hydrogenation of aldehydes, since insertion of CO2 into the metal–hydride bond was also found to proceed via a nucleophilic attack in the outer coordination sphere of the catalyst. It has to be mentioned that Fe1-NH was inactive in EtOH, since the formation of the respective dihydride is effectively suppressed in this solvent (Scheme 12). However, reasonable catalytic activity was found for the hydrogenation of formate when the reaction was carried out in THF/water mixtures. On the other hand, with Fe1-NMe the best results for the hydrogenation of CO2 were observed by using EtOH as the solvent in the presence of stoichiometric amounts of DBU, reaching TONs of more than 10 000 and high yields of formate.
Scheme 11. Iron-Catalyzed Hydrogenation of CO2 and NaHCO3.
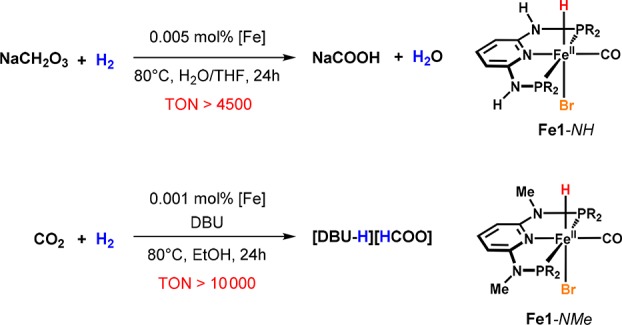
Scheme 12. Formation of Fe3-NH and Fe3-NMe and Reaction with CO2.
With Mn1-NH we reported the first example of Mn(I)-catalyzed hydrogenation of CO2 to give HCOOH (Scheme 13).38 The catalytic performance of this system is quite similar to that of the iron dihydride complex trans-Fe3-NMe, reaching TONs of up to 10 000, and quantitative yields were obtained after 24 h using DBU as the base at 80 °C and a total pressure of 80 bar. Mn1-NH showed improved long-term stability under the applied reaction conditions. In contrast to iron, we could accomplish a significant increase of the reaction rate by employing lithium triflate as a cocatalyst,39−41 yielding TONs of more than 30 000, which are among the highest reported for base-metal-catalyzed CO2 hydrogenations to date.
Scheme 13. Manganese-Catalyzed Hydrogenation of CO2.
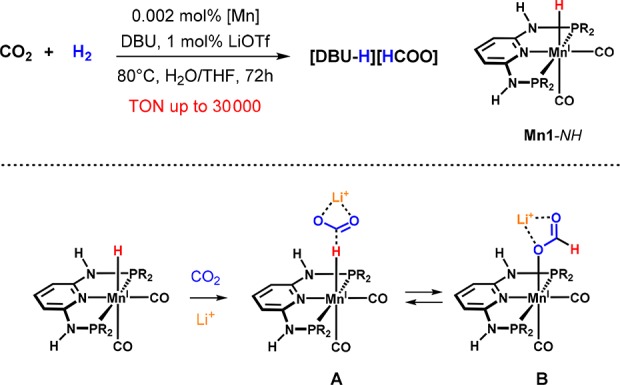
Analogously to our mechanistic studies on iron, we recognized that nucleophilic attack of the hydride on CO2 in the second coordination sphere of the metal constitutes the key step of the reaction. Moreover, this step leading to the formation of the corresponding formate species constitutes a convenient model illustrating the basicity of the hydride complex. Accordingly, we could compare the natures of the M–H bonds in these Fe(II) and Mn(I) hydride complexes by means of DFT calculations. These data also show that insertion into the metal–hydride bond is thermodynamically unfavorable in the case of Mn1-NMe featuring NMe linkers. In fact, this catalyst displayed only poor catalytic activity. Finally, the beneficial effect of LiOTf might be related to the stabilization of the κ1-H-bound formate in A, since B represents a resting state that is outside the catalytic cycle.
Acceptorless Alcohol Dehydrogenation and Hydrogen Autotransfer Reactions
Our results obtained from hydrogenation reactions nicely illustrated the similarities between iron and manganese and substantially contributed our knowledge about the main factors that influence the basic reactivities of these catalysts. However, an aspect that appears to be at least equally important for further catalytic applications is the reversibility of these processes. Therefore, as the formally reverse process, we also focused on dehydrogenation reactions, which in contrast to hydrogenations unveiled significant differences between iron and manganese.
While iron complexes supported by aliphatic PNP pincer ligands were shown to catalyze not only the hydrogenation of unsaturated compounds but also the acceptorless dehydrogenation of saturated substrates,42,43 pyridine-based systems have not yet been found to promote such a reaction. This difference may be attributed to the fact that unsaturated 16-electron intermediates formed in the course of H2 liberation via metal–ligand cooperation are efficiently stabilized by the deprotonated nitrogen atom, which acts as both a strong σ and π donor. In contrast, pyridine-based pincer systems are only weak π donors and thus are unable to stabilize 16-electron intermediates (Figure 7). Accordingly, complexes with aliphatic PNP amido ligands could even be isolated and structurally characterized.39,43 The deprotonated 16-electron species A in the case of Fe1-NH could not be observed directly but could be trapped in the presence of CO in the form of [Fe(PNPH-iPr)(H)(CO)2] (B), which exhibits a characteristic AB pattern in the 31P{1H} NMR spectrum (Scheme 14). Together with B, the formation of the Fe(0) complex [Fe(PNP-iPr)(CO)2] (C),22 free PNP ligand, and intractable materials were also observed. Scrambling of the CO ligands to form C represents a major decomposition and catalyst deactivation pathway.
Scheme 14. Trapping of B and Detection by 31P{1H} NMR Spectroscopy.

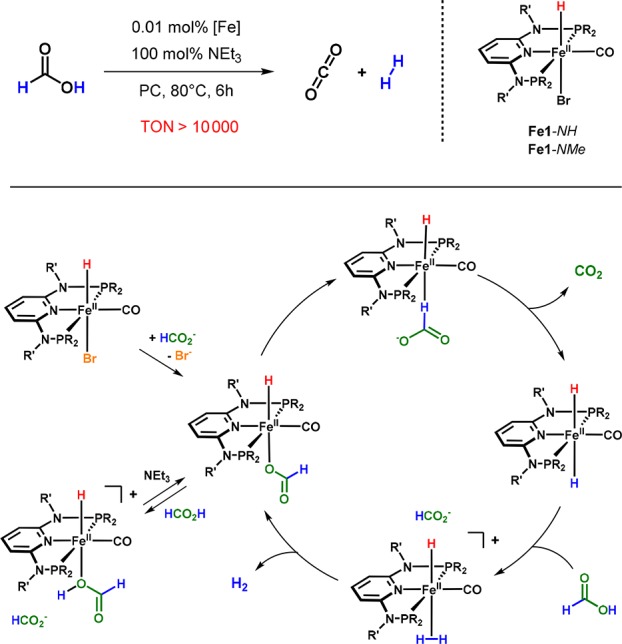
Dehydrogenation of formic acid, which was found to be catalyzed by the iron complexes Fe1-NH and Fe1-NMe, constitutes the only exception, since this reaction, just as the hydrogenation of CO2 mentioned before, does not proceed via a bifunctional mechanism (Scheme 15).44
Scheme 15. Iron-Catalyzed Dehydrogenation of Formic Acid.
In contrast to iron, manganese PNP pincer complexes are much more suitable for acceptorless dehydrogenations, particularly considering dehydrogenative coupling reactions. For example, we found that Mn1 is able to promote the catalytic coupling of alcohols and amines to yield imines, thereby generating water and molecular hydrogen as the sole byproducts (Scheme 16).45 For comparison, we also tested the iron complex Fe1-NH in this reaction. Surprisingly, this complex was catalytically active as well but afforded secondary amines instead. It has to be mentioned that this divergent reactivity between the Fe and Mn catalysts is a particular property of pyridine-derived PNP complexes. While Milstein and co-workers also reported on the dehydrogenative coupling of alcohols and amines catalyzed by a lutidine-based system,29 Mn catalysts based on aliphatic PNP pincer ligands were found to promote coupling reactions that proceed via a hydrogen autotransfer mechanism and do not liberate dihydrogen.46−48
Scheme 16. Coupling of Alcohols and Amines Catalyzed by Mn(I) and Fe(II) PNP Pincer Complexes.
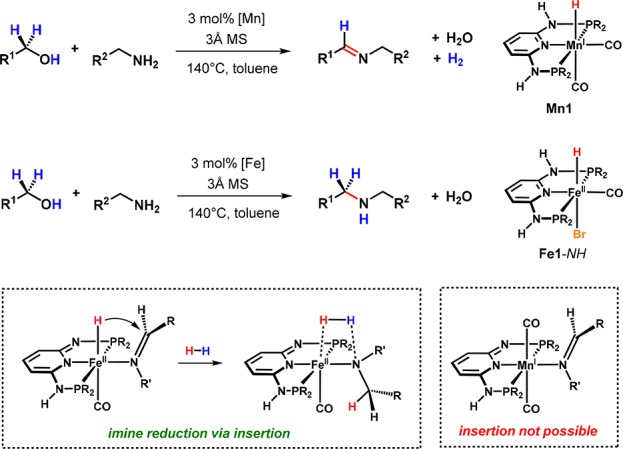
Mechanistic consideration suggests that in both cases the O–H bond of the alcohol is cleaved between the metal center and the deprotonated side arm of the ligand to form an alkoxide intermediate, which is converted to the respective aldehyde via a nonclassical β-hydride elimination. The resulting hydride and dihydride complexes, respectively, release dihydrogen in a bifunctional manner, recovering the initial deprotonated 16-electron species again. However, while Mn just reenters this catalytic cycle, it seems obvious that iron allows for the insertion of the imine that has been formed by condensation of the aldehyde and an amine to finally yield an amine in the presence of the previously released H2. This separate reduction cycle proceeds analogously to the mechanism that we proposed for the hydrogenation of ketones. Moreover, it gives a reasonable explanation for the divergence between the two metals, as Mn is not capable of following an insertion pathway.
We recently also discovered that instead of the rather sensitive catalyst Fe1-NH, even the respective iron dibromide PNP complexes and related triazine-based dibromide complexes, which are more easily accessible and air-stable, can be used as precatalysts for the alkylation of amines with alcohols.49 Systematic variations of the ligand scaffold can be accomplished, giving valuable information on structure–activity relationships (Scheme 17).
Scheme 17. Alkylation of Amines with Alcohols Catalyzed by Air-Stable Precatalysts.
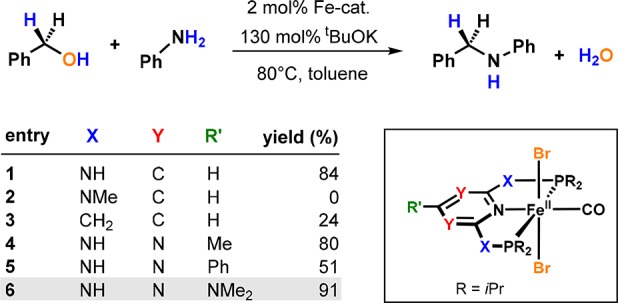
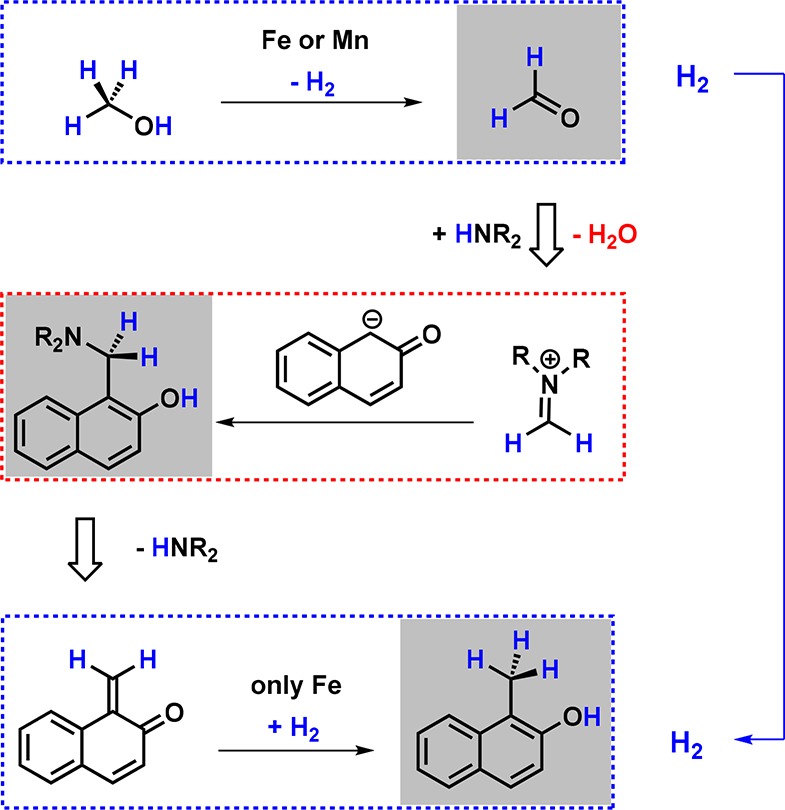
Divergence was also observed when Mn1-NH was applied for the catalytic aminomethylation of phenols and heteroaromatic compounds utilizing amines and MeOH as a C1 building block (Scheme 18).50 From a mechanistic perspective, this represents a Mannich-type reaction requiring formaldehyde as a reactant, which is generated in situ through the catalytic dehydrogenation of MeOH. In a preliminary study, Fe1-NH was shown to catalyze the methylation of 2-naphthol rather than its aminomethylation, underlining the divergent reactivity of isoelectronic Mn(I) and Fe(II) PNP pincer systems (Scheme 19).
Scheme 18. Manganese-Catalyzed Aminomethylation of Aromatic Compounds.

Scheme 19. Simplified Mechanism of the Mn- and Fe-Catalyzed Aminomethylation of 2-Naphthol.
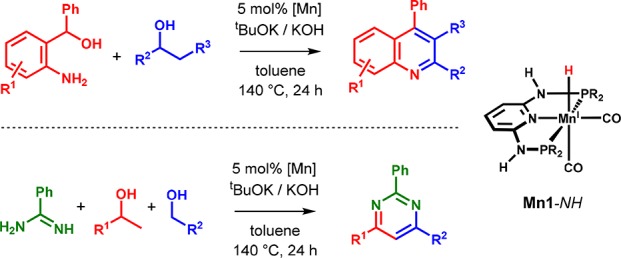
Finally, the ability of Mn(I) PNP complexes to promote a sequence of dehydrogenation and condensation steps that gives rise to selective C–C and C–N bond formations could even be extended to more complex structures. For example, Mn1-NH was successfully employed in the synthesis of substituted quinolones and pyrimidines using combinations of 2-aminobenzyl alcohols and alcohols as well as benzamidine and two different alcohols, respectively (Scheme 20).51 The Kempe group even extended this concept to pyrroles and tetrasubstituted pyrimidines by using analogous triazine-based catalysts.52,53
Scheme 20. Manganese-Catalyzed Multicomponent Reactions To Yield Quinolines and Pyrimidines.
Summary and Outlook
We have described the catalytic activity of well-defined isoelectronic Fe(II) and Mn(I) PNP pincer complexes based on the 2,6-diaminopyridine scaffold or derivatives thereof that have been developed in our group. Several of these compounds are active catalysts for the hydrogenation of carbonyl compounds as well as for the acceptorless dehydrogenation of alcohols. The key aspects of these catalysts have been the recognition of decisive structural features, i.e., enabling and blocking of bifunctional behavior by variations of the amine linkers (NH vs NR) as well as the determination and optimization of proper reaction conditions that permit high catalytic turnovers. Mechanistic insights, including catalyst activation and the identification of the reactive species, revealed specific similarities and differences between the described systems. The current knowledge about the nature and characteristics of active iron- and manganese-based systems has paved the way for conceptually and mechanistically well-founded research that may lead to further developments and the discovery of novel catalysts extending the scope and limitations of reactivity. One aspect is the replacement of CO by other π-accepting ligands such as NO+, CN–, or isocyanides or the partial or complete removal of CO ligands, which may provide new reactivities as already accomplished with Fe(II) polyhydride complexes, e.g., [Fe(PNPMe-iPr)(H)2(η2-H2)].54 Challenging in Mn(I) chemistry also will be the development of catalysts that originate from inexpensive Mn(II) materials, as Mn(I) catalyst formation is currently restricted to the expensive precursor [Mn(CO)5X] (X = Cl, Br). In view of the many inspiring discoveries and achievements in homogeneous manganese and iron catalysis in the past few years, it seems that research into this field is merely at the beginning of its development and that many breakthroughs are still ahead, rewarding those who do not mind great challenges.
Acknowledgments
Financial support by the Austrian Science Fund (FWF) is gratefully acknowledged (Projects P29584-N28 and P28866-N34). We also thank the present and former group members, colleagues, and collaborators.
Biographies
Nikolaus Gorgas (born in 1987 in Vienna, Austria) received his M.S. degree in 2012 from the University of Vienna, working with Prof. Walter Weissensteiner. Afterward he joined the research group of Prof. Karl Kirchner at the Technical University of Vienna (TU Wien), where he obtained his Ph.D. in 2017. He is currently a postdoctoral fellow in the same group. His present research interests include base metal polyhydrides and σ-bond complexes and their applications in homogeneous catalysis.
Karl Kirchner (born in 1960 in Wiener Neustadt, Austria) obtained his diploma (1984) and his doctoral degree (1987) from TU Wien, working with Prof. Roland Schmid. After a two-year postdoctoral stay at Washington State University with Prof. John P. Hunt and an additional postdoctoral year with Nobel Laureate Prof. Henry Taube at Stanford University, he returned to TU Wien and completed his Habilitation with Prof. Roland Schmid (1994). He is presently Professor of Organometallic Chemistry and Head of the Division of Inorganic Chemistry. His expertise is centered around the themes of ligand design, synthesis of well-defined nonprecious transition metal complexes, and their applications in homogeneous catalysis.
The authors declare no competing financial interest.
References
- Catalysis without Precious Metals; Bullock R. M., Ed.; Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Gunanathan C.; Milstein D. Applications of Acceptorless Dehydrogenation and Related Transformations in Chemical Synthesis. Science 2013, 341, 1229712. 10.1126/science.1229712. [DOI] [PubMed] [Google Scholar]
- The Handbook of Homogeneous Hydrogenation; de Vries J. G., Elsevier C. J., Eds.; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Enthaler S.; Junge K.; Beller M. Sustainable metal catalysis with iron: from rust to a rising star?. Angew. Chem., Int. Ed. 2008, 47, 3317–3321. 10.1002/anie.200800012. [DOI] [PubMed] [Google Scholar]
- Misal Castro L. C.; Li H.; Sortais J.-B.; Darcel C. When Iron Met Phosphines: A Happy Marriage for Reduction Catalysis. Green Chem. 2015, 17, 2283–2303. 10.1039/C4GC01866D. [DOI] [Google Scholar]
- Bauer I.; Knölker H.-J. Iron Catalysis in Organic Synthesis. Chem. Rev. 2015, 115, 3170–3387. 10.1021/cr500425u. [DOI] [PubMed] [Google Scholar]
- Zell T.; Milstein D. Hydrogenation and Dehydrogenation Iron Pincer Catalysts Capable of Metal-Ligand Cooperation by Aromatization/Dearomatization. Acc. Chem. Res. 2015, 48, 1979–1994. 10.1021/acs.accounts.5b00027. [DOI] [PubMed] [Google Scholar]
- Chakraborty S.; Bhattacharya P.; Dai H.; Guan H. Nickel and Iron Pincer Complexes as Catalysts for the Reduction of Carbonyl Compounds. Acc. Chem. Res. 2015, 48, 1995–2003. 10.1021/acs.accounts.5b00055. [DOI] [PubMed] [Google Scholar]
- Garbe M.; Junge K.; Beller M. Homogeneous Catalysis by Manganese-Based Pincer Complexes. Eur. J. Org. Chem. 2017, 2017, 4344–4362. 10.1002/ejoc.201700376. [DOI] [Google Scholar]
- Maji B.; Barman M. Recent Developments of Manganese Complexes for Catalytic Hydrogenation and Dehydrogenation Reactions. Synthesis 2017, 49, 3377–3393. 10.1055/s-0036-1590818. [DOI] [Google Scholar]
- Kallmeier F.; Kempe R. Manganese Complexes for (De)Hydrogenation Catalysis: A Comparison to Cobalt and Iron Catalysts. Angew. Chem., Int. Ed. 2018, 57, 46–60. 10.1002/anie.201709010. [DOI] [PubMed] [Google Scholar]
- Filonenko G. A.; van Putten R.; Hensen E. J. M.; Pidko E. A. Catalytic (De)hydrogenation Promoted by Non-Precious Metals - Co, Fe and Mn: Recent Advances in an Emerging Field. Chem. Soc. Rev. 2018, 47, 1459–1483. 10.1039/C7CS00334J. [DOI] [PubMed] [Google Scholar]
- Bauer G.; Kirchner K. A. Well-defined Bifunctional Iron Catalysts for the Hydrogenation of Ketones - Iron, the New Ruthenium. Angew. Chem., Int. Ed. 2011, 50, 5798–5800. 10.1002/anie.201101579. [DOI] [PubMed] [Google Scholar]
- Fürstner A. Iron Catalysis in Organic Synthesis: A Critical Assessment of What It Takes To Make This Base Metal a Multitasking Champion. ACS Cent. Sci. 2016, 2, 778–789. 10.1021/acscentsci.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zell T.; Langer R. From Ruthenium to Iron and Manganese - a Mechanistic View on Challenges and Design Principles of Base Metal Hydrogenation Catalysts. ChemCatChem 2018, 10, 1930–1940. 10.1002/cctc.201701722. [DOI] [Google Scholar]
- Benito-Garagorri D.; Becker E.; Wiedermann J.; Lackner W.; Pollak M.; Mereiter K.; Kisala J.; Kirchner K. Achiral and Chiral Transition Metal Complexes with Modularly Designed Tridentate PNP Pincer-Type Ligands Based on N-Heterocyclic Diamines. Organometallics 2006, 25, 1900–1913. 10.1021/om0600644. [DOI] [Google Scholar]
- Glatz M.; Holzhacker C.; Bichler B.; Mastalir M.; Stöger B.; Mereiter K.; Weil M.; Veiros L. F.; Mösch-Zanetti N. C.; Kirchner K. FeII Carbonyl Complexes Featuring Small to Bulky PNP Pincer Ligands - Facile Substitution of κ2P,N-Bound PNP Ligands by Carbon Monoxide. Eur. J. Inorg. Chem. 2015, 2015, 5053–5065. 10.1002/ejic.201500646. [DOI] [Google Scholar]
- Holzhacker C.; Stöger B.; Carvalho M. D.; Ferreira L. P.; Pittenauer E.; Allmaier G.; Veiros L. F.; Realista S.; Gil A.; Calhorda M. J.; Müller D.; Kirchner K. Synthesis and Reactivity of Taddol-Based Chiral Fe(II) PNP Pincer Complexes - Solution Equilibria between κ2P,N- and κ3P,N,P-Bound PNP Pincer Ligands. Dalton Trans. 2015, 44, 13071–13086. 10.1039/C5DT00832H. [DOI] [PubMed] [Google Scholar]
- Werkmeister S.; Neumann J.; Junge K.; Beller M. Pincer-Type Complexes for Catalytic (De)Hydrogenation and Transfer (De)Hydrogenation Reactions: Recent Progress. Chem. - Eur. J. 2015, 21, 12226–12250. 10.1002/chem.201500937. [DOI] [PubMed] [Google Scholar]
- Khusnutdinova J. R.; Milstein D. Metal-Ligand Cooperation. Angew. Chem., Int. Ed. 2015, 54, 12236–12273. 10.1002/anie.201503873. [DOI] [PubMed] [Google Scholar]
- Benito-Garagorri D.; Kirchner K. Modularly Designed Transition Metal PNP and PCP Pincer Complexes based on Aminophosphines - Synthesis and Catalytic Applications. Acc. Chem. Res. 2008, 41, 201–213. 10.1021/ar700129q. [DOI] [PubMed] [Google Scholar]
- Bichler B.; Holzhacker C.; Stöger B.; Puchberger M.; Veiros L. F.; Kirchner K. Heterolytic Cleavage of Dihydrogen by an Iron(II) PNP Pincer Complex via Metal-Ligand Cooperation. Organometallics 2013, 32, 4114–4121. 10.1021/om400241x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer R.; Leitus G.; Ben-David Y.; Milstein D. Efficient Hydrogenation of Ketones Catalyzed by an Iron Pincer Complex. Angew. Chem., Int. Ed. 2011, 50, 2120–2124. 10.1002/anie.201007406. [DOI] [PubMed] [Google Scholar]
- Langer R.; Iron M. A.; Konstantinovski L.; Diskin-Posner Y.; Leitus G.; Ben-David Y.; Milstein D. Iron Borohydride Pincer Complexes for the Efficient Hydrogenation of Ketones under Mild, Base-Free Conditions: Synthesis and Mechanistic Insight. Chem. - Eur. J. 2012, 18, 7196–7209. 10.1002/chem.201200159. [DOI] [PubMed] [Google Scholar]
- Gorgas N.; Stöger B.; Veiros L. F.; Pittenauer E.; Allmaier G.; Kirchner K. Efficient Hydrogenation of Ketones and Aldehydes Catalyzed by Well-Defined Fe(II) PNP Pincer Complexes: Evidence for an Insertion Mechanism. Organometallics 2014, 33, 6905–6914. 10.1021/om5009814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgas N.; Stöger B.; Veiros L. F.; Kirchner K. Highly Efficient and Selective Hydrogenation of Aldehydes: A Well-Defined Fe(II) Catalyst Exhibits Noble-Metal Activity. ACS Catal. 2016, 6, 2664–2672. 10.1021/acscatal.6b00436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morello G. R.; Hopmann K. H. A Dihydride Mechanism Can Explain the Intriguing Substrate Selectivity of Iron-PNP-Mediated Hydrogenation. ACS Catal. 2017, 7, 5847–5855. 10.1021/acscatal.7b00764. [DOI] [Google Scholar]
- Brünig J.; Csendes Z.; Weber S.; Gorgas N.; Bittner R. W.; Limbeck A.; Bica K.; Hoffmann H.; Kirchner K. Chemoselective Supported Ionic-Liquid-Phase (SILP) Aldehyde Hydrogenation Catalyzed by an Fe(II) PNP Pincer Complex. ACS Catal. 2018, 8, 1048–1051. 10.1021/acscatal.7b04149. [DOI] [Google Scholar]
- Mukherjee A.; Nerush A.; Leitus G.; Shimon L. J. W.; Ben David Y.; Espinosa Jalapa N. A.; Milstein D. Manganese-Catalyzed Environmentally Benign Dehydrogenative Coupling of Alcohols and Amines to Form Aldimines and H2: A Catalytic and Mechanistic Study. J. Am. Chem. Soc. 2016, 138, 4298–4301. 10.1021/jacs.5b13519. [DOI] [PubMed] [Google Scholar]
- Elangovan S.; Topf C.; Fischer S.; Jiao H.; Spannenberg A.; Baumann W.; Ludwig R.; Junge K.; Beller M. Selective Catalytic Hydrogenations of Nitriles, Ketones, and Aldehydes by Well-Defined Manganese Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 8809–8814. 10.1021/jacs.6b03709. [DOI] [PubMed] [Google Scholar]
- Elangovan S.; Garbe M.; Jiao H.; Spannenberg A.; Junge K.; Beller M. Hydrogenation of Esters to Alcohols Catalyzed by Defined Manganese Pincer Complexes. Angew. Chem., Int. Ed. 2016, 55, 15364–15368. 10.1002/anie.201607233. [DOI] [PubMed] [Google Scholar]
- Andérez-Fernández M.; Vogt L. K.; Fischer S.; Zhou W.; Jiao H.; Garbe M.; Elangovan S.; Junge K.; Junge H.; Ludwig R.; Beller M. A Stable Manganese Pincer Catalyst for the Selective Dehydrogenation of Methanol. Angew. Chem., Int. Ed. 2017, 56, 559–562. 10.1002/anie.201610182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D. H.; Trivelli X.; Capet F.; Paul J.-F.; Dumeignil F.; Gauvin R. M. Manganese Pincer Complexes for the Base-Free, Acceptorless Dehydrogenative Coupling of Alcohols to Esters: Development, Scope, and Understanding. ACS Catal. 2017, 7, 2022–2032. 10.1021/acscatal.6b03554. [DOI] [Google Scholar]
- Bertini F.; Glatz M.; Gorgas N.; Stöger B.; Peruzzini M.; Veiros L. F.; Kirchner K.; Gonsalvi L. Carbon Dioxide Hydrogenation Catalyzed by Well-Defined Mn(I) PNP Pincer Hydride Complexes. Chem. Sci. 2017, 8, 5024–5029. 10.1039/C7SC00209B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatz M.; Stöger B.; Himmelbauer D.; Veiros L. F.; Kirchner K. Chemoselective Hydrogenation of Aldehydes under Mild and Base-Free Conditions—Manganese Outperforms Rhenium. ACS Catal. 2018, 8, 4009–4016. 10.1021/acscatal.8b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau-Voisine A.; Wang D.; Roisnel T.; Darcel C.; Sortais J.-B. Hydrogenation of Ketones with a Manganese PN3P Pincer Pre-Catalyst. Catal. Commun. 2017, 92, 1–4. 10.1016/j.catcom.2016.12.017. [DOI] [Google Scholar]
- Kallmeier F.; Irrgang T.; Dietel T.; Kempe R. Highly Active and Selective Manganese C=O Bond Hydrogenation Catalysts: The Importance of the Multidentate Ligand, the Ancillary Ligands, and the Oxidation State. Angew. Chem., Int. Ed. 2016, 55, 11806–11809. 10.1002/anie.201606218. [DOI] [PubMed] [Google Scholar]
- Bertini F.; Gorgas N.; Stöger B.; Peruzzini M.; Veiros L. F.; Kirchner K.; Gonsalvi L. Efficient and Mild Carbon Dioxide Hydrogenation to Formate Catalyzed by Fe(II) Hydrido Carbonyl Complexes Bearing 2,6-(Diaminopyridyl)diphosphine Pincer Ligands. ACS Catal. 2016, 6, 2889–2893. 10.1021/acscatal.6b00416. [DOI] [Google Scholar]
- Bielinski E. A.; Lagaditis P. O.; Zhang Y.; Mercado B. Q.; Würtele C.; Bernskoetter W. H.; Hazari N.; Schneider S. Lewis Acid-Assisted Formic Acid Dehydrogenation Using a Pincer-Supported Iron Catalyst. J. Am. Chem. Soc. 2014, 136, 10234–10237. 10.1021/ja505241x. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; MacIntosh A. D.; Wong J. L.; Bielinski E. A.; Williard P. G.; Mercado B. Q.; Hazari N.; Bernskoetter W. H. Iron Catalyzed CO2 Hydrogenation to Formate Enhanced by Lewis Acid Co-Catalysts. Chem. Sci. 2015, 6, 4291–4299. 10.1039/C5SC01467K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielinski E. A.; Förster M.; Zhang Y.; Bernskoetter W. H.; Hazari N.; Holthausen M. C. Base-Free Methanol Dehydrogenation Using a Pincer-Supported Iron Compound and Lewis Acid Co-Catalyst. ACS Catal. 2015, 5, 2404–2415. 10.1021/acscatal.5b00137. [DOI] [Google Scholar]
- Chakraborty S.; Lagaditis P. O.; Förster M.; Bielinski E. A.; Hazari N.; Holthausen M. C.; Jones W. D.; Schneider S. Well-Defined Iron Catalysts for the Acceptorless Reversible Dehydrogenation-Hydrogenation of Alcohols and Ketones. ACS Catal. 2014, 4, 3994–4003. 10.1021/cs5009656. [DOI] [Google Scholar]
- Chakraborty S.; Brennessel W. W.; Jones W. D. A Molecular Iron Catalyst for the Acceptorless Dehydrogenation and Hydrogenation of N-Heterocycles. J. Am. Chem. Soc. 2014, 136, 8564–8567. 10.1021/ja504523b. [DOI] [PubMed] [Google Scholar]
- Mellone I.; Gorgas N.; Bertini F.; Peruzzini M.; Kirchner K.; Gonsalvi L. Selective Formic Acid Dehydrogenation Catalyzed by Fe-PNP Pincer Complexes Based on the 2,6-Diaminopyridine Scaffold. Organometallics 2016, 35, 3344–3349. 10.1021/acs.organomet.6b00551. [DOI] [Google Scholar]
- Mastalir M.; Glatz M.; Gorgas N.; Stöger B.; Pittenauer E.; Allmaier G.; Veiros L. F.; Kirchner K. Divergent Coupling of Alcohols and Amines Catalyzed by Isoelectronic Hydride MnI and FeII PNP Pincer Complexes. Chem. - Eur. J. 2016, 22, 12316–12320. 10.1002/chem.201603148. [DOI] [PubMed] [Google Scholar]
- Elangovan S.; Neumann J.; Sortais J.-B.; Junge K.; Darcel C.; Beller M. Efficient and Selective N-Alkylation of Amines with Alcohols Catalysed by Manganese Pincer Complexes. Nat. Commun. 2016, 7, 12641. 10.1038/ncomms12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña-López M.; Piehl P.; Elangovan S.; Neumann H.; Beller M. Manganese-Catalyzed Hydrogen-Autotransfer C-C Bond Formation: α-Alkylation of Ketones with Primary Alcohols. Angew. Chem., Int. Ed. 2016, 55, 14967–14971. 10.1002/anie.201607072. [DOI] [PubMed] [Google Scholar]
- Fu S.; Shao Z.; Wang Y.; Liu Q. Manganese-Catalyzed Upgrading of Ethanol into 1-Butanol. J. Am. Chem. Soc. 2017, 139, 11941–11948. 10.1021/jacs.7b05939. [DOI] [PubMed] [Google Scholar]
- Mastalir M.; Stöger B.; Pittenauer E.; Puchberger M.; Allmaier G.; Kirchner K. Air Stable Iron(II) PNP Pincer Complexes as Efficient Catalysts for the Selective Alkylation of Amines with Alcohols. Adv. Synth. Catal. 2016, 358, 3824–3831. 10.1002/adsc.201600689. [DOI] [Google Scholar]
- Mastalir M.; Pittenauer E.; Allmaier G.; Kirchner K. Manganese-Catalyzed Aminomethylation of Aromatic Compounds with Methanol as Sustainable C1 Building Block. J. Am. Chem. Soc. 2017, 139, 8812–8815. 10.1021/jacs.7b05253. [DOI] [PubMed] [Google Scholar]
- Mastalir M.; Glatz M.; Pittenauer E.; Allmaier G.; Kirchner K. Sustainable Synthesis of Quinolines and Pyrimidines Catalyzed by Manganese PNP Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 15543–15546. 10.1021/jacs.6b10433. [DOI] [PubMed] [Google Scholar]
- Deibl N.; Kempe R. Manganese-Catalyzed Multicomponent Synthesis of Pyrimidines from Alcohols and Amidines. Angew. Chem., Int. Ed. 2017, 56, 1663–1666. 10.1002/anie.201611318. [DOI] [PubMed] [Google Scholar]
- Kallmeier F.; Dudziec B.; Irrgang T.; Kempe R. Manganese-Catalyzed Sustainable Synthesis of Pyrroles from Alcohols and Amino Alcohols. Angew. Chem., Int. Ed. 2017, 56, 7261–7265. 10.1002/anie.201702543. [DOI] [PubMed] [Google Scholar]
- Gorgas N.; Alves L. G.; Stöger B.; Martins A. M.; Veiros L. F.; Kirchner K. Stable, Yet Highly Reactive Nonclassical Iron(II) Polyhydride Pincer Complexes: Z -Selective Dimerization and Hydroboration of Terminal Alkynes. J. Am. Chem. Soc. 2017, 139, 8130–8133. 10.1021/jacs.7b05051. [DOI] [PubMed] [Google Scholar]