

Abstract
Backgound
The inability to detect premature atherosclerosis significantly hinders implementation of personalized therapy to prevent coronary heart disease. A comprehensive understanding of arterial protein networks and how they change in early atherosclerosis could identify new biomarkers for disease detection and improved therapeutic targets.
Methods
Here we describe the human arterial proteome and proteomic features strongly associated with early atherosclerosis based on mass-spectrometry analysis of coronary artery and aortic specimens from 100 autopsied young adults (200 arterial specimens). Convex analysis of mixtures, differential dependent network modeling and bioinformatic analyses defined the composition, network re-wiring and likely regulatory features of the protein networks associated with early atherosclerosis and how they vary across two anatomic distributions.
Results
The data document significant differences in mitochondrial protein abundance between coronary and aortic samples (coronary≫aortic), and between atherosclerotic and normal tissues (atherosclerotic≪normal), as well as major alterations in TNF, insulin receptor, PPAR-α and PPAR-γ protein networks in the setting of early disease. In addition, a subset of tissue protein biomarkers indicative of early atherosclerosis was shown to predict anatomically defined coronary atherosclerosis when measured in plasma samples in a separate clinical cohort (AUC = 0.92(0.83-0.96)) – thereby validating the use of human tissue proteomics to discover relevant plasma biomarkers for clinical applications. In addition to the specific proteins and pathways identified here, the publicly available data resource and the employed analysis pipeline illustrate a strategy for interrogating and interpreting the proteomic architecture of tissues that may be relevant for other chronic diseases characterized by multi-cellular tissue phenotypes.
Conclusions
The human arterial proteome can be viewed as a complex network whose architectural features vary considerably as a function of anatomic location and the presence or absence of atherosclerosis. The data suggest important reductions in mitochondrial protein abundance in early atherosclerosis and also identify a subset of plasma proteins that are highly predictive of angiographically defined coronary disease.
Keywords: atherosclerosis, proteomics, mitochondria, complex networks
Introduction
At the molecular level atherosclerosis can be defined as an assembly of hundreds of intra- and extra-cellular proteins that jointly alter cellular processes and produce characteristic remodeling of the local vascular environment. Ultimately, these proteomic changes produce the lesions responsible for most ischemic cardiovascular events. Unfortunately, current methods to treat and prevent cardiovascular disease focus on antecedent risk factors that are not deterministic of these changes, or on anatomic manifestations of disease that are not clinically evident until long after these proteomic changes are underway. To improve early disease detection, and to interrupt the disease process before clinical consequences occur, it is necessary to recognize the specific patterns and dynamic features of arterial protein networks that constitute the molecular signatures of healthy and atherosclerotic arterial tissues.
Previous studies have described features of the arterial proteome in murine1-3 and cell models of atherosclerosis4, 5 and in limited numbers of human arterial samples with and without atherosclerosis6-19. The most common source of human arterial samples has been carotid endarterectomy explants from older individuals with late stage carotid stenosis. To date there has not been a comprehensive survey of the human arterial proteome based on a large number of human coronary and aortic samples, using contemporary LC-MS/MS technology and subsequent identification of the proteins that signify presence of pre-clinical atherosclerosis. Accordingly, we established a tissue acquisition, mass-spectrometry analysis and statistical and bioinformatic pipeline to characterize the human coronary and distal aortic arterial proteome and to identify those proteins, networks and pathways most strongly associated with early atherosclerotic lesions. Detailed analyses of the detected proteins reveal several key features of the coronary and aortic proteome in health and disease and identify a subset of proteins that are also highly informative plasma biomarkers for presence of coronary atherosclerosis in the clinical setting.
Methods
The data, analytic methods, and study materials are available to other researchers for purposes of reproducing the results or replicating procedures. Specifically, the DDA MS and the MRM MS data along with the Skyline document have been uploaded to Peptide Atlas. The Spectral Library used for DIA MS quantitation has also been uploaded to Peptide Atlas. The data can be accessed at http://www.peptideatlas.org/PASS/PASS01066 or via FTP using the following credentials: Servername: ftp.peptideatlas.org; Username: PASS01066; Password: KV454u. A detailed description of all the methods can be found in Online Extended Methods.
Arterial Sample Acquisition and Pathology Grading Methods
Male and female coroner's cases of any race, aged 18-50 years (men) or 18-60 years (women) with no ante mortem clinical suspicion of coronary disease autopsied < 24 hours of death were eligible for inclusion. This report includes data from the first 100 autopsies included in the study (age range: 15-55 yrs., 75% males, 67% White, 26% Black, 7% Other). The Medico-Legal Death Investigators obtain signed family consent for retrieval of anatomic specimens prior to the autopsy using a protocol approved by the Lousiana State Univeristy IRB. During the autopsy the pathologist dissected the aorta (ligamentum arteriosum to aortic bifurcation) and LAD and removed branching arteries and adventitial or epicardial adipose tissue (Supplemental Fig. 1). A 5mm segment of the mid-LAD and 10mm segnment of the distal abdominal aorta (AA) were graded by the pathologist for % intimal surface involvement of the following atherosclerotic changes: a) fatty streaks (FS); b) fibrous plaques (FP); c) complicated lesions (CL); and d) calcified lesions (CO), and subsequently submitted for proteomic analysis.
Protein Extraction, MS Analysis and Protein Identification
Arterial samples were pulverized in liquid nitrogen, homogenized and digested with trypsin (1:20). A total of 2.0 μg of peptides per sample were analyzed using label-free quantification on a reversed-phase liquid chromatography tandem mass spectrometry (RPLC–MS/MS) online with an Orbitrap Elite mass spectrometer (Thermo Scientific, USA) coupled to an Easy-nLC 1000 system (Thermo Scientific, USA). The analysis was operated in a data-dependent acquisition (DDA) mode in the Orbitrap analyzer, followed by tandem mass spectra of the 20 most abundant peaks in the linear ion trap. One LAD and one AA sample (from different subjects) were excluded because of poor protein yield leaving n=99 samples from each territory for analysis.
The MS/MS data was searched against the concatenated target/decoy 20 Human Uniprot 21 database as of July 24, 2015, with only reviewed and canonical sequences used. ProteinProphet 22 was then used to infer protein identifications. Label-free quantification and subsequent median normalization of each protein was performed using weighted spectral counting 23.
Statistical Analyses
Post-processing Quality Control and Data Imputation
A total of 1925 unambiguous proteins were detected in one or more of the 99 LAD samples. Of these proteins 944 had ≤50% missingness and 375 had no missingness in all 99 samples (Supplemental Fig. 2). Missing values for the 944 proteins with ≤ 50% missingness were imputed using a low rank approximation derived from non-linear iterative partial least squares (NIPALS) PCA 24 (Supplemental Fig. 2). In a similar manner the AA samples revealed 1495 unambiguous proteins including 725 with ≤50% missingness.
Weighted Co-Expression Network Modelling
The WGCNA package in R was used to identify distinct protein modules among the 944 proteins used for analysis 25. The power parameter was selected such that the topological overlap connectivity (k) of the entire network approximated a scale-free topology. To assess stability of module assignments, 50 bootstrap samples of the data were created and each one interrogated using identical WCGNA parameters.
Analysis of Association with Extent of Atherosclerosis or Anatomic Location
Regression Models
Both MANOVA and generalized linear models (GLM) with adjustment for age, sex, and race were used to model the distributions of % intimal surface demonstrating fibrous plaque (FP), fatty streaks (FS), and normal (NL) intima as a function of each individual protein. In a similar manner ordinal regression was used to examine the association between individual proteins and % FP treated as three level ordinal variables (FP: 0%, 1-59%, ≥60%).
When modeling the association between proteins identifed using data-independent acquisition (DIA-SWATH) and disease or location we also adjusted for for several housekeeping proteins (Proteasome subunit beta type-2, Small nuclear ribonucleoprotein Sm D3, Receptor expression-enhancing protein 5, Ras-related protein Rab-7a) and Myosin-11 (a vascular smooth muscle cell marker protein) to account for possible differences in tissue sample volumes, cellular composition, or protein yields.
Convex Analysis of Mixtures and Protein Expression Differences in Complex Tissues Analysis
Tissue heterogeneity, where multiple tissue types are variably mixed in each individual sample, represents a major confounder when seeking to identify tissue-specific disease markers 26-30. Accordingly, we used Convex Analysis of Mixtures (CAM) 31 to perform a fully unsupervised data deconvolution to define the optimal data-derived tissue types present in the heterogeneous samples and to characterize the molecular markers whose expressions are maximally enriched in each empirically defined tissue.
GO Term and Pathway Enrichment Analysis
GO Term and pathway enrichment analyses using GO Term Finder 32, and IPA (Ingenuity Pathway Analysis, Ingenuity Systems, Redwood City, CA) tools were used to characterize the evaluable proteins obtained from the arterial samples. For the analyses of selected normal and atherosclerosis enriched samples, the significant differentially expressed proteins with |fold change| ≥1.7 and the false discovery rate (FDR) corrected p-value of 0.05 were analyzed with IPA for pathways, upstream regulators, and associated diseases and biological functions. The fold change cut-point of ≥1.7 was chosen to yield approximately 100 proteins for further analysis. Sensitivity analyses were also performed using proteins selected purely on the basis of q-value < 0.01 for FP vs NL.
Differential Dependent Network Analyses
Significant rewiring of biologic networks provides a unique perspective on phenotypic transitions that can occur in biological systems33-35. We used knowledge-fused differential dependency networks (kDDN) 36-38 to systematically characterize significant network rewiring in the arterial proteome between normal and fibrous plaque samples. We then used permutation-based significance tests to estimate p-values of detected differential dependence edges 36, 38
Multiple Reaction Monitoring Assay Design
The 38 proteins identified by DDA-MS analysis to be most highly associated with fibrous plaques were selected as candidate circulating biomarker proteins. The top three to four performing peptides from each protein were included for downstream MRM-method building. Five proteins (DRB1, LCP1, RNASE1, MTHFD1, and CALU) were excluded at this step for absence of any detectable peptides in a reference plasma sample. Unscheduled MRM methods were developed against 323 fragments from 86 peptides for the remaining 33 putative FP marker proteins. The MRM data for plasma samples from the case and control CAD validation cohort were acquired on a SCIEX QTRAP6500 using multiple reaction monitoring (MRM) scanning in positive mode (Framingham, MA). Manual filtering eliminated an additional eight proteins from further analysis due to low or undetectable signal-to-noise ratios (CATB, TSP1, NNMT, S100A9, ITIH2, POSTN, CORO1A, and HTRA). The final 25 candidate proteins were processed to generate protein-level abundance data from the endogenous peptides.
Prediction of CAD from the Candidate Plasma Fibrous Plaque Biomarkers
An adaptive elastic net model (α=0.9)39-41 optimized by leave-one-out validation was used to select among the 25 candidate biomarkers for prediction of case status. Bias-corrected bootstrap sampling (n=10,000) was used to evaluate overall performance of the optimized model based on median (95% CI) AUC from the bootstrap samples 42.
Results
Global Analysis of Coronary and Abdominal Aortic Proteomes Identifies Novel Arterial Proteins and Scale-Free Network Topologies
Based on stringent quality control and calling criteria a total of 1925 unambiguous protein groups (hereinafter referred to as “proteins”) were identified in one or more left anterior descending (LAD) coronary artery or distal abdominal aorta (AA) samples, including 974 proteins present in 50% or more of the LAD or AA samples (Supplemental Table 1). The 974 proteins represent a wide range of biological processes, molecular functions, cellular components and canonical pathways43 (Supplemental Figs. 3-5 and Supplemental Tables 2-9). Of the 1925 proteins, 43 have not been previously described in thirteen prior studies that directly analyzed protein content of human arterial tissues (Supplemental Table 1). All but eight of these proteins were nevertheless predicted based on RNASeq analysis of human coronary oraortic samples from the GTEX Consortium 44.
The 944 proteins unique to the LAD exhibited a scale-free network topology typically seen in complex adaptive networks of molecular or cellular constituents from a variety of living organisms 45-47 (Fig. 1). Furthermore, these proteins included several distinct and reproducible co-expression modules that roughly correlated with specific cellular functions and locations such as mitochondrial proteins involved with cellular respiration (Red module), nuclear proteins involved with chromatin assembly and organization (Turquoise module), and extra cellular matrix proteins (Brown module). A similar scale-free topology and functional modular structure was also evident in the protein data from the AA (Supplemental Fig. 6.) A scale-free topology suggests that the arterial proteome may arise from a complex adaptive system with properties such as self-organized criticality, emergence, and resilience.46, 48, 49 Complex adaptive systems are uniquely well-suited for description using graph theory and network or non-linear dynamic modelling to reveal functional insights that may be less evident from more conventional linear conceptual models and methods33, 50, 51 (see below). The number and diversity of proteins detected and the fact that they exhibit a scale-free topology lends internal validity to our protein extraction and measurement approach and suggests that features of a complex adaptive system that are evident in gene transcript data remain after translation and protein catabolism.
Figure 1. Weighted Co-Expression Network Analysis of Human LAD Proteins.
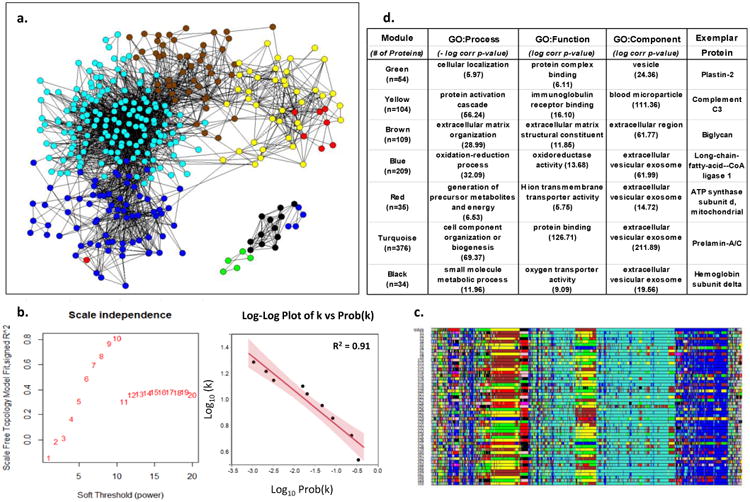
a. Adjacency map of LAD proteins color coded by module assignment based on hierarchical clustering of the topological overlap matrix (TOM)-based dissimilarity measure. For clarity of presentation only nodes (proteins) with at least one edge (adjacency measure (k)) > 97.5%tile are shown (n-544). b. Left panel shows that scale-free topology is best approximated when the adjacency power parameter β= 10. Right panel shows the log-log plot of adjacency (k) vs prob(k) with β= 10, confirming the power-law relationship in the connectivity of the expressed proteins. c. Module assignment for fifty 90% random samples of the data illustrating the overall stability of the modular structure of the protein expression patterns. Colors are assigned according cluster size which may vary with each random sample. As a result actual color assignment may vary from run to run, but module membership remains relatively stable. d. Top non-redundant GO Terms with Bonferroni corrected p-values and an exemplar protein for each module.
Comparison of Normal Coronary and Aortic Proteomes Reveal Significant Differences in Mitochondrial Protein Mass
Several hundred proteins were detected in the LAD but not in the AA samples (Supplemental Fig. 7). This pattern was observed when limiting the analysis to completely normal samples (n=30 in each territory) or when limiting the proteins to those present in ≥ 50% of the LAD and/or AA samples. GO term analysis of the proteins exclusively detected in the LAD indicated significant enrichment of mitochondrial proteins (p-value range 1.7×10-6 to 1.8×10-28).
To confirm this apparent differential abundance of mitochondrial proteins between LAD and AA samples we performed a more sensitive data independent acquisition MS (DIA-MS also known as SWATH) analysis of entirely normal samples (n=30 in each anatomic location), focusing on 114 quantified mitochondrial proteins involved with fatty acid metabolism, oxidative phosphorylation, tricarboxylic acid (TCA) cycle and mitochondrial biogenesis. To account for possible site and sample differences in cellular material or protein extraction yields the quantitative results were adjusted for several smooth muscle cell specific housekeeping proteins and age and sex of the autopsied cases. Overall, mitochondrial proteins were 1.98-fold more abundant in the LAD compared with the AA (p<0.001) including a 2.25 fold increase in oxidative phosphorylation proteins (p<0.001) and anisolated >10-fold excess of inorganic pyrophosphatase (p<0.001; Fig. 2). A similar comparison of soluble ECM proteins revealed only a small, albeit statistically significant (p=0.01) 6% excess in soluble ECM proteins in the AA samples compared with the LAD (Supplemental Fig. 8). A notable exception was tenascin which had a >10-fold excess in AA samples compared with LAD (p<0.0001).
Figure 2. Comparison of Mitochondrial Proteins in Normal LAD and AA Samples.
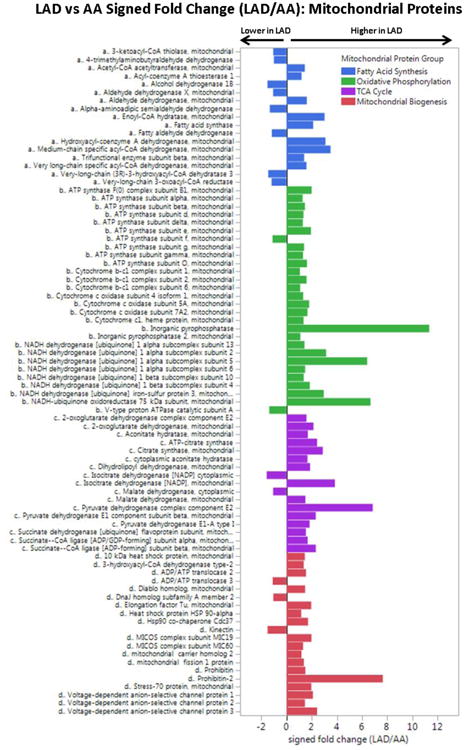
DIA-MS analysis of completely normal LAD and AA samples (n = 30 from each anatomic region) with adjustment for age, sex, MYH11, RABA7A, TERA, G6PI. LAD vs AA MANOVA p-values by mitochondrial protein group: fatty acid metabolism, p = 0.04; oxidative phosphorylation, p < 0.0001; TCA, p < 0.0001; mito biogenesis, p < 0.0001.
These data suggest fundamental differences in mitochondrial mass and potential aerobic capacity between LAD and AA tissues, possibly reflecting the differing energy requirements of these two arterial tissue types. This heterogeneity in the proteomic profile of two arterial tissues emphasize the need for arterial anatomic specificity when characterizing the proteomics and functional biology of arterial tissues – especially if considering mechanisms or interventions that involve metabolic pathways (see below).
Atherosclerotic tissues in both the LAD and AA present a proteomic profile consistent with TNF-α activation; however, the LAD also provides evidence of inhibition of PPAR-α, PPAR-γ, and insulin receptor regulated proteins - a pattern that is not evident in the AA
To define the proteomic profile of early atherosclerosis two complementary phenotyping strategies were used. First, each sample was graded by a vascular histopathologist according to %surface area involvement of normal intima (NL), fatty streak (FS) or fibrous plaque (FP). Extensive regression analyses (MANOVA, GLM, Ordinal Regression, and Elastic Net) were performed to identify proteins individually or jointly associated with %FP in the LAD and AA samples (Supplemental Tables 10-11, Supplemental Fig. 9). The internal validity of these data is supported by the presence and consistency across anatomic locations of several known protein markers of atherosclerosis from model systems among the top hits (e.g. Apo B-100, Ig mu chain C, CD5 antigen-like, plastin-2, tenascin, thrombospondin-1, cathepsin B, and vitronectin.)1
Second, convex analysis of mixtures52 (CAM) was used to de-convolve the global protein profiles from individual samples into unsupervised data-derived tissue phenotypes and to identify marker proteins associated with each phenotype (LAD: Fig. 3; AA: Supplemental Figures 10-11). This approach has the advantage of generating information about tissue phenotype from global protein profiles independent of the pathologist visual inspection of the arterial samples.
Figure 3. Convex Analysis of Mixtures of LAD Protein Data.
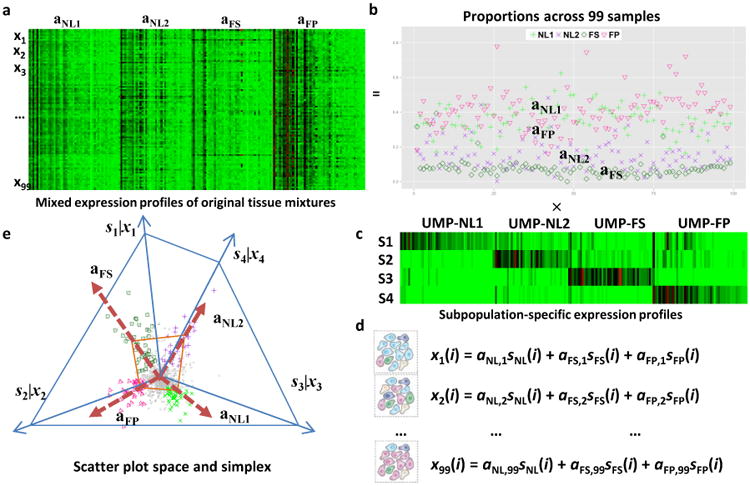
a. Heatmap of mixed expressions of upregulated marker proteins (UMPs) in 99 LAD samples. b. Estimated proportions of NL1, NL2, FS, and FP across 99 LAD samples. c. Heatmap of subpopulation-specific expressions of UMPs. d. Mathematical description on the i-th protein expression readout ‘x’ as a weighted sum of the protein expressions in the distinctive tissue types ‘s’ present in the heterogeneous samples, weighted by the mixing proportions ‘a’. e. Geometry of the mixing operation in scatter space that produces a compressed and rotated scatter simplex whose vertices host subpopulation-specific UMPs and correspond to mixing proportions.
To take full advantage of both phenotyping strategies we used principal component analysis and hierarchical clustering of the pathologist- and CAM-derived phenotype data to produce a patho-proteomic classification for each LAD sample (Fig. 4). Clusters at the extremes of the first principle component identified samples highly enriched with FP or NL tissue (FP: n=15; NL: n=30) with little or no confounding from fatty streaks from either a gross pathology or global proteomic perspective.
Figure 4. Patho-Proteomic Phenotyping.
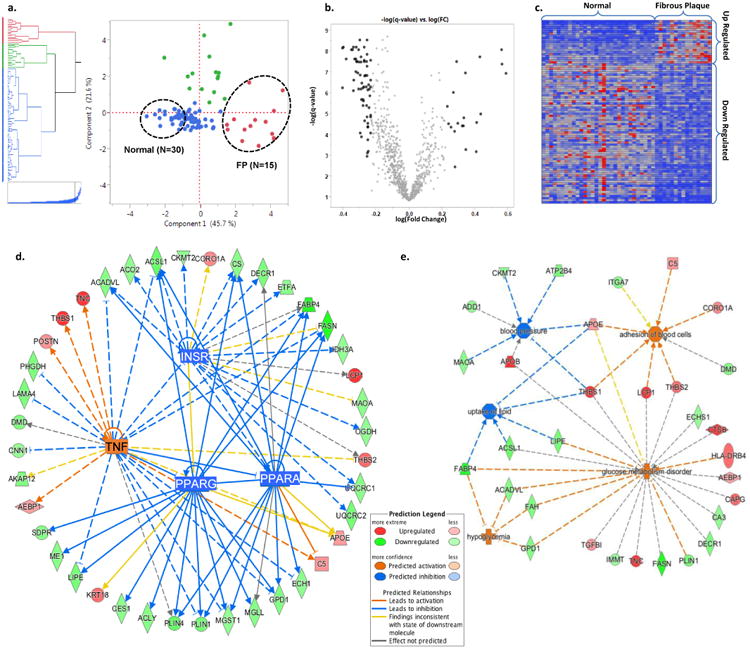
a. Hierarchical clustering and principal component score plot of the pathologist grading of extent of FP, FS, and normal tissue combined with CAM-derived estimates of proportion of four different empirical tissue types. Dashed circles indicate samples (2:1) from the extremes of the first principal component which separates fibrous plaques and normal arterial tissue. b. Volcano plot of log(fold change:FP/Normal) vs. -log(t-test q-value) for 944 arterial proteins. Black data points indicate proteins with a fold-change of >1.7 (or < 1/1.7) and a q-value of <0.05. (Two proteins with –log(q-value)>10 not shown.) c. Heatmap of the spectral count values for the N=88 proteins meeting the fold-change and q-value criteria noted in panel b. The individual proteins are listed in Supplementary Tables 8 and 9. d. A combined network map of the significantly up- and down-regulated proteins that are consistent with effects of an upstream master regulator. The observed pattern of proteins in FP are highly suggestive of TNF activation (p=1.64E-6, z-score=3.19), and inhibition of PPAR-α (p=3.55E-10, z-score=-3.06), PPAR-γ (p=2.87E-10, z-score=-3.02), and the insulin receptor (1.41E-12, z-score=-2.16). PPAR-α, PPAR-γ, and the insulin receptor pathways are themselves inhibited by TNF activation. When using >1.5 fold-change criteria, several additional regulatory networks were also identified (e.g. TGFB1, TP53, SP1, MYC). e. Predicted disease processes and their affiliated proteins significantly overrepresented by the up- and down-regulated proteins in fibrous plaques. The data are consistent with major alterations in cell-cell adhesion/interaction (p = 3.64E-03), lipid uptake (p = 1.67E-03), glucose homeostasis (p = 2.83E-06) and blood pressure regulation (p = 3.68E-03).
Comparing these FP-, and NL-enriched LAD samples identified eighty-nine (n=89) individual proteins with ≥(+/-) 1.7 fold-difference and a t-test q-value of ≤0.05 for FP vs NL (Supplemental Tables 12 and 13). Bioinformatic functional analysis of these atherosclerosis associated proteins revealed a pattern consistent with activation of the TNF-α pathway (p=2.64E-07), but also inhibition of insulin receptor, PPAR-α and PPAR-γ pathways (p=4.22E-10, 2.42E-13, 8.56E-16 respectively) (Fig. 4, Supplemental Table 14 and 15). A similar analysis of the atherosclerosis proteins in the AA samples (FP: n= 9, NL: N= 18) also produced a pattern consistent with TNF activation (p=1.92E-05) similar to the LAD (Supplemental Tables 16 and 17) and highlighted a core group of n=19 early atherosclerosis-associated proteins that were shared across both anatomic territories (Supplemental Table 18). However, in the AA sample proteomes there was no compelling evidence of inhibition of the insulin receptor, PPAR-α or PPAR-γ pathways (Supplemental Tables 14). In sensitivity analyses using only a t-test q-value of ≤0.01 for FP vs NL samples, qualitively similar results concerning upstream regulators and regulated pathways were observed (Supplemental Table 19). The apparent inhibition of the insulin receptor, PPAR-α or PPAR-γ pathways in atherosclerotic samples from the LAD and AA suggests that there are fundamental metabolic derangements
Differential network analysis of coronary and aortic proteomes indicate divergent mitochondrial dynamics in the setting of atherosclerosis characterized by reduced mitochondrial mass in coronary arteries that is not evident in in the distal abdominal aorta
An important feature of complex adaptive systems is the potential for network topologies to change under different conditions. Accordingly, we used differential dependent network (DDN) analysis of the FP-associated proteins identified above to select proteins pivotal in the re-wiring of the network structure between NL and FP in the LAD samples (Fig. 5). Analysis of n=26 re-wiring hub proteins revealed significant enrichment of TCA proteins (p=4.8×10-6). Subsequent analysis of individual TCA proteins documented an average 60% reduction in TCA proteins in FP enriched samples vs. NL.
Figure 5. Differential Dependent Network Analysis of LAD Fibrous Plaque Proteins.
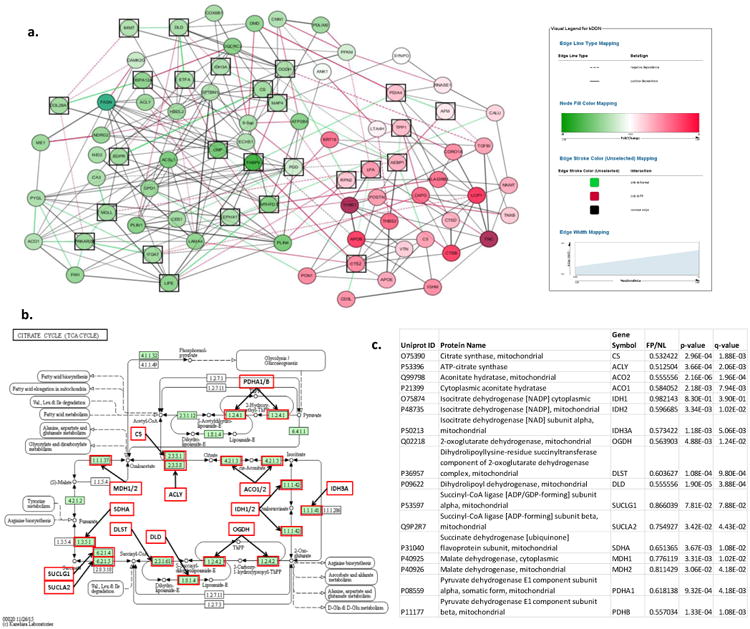
a. Plot depicts re-wiring of the protein network between FP and normal samples. Green nodes are down-regulated and red nodes are up-regulated in FP samples. Green edges indicate significant correlation in normal samples, but not FP samples. Red edges indicate significant correlation in FP samples. Black squares indicate differential network “hub proteins” (ie. proteins with different couplings to network partners in FP and normal samples.) GO term analysis of the differential network hub proteins indicated significant enrichment of TCA proteins (p=4.8×10-6). b. TCA cycle proteins with MS data available for additional anlaysis. Every protein indicated by a red box was quantitatively lower in FP samples than in normal samples after adjustment for housekeeping proteins, age and sex. c. Statistical comparison of TCA proteins in FP vs normal LAD samples.
DIA-MS analysis of the same broad-based panel of n=114 mitochondrial proteins used previously was performed comparing FP vs NL samples from the LAD (Fig. 6). The results document a consistent reduction of a wide range of mitochondrial proteins in the FP samples compared to NL samples after adjustment for vascular smooth muscle specific housekeeping proteins, age and sex. In contrast, a similar analysis of the same proteins in AA samples revealed a much less consistent and non-statistically significant pattern of mitochondrial protein suppression. To determine if this was a mitochondrial specific phenomenon we also performed targeted DIA-MS analysis of a targeted panel of soluble ECM proteins (n=77) and found a modest atherosclerosis-associated increase of soluble ECM proteins in both territories (LAD mean fold-increase = 1.25, MANOVA p-value= 0.02; AA mean fold increase = 1.78 fold increase, p-value = 0.017); although there were specific examples of anatomic discordance that deserve further study (e.g. laminins and nidogens) (Supplemental Fig. 12). Collectively, these data demonstrate how the concept of network re-wiring can produce biologically coherent insights that may not be evident from conventional statistical or pathway enrichment analysis strategies.
Figure 6. Comparison of Mitochondrial Proteins in FP vs NL Samples from the LAD and AA.
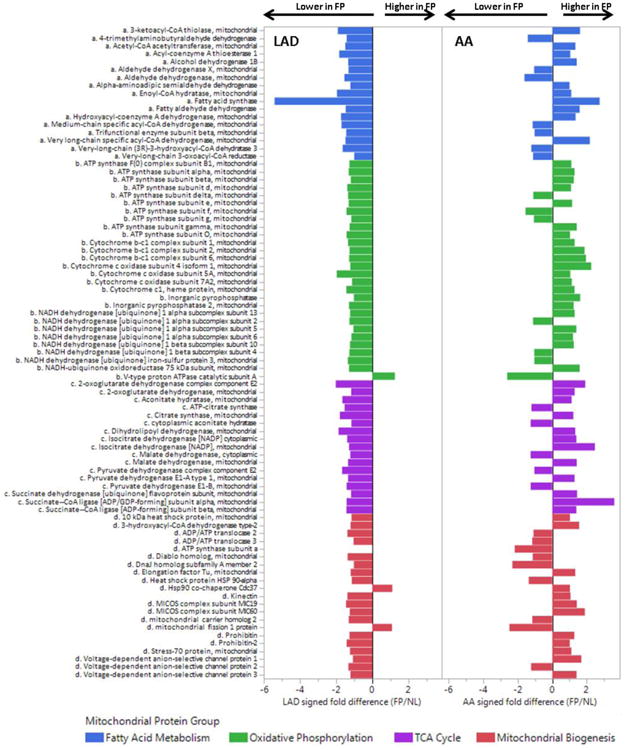
DIA-MS analysis was used to compare a targeted set of mitochondrial proteins in n=15 FP and n=30 NL LAD samples after adjustment for age, sex, MYH11, RABA7A, TERA, G6PI. Histogram bars indicate relative difference ebetween FP and NL samples in each anaotomic location. LAD MANOVA p-values for each mitochondrial protein group: p < 0.0001 for each group; AA MANOVA p-values for each mitochondrial protein group: p= n.s for each group.
Clinical Validation of the Plasma Multiplex Atherosclerosis Biomarker Panel
To establish that fibrous plaque proteins discovered using human arterial samples could serve as informative plasma biomarkers of coronary atherosclerosis we developed a highly reproducible multiple reaction monitoring (MRM) assay for a subset of 25 of these proteins that were measurable in plasma (Supplemental Figure 13). Subsequently, we compared fasting levels of these proteins in 45 women with angiographically verified coronary atherosclerosis (cases) and 41 similar- aged women who were free of signs, symptoms or risk factors for coronary disease (controls) (Methods). An elastic net model identified 13 proteins that jointly contributed to prediction of case status while also avoiding overfitting (Fig. 7: A and B). The bias corrected median (95%CI) AUC for this model based on 10,000 bootstrap samples was 0.92 (0.83-0.96) and the median misclassification rate was 0.13. Among individual proteins with the largest effects sizes vitronectin, TGF-beta-induced protein, complement factor 7 and apo B were positively associated with presence of coronary atherosclerosis and tripeptidyl-peptidase 1, ITI heavy chain H1, and leukotriene A-4 hydrolase were negatively associated.
Figure 7. Clinical Validation of Fibrous Plaque Proteins.
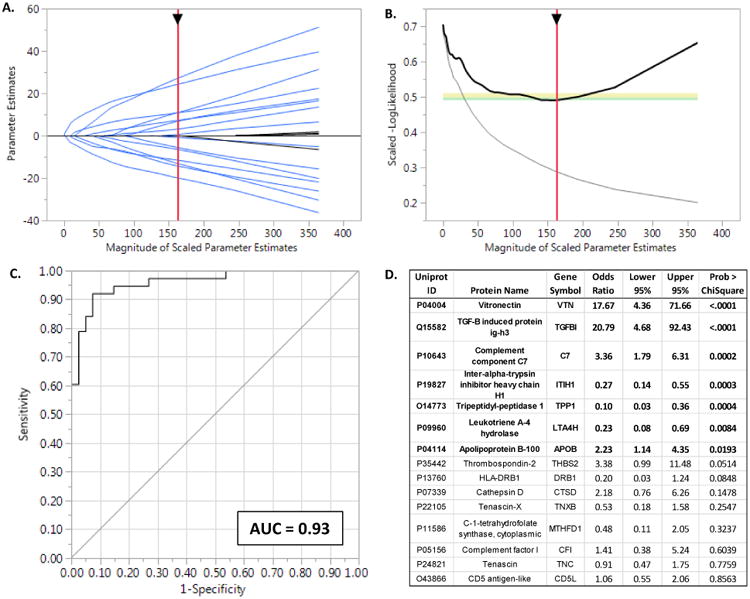
A. Solution path for the elastic net model (α = 0.9) with parameter estimates as a function of increasing number of variables entered into the model as reflected by the sum of the Scaled Parameter Estimates. The generally monotonically increasing solution paths suggest relative stability and no major interactions among the proteins included in the model. B. The-LogLikelihood derived from the leave-one-out validation samples as a function of increasing number of variables entered into the model. The negative inflection point indicates the optimal feature selection after which adding additional variables lead to worsening (increasing) –LogLikelihoods due to overfitting of the data. C. The ROC curve for the optimal model predicting presence of angiographically proven coronary disease. Bootstrap estimates of the AUC and its 95% CI based on 10,000 samples were 0.93 and 0.85-0.97 respectively. D. Parameter estimates for proteins included in the plasma protein biomarker panel. Six proteins (indicated in bold) had the strongest evidence for contributing to model performance. However, elastic net models permit inclusion of additional variables that may be correlated with other terms in the model. These proteins may not individually contribute greatly to the overall prediction performance, but nevertheless participate in underlying mechanistic pathways worthy of further consideration.
Discussion
The results here provide a comprehensive survey of human coronary and aortic proteins and identify individual proteins, protein networks and regulatory pathways that differ between arterial beds or that are indicative of early atherosclerosis. These data can be distinguished from prior work because of the inclusion of human coronary arteries which are responsible for the majority of acute ischemic vascular disease morbidity and mortality, the number and diversity of proteins identified, and the fact that these proteins were obtained from a large number of samples where the effects of local tissue context and natural biologic variation are manifest. Several analytic methods were used to illuminate complex networks involving many proteins that jointly signify arterial health and disease. Importantly, these data also show that proteins discovered using comprehensive tissue proteomics can be used to develop highly informative multiplex plasma biomarker assays with direct clinical utility. The publicly accessible data generated by this work, and the analytic methods depicted here represent additional resources that may lead to a wide range of future translational research concerning human arterial protein biology.
There are several notable findings among the many results reported here. First, the human arterial proteome exhibits features consistent with a complex adaptive network, reiterating the concept that complex adaptive networks are an overarching organizational feature common to many biologic systems53. Transcriptional profiles are well known to exhibit features of a complex adaptive system54. To our knowledge this has not been demonstrated at the protein level in human arterial tissue in such a large number of samples as described here; although others have described features of a complex adaptive system in proteomic analysis of human ovarian tumors55. The fact that our data do indeed resemble a complex adaptive network lends internal validity to our rendering of the arterial proteome. Looking at the arterial proteome through the lens of complexity theory including concepts such as scale-invariance (as documented here), self-organized criticality, emergence, and resiliency46, 48, 49 may produce insights that have escaped more narrowly focused investigations of a smaller number of proteins or a more linear and deterministic framework. The differential dependent networks presented here emphasize this point by defining dynamic features of complex protein networks that signify normal versus atherosclerotic arteries and by using key elements of these dynamic networks to discover important aspects of the proteome in atherosclerosis that were not easily evident by other means.
Second, the data reveal significant anatomic variation in the abundance of mitochondrial proteins in normal arterial samples, suggesting that coronary arteries likely have considerably greater aerobic capacity than the distal aorta. This fits our understanding of the different embryology28 and normal physiologic roles of these distinct regions of the arterial system with the coronary arteries having greater energy requirements associated with regulation of coronary blood flow compared to the distal aorta which serves primarily as passive conduit for blood delivery to the lower extremities. To our knowledge this substantial variation in mitochondrial protein mass between coronary and aortic tissues in humans has not been previously documented.
Third, the data indicate profound anatomic differences in key metabolic regulatory pathways and mitochondrial dynamics in the setting of atherosclerosis. Specifically, the data reveal a broad-based reduction of mitochondrial protein mass and a proteomic pattern consistent with inhibition of PPAR-α, PPAR-γ, and insulin receptor regulated pathways in atherosclerotic coronary arteries, but not in similarly diseased distal abdominal aortic samples. These data provide critical anatomic specificity to the growing body of evidence that mitochondrial dysfunction and altered mitochondrial dynamics are also central features of atherosclerosis 56,15,57, and may provide a mechanistic framework to explain the anatomic dimorphism between coronary and peripheral arterial disease with respect to conventional and genetic risk factors for atherosclerosis.58, 59 It remains to be determined whether the reduction in mitochondrial protein mass in coronary atherosclerosis is a consequence of oxidative, or Ca++-mediated mitochondrial damage and mitophagy, impaired mitochondrial biogenesis, or both. It is interesting to note that the concurrent increases in the lysosomal cathepsins in the atherosclerotic samples may signify enhanced lysosomal biogenesis useful for targeted degradation of damaged mitochondria.56
The data also reassuringly highlight numerous individual proteins and a global proteomic pattern of TNF-α activation in the FP samples consistent with several decades of research documenting the role of inflammation in the pathogenesis of atherosclerosis. 60 However, even the more familiar indicators of TNF-α activation and inflammation exhibited substantial anatomic variation from LAD to distal aorta in our data.
Recognizing the anatomic heterogeneity with respect to mitochondrial and metabolic enzyme profiles has important implications for research on arterial health and disease. Many of the pathogenic mechanisms and targeted therapies for atherosclerosis are directly related to fatty acid metabolism or aerobic energy biosynthesis and redox homeostasis61. Unfortunately, a great deal of our understanding of arterial biology has been generated without regard to anatomic distribution. For instance many animal models of atherosclerosis rely on aortic, femoral or carotid manifestations of disease with the assumption that findings are generalizable to human coronary arteries. Likewise the overwhelming majority of human arterial proteomic data have been generated from carotid arteries – typically severely diseased endarterectomy specimens. The current data suggest that a complete understanding of the pathogenesis and prevention of human arterial atherosclerosis may require more anatomically specific methods of interrogation.
Finally, the data demonstrate the translational potential of proteomics from complex human tissues to identify clinically informative plasma biomarkers. While significant portions of the arterial proteome reside in compartments inaccessible to the plasma blood pool, there are many others that are secreted or released into the circulation, and the plasma concentrations for some of these, such as vitronectin, are uniquely informative about the presence of early atherosclerosis. The prominent contribution of vitronectin as an atherosclerosis biomarker is plausible based on accumulating evidence that this polyfunctional matricellular protein plays a critical role in organizing the earliest response to tissue injury62. Similarly compelling arguments can be made in support of many of the other atherosclerosis associated proteins (e.g. CD5 antigen-like, complement factor I, complement component C7, etc). However, based on the data presented here, early atherosclerosis is likely best revealed through changes in a complex array of many proteins rather than any single sentinel protein. Fortunately, recent advances in mass spectrometry technology now permit sophisticated multiplex assays to measure many more proteins with very small plasma volumes – making multidimensional proteomics assays such as the one described here a clinical possibility in the near future.
Several limitations in the current work should be considered. First, although a comprehensive analysis of 200 human arterial specimens is a considerable technical accomplishment, and despite the fact that we used stringent statistical methods and other information theoretical considerations to minimize false positives (generally to ≤ 5%), there remains the possibility of residual confounding or limited statistical power which could obscure both positive and negative associations. The fact that many of the sentinel findings generated by the initial data dependent MS approach were subsequently confirmed and strengthened by a separate data independent acquisition MS method (which uses different peptides for protein identification) provides further support for the validity of the findings. Second, highly crosslinked ECM proteins can be relatively refractory to extraction without specialized solubilization strategies 63. Accordingly, there may be differences in tissue ECM composition by anatomic location or disease status that are not evident based on the soluble fraction examined here. This limitation not withstanding, we were able to achieve excellent coverage excellent ECM coverage including collagens, fibronectin, elastins, lamins including feparin sulfate and chondroitin sulfate proteoglycans (see Supplemental Table 1). We attribute this to the fact that we used both DDA and DIA acquisitions to maximize peptide detection and protein identification. Third, the fact that the samples were all collected post-mortem raises questions about possible effects of death on the stability of individual proteins or specific cellular functional processes. If such effects are differentially manifest in normal versus atherosclerotic tissues, this could give the appearance of differences by disease status that are not present during life. It is reassuring that many of the individual proteins identified here overlap with findings from animal models of atherosclerosis and in-vitro studies where the effects of post-mortem hypoxia and cessation of cellular metabolic activity are more easily minimized. Fourth, the observed differences in the proteome between normal and diseased samples represent a unique, albeit macroscopic view of the proteomic architecture of human coronary and distal aortic atherosclerosis. More work is required to clarify the functional role of the specific proteins, protein networks and pathways identified here, and to establish if there is therapeutic value in manipulating them. Nevertheless the data already reveal the potential clinical utility of a diverse array of plasma proteins as atherosclerosis biomarkers even as their mechanistic underpinning continue to be explored.
In summary, these data represent the most comprehensive description of the human coronary and aortic proteome to date and reveal numerous proteins, networks and pathways that are strongly indicative of early atherosclerosis. They also indicate fundamental differences in mitochondrial dynamics between the coronary artery and the distal aorta in both normal and atherosclerotic conditions. The data highlight the value of new methods to extricate tissue phenotypes from heterogeneous tissue samples and depict dynamic features of protein networks that vary as a function of disease state and anatomic location. Finally the results illustrate the potential utility of using tissue proteomics to identify plasma biomarker and provide evidence of the clinical utility or a multiplex biomarker panel to indicate the presence of coronary atherosclerosis. Altogether these data and methods establish a new foundation for research to better understand the human arterial proteomic architecture in health and disease.
Supplementary Material
Clinical Perspective.
What is new?
Proteomic analysis of human coronary arteries (n=100) and aortas (n=100) identified hundereds of proteins (n=1925), and numerous networks and pathways that are associated with early atherosclerosis.
Atherosclerotic samples had significant reductions in mitochondrial protein abundance.
Atherosclerostic sample proteins indicated inhibition of the insulin receptor, PPAR-α, and PPAR-γ regulated pathways and activation of the TNF regulated pathway.
A 13-protein plasma multiplex assay based on the most promising atherosclerosis-associated proteins was strongly predictive of angiographically defined CAD in an independent clinical cohort (AUC =0.92; 95% CI:0.83-0.96).
What are the clinical implications?
High resolution mass-spec proteomics and network analyses can produce new mechanistic insights and indentify novel biomarkers for presence of subclinical atherosclerosis.
Advances in sample collection and high-throughput processing mean that multidimensional proteomics assays, including proteins identified here will be available to support precision medicine treatment and prevention in the near future.
Acknowledgments
N/A
Sources of Funding: This work was supported by 1R01HL111362 from the, National Heart, Lung and Blood Institute of the National Institutes of Health
Footnotes
Disclosures: None
References
- 1.Hanzawa H, Sakamoto T, Kaneko A, Manri N, Zhao Y, Zhao S, Tamaki N, Kuge Y. Combined Plasma and Tissue Proteomic Study of Atherogenic Model Mouse: Approach To Elucidate Molecular Determinants in Atherosclerosis Development. J Proteome Res. 2015;14:4257–4269. doi: 10.1021/acs.jproteome.5b00405. [DOI] [PubMed] [Google Scholar]
- 2.Jing L, Parker CE, Seo D, Hines MW, Dicheva N, Yu Y, Schwinn D, Ginsburg GS, Chen X. Discovery of biomarker candidates for coronary artery disease from an APOE-knock out mouse model using iTRAQ-based multiplex quantitative proteomics. Proteomics. 2011;11:2763–2776. doi: 10.1002/pmic.201000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mayr M, Chung YL, Mayr U, Yin X, Ly L, Troy H, Fredericks S, Hu Y, Griffiths JR, Xu Q. Proteomic and metabolomic analyses of atherosclerotic vessels from apolipoprotein E-deficient mice reveal alterations in inflammation, oxidative stress, and energy metabolism. Arterioscler Thromb Vasc Biol. 2005;25:2135–2142. doi: 10.1161/01.ATV.0000183928.25844.f6. [DOI] [PubMed] [Google Scholar]
- 4.Fach EM, Garulacan LA, Gao J, Xiao Q, Storm SM, Dubaquie YP, Hefta SA, Opiteck GJ. In vitro biomarker discovery for atherosclerosis by proteomics. Mol Cell Proteomics. 2004;3:1200–1210. doi: 10.1074/mcp.M400160-MCP200. [DOI] [PubMed] [Google Scholar]
- 5.Conway JP, Kinter M. Proteomic and transcriptomic analyses of macrophages with an increased resistance to oxidized low density lipoprotein (oxLDL)-induced cytotoxicity generated by chronic exposure to oxLDL. Mol Cell Proteomics. 2005;4:1522–1540. doi: 10.1074/mcp.M500111-MCP200. [DOI] [PubMed] [Google Scholar]
- 6.Olson FJ, Sihlbom C, Davidsson P, Hulthe J, Fagerberg B, Bergstrom G. Consistent differences in protein distribution along the longitudinal axis in symptomatic carotid atherosclerotic plaques. Biochem Biophys Res Commun. 2010;401:574–580. doi: 10.1016/j.bbrc.2010.09.103. [DOI] [PubMed] [Google Scholar]
- 7.Liang W, Ward LJ, Karlsson H, Ljunggren SA, Li W, Lindahl M, Yuan XM. Distinctive proteomic profiles among different regions of human carotid plaques in men and women. Sci Rep. 2016;6:26231. doi: 10.1038/srep26231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Didangelos A, Yin X, Mandal K, Saje A, Smith A, Xu Q, Jahangiri M, Mayr M. Extracellular matrix composition and remodeling in human abdominal aortic aneurysms: a proteomics approach. Mol Cell Proteomics. 2011;10:M111.008128. doi: 10.1074/mcp.M111.008128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porcelli B, Ciari I, Felici C, Pagani R, Banfi C, Brioschi M, Giubbolini M, de Donato G, Setacci C, Terzuoli L. Proteomic analysis of atherosclerotic plaque. Biomed Pharmacother. 2010;64:369–372. doi: 10.1016/j.biopha.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 10.Lepedda AJ, Cigliano A, Cherchi GM, Spirito R, Maggioni M, Carta F, Turrini F, Edelstein C, Scanu AM, Formato M. A proteomic approach to differentiate histologically classified stable and unstable plaques from human carotid arteries. Atherosclerosis. 2009;203:112–118. doi: 10.1016/j.atherosclerosis.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de la Cuesta F, Alvarez-Llamas G, Maroto AS, Donado A, Zubiri I, Posada M, Padial LR, Pinto AG, Barderas MG, Vivanco F. A proteomic focus on the alterations occurring at the human atherosclerotic coronary intima. Mol Cell Proteomics. 2011;10:M110.003517. doi: 10.1074/mcp.M110.003517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bagnato C, Thumar J, Mayya V, Hwang SI, Zebroski H, Claffey KP, Haudenschild C, Eng JK, Lundgren DH, Han DK. Proteomics analysis of human coronary atherosclerotic plaque: a feasibility study of direct tissue proteomics by liquid chromatography and tandem mass spectrometry. Mol Cell Proteomics. 2007;6:1088–1102. doi: 10.1074/mcp.M600259-MCP200. [DOI] [PubMed] [Google Scholar]
- 13.Didangelos A, Yin X, Mandal K, Baumert M, Jahangiri M, Mayr M. Proteomics characterization of extracellular space components in the human aorta. Mol Cell Proteomics. 2010;9:2048–2062. doi: 10.1074/mcp.M110.001693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mayr M, Grainger D, Mayr U, Leroyer AS, Leseche G, Sidibe A, Herbin O, Yin X, Gomes A, Madhu B, Griffiths JR, Xu Q, Tedgui A, Boulanger CM. Proteomics, metabolomics, and immunomics on microparticles derived from human atherosclerotic plaques. Circ Cardiovasc Genet. 2009;2:379–388. doi: 10.1161/CIRCGENETICS.108.842849. [DOI] [PubMed] [Google Scholar]
- 15.Viiri LE, Full LE, Navin TJ, Begum S, Didangelos A, Astola N, Berge RK, Seppala I, Shalhoub J, Franklin IJ, Perretti M, Lehtimaki T, Davies AH, Wait R, Monaco C. Smooth muscle cells in human atherosclerosis: proteomic profiling reveals differences in expression of Annexin A1 and mitochondrial proteins in carotid disease. J Mol Cell Cardiol. 2013;54:65–72. doi: 10.1016/j.yjmcc.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 16.Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, Connolly ES, Solomon R, Jones DM, Heyer EJ, Spite M, Tabas I. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016;7:12859. doi: 10.1038/ncomms12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Preil SA, Kristensen LP, Beck HC, Jensen PS, Nielsen PS, Steiniche T, Bjorling-Poulsen M, Larsen MR, Hansen ML, Rasmussen LM. Quantitative Proteome Analysis Reveals Increased Content of Basement Membrane Proteins in Arteries From Patients With Type 2 Diabetes Mellitus and Lower Levels Among Metformin Users. Circ Cardiovasc Genet. 2015;8:727–735. doi: 10.1161/CIRCGENETICS.115.001165. [DOI] [PubMed] [Google Scholar]
- 18.Maleki S, Kjellqvist S, Paloschi V, Magne J, Branca RM, Du L, Hultenby K, Petrini J, Fuxe J, Lehtio J, Franco-Cereceda A, Eriksson P, Bjorck HM. Mesenchymal state of intimal cells may explain higher propensity to ascending aortic aneurysm in bicuspid aortic valves. Sci Rep. 2016;6:35712. doi: 10.1038/srep35712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doll S, Dressen M, Geyer PE, Itzhak DN, Braun C, Doppler SA, Meier F, Deutsch MA, Lahm H, Lange R, Krane M, Mann M. Region and cell-type resolved quantitative proteomic map of the human heart. Nat Commun. 2017;8:1469. doi: 10.1038/s41467-017-01747-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 21.Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O'Donovan C, Redaschi N, Yeh LS. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004;32:D115–119. doi: 10.1093/nar/gkh131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 23.Vogel C, Marcotte EM. Label-free protein quantitation using weighted spectral counting. Methods Mol Biol. 2012;893:321–341. doi: 10.1007/978-1-61779-885-6_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Candès E, Recht B. Exact Matrix Completion via Convex Optimization. Foundations of Computational mathematics. 2009;9.6:717–772. [Google Scholar]
- 25.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 26.Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, Teichmann SA, Marioni JC, Stegle O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol. 2015;33:155–160. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- 27.Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, Wildberg A, Gao D, Fung HL, Chen S, Vijayaraghavan R, Wong J, Chen A, Sheng X, Kaper F, Shen R, Ronaghi M, Fan JB, Wang W, Chun J, Zhang K. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352:1586–1590. doi: 10.1126/science.aaf1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guintivano J, Aryee MJ, Kaminsky ZA. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013;8:290–302. doi: 10.4161/epi.23924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuhn A, Thu D, Waldvogel HJ, Faull RL, Luthi-Carter R. Population-specific expression analysis (PSEA) reveals molecular changes in diseased brain. Nat Methods. 2011;8:945–947. doi: 10.1038/nmeth.1710. [DOI] [PubMed] [Google Scholar]
- 30.Rahmani E, Zaitlen N, Baran Y, Eng C, Hu D, Galanter J, Oh S, Burchard EG, Eskin E, Zou J, Halperin E. Sparse PCA corrects for cell type heterogeneity in epigenome-wide association studies. Nat Methods. 2016;13:443–445. doi: 10.1038/nmeth.3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang N, Hoffman EP, Chen L, Chen L, Zhang Z, Liu C, Yu G, Herrington DM, Clarke R, Wang Y. Mathematical modelling of transcriptional heterogeneity identifies novel markers and subpopulations in complex tissues. Sci Rep. 2016;6:18909. doi: 10.1038/srep18909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. GO∷TermFinder--open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20:3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitra K, Carvunis AR, Ramesh SK, Ideker T. Integrative approaches for finding modular structure in biological networks. Nat Rev Genet. 2013;14:719–732. doi: 10.1038/nrg3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu JX, Thomas CE, Brunak S. Network biology concepts in complex disease comorbidities. Nat Rev Genet. 2016;17:615–629. doi: 10.1038/nrg.2016.87. [DOI] [PubMed] [Google Scholar]
- 35.Hudson NJ, Dalrymple BP, Reverter A. Beyond differential expression: the quest for causal mutations and effector molecules. BMC Genomics. 2012;13:356. doi: 10.1186/1471-2164-13-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang B, Tian Y, Jin L, Li H, Shih Ie M, Madhavan S, Clarke R, Hoffman EP, Xuan J, Hilakivi-Clarke L, Wang Y. DDN: a caBIG(R) analytical tool for differential network analysis. Bioinformatics. 2011;27:1036–1038. doi: 10.1093/bioinformatics/btr052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian Y, Zhang B, Hoffman EP, Clarke R, Zhang Z, Shih Ie M, Xuan J, Herrington DM, Wang Y. KDDN: an open-source Cytoscape app for constructing differential dependency networks with significant rewiring. Bioinformatics. 2015;31:287–289. doi: 10.1093/bioinformatics/btu632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian Y, Zhang B, Hoffman EP, Clarke R, Zhang Z, Shih IeM, Xuan J, Herrington DM, Wang Y. Knowledge-fused differential dependency network models for detecting significant rewiring in biological networks. BMC Syst Biol. 2014;8:87. doi: 10.1186/s12918-014-0087-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tibshirani R. Regression Shrinkage and Selection via the Lasso. (B).J R Stat Soc. 1996;58:267–288. [Google Scholar]
- 40.Zou H, Hastie T. Regularization and Variable Selection via the Elastic Net. (B).J R Stat Soc. 2005;67:301–320. [Google Scholar]
- 41.Zou H, Zhang H. On the Adaptive Elastic-Net with A Diverging Number of Parameters. Ann Stat. 2009;37:1733–1751. doi: 10.1214/08-AOS625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Efron B. Nonparametric Standard Errors and Confidence Intervals. Can J Stat. 1981;9:139–158. [Google Scholar]
- 43.Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43:D1049–1056. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. doi: 10.1038/nature24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carter SL, Brechbuhler CM, Griffin M, Bond AT. Gene co-expression network topology provides a framework for molecular characterization of cellular state. Bioinformatics. 2004;20:2242–2250. doi: 10.1093/bioinformatics/bth234. [DOI] [PubMed] [Google Scholar]
- 46.Jeong H, Tombor B, Albert R, Oltvai ZN, Barabasi AL. The large-scale organization of metabolic networks. Nature. 2000;407:651–654. doi: 10.1038/35036627. [DOI] [PubMed] [Google Scholar]
- 47.Bergmann S, Ihmels J, Barkai N. Similarities and differences in genome-wide expression data of six organisms. PLoS Biol. 2004;2:E9. doi: 10.1371/journal.pbio.0020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitchell M. Complexity: A Guided Tour New York. New York: Oxford University Press; 2009. [Google Scholar]
- 49.Gao J, Barzel B, Barabasi AL. Universal resilience patterns in complex networks. Nature. 2016;536:238. doi: 10.1038/nature18019. [DOI] [PubMed] [Google Scholar]
- 50.Creixell P, Schoof EM, Erler JT, Linding R. Navigating cancer network attractors for tumor-specific therapy. Nat Biotechnol. 2012;30:842–848. doi: 10.1038/nbt.2345. [DOI] [PubMed] [Google Scholar]
- 51.Ideker T, Krogan NJ. Differential network biology. Mol Syst Biol. 2012;8:565. doi: 10.1038/msb.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang N, Meng F, Chen L, Madhavan S, Clarke R, Hoffman E, JXuan J, Wang Y. The CAM Software for Nonnegative Blind Source Separation in R-Java. J Mach Learn Res. 2013;14:2899–2903. [Google Scholar]
- 53.Boccaletti S, Latora V, Moreno Y, Chavez M, Hwang D. Complex Networks: Structure and Dynamics. Phys Rep. 2006;424:175–308. [Google Scholar]
- 54.Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- 55.Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou JY, Petyuk VA, Chen L, Ray D, Sun S, Yang F, Chen L, Wang J, Shah P, Cha SW, Aiyetan P, Woo S, Tian Y, Gritsenko MA, Clauss TR, Choi C, Monroe ME, Thomas S, Nie S, Wu C, Moore RJ, Yu KH, Tabb DL, Fenyo D, Bafna V, Wang Y, Rodriguez H, Boja ES, Hiltke T, Rivers RC, Sokoll L, Zhu H, Shih IM, Cope L, Pandey A, Zhang B, Snyder MP, Levine DA, Smith RD, Chan DW, Rodland KD. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell. 2016;166:755–765. doi: 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mercer JR, Cheng KK, Figg N, Gorenne I, Mahmoudi M, Griffin J, Vidal-Puig A, Logan A, Murphy MP, Bennett M. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ Res. 2010;107:1021–1031. doi: 10.1161/CIRCRESAHA.110.218966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sergin I, Evans TD, Zhang X, Bhattacharya S, Stokes CJ, Song E, Ali S, Dehestani B, Holloway KB, Micevych PS, Javaheri A, Crowley JR, Ballabio A, Schilling JD, Epelman S, Weihl CC, Diwan A, Fan D, Zayed MA, Razani B. Exploiting macrophage autophagy-lysosomal biogenesis as a therapy for atherosclerosis. Nat Commun. 2017;8:15750. doi: 10.1038/ncomms15750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen Q, Smith CY, Bailey KR, Wennberg PW, Kullo IJ. Disease location is associated with survival in patients with peripheral arterial disease. J Am Heart Assoc. 2013;2:e000304. doi: 10.1161/JAHA.113.000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kullo IJ, Leeper NJ. The genetic basis of peripheral arterial disease: current knowledge, challenges, and future directions. Circ Res. 2015;116:1551–1560. doi: 10.1161/CIRCRESAHA.116.303518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 61.Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107:839–850. doi: 10.1161/CIRCRESAHA.110.224766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leavesley D, Kashyap A, Croll T, Sivaramakrishnan M, Shokoohmand A, Hollier B, Upton Z. Vitronectin—Master Controller or Micromanager? Internat Union Biochem Mol Biol. 2013;65:807–818. doi: 10.1002/iub.1203. [DOI] [PubMed] [Google Scholar]
- 63.Suna G, Wojakowski W, Lynch M, Barallobre-Barreiro J, Yin X, Mayr U, Baig F, Lu R, Fava M, Hayward R, Molenaar C, White SJ, Roleder T, Milewski KP, Gasior P, Buszman PP, Buszman P, Jahangiri M, Shanahan CM, Hill J, Mayr M. Extracellular Matrix Proteomics Reveals Interplay of Aggrecan and Aggrecanases in Vascular Remodeling of Stented Coronary Arteries. Circulation. 2018;137:166–183. doi: 10.1161/CIRCULATIONAHA.116.023381. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.