

Abstract
Purpose
Patients who inherit a pathogenic loss-of-function genetic variant involving one of the four succinate dehydrogenase (SDH) subunit genes have up to an 86% chance of developing one or more cancers by the age of 50. If tumors are identified and removed early in these high-risk patients, they have a higher potential for cure. Unfortunately, many alterations identified in these genes are variants of unknown significance (VUS), confounding the identification of high-risk patients. If we could identify misclassified SDH VUS as benign or pathogenic SDH mutations, we could better select patients for cancer screening procedures and remove tumors at earlier stages.
Experimental Design
In this study, we combine data from clinical observations, a functional yeast model, and a computational model to determine the pathogenicity of 22 SDHA VUS. We gathered SDHA VUS from two primary sources: The OHSU Knight Diagnostics Laboratory and the literature. We used a yeast model to identify the functional effect of a VUS on mitochondrial function with a variety of biochemical assays. The computational model was used to visualize variants’ effect on protein structure.
Results
We were able to draw conclusions on functional effects of variants using our three-prong approach to understanding VUS. We determined that 16 (73%) of the alterations are actually pathogenic, causing loss of SDH function, and six (27%) have no effect upon SDH function.
Conclusions
We thus report the reclassification of the majority of the VUS tested as pathogenic, and highlight the need for more thorough functional assessment of inherited SDH variants.
Keywords: GI stromal tumor, succinate dehydrogenase, variants of unknown significance, mutation, paraganglioma
Introduction
SDH, succinate dehydrogenase, also known as complex II of the electron transport chain (ETC), is a four-subunit complex encoded by nuclear genes (SDHA, SDHB, SDHC, and SDHD, collectively referred to as SDHx). The assembled SDH complex localizes to the inner membrane of the mitochondria and links the tricarboxylic acid (TCA) cycle to the ETC, making SDH function critical for aerobic respiration (1).
Loss-of-function mutations affecting the SDH complex predispose patients to develop multiple cancers, including gastrointestinal stromal tumor (GIST), paraganglioma, pheochromocytoma, renal cell carcinoma, Hodgkin lymphoma, chronic lymphocytic leukemia, thyroid cancer, pituitary adenomas, and neuroendocrine tumors of the pancreas (2-6). Tumor formation due to SDH-deficiency requires the complete loss of function of at least one SDHx subunit (e.g. A, B, C, or D), causing destabilization and loss of enzymatic function of the entire SDH complex (7). There are several genetic mechanisms that can lead to SDH-deficiency. Typically, loss of function of an SDHx subunit is the result of a combination of an inactivating germline mutation (first hit) with a somatic loss of heterozygosity or other inactivating mutation affecting the other allele (second hit). Less commonly, loss of SDH complex occurs due to somatic inactivation of both alleles of a given complex subunit or SDH assembly factor. Finally, SDH-deficiency can be caused by an SDHC epimutation, defined as hypermethylation of the SDHC promoter, which leads to repression of SDHC transcription and depletion of SDHC protein levels, without a known underlying heritable cause (8).
Importantly, germline loss-of-function genetic SDHx variants are associated with a high lifetime risk of developing the aforementioned malignancies. For example, the chance of a patient with germline loss-of-function SDHD variant of developing one or more primary tumors by the age of 50 was reported to be 86% (9,10). Therefore, if we could identify high-risk patients through genetic testing and follow them serially with specialized screening tests, early tumor detection may lead to curative surgical resection before the tumors are metastatic/incurable. Early detection is crucial since there are no effective medical treatments for patients with advanced SDH-deficient cancers.
Currently, an SDH-deficient tumor is identified by measuring SDHB protein abundance using immunohistochemistry (IHC); absence of SDHB protein is indicative of loss of SDH function. However, it can be challenging to determine the underlying cause of SDH-deficiency in a tumor lacking SDHB expression. Clinical sequencing panels may turn up missense mutations in SDHx genes, but many of these are VUS. In addition, such panels can miss large intragenic deletions, and are not designed to identify epigenetic silencing of the SDHC promoter. Some SDHB, C, and D VUS have been functionally characterized to determine their effect on function, and thus their pathogenicity in tumors like paraganglioma and pheochromocytoma. However, the study of the functional consequences of SDHA VUS has lagged behind that of other SDHx subunits (7).
GIST is a heterogeneous group of tumors that arise from the interstitial cells of Cajal (ICC). However there are several different driver genes that when mutated give rise to GIST (11). The molecular classification of GIST is especially important because of the treatment implications of the different genetic drivers. The majority of GISTs have an activating receptor tyrosine kinase (RTK) mutation but about 13% of GIST lack RTK mutations (RTK-WT). Most RTK-WT GIST are SDH-deficient as assessed by immunohistochemistry (IHC) for SDHB. SDHA pathogenic variants are found in 47% of SDH-deficient GIST, and the majority of these SDHA mutations are germline, and thus heritable, variants (12). However, some the SDHA variants we find in GIST are VUS. A universal problem in the field is that these variants are rarely seen with a complete clinical and pathogenic annotation making it difficult to draw conclusions on functional effect from clinical data alone. Our study gathered these VUS from two primary sources; OHSU and the literature (12,13). We then combine data from clinical observations, a functional yeast model, and a computational model to understand the effects of SDHA VUS identified in GIST specimens on SDH complex function. Historically, yeast have been a robust system for identifying the assembly and enzymatic activity of SDH (14-16). In addition, yeast have proven to be an ideal model to study the functional effect of SDHB/C/D variants on SDH complex activity (17-20). Yeast are able to survive without functional mitochondria (e.g. lacking SDH complex activity) if they are provided a fermentable carbon source; thus, they provide a unique model system to study the biochemical effects of SDHA VUS (21).
Based on our findings, we discriminated between SDHA VUS that affected or did not affect SDH complex activity, and thus, their potential for pathogenicity. These data will aid clinicians’ ability to provide genetic counseling and tumor surveillance to patients with germline inheritance of these specific SDHA variants.
Materials and methods
Yeast strains and vectors
All Saccharomyces cerevisiae strains used in this study were derivatives of BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0). The SDH1 (yeast homolog to SDHA) deletion strain (sdh1Δ) was purchased from ATCC (catalog #4004998). The sdh1Δ was constructed as part of the Saccharomyces Genome Deletion Project by homologous recombination using the KanMX4 cassette (22). We verified the deletion of SDH1 using PCR mapping of the SDH1 locus with primer pairs recommended by the Saccharomyces Genome Deletion Project.
An amplicon containing the WT SDH1 (including the native SDH1 promoter and 3′ UTR) was generated from WT BY4741 and cloned into the pRS416 plasmid (23) (ATCC) and expressed in the sdh1Δ strain. The SDH1 point mutations were introduced by QuikChange mutagenesis PCR system (Agilent Technology, cat #200521). All mutations were confirmed by Sanger sequencing. Yeast strains were transformed using Frozen EZ Yeast Transformation II (Zymo Research, catalog # T2001). Strains were grown in synthetic complete medium lacking uracil to maintain plasmid selection with either 2% glucose or 3% glycerol as the carbon source.
Alignment of multiple species’ succinate dehydrogenase flavoprotein subunit
Cluster Omega was used to align the protein sequences of SDH flavoproteins including E. Coli (P0AC41), yeast (Q00711), human (P31040), and pig (Q0QF01).
Immunoblotting
Intact mitochondria were isolated using a previously described method (24). Steady-state levels of mitochondrial proteins were resolved on SDS-PAGE, transferred to nitrocellulose membrane, probed using the indicated primary antibodies and visualized using Amersham enhanced chemiluminescence (ECL) Western Blotting Detection Reagent (GE life sciences, catalog #RPN2106) with horseradish peroxidase-conjugated secondary antibodies (BioRad, catalog #1662408EDU). We used previously described polyclonal rabbit antibodies for immunodetection of Sdh1 and Sdh2 (25). Anti-porin was purchased from ThermoFisher Scientific (catalog # 459500).
Analysis of Sdh1-bound flavin adenine dinucleotide (FAD) and total mitochondrial FAD
Levels of FAD covalently bound to Sdh1 were analyzed as previously described (14,26). Briefly, mitochondrial proteins were resolved on SDS-PAGE and the gel was placed in a 10% acetic acid solution for 20 minutes to oxidize flavin. FAD was visualized upon exposure to UV light using a Bio-Rad ChemiDoc MP Imaging System.
Oxygen consumption assay
Yeast strains were grown to confluency in glucose-based media and then switched to glycerol-based media for 12 hours. Two million cells were plated onto commercially available microplates with oxygen sensors (Oxoplate; PreSens catalog #OP96U) (27). Kinetic reading of oxygen consumption was measured using a spectrofluorometer. A Student’s t-test was used to determine statistical significance between a variant of interest and yeast complemented with WT Sdh1.
Computational modeling of Sdh1 variants
A model of yeast Sdh1 (Q00711) from the Swiss Model repository was refined using Yasara Homology Modelling to include the liganded flavin adenine dinucleotide (FAD) interactions with the peptide chain, followed by optimization of the loop and side-chains interactions. Side-chain rotamers were fine-tuned considering electrostatic and knowledge-based packing interactions as well as solvation effects. An unrestrained high-resolution refinement with explicit solvent molecules was run, using YAMBER, a second-generation self-parameterizing force field derived from the AMBER force field. Clinically identified variants were introduced in the homology model, and the structures were minimized as described earlier. The variants were compared with the wild-type (WT) model of E. coli or yeast Sdh1 using PYMOL.
FoldX analysis
FoldX was used to measure the effect of point mutations on the stability of the Sdh1 yeast model (28). Except as noted, we used the default software settings. The move neighbors setting was turned off, and the average ΔΔG was calculated after three runs.
Mutation analysis
We gathered SDHA VUS from two primary sources: The OHSU Knight Diagnostics Laboratory (Portland, OR) and the literature(12,13). The majority of the variants pulled from the literature were found in a review highlighting the need for a functional model to characterize SDHx variants of unknown significance (12). The remaining literature variants are from a paper identifying novel causes of GIST that were WT for any known oncogenic driver of GIST (13). A collaboration with the OHSU Knight Diagnostics Laboratory, which offers a targeted exome panel to identify genetic drivers in GIST which includes all four SDHx subunits (https://www.ohsu.edu/custom/knight-diagnostic-labs/home/test-details?id=GeneTrails+GIST+Genotyping+Panel), lead to identification of several novel variants. All of the variants came from tumor samples that lacked any known oncogenic driver of GIST (e.g. no KIT mutations).
Results
Clinical analysis
All the known clinical information on the SDHA VUS identified in this study including SDHA/B IHC, tumor mutant allele fraction, other disease references, population data from the ExAC database (29), and ClinVar clinical significance (30) is listed in Table 1. Unfortunately, there were few variants with complete clinical and pathological annotation, emphasizing the challenge of trying to understand the functional effects of these variants from available clinical data alone. Currently, SDHx subunit testing remains uncommon for GIST and only a limited number of reference laboratories offer this testing. However, these laboratories usually do not have access to full clinical annotation and/or prior SDHB IHC testing results. This limitation applies to the published cases as well as the cases from the Knight Diagnostic laboratory. We listed all available additional clinical information for tumors with VUS in supplemental table 2.
Table 1. Clinical variants of human SDHA with unknown significance.
All of the genetic variants were found in GIST, except for hG260R which was found in a paraganglioma. A summary of available clinical data for each variant is listed, including source of the variant, clinical IHC results for SDHB/SDHA, frequency of the variant in tumor, other SDHA variants found in the same tumor, information on functional effect of other variants in the tumor, other disease references to the variant of interest in the literature, population allele frequency, and results from our yeast model. The no effect variants are highlighted in grey for clarity. Loss of function (LOF); Not available (N/A); Not Done (ND); neg (negative-SDHx absent); pos (positive-SDHx expression).
| hSDHA mutation | ySdh1 mutation | Source | SDHB/SDHA IHC | Tumor mutant allele fraction | Other disease references | Allele frequency from EXAC database | ClinVar clinical significance | Conclusions drawn |
| R31X | R19X | Control | Neg/neg (38) | N/A | GIST (38,39) Paraganglioma (40) | 1.647 × 10−4 | Pathogenic/Likely pathogenic | LOF (control) |
| H99S | H90S | Control (14) | N/A | N/A | None | N/A | N/A (H99Y Likely pathogenic) | LOF (control) |
| G106R | G97R | OHSU | N/A | 92% | Novel | N/A | N/A | LOF |
| N118S | N109S | OHSU | N/A | 50.7% | Novel | N/A | N/A | No effect |
| T143M | T134M | Literature (13) | N/A | 22% | Novel | N/A (T143R reported 8.31 × 10−6) | Uncertain significance | LOF |
| R171H | R162H | OHSU | N/A | 34% | Novel | 8.24 × 10−6 | Uncertain significance | No effect |
| R171H | R162H | OHSU | N/A | 40.3% | Novel | 8.24 × 10−6 | Uncertain significance | No effect |
| R188W | R179W | Literature (12) | Neg/pos [30] | N/A | None | N/A (R188Q reported 1.65 × 10−5) | N/A | LOF |
| R195W | R186W | OHSU | Neg/ND | 66.7% | GIST (37) | N/A (R195Q reported 8.23 × 10−6) | N/A | No effect |
| G260R | G251R | Literature (12) | Pos/pos [32] | N/A | None | N/A | Uncertain significance | LOF |
| H296Y | H287Y | Literature (13) | N/A | 87% | Novel | N/A | N/A | LOF |
| R312C | R303C | OHSU | N/A | 34.5% | Novel | N/A | N/A (R312P Uncertain significance) | LOF |
| R408C | Y399C | OHSU | N/A | 52.8% | GIST(41); late onset neurodegenerative disease (42) | N/A | N/A | LOF |
| G419R | G410R | OHSU | N/A | 77% | GIST (43) | N/A | N/A | LOF |
| C438F | C431F | OHSU | N/A | 48.5% | Novel | N/A | N/A | LOF |
| G439E | G432E | OHSU | N/A | 87.6% | Novel | N/A | N/A | LOF |
| R451C | R444C | Literature (13) | N/A | 18% | Complex II deficiency (42) | N/A (R451H reported 8.23 × 10−6) | N/A | LOF |
| R451H | R444H | OHSU | N/A | 47.5% | Novel | 8.23 × 10−6 | N/A | LOF |
| R451H | R444H | OHSU | N/A | 39.5% | Novel | 8.23 × 10−6 | N/A | LOF |
| A454E | A447E | Literature (12) | Neg/pos [31] | N/A | None | N/A | N/A (A454T Uncertain significance) | LOF |
| R465W | R458W | OHSU | N/A | 50% | Novel | 8.23 × 10−6 | Uncertain significance | LOF |
| T508I | T501I | OHSU | N/A | 50.6% | Cardiomyopathy and Leukodystrophy (44) | 7.60 × 10−4 | Conflicting interpretations of pathogenicity | No effect |
| R589G | R582G | OHSU | N/A | 40.2% | Paraganglioma (45) | 8.29 × 10−6 | Uncertain significance | LOF |
| H625W | H601Y | Literature (12) | Neg/neg [39] | N/A | Pituitary adenoma and Pheochromocytoma/paraganglioma | N/A | N/A | LOF |
| Y629F | W605F | OHSU | N/A | 99.6% | 1.52 × 10−1 | Benign/likely benign | No effect | |
| V657I | V633I | OHSU | N/A | 95.1% | Not pathogenic (46) | 1.30 × 10−1 | Likely benign | No effect |
Functional studies in yeast
To understand the functional consequences of VUS in human SDHA, we used complementation studies in a yeast strain lacking Sdh1 (sdh1Δ). The flavoprotein subunit of the SDH complex is highly conserved across all species, including yeast Sdh1 (ySdh1) and human SDHA (hSDHA) (Supplemental Figure 1). For simplicity, we use yeast nomenclature throughout this study and reference the corresponding human variant in all of the tables.
As a positive control, we generated a WT SDH1 amplicon from BY4741 that included the promoter and the 3′UTR which was cloned into a pRS416 plasmid and expressed in sdh1Δ yeast. We used sdh1Δ as our negative control. Sdh1Δ do not have a functional SDH complex and were unable to utilize their mitochondria for canonical TCA or ETC pathways, allowing growth on a fermentable carbon source (glucose), but preventing growth with a non-fermentable carbon source such as glycerol (Figure 1). Complementation with WT SDH1 (+Sdh1) restored growth on glycerol (Figure 1). Using this strategy, we tested the ability of plasmids encoding individual VUS to complement the sdh1Δ strain and restore a normal growth phenotype. The two loss-of-function controls (yR19X and yH90S), as well as 16 of the variants shown in Figure 1 (yG97R, yT134M, yR179W, yG251R, yH287Y, yR303C, yY399C, yG410R, yC431F, yG432E, yR444C, yR444H, yA447E, yR458W, yR582G, yH601Y) were unable to grow on glycerol, indicating that these variants result in loss of oxidative phosphorylation (Figure 1A). In contrast, six of the variants (yN109S, yR162H, yR186W, yT501I, yW605FF, yV633I) were able to grow on glycerol, indicating these variants had no effect on the Sdh1 function as their phenotype was the same as WT Sdh1 (Figure 1B).
Figure 1. ySdh1 variants causing loss of function are unable to use glycerol as a sole carbon source or consume oxygen.
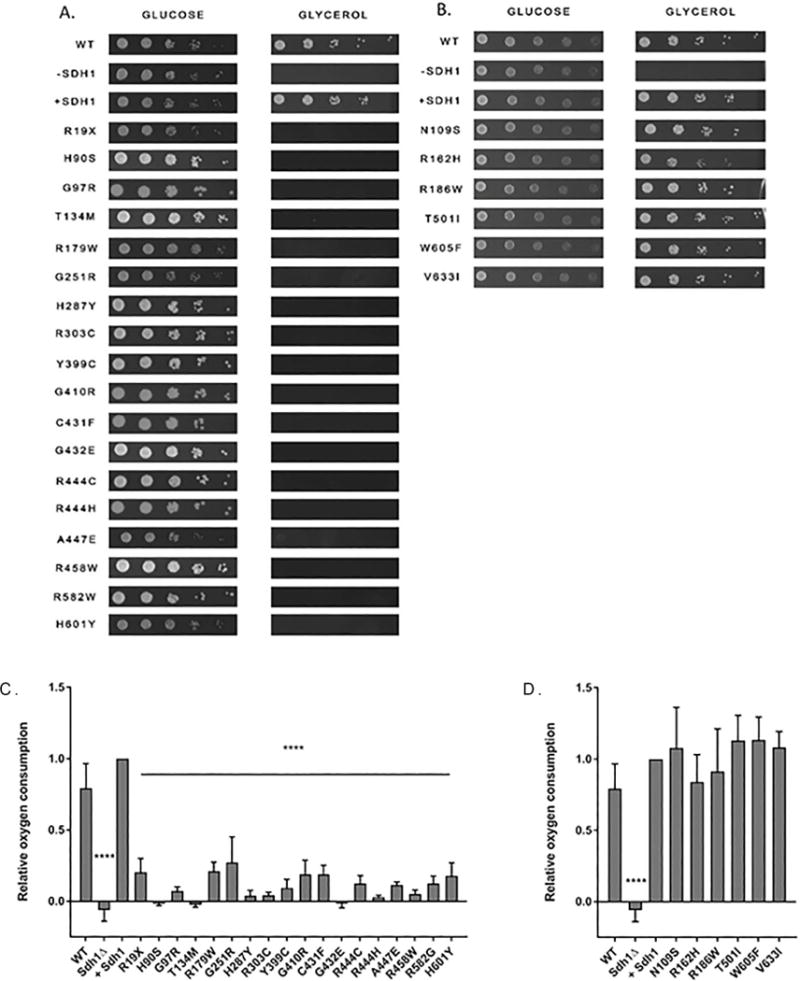
Yeast were grown to a confluency of 2 × 107/mL and then serially diluted and plated onto either glucose or glycerol media plates (Left to right: highest to lowest). All variants, regardless of SDH complex function, can grow on glucose but only those variants that complement sdh1Δ and restore SDH complex activity are able to grow on glycerol and consume oxygen. A. Loss-of-function variants that are unable to grow with glycerol as their sole carbon source. B. Variants with no effect are able to grow with glycerol as their sole carbon source. C. Sdh1 variants that consume significantly less oxygen than cells complemented with WT Sdh1 (+Sdh1 control). D. Variants that consume oxygen at a similar rate to the +Sdh1 control. **** p-value <0.0001
As a secondary measure of SDH complex activity, we measured oxygen consumption to assess oxidative phosphorylation, a surrogate for measuring the ability of the SDH complex to transfer electrons to the ETC. In concordance with our previous results, the same 18 variants that were unable to grow on glycerol consumed significantly less (p<0.0001) oxygen than WT Sdh1 (Figure 1C). Variants that grew on glycerol had similar oxygen consumption to WT Sdh1, confirming that these variants do not disrupt the ETC (Figure 1D). To ensure oxygen consumption was due to the ETC, azide was added to each cell strain, and this promptly abolished oxygen consumption for all controls and variants.
To better understand how each variant affects Sdh1 protein function, we measured the covalent attachment of FAD to Sdh1, a process known as flavination. We also measured the protein abundance of Sdh1 and Sdh2 (Figure 2). Sdh1 flavination is critical for the catalytic activity of Sdh1; without covalent attachment of flavin, the Sdh1 protein is unable to oxidize succinate (14). As a positive control for a variant that inhibited insertion of FAD into Sdh1, we used the previously described Sdh1 H90S variant (14). Variants that complemented sdh1Δ yeast in our functional assays had similar levels of flavinated Sdh1, total Sdh1, and total Sdh2 as yeast complemented with WT Sdh1 (Figure 2D). However, variants that were unable to perform oxidative phosphorylation had differential effects on the levels of flavinated Sdh1 and total Sdh1 protein (Figure 2 A–C). Some loss-of-function variants inhibited flavination without affecting the abundance of Sdh1 (H90S, G97R, T134M, R444C, H601Y) while others caused a marked decrease in Sdh1 protein abundance (R179W, G251R, H287Y, R303C, Y399C, G410R, C431F, G432E, R444H, A447E, R458W, R582G). All of the loss-of-function variants resulted in a decrease in Sdh2 protein abundance, consistent with loss of a functional SDH complex as described previously (25). In a subset of no effect and loss-of-function variants, we confirmed that Sdh1 mRNA expression was not significantly different from WT.
Figure 2. Abundance of flavinated Sdh1, total Sdh1, and total Sdh2 in mitochondrial lysates from yeast expressing each variant.
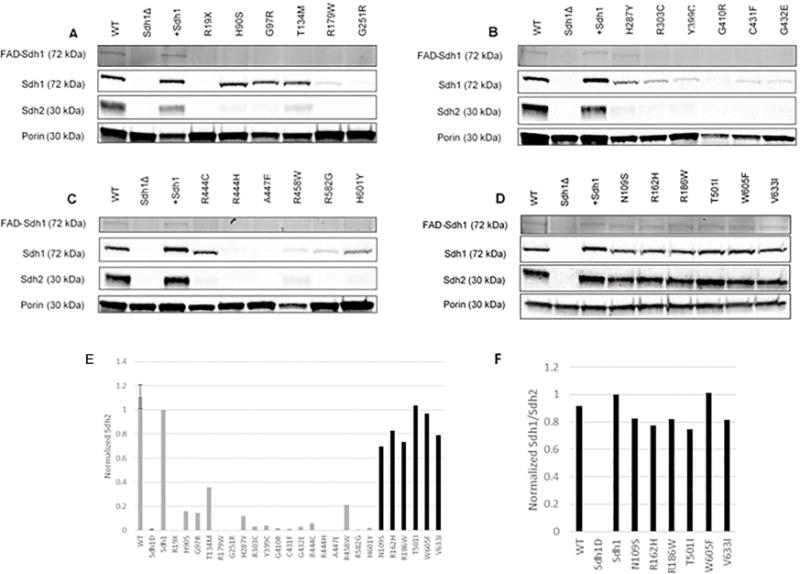
Western blotting of mitochondrial lysates from each variant are shown. Positive and negative control lanes are included on each western blot (WT, SDH1delta, +SDH1). WT and +Sdh1 show bands at expected size for each protein. Sdh1Δ does not show bands for Sdh1-FAD, Sdh1, or Sdh2, but does have normal expression of the mitochondrial marker, porin. A-C. Loss-of-function variants are shown in numerical order by altered amino acid residue. D. No effect variants are shown in numerical order by altered amino acid residue and have with no reduction in flavin, Sdh1, or Sdh2 expression compared to WT controls. E. Sdh2 protein abundance was calculated relative to Sdh1 and then normalized to porin (control for mitochondrial protein loading). The no effect variants are labeled in black, while the loss-of-function variants are in gray. No effect variants have Sdh2 protein abundance more similar to complemented Sdh1 and loss-of-function variants have decreased Sdh2 similar to sdh1Δ. F. Ratio of Sdh1/Sdh2 protein abundance in no effect variants.
Based on all of the above results, we characterized 16 variants (73%) as causing loss of function and six variants (27%) as having no effect on SDH function (Table 2). All the loss-of-function variants were unable to grow on glycerol, had decreased oxygen consumption, and showed decreased Sdh2 protein abundance. In contrast, the no effect group had similar results to cells complemented with WT Sdh1 protein in these assays.
Table 2. Consistent findings across multiple functional assays for each variant in our yeast model.
A summary of results including the growth on glycerol, oxygen consumption, Sdh2 protein abundance, consequences of changing the WT amino acid using computational modeling and literature search for studies in other species’ flavoprotein are tabulated along with our classification of each variant based on our yeast model. The no effect variants are highlighted in grey for clarity. Numerical values for oxygen consumption were taken from Figure 1C and D. Numerical values for Sdh2 protein were taken from Figure 2E.
| hSDHA mutation | ySdh1 mutation | Growth phenotype on glycerol | Oxygen consumption (% of complemented) | Sdh2 protein (% of complemented) | Structural implications of variants | Group name |
|---|---|---|---|---|---|---|
| R31X (control) | R19X (control) | Unable to grow | Decreased (20%) | Decreased (0%) | Truncates mitochondrial targeting sequence (47) | Loss-of-function (control) |
| H99S (control) | H90S (control) | Unable to grow | Decreased (−2%) | Decreased (15%) | Inhibits covalent bond to FAD (14) | Loss-of-function (control) |
| G106R | G97R | Unable to grow | Decreased (7%) | Decreased (14%) | Distorts the active site (31) | Loss-of-function |
| N118S | N109S | Growth | Similar (108%) | Similar (83%) | Surface of protein | No effect |
| T143M | T134M | Unable to grow | Decreased (3%) | Decreased (35%) | No obvious disturbances but causes loss of function | Loss-of-function |
| R171H | R162H | Growth | Similar (84%) | Similar (78%) | Surface of protein | No effect |
| R188W | R179W | Unable to grow | Decreased (21%) | Decreased (0%) | Disrupts salt bridge with D108 | Loss-of-function |
| R195W | R186W | Growth | Similar (91%) | Similar (82%) | Surface of protein | No effect |
| G260R | G251R | Unable to grow | Decreased (27%) | Decreased (0%) | Obstructs flavin binding pocket | Loss-of-function |
| H296Y | H287Y | Unable to grow | Decreased (4%) | Decreased (11%) | Inhibits succinate binding (31) | Loss-of-function |
| R312C | R303C | Unable to grow | Decreased (4%) | Decreased (3%) | Contributes to proper orientation and activation of the flavin isoalloxazine ring to facilitate formation of the covalent FAD bond and disrupts salt bridge (48) | Loss-of-function |
| R408C | Y399C | Unable to grow | Decreased (9%) | Decreased (4%) | In flavin binding site | Loss-of-function |
| G419R | G410R | Unable to grow | Decreased (19%) | Decreased (2%) | Distorts helix | Loss-of-function |
| C438F | C431F | Unable to grow | Decreased (19%) | Decreased (2%) | Bulky change disrupts helix | Loss-of-function |
| G439E | G432E | Unable to grow | Decreased (−2%) | Decreased (3%) | Obstructs flavin binding pocket | Loss-of-function |
| R451C | R444C | Unable to grow | Decreased (12%) | Decreased (6%) | Disrupts flavin binding, succinate binding and proton shuttle necessary for catalytic activity (31) | Loss-of-function |
| R451H | R444H | Unable to grow | Decreased (3%) | Decreased (0%) | Disrupts flavin binding, succinate binding and proton shuttle necessary for catalytic activity (31) | Loss-of-function |
| A454E | A447E | Unable to grow | Decreased (11%) | Decreased (0%) | Lines succinate binding site (31) | Loss-of-function |
| R465W | R458W | Unable to grow | Decreased (5%) | Decreased (21%) | Disrupts salt bridge with E136 | Loss-of-function |
| T508I | T501I | Growth | Similar (113%) | Similar (75%) | Surface of protein | No effect |
| R589G | R582G | Unable to grow | Decreased (12%) | Decreased (0%) | Inhibits C-terminal flavination (25) | Loss-of-function |
| H625W | H601Y | Unable to grow | Decreased (18%) | Decreased (2%) | Inhibits C-terminal flavination | Loss-of-function |
| Y629F | W605F | Growth | Similar (113%) | Similar (101%) | Surface of protein | No effect |
| V657I | V633I | Growth | Similar (108%) | Similar (82%) | Surface of protein | No effect |
Computational modeling
We next performed computational modeling to predict the potential structural implications of each variant. Using the two groups (loss-of-function and no effect) identified in our yeast model, we further characterized each variant by visualizing the location in the Sdh1 protein. Both groups, loss-of-function and no effect, were located throughout the four domains of Sdh1, suggesting that there are not “hotspot” areas or domains for pathogenic variants, unlike the situation for some proteins. Within the loss-of-function group, 12 variants (yH90S (loss-of-function control), yG97R, yG251R, yH287Y, yR303C, yY399C, yG432E, yR444C, yR444H, yA447E, yR582G, yH601Y) were identified as being involved in cofactor (FAD) or substrate (succinate) binding in the Sdh1 active site (Figure 3, Table 2, and Supplemental Video 1). Structural studies from the homologous protein quinol:fumarate reductase (QFR) in E. coli suggests an important catalytic function for most of these amino acids (31). In addition to the amino acids directly interacting with the flavin binding pocket, it has been previously shown that the C-terminal domain of Sdh1 is crucial for the flavination of Sdh1 (25). Two variants located in the C-terminal domain of Sdh1, yR582G and yH601Y, both inhibited flavination of Sdh1. To visualize the substrate (succinate) and cofactor (FAD), which were not visualized using the yeast structure, these Sdh1 variants were modeled using the E. Coli crystal structure of Sdh1.
Figure 3. Variants involving the active site cause loss of Sdh1 function.
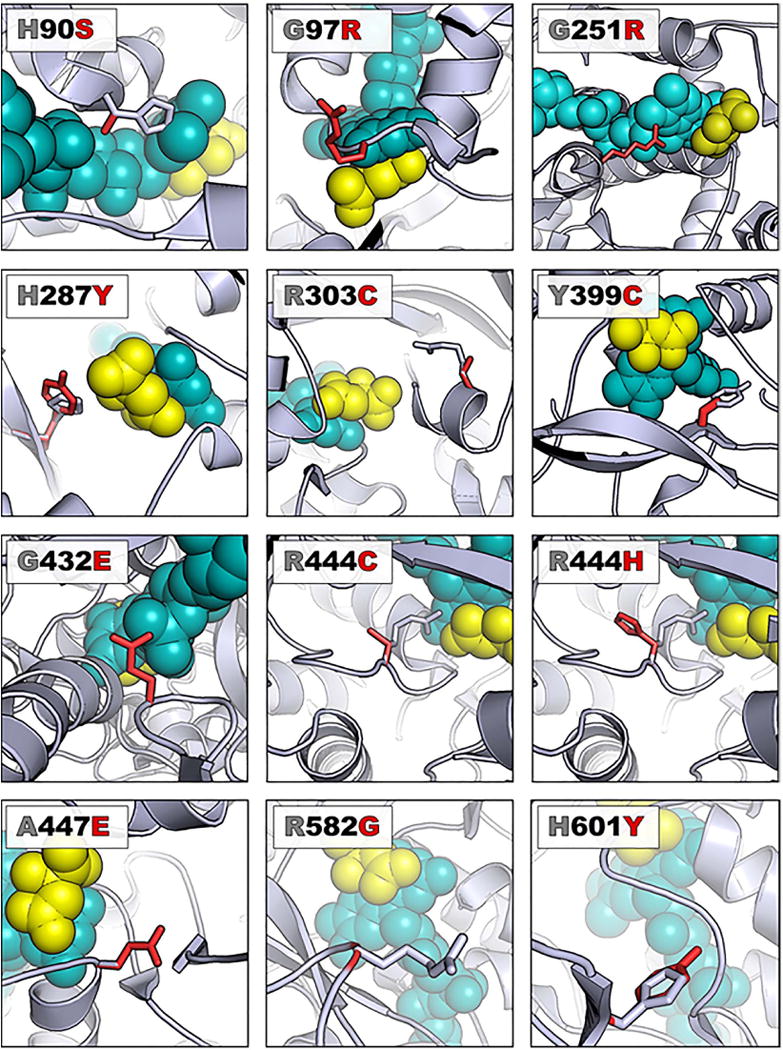
A ribbon representation of Sdh1 (E. coli model PBD file 2WP9) is shown with WT protein carbons colored gray and the variant carbons colored red. The FAD is shown as teal-colored spheres while the dicarboxylic acid substrate (succinate analog) is depicted with yellow-colored spheres. In each case, the view has been rotated so that residues of interest are clearly observed. References detailing structural implications of each variant can be found in Table 2.
The other six loss-of-function variants (yR19X (control-not picture in computational models because it is an early truncation of the protein), yT134M, yR179W, yG410R, yG431F, yR458W) are not located in the active site of Sdh1. Changes in protein structure based on each of these variants are visualized in Figure 4A and Supplemental Video 1. Further, FoldX analyses of the variants compared to WT provided insight on how the variants affect stability (Supplemental Table 2) (28). yT134M, yR179W, yG410R, and yG431F all have positive ΔΔG (change in free energy) indicating these changes are highly destabilizing. Some of these also were associated with decreased Sdh1 protein abundance (Figure 2).
Figure 4. Structural consequences of variants visualized in a yeast model (yeast model PBD file 1ORZ).
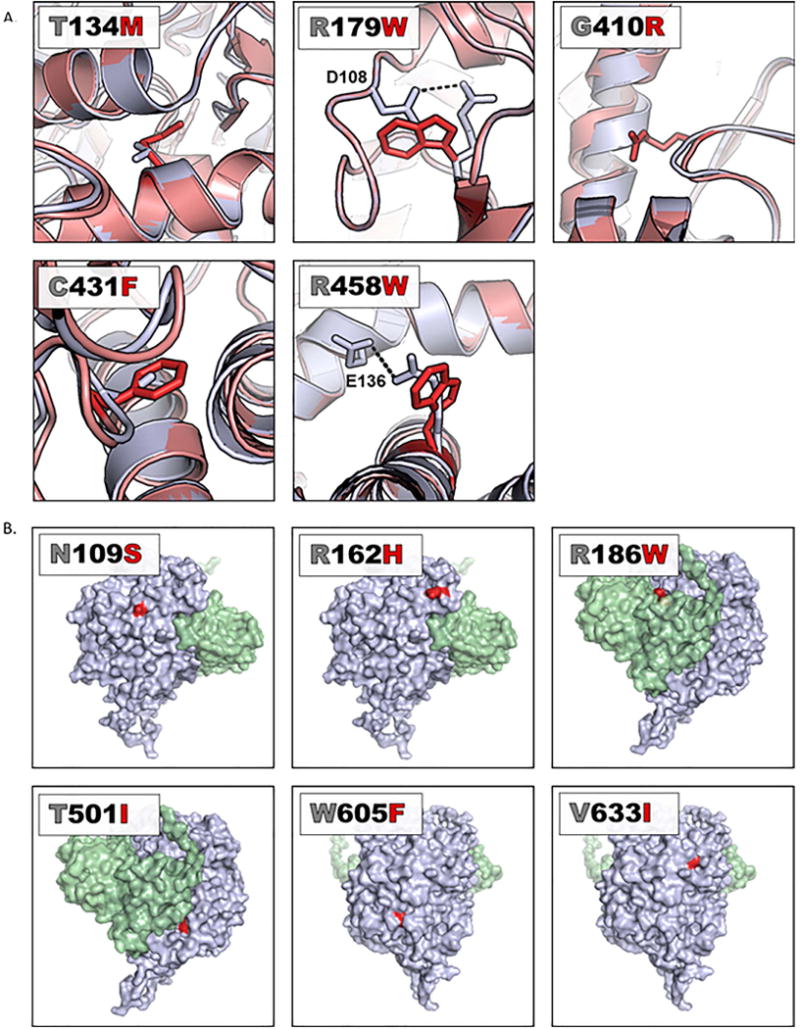
A. Variants not in the active site causing loss-of-function. WT Sdh1 protein is represented as a gray ribbon with the amino acid of interest labeled as a gray stick and the re-folded variant Sdh1 protein as a red ribbon with the amino acid of interest indicated by a red stick. In each case, the view has been rotated such that residues of interest are clearly observed. Potential structural implications of each variant can be found in Table 2. B. Variants that do not affect protein function are located on the surface of Sdh1. The surface area of WT Sdh1 is shown in gray, the variant residue is labeled red, and the surface area of Sdh2 is labeled in green. In each case, the view has been rotated such that residues of interest are clearly observed.
Interestingly, all the variants with no functional effects were localized to the surface of the protein (Figure 4B, Supplemental Video 1) where it would be predicted that they would be less likely to affect protein structure and function. Notably, these mutations did not map into any of the known critical domains of Sdh1 or predicted Sdh2 interaction sites.
Discussion
Molecular testing plays an important role in the clinical management of GIST, including decision making about appropriate medical and surgical therapy. SDH deficiency is a potential cause of GIST in tumors lacking known oncogenic drivers such as gain-of-function KIT mutations. To attempt to identify the cause of SDH deficiency in such GIST, it is necessary to use multi-gene sequencing panels to search for SDHx pathogenic mutations, however, as the routine use of multi-gene sequencing panels has expanded, there has also been an increase in reported VUS. Due to the multiple potential causes of SDH-deficiency, it is difficult to determine if a newly discovered VUS is responsible for the defect, or if a different genetic mechanism is responsible for the loss of SDH complex activity. This problem is exacerbated in GIST as loss of SDHA protein is the most common cause of SDH deficiency in GIST, but SDHA variants have not been extensively characterized at the functional level. Finally, many SDHA VUS are also found in the germline, meaning that they can be inherited. Knowing a patient has a loss-of-function SDHA variant in a tumor dramatically changes clinical decision making concerning screening recommendations for tumor syndromes, which affects both the patient and other family members. Since there are no effective medical treatments for advanced SDH-deficient tumors, early detection of tumors allowing curative surgical resection is crucial.
A major problem in the field has been the lack of a validated model system to assess the biochemical function of SDHA VUS. Currently, there are no human SDHA-deficient cell lines and assessing SDH complex activity in tumor samples is difficult. Previously it has been shown that a yeast model can be used to evaluate the pathogenic significance of SDHB mutations (17,18). Extending these studies, we report the successful use of a yeast model to characterize SDHA variants found in GIST tumors as either causing loss of function or having no biochemical consequence. Together with computational modeling, structural homology, and patient data, we have drawn conclusions on how and why the SDHA variants we studied affect SDH function, providing clinicians with information to guide genetic counseling of patients and family members who harbor one of these germline VUS.
Using our yeast model, we characterized variants as either causing loss of protein function or having no effect. All variants in the loss-of-function group were unable to grow on a non-fermentable carbon source (glycerol). In addition, these variants also failed to consume oxygen, indicating defective oxidative phosphorylation. Interestingly, these variants had differential effects on Sdh1 flavination and/or protein abundance. As a group, all of the loss-of-function variants were associated with a dramatic decrease in Sdh2 protein abundance, which is in concordance with current clinical testing to determine SDH-deficiency in tumors—assessment of SDHB (human equivalent to ySdh2) expression using IHC (2,32-36). Our data provides further evidence supporting the role of SDHB IHC as the most reliable clinical test to identify SDH deficiency in a tumor. Notably, we identified four novel loss-of-function variants that did not affect Sdh1 protein abundance (yG97R, yT134M, yR444C, yH601Y). These variants prevent flavination of Sdh1 making Sdh1 dysfunctional but still expressed at normal levels. Our findings are consistent with previous reports where SDHA expression was retained in some SDHA-mutant tumors that lack SDHB expression (37), confirming that not all dysfunctional SDHA (or Sdh1) variants lead to decreased protein expression of this subunit. There are three clinical samples (R188W, G260R, A454E) that we characterized as loss-of-function variants but are associated with normal sdh1 in our yeast model. Based on these results and coupled with the fact the SDHA IHC is not universally available, we advocate for the use of SDHB IHC to identify tumors that are SDH-deficient.
The conclusions drawn from our yeast model are largely supported by our computational modeling results. For many variants, we could find confirmatory structural modeling evidence that the amino acids affected by these mutations played a role in the catalytic site of Sdh1. The computational model and energy minimization also allowed us to explain why some variants could reduce Sdh1 protein abundance. Furthermore, all the variants that did not affect protein function were on the surface of the protein as visualized by our computational model, suggesting that they would not interact with the catalytic mechanism of Sdh1.
Based on our biochemical data, there are still several tumors that have no known mechanism for loss of SDH activity (Table 1). One of these tumors had the hR195W (yR186W) variant with an allele frequency of 66.7% and negative SDHB IHC. This tumor is likely driven by a different mechanism of SDH-deficiency. The rest of the unexplained tumors had no IHC data available, so it is possible that these GISTs are not driven by SDH-deficiency and instead belong to a different subtype of GIST (11). Alternatively, these GISTs may be SDH-deficient by a different genetic mechanism than the SDHA variant identified using targeted exome sequencing. There are several weaknesses of various targeted exome sequencing panels including potential lack of coverage for regions of certain SDHx genes, failure to detect large genomic deletions involving SDHx subunits, as well as the inability to measure hypermethylation of the SDHC promoter. In addition, mutations that inactivate newly identified SDH assembly factors (SDHAF3, 4) are typically not included in clinical cancer gene sequencing panels (15,16). Given the limitations of using de-identified results from clinical testing, we did not have access to residual tumor samples to perform additional IHC or genomic testing.
We used a yeast model to characterize 22 SDHA VUS. This data revealed 16 (73%) of SDHA VUS as loss-of-function (and therefore pathogenic), highlighting the importance of understanding such variants to provide better clinical recommendations for genetic counselors concerning family screening and early detection protocols. However, our approach using a functional yeast model paired with computational modeling can distinguish between SDHA VUS that cause loss of function and those that have no biochemical effect, allowing us to discriminate between pathogenic and non-pathogenic variants.
Supplementary Material
Translational relevance.
Molecular testing plays an important role in the clinical management of gastrointestinal stromal tumors (GIST), including decision making about the most appropriate medical or surgical therapy. As the routine use of multi-gene sequencing panels has expanded, there has also been an increase in reported variants of unknown significance (VUS) of the SDHA gene. In many cases, these SDHA VUS are present in the germline and are therefore potentially heritable by other family members. In order to understand the functional consequences of these variants, we combined clinical observations, data from a functional yeast model, and computational modeling to classify these SDHA VUS as having no effect or causing loss of function. These results will be helpful for appropriate genetic counseling of individuals with these germline variants. In addition, our data highlight the limitations of SDHA immunohistochemistry in clinical testing of tumors with SDHA VUS.
Acknowledgments
Thank you to Arin McKinley, Diana Griffith, Janice Patterson, and Ashley Young for their constant support during the completion of this project. The advice and feedback from Amanda McCullough, Maureen Hoatlin, and David Koeller were extremely beneficial for experimental design and interpretation of results. Also, thank you to Dennis Winge for his generous gift of primary antibodies for Sdh1 and Sdh2.
Financial support
Amber E Bannon-The author acknowledge of the support the OHSU OCTRI TL1TR000129
Oliver Harismendy-The authors acknowledge the support of NIH R21CA177519, U01CA196406
Jason K. Sicklick-The authors acknowledge the support of NIH K08CA168999, R21CA192072, and P30CA023100
Michael C. Heinrich-GIST Cancer Research Fund (MCH), Life Raft Group (MCH), and a Merit Review Grant from the Department of Veterans Affairs (MCH, 2I01BX000338-05)
Contributor Information
Amber E Bannon, Cell and Developmental Biology, Heinrich Lab, Oregon Health and Science University, Portland, Oregon.
Jason Kent, Cell and Developmental Biology, Heinrich Lab, Oregon Health and Science University, Portland, Oregon.
Isaac Forquer, Portland VA Medical Center and Oregon Health & Science University.
Ajia Town, Heinrich Lab, Oregon Health and Science University, Portland, Oregon.
Lillian R. Klug, Cancer Biology, Heinrich Lab, Oregon Health and Science University, Portland, Oregon
Kelly McCann, Department of Medicine, Division of Hematology-Oncology, University of California Los Angeles David Geffen School of Medicine.
Carol Beadling, Oregon Health and Science University, Portland, Oregon.
Oliver Harismendy, Division of Biomedical Informatics, Department of Medicine, Moores UCSD Cancer Center, University of California San Diego, La Jolla, CA, USA.
Jason K. Sicklick, Division of Surgical Oncology, Department of Surgery, Moores UCSD Cancer Center, University of California San Diego, La Jolla, CA, USA
Christopher Corless, Knight Cancer Institute and Department of Pathology, Oregon Health and Science University, Portland, Oregon.
Ujwal Shinde, Department of Biochemistry and Molecular Biology, MRB 631, Oregon Health and Science University, Portland, Oregon.
Michael C. Heinrich, Department of Medicine; Department of Cell, Developmental, and Cancer Biology, Portland VA Health Care System and OHSU Knight Cancer Institute, Portland, OR
References
- 1.Bezawork-Geleta A, Rohlena J, Dong L, Pacak K, Neuzil J. Mitochondrial Complex II: At the Crossroads. Trends in biochemical sciences. 2017 doi: 10.1016/j.tibs.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Janeway KA, Kim SY, Lodish M, Nose V, Rustin P, Gaal J, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(1):314–8. doi: 10.1073/pnas.1009199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burnichon N, Abermil N, Buffet A, Favier J, Gimenez-Roqueplo AP. The genetics of paragangliomas. European annals of otorhinolaryngology, head and neck diseases. 2012;129(6):315–8. doi: 10.1016/j.anorl.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 4.Baysal BE. A recurrent stop-codon mutation in succinate dehydrogenase subunit B gene in normal peripheral blood and childhood T-cell acute leukemia. PLoS One. 2007;2(5):e436. doi: 10.1371/journal.pone.0000436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beamer LC. Cowden syndrome: what oncology nurses need to know about increased risk of developing certain cancers. Oncol Nurs Forum. 2014;41(5):555–7. doi: 10.1188/14.ONF.555-557. [DOI] [PubMed] [Google Scholar]
- 6.Niemeijer ND, Papathomas TG, Korpershoek E, de Krijger RR, Oudijk L, Morreau H, et al. Succinate Dehydrogenase (SDH)-Deficient Pancreatic Neuroendocrine Tumor Expands the SDH-Related Tumor Spectrum. The Journal of clinical endocrinology and metabolism. 2015;100(10):E1386–93. doi: 10.1210/jc.2015-2689. [DOI] [PubMed] [Google Scholar]
- 7.van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EM, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. The Lancet Oncology. 2009;10(8):764–71. doi: 10.1016/s1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Killian JK, Miettinen M, Walker RL, Wang Y, Zhu YJ, Waterfall JJ, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Science translational medicine. 2014;6(268):268ra177. doi: 10.1126/scitranslmed.3009961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ricketts CJ, Forman JR, Rattenberry E, Bradshaw N, Lalloo F, Izatt L, et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Human mutation. 2010;31(1):41–51. doi: 10.1002/humu.21136. [DOI] [PubMed] [Google Scholar]
- 10.Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. Jama. 2004;292(8):943–51. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- 11.Bannon AE, Klug LR, Corless CL, Heinrich MC. Using molecular diagnostic testing to personalize the treatment of patients with gastrointestinal stromal tumors. Expert review of molecular diagnostics. 2017 doi: 10.1080/14737159.2017.1308826. [DOI] [PubMed] [Google Scholar]
- 12.Evenepoel L, Papathomas TG, Krol N, Korpershoek E, de Krijger RR, Persu A, et al. Toward an improved definition of the genetic and tumor spectrum associated with SDH germ-line mutations. Genetics in medicine : official journal of the American College of Medical Genetics. 2015;17(8):610–20. doi: 10.1038/gim.2014.162. [DOI] [PubMed] [Google Scholar]
- 13.Shi E, Chmielecki J, Tang CM, Wang K, Heinrich MC, Kang G, et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. Journal of translational medicine. 2016;14(1):339. doi: 10.1186/s12967-016-1075-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325(5944):1139–42. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Vranken JG, Bricker DK, Dephoure N, Gygi SP, Cox JE, Thummel CS, et al. SDHAF4 promotes mitochondrial succinate dehydrogenase activity and prevents neurodegeneration. Cell metabolism. 2014;20(2):241–52. doi: 10.1016/j.cmet.2014.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Na U, Yu W, Cox J, Bricker DK, Brockmann K, Rutter J, et al. The LYR factors SDHAF1 and SDHAF3 mediate maturation of the iron-sulfur subunit of succinate dehydrogenase. Cell metabolism. 2014;20(2):253–66. doi: 10.1016/j.cmet.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Panizza E, Ercolino T, Mori L, Rapizzi E, Castellano M, Opocher G, et al. Yeast model for evaluating the pathogenic significance of SDHB, SDHC and SDHD mutations in PHEO-PGL syndrome. Human molecular genetics. 2013;22(4):804–15. doi: 10.1093/hmg/dds487. [DOI] [PubMed] [Google Scholar]
- 18.Goffrini P, Ercolino T, Panizza E, Giache V, Cavone L, Chiarugi A, et al. Functional study in a yeast model of a novel succinate dehydrogenase subunit B gene germline missense mutation (C191Y) diagnosed in a patient affected by a glomus tumor. Human molecular genetics. 2009;18(10):1860–8. doi: 10.1093/hmg/ddp102. [DOI] [PubMed] [Google Scholar]
- 19.Szeto SS, Reinke SN, Sykes BD, Lemire BD. Ubiquinone-binding site mutations in the Saccharomyces cerevisiae succinate dehydrogenase generate superoxide and lead to the accumulation of succinate. The Journal of biological chemistry. 2007;282(37):27518–26. doi: 10.1074/jbc.M700601200. [DOI] [PubMed] [Google Scholar]
- 20.Alston CL, Ceccatelli Berti C, Blakely EL, Olahova M, He L, McMahon CJ, et al. A recessive homozygous p.Asp92Gly SDHD mutation causes prenatal cardiomyopathy and a severe mitochondrial complex II deficiency. Human genetics. 2015;134(8):869–79. doi: 10.1007/s00439-015-1568-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fontanesi F, Diaz F, Barrientos A. Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using yeast models of OXPHOS deficiencies. Current protocols in human genetics. 2009 doi: 10.1002/0471142905.hg1905s63. Chapter 19:Unit19.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, et al. Functional characterization of the Scerevisiae genome by gene deletion and parallel analysis. Science. 1999;285(5429):901–6. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 23.Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122(1):19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muller F, Crofts AR, Kramer DM. Multiple Q-cycle bypass reactions at the Qo site of the cytochrome bc1 complex. Biochemistry. 2002;41(25):7866–74. doi: 10.1021/bi025581e. [DOI] [PubMed] [Google Scholar]
- 25.Kim HJ, Jeong MY, Na U, Winge DR. Flavinylation and assembly of succinate dehydrogenase are dependent on the C-terminal tail of the flavoprotein subunit. The Journal of biological chemistry. 2012;287(48):40670–9. doi: 10.1074/jbc.M112.405704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bafunno V, Giancaspero TA, Brizio C, Bufano D, Passarella S, Boles E, et al. Riboflavin uptake and FAD synthesis in Saccharomyces cerevisiae mitochondria: involvement of the Flx1p carrier in FAD export. The Journal of biological chemistry. 2004;279(1):95–102. doi: 10.1074/jbc.M308230200. [DOI] [PubMed] [Google Scholar]
- 27.Kitanovic A, Bonowski F, Heigwer F, Ruoff P, Kitanovic I, Ungewiss C, et al. Acetic acid treatment in S. cerevisiae creates significant energy deficiency and nutrient starvation that is dependent on the activity of the mitochondrial transcriptional complex Hap2-3-4-5. Frontiers in oncology. 2012;2:118. doi: 10.3389/fonc.2012.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic acids research. 2005;33:W382–8. doi: 10.1093/nar/gki387. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic acids research. 2016;44(D1):D862–8. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iverson TM. Catalytic mechanisms of complex II enzymes: a structural perspective. Biochimica et biophysica acta. 2013;1827(5):648–57. doi: 10.1016/j.bbabio.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boikos SA, Pappo AS, Killian JK, LaQuaglia MP, Weldon CB, George S, et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report From the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA oncology. 2016;2(7):922–8. doi: 10.1001/jamaoncol.2016.0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gill AJ, Chou A, Vilain RE, Clifton-Bligh RJ. “Pediatric-type” gastrointestinal stromal tumors are SDHB negative (“type 2”) GISTs. The American journal of surgical pathology. 2011;35(8):1245–7. doi: 10.1097/PAS.0b013e3182217b93. author reply 7–8. [DOI] [PubMed] [Google Scholar]
- 34.Gill AJ, Benn DE, Chou A, Clarkson A, Muljono A, Meyer-Rochow GY, et al. Immunohistochemistry for SDHB triages genetic testing of SDHB, SDHC, and SDHD in paraganglioma-pheochromocytoma syndromes. Human pathology. 2010;41(6):805–14. doi: 10.1016/j.humpath.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 35.Gaal J, Stratakis CA, Carney JA, Ball ER, Korpershoek E, Lodish MB, et al. SDHB immunohistochemistry: a useful tool in the diagnosis of Carney-Stratakis and Carney triad gastrointestinal stromal tumors. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2011;24(1):147–51. doi: 10.1038/modpathol.2010.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doyle LA, Nelson D, Heinrich MC, Corless CL, Hornick JL. Loss of succinate dehydrogenase subunit B (SDHB) expression is limited to a distinctive subset of gastric wild-type gastrointestinal stromal tumours: a comprehensive genotype-phenotype correlation study. Histopathology. 2012;61(5):801–9. doi: 10.1111/j.1365-2559.2012.04300.x. [DOI] [PubMed] [Google Scholar]
- 37.Miettinen M, Killian JK, Wang ZF, Lasota J, Lau C, Jones L, et al. Immunohistochemical loss of succinate dehydrogenase subunit A (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation. The American journal of surgical pathology. 2013;37(2):234–40. doi: 10.1097/PAS.0b013e3182671178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Italiano A, Chen CL, Sung YS, Singer S, DeMatteo RP, LaQuaglia MP, et al. SDHA loss of function mutations in a subset of young adult wild-type gastrointestinal stromal tumors. BMC cancer. 2012;12:408. doi: 10.1186/1471-2407-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pantaleo MA, Nannini M, Astolfi A, Biasco G. A distinct pediatric-type gastrointestinal stromal tumor in adults: potential role of succinate dehydrogenase subunit A mutations. The American journal of surgical pathology. 2011;35(11):1750–2. doi: 10.1097/PAS.0b013e318230a523. [DOI] [PubMed] [Google Scholar]
- 40.Korpershoek E, Favier J, Gaal J, Burnichon N, van Gessel B, Oudijk L, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. The Journal of clinical endocrinology and metabolism. 2011;96(9):E1472–6. doi: 10.1210/jc.2011-1043. [DOI] [PubMed] [Google Scholar]
- 41.Heinrich M, Rankin C, Blanke CD, Demetri GD, Borden EC, Ryan CW, et al. Correlation of Long-term Results of Imatinib in Advanced Gastrointestinal Stromal Tumors With Next-Generation Sequencing Results: Analysis of Phase 3 SWOG Intergroup Trial S0033. JAMA oncology. 2017 doi: 10.1001/jamaoncol.2016.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Birch-Machin MA, Taylor RW, Cochran B, Ackrell BA, Turnbull DM. Late-onset optic atrophy, ataxia, and myopathy associated with a mutation of a complex II gene. Annals of neurology. 2000;48(3):330–5. [PubMed] [Google Scholar]
- 43.Pantaleo MA, Astolfi A, Urbini M, Nannini M, Paterini P, Indio V, et al. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. European journal of human genetics : EJHG. 2014;22(1):32–9. doi: 10.1038/ejhg.2013.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alston CL, Davison JE, Meloni F, van der Westhuizen FH, He L, Hornig-Do HT, et al. Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. Journal of medical genetics. 2012;49(9):569–77. doi: 10.1136/jmedgenet-2012-101146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, et al. SDHA is a tumor suppressor gene causing paraganglioma. Human molecular genetics. 2010;19(15):3011–20. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baysal BE, Lawrence EC, Ferrell RE. Sequence variation in human succinate dehydrogenase genes: evidence for long-term balancing selection on SDHA. BMC biology. 2007;5:12. doi: 10.1186/1741-7007-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chapman KB, Solomon SD, Boeke JD. SDH1, the gene encoding the succinate dehydrogenase flavoprotein subunit from Saccharomyces cerevisiae. Gene. 1992;118(1):131–6. doi: 10.1016/0378-1119(92)90260-v. [DOI] [PubMed] [Google Scholar]
- 48.Cecchini G, Schroder I, Gunsalus RP, Maklashina E. Succinate dehydrogenase and fumarate reductase from Escherichia coli. Biochimica et biophysica acta. 2002;1553(1–2):140–57. doi: 10.1016/s0005-2728(01)00238-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.