

Abstract
Activated macrophages are well known to exhibit anti-tumor properties. However, certain cell types show intrinsic resistance. Searching for a mechanism that could explain this phenomenon, we observed that the supernatant of resistant cells could confer resistance to otherwise sensitive tumor cells, suggesting the presence of a secreted suppressor factor. The effect was abolished upon dialysis, indicating that the suppressor factor has a low molecular weight. Further studies showed that prostaglandin E2 (PGE2) is secreted by the resistant tumor cells and that inhibition of PGE2 production by indomethacin, a cyclooxygenase (COX) inhibitor, eliminated the macrophage suppression factor from the supernatant, and sensitized the resistant tumor cells to macrophage cytotoxicity. This study emphasizes the important role of tumor-secreted PGE2 in escaping macrophage surveillance and justifies the use of COX inhibitors as an adjuvant for improving tumor immunotherapy.
Keywords: Indomethacin, PGE2, Tumor cell resistance, Macrophage cytotoxicity, TNFα
1. Introduction
Activated macrophages have the propensity to kill tumor cells both in vitro and in vivo in virtue of their ability to produce tumor necrosis factor-α (TNFα), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and nitric oxide [1–3]. Usually the macrophages need to be activated in order to kill tumor cells. An exception is the ability of macrophages to eliminate teratocarcinoma cells and embryonic stem cells without the need for an external activation signal [4]. Several activation agents can increase the tumoricidal activity of macrophages, including the cytokines IFN7, IL-12 and TNFα, whole bacteria such as Mycobacterium bovis Bacille Calmette-Guérin (BCG) and mycoplasma, and bacterial and yeast components such as lipopolysaccharide (LPS), zymosan and β-glucans [2,5–7].
The tumor microenvironment is comprised of a variety of non-malignant cells such as fibroblasts, stromal cells, endothelial cells, pericytes and a variety of immune cells that can make up to 90% of the total tumor volume [8]. Within the tumor microenvironment, the macrophages are exposed to a wide range of tumor-secreted factors as well as cytokines and chemokines secreted by immune cells, which modulate macrophage functions. Among these factors, TGFβ has drawn much attention, being produced by the stimulated macrophages themselves and various tumor cells. TGFβ promotes macrophage polarization from an anti-tumor M1 to a pro-tumor M2 phenotype [9,10]. Other factors that can contribute to this polarization are the cytokines IL-4, IL-13 and IL-10 [10,11] and repeated activation of macrophages, which leads to loss of TNFα secretion and acquisition of high iNOS activity (our unpublished data). Indeed, the central macrophage product TNFα promotes the generation of immature myeloid-derived suppressor cells (MDSCs) [12], providing a negative feedback mechanism to tune the immune response. Alternatively activated M2 macrophages have a strikingly different gene expression profile compared with M1 macrophages and express a different combination of surface receptors (e.g., CD163), cytokines (e.g., IL-10), tumorgrowth factors (e.g., EGF, FGF1,TGFβ1), pro-angiogenic factors (e.g., VEGF), matrix remodeling factors (e.g., fibrin and matrix metallopeptidases) and chemokines (e.g., CCL17/TARC, CCL22/MDC and CCL24/Eotaxin-2) [10,11]. In addition, M2 macrophages produce lower levels of ROS, but express higher levels of arginase I and indoleamine 2,3-dioxygenase (IDO) [10,11]. Additional subtypes of macrophages have also been identified [13], suggesting a broad spectrum of macrophage activation stages [6].
We have observed that while some tumor cells are susceptible to macrophage cytotoxicity, others are resistant. The aim of our study was to characterize the mechanisms involved in conferring macrophage resistance upon tumor cells. This study shows that a low molecular weight factor secreted by tumor cells, defined as prostaglandin E2 (PGE2), prevents macrophage activation required for tumor cytotoxicity. Inhibition of PGE2 production, using the non-steroid anti-inflammatory drug (NSAID) indomethacin, not only restored macrophage activation, but also conferred sensitivity of the otherwise resistant tumor cells to macrophage cytotoxicity.
2. Material and methods
2.1. Mice
C57BL/6 mice were obtained from the Animal Breeding Farm, Hebrew University-Hadassah Medical School of Jerusalem. All experiments involving animals were approved by the Hebrew University’s Institutional Animal Care and Use Committee.[23]
2.2. Cell cultures
Bone-marrow-derived macrophages (BMMΦ) were obtained from bone marrow cells (usually ~30 ×106 cells per mouse) harvested from the femur and tibia of 6–8-week old female C57BL/6 mice, which were cultivated in DMEM supplemented with 15% heat-inactivated fetal calf serum (FCS), 5% heat-inactivated horse serum, 30% L929 cell conditioned medium (LCM), 2 mM L-glutamine, 10 mM HEPES, 100U/ml penicillin and 100 μg/ml streptomycin. The macrophages were cultivated on 9 cm diameter bacteriological grade culture dishes (Miniplast, Ein Shemer, Israel) and were used as effectors 10–21 days after bone marrow seeding. LCM was prepared by seeding 106 L929 cells in 20 ml DMEM supplemented with 10% FCS in a 75 cm2 tissue culture flask (Nunclon, Denmark). Following 4–5 days incubation, when a monolayer had been reached, the supernatant was collected and sterile filtered.
A9 fibrosarcoma cells (a C3H fibrosarcoma derived from L929 cells), L929 fibrosarcoma cells, NIH3T3 mouse embryonic fibroblast-like cells and M109 Madison lung carcinoma cells were grown in DMEM supplemented with 5% heat-inactivated FCS, 2 mM L-glutamine, 10 mM HEPES and antibiotics. The FCS used was selected from batches that did not pre-activate macrophages. All cell cultures were incubated at 37°C in a humidified incubator containing 5% CO2. All cultures were routinely tested for being mycoplasma-free.
2.3. Cytotoxicity assay
Cytotoxicity assay was performed as described previously [4]. Briefly, target cells in the log phase of growth were pulsed with 1 μCi/ml of [3H]thymidine (sp. Act 5 Ci/mM; American Radiolabeled Chemicals, Inc.) for 24 h, washed in PBS, trypsinized and resuspended in DMEM with 10% FCS, 2 mM L-glutamine, and 10 mM HEPES. Ten thousand target cells were added to 1 × 105 BMMΦ in 96 flat-bottomed microwells (Nunc, Denmark) in 300 μl DMEM supplemented with 10% heat-inactivated FCS in the absence or presence of 1 μg/ml LPS (Escherichia coli o55:B5, Bacto®, Difco). Following 72 h, the supernatants (300 μl) were harvested from the microwells, diluted with Insta-Gel (Packard, Downers Grove, Illinois) and the radioactivity counted. The samples were kept at 4°C for 24 h prior to counting. Percentage specific cytolysis was calculated by the following formula: %Cytolysis=[(E-SR)/(T-SR)] × 100% where E is the d.p.m. of the supernatant from co-culture of target cells and macrophages, SR the spontaneous release in d.p.m. of an equal number of target cells in medium without macrophages, and T the total d.p.m. uptake of target cells.
For determination of target cell sensitivity to TNFα, 5000 [3H]thymidine-labeled cells were seeded in 100 μl DMEM with 5% FCS in each well of a 96-well plate. At the following day, 50 μl of various concentrations of TNFα (Genentech Inc., San Francisco) were added, followed by a 3-day incubation at 37°C. The extent of cell death was determined by measuring the released radioactivity as described above. Control wells got 50 μl of medium. Alternatively, cells were incubated with TNFα in the presence of 2 μg/ml Actinomycin D (Sigma), and the extent of cell death determined 18 h later.
2.4. Production of TNFα by activated macrophages and determination of TNFα titer
One hundred thousand BMMΦ were added to each of the 96 flat-bottomed microwells in 100 μl DMEM with 10% heat-inactivated FCS, 18 h prior to activation. The activation step was performed by changing the medium to DMEM without FCS or cell culture supernatant in the same medium, either in the absence or presence of μg/ml LPS, followed by incubation for 24 h. Macrophage supernatants were assayed for TNFα by bioassay as described previously [4]. Briefly, 4 × 104 Cl-7 cells were plated per 96 flat-bottomed microwell in 100 μl DMEM with 5% FCS. On the following day, 3fold dilutions of test supernatants and control media were made in the wells, followed by immediate addition of actinomycin D (Sigma; 2 μg/ml, final concentration). The cultures were incubated for 20 h at 37°C, and the survived Cl-7 cells were stained for 10 min with crystal violet (0.2% in 2% ethanol), washed with running tap water and allowed to dry. The destruction of the Cl-7 monolayer was determined by the amount of light (at 550 nm) absorbed by the residual stained cells in the wells using a Dynatech MicroElisa Reader (Artek, Farmingdale, NY). The S50 titer of TNFα was defined as the reciprocal of the dilution of the test solution required to destroy 50% of the target cell monolayer, as compared to control samples.
2.5. Conditioned medium of cultured cells
2 × 105 cells were seeded per well in a 24-well culture plate (Nunclon, Denmark) in 1.5 ml DMEM with 5% FCS. After 24 h, the medium was exchanged to fresh medium, and the supernatant collected 24 h later.The supernatants were centrifuged at 1200 rpm for 15 min, and kept at 4°C until use.
2.6. Surface TNF receptor binding assay
The assay was performed in accordance to Holtmann & Wallach [14]. TNFα was labeled with 125I by the chloramine-T method to a specific radioactivity of 1500 Ci/mmol. One million target cells were seeded in growth medium in tissue culture plates the day before assay. On the following day, the cells were washes and incubated on ice for 2h with 0.5 nM 125I-TNFα in the absence or presence of excess unlabeled TNFα (20 μM) in PBS containing 140 mM NaCl, 1.5 mM KH2PO4,8 mM Na2HPO4,2.7 mM KCl, 0.5 mM MgCl2, 0.9 mM CaCl2,0.5% bovine serum albumin (BSA) and 15 mM sodium azide. Thereafter, the cells were washed three times in the binding buffer, detached in Ca2+- and Mg2+-free PBS containing 5 mM EDTA and transferred to vials for radioactivity measurements. Specific binding of TNFα was calculated by subtracting the values of binding observed in the presence of an excess of unlabeled TNFα from the value of binding observed with 127I-TNFα alone.
2.7. Determination of prostaglandin E2 (PGE2) concentration
The PGE2 concentrations in cell supernatants were determined by radioimmunoassay. 100 μl of sample, buffer alone or PGE2 standard (0.15–10 ng/ml; Sigma) were mixed with 100 μl 0.01 M sodium phosphate buffer pH 7.4 containing 0.15 M NaCl, 0.1% BSA, 0.1% NaN3 and 500 μl anti-serum to PGE2 (Bio-Makor; diluted 1:10). Following a 30min incubation at 4°C, 100 μl of [5,6,8,11,12,14,15-3H(N)]-PGE2 (0.1mCi/ml, Amersham, England) was added at a dilution giving 50,000–100,000 d.p.m. After 60 min incubation at 4°C, 200 μl of a dextran coated charcoal solution [1% charcoal with 0.1% dextran (MW 35,000–45,000; Sigma)] in sodium phosphate buffer were added, except for samples intended for total radioactive read. After vigorous mixing, the samples were incubated for 10 min at 4°C, followed by centrifugation at 3000 rpm for 15 min at 4°C. 250 μl of the supernatant was mixed with 3 ml of Instagel, and the radioactivity was measured in a β-counter. No antibody was added to the blank samples. The percentage of bound radioactive PGE2 was calculated according to [(S - B)/(T - B)] × 100%, where S is the d.p.m of the sample, B the d.p.m of the blank sample and T the total radioactive amount. The limitation of this assay was 5 pg/ml PGE2.
2.8. Statistical analysis
Experiments were repeated 3–5 times. The arithmetic average of all experiments performed is given. Statistical significance was assessed by the one-tail distribution-free Mann-Whitney U test. Error bars represent standard error. Differences were considered significant when the p value was 0.05 or less.
3. Results
3.1. Differential sensitivity of transformed cells to macrophage cytotoxicity
The aim of this study was to understand why some transformed cells are resistant to macrophage-mediated killing. We observed that M109 lung carcinoma cells and NIH3T3 fibroblast-like cells were relatively resistant, whereas A9 and L929 fibroblast-like cells were highly sensitive to killing by activated macrophages (Fig. 1A). None of these cells were killed by non-activated macrophages, as expected. The resistance could be due to intrinsic resistance to TNFα-mediated killing, the major mediator of macrophage cytotoxicity, or the secretion of a macrophage suppressor factor by the tumor cells. We first analyzed whether the NIH3T3 and M109 cells express TNF receptors by using the TNFα binding assay [14]. We found that NIH3T3 and M109 bound even more 127 I TNFα per cell than the macrophage-sensitive L929 cells (Table 1), thus excluding the lack of TNF receptors as the reason for their resistance to macrophage cytotoxicity. It could be that the cells are resistant to the cytotoxic effect of TNFα. To test this possibility, the cells were incubated in various dilutions of activated macrophage-conditioned medium, which contained an active TNFα S50 titer of 99,170 when analyzed on Cl-7 cells in the presence of actinomycin D. This assay shows that both NIH3T3 and M109 responded to the macrophage-conditioned medium by cell death (Fig. 1B).
Fig. 1.
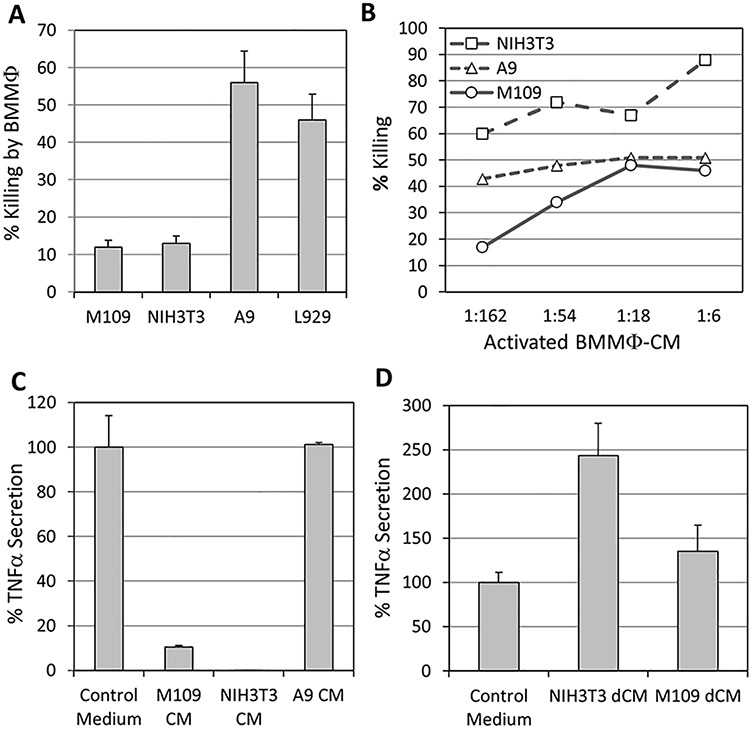
(A) The sensitivity of various transformed cells to activated macrophage cytotoxicity. 1 × 104 target cells were incubated with 1 × 105 BMMΦ in the presence of 1 μg/ml LPS for 3 days. In the absence of LPS, there was no killing of any cells. p<0.05 for M109/NIH3T3 versus A9/L929. (B) Cell sensitivity to TNFα. NIH3T3, M109 and A9 cells were incubated in various dilutions of activated macrophage-conditioned medium (BMMΦ-CM) in the presence of 2 μg/ml actinomycin D, and the extent of cell death determined 18 h later. (C) Inhibition of TNFα secretion by the conditioned media of transformed cells. One hundred thousand macrophages were incubated in serum-free medium or in conditioned media (CM) collected from NIH3T3, M109 or A9 cells, in the absence or presence of 1 μg/ml LPS for 24 h, and the amount of active TNFα secreted was determined as described in the Section 2.4. The relative TNFα secretion by macrophages in the presence of conditioned medium is presented in comparison to control medium. p<0.05 for M109 CM and NIH3T3 CM versus control medium and A9 CM. (D) The suppressor factor is lost upon dialysis of the conditioned media. Conditioned media were dialyzed for 48 h against PBS, and then against DMEM, using a dialysis tube with a cut-off of 10,000 Da. The effect of the dialyzed conditioned media (dCM) on LPS-induced TNFα secretion by macrophages was analyzed. The data are presented as relative TNFα secretion by macrophages in the presence of dialyzed conditioned medium in comparison to dialyzed control medium. p<0.05 for NIH3T3 dCM and M109 dCM versus dialyzed control medium.
Table 1.
Binding of 125I-TNFα to the different cell lines. The numbers represents the amount of radioactive TNFα bound to 1 × 106 cells after subtraction of the non-specific radioactive binding observed in the presence of excess unlabeled TNFα. The input 125I-TNFα was 2 × 105 cpm.
| Cell lines | Specific binding (cpm) |
|---|---|
| NIH3T3 | 10,427 |
| M109 | 12,778 |
| L929 | 4045 |
We next studied whether the resistant tumors secrete a factor that prevents macrophage activation. For this purpose, macrophages were incubated with the conditioned medium of the transformed cells in the absence or presence of 1 μg/ml LPS, and following a 24 h-incubation at 37°C, the amount of TNFα secreted by the macrophages was determined. We observed that, while the conditioned medium of A9 cells did not interfere with the macrophages’ ability to secrete TNFα in response to LPS, the conditioned media of NIH3T3 and M109 cells almost completely blocked TNFα secretion by macrophages (90–98% inhibition; Fig. 1C). These data suggest that the transformed cells indeed secrete a macrophage inhibitory factor.
In order to study whether the inhibitory factor has a low or high molecular weight, we dialyzed the cell conditioned media or control medium against PBS for 48 h and then against DMEM to restore essential nutrients using a dialysis tube with an MW cutoff of 10,000 Da. The dialyzed conditioned media were analyzed for their effects on LPS-induced TNFα secretion by macrophages. After dialysis, the conditioned media of NIH3T3 and M109 had lost their ability to inhibit TNFα secretion, and they even enhanced its production (Fig. 1D). This indicates the presence of a low molecular weight inhibitory factor in the conditioned media.
3.2. Identification of PGE2 as the macrophage inhibitory factor
Since it has been reported that the low-molecular weight biochemical compound prostaglandin E2 (PGE2) can inhibit TNFα secretion from macrophages [15], we wondered whether our inhibitory factor is PGE2. We first analyzed the presence of PGE2 in the conditioned media, and observed that both NIH3T3 and M109 cells produced high levels of PGE2, while PGE2 couldn’t be detected in the conditioned media of A9 cells (Table 2). We then analyzed the effect of various concentrations of PGE2 on TNFα secretion by macrophages, and surprisingly observed a dose-dependent effect where high PGE2 concentrations (from 1 nM-1 μM) strongly inhibited TNFα secretion, while low concentrations (especially at 0.1–1 pM) strongly enhanced it (Fig. 2A). The synergistic effect of low PGE2 concentrations on TNFα secretion might explain why the dialyzed conditioned medium even enhanced its secretion by LPS-stimulated macrophages (Fig. 1D). In order to validate that the PGE2 secreted from NIH3T3 and M109 is the factor that inhibited macrophage activation, the cells were treated with the cyclooxygenase (COX) inhibitor indomethacin, and the resulting conditioned medium was analyzed for their ability to affect TNFα production. Indeed, indomethacin abolished the macrophage inhibitory effect of the conditioned medium and even enhanced TNFα secretion (Fig. 2B). The PGE2 concentration in the conditioned medium was negligible in the presence of indomethacin (undetectable in the NIH3T3 conditioned media, while 10 ng/ml in the M109 conditioned media). In light of these encouraging data, it was intriguing to determine whether indomethacin could sensitize the resistant cells to macrophage cytotoxicity. For this purpose, NIH3T3, M109 and A9 cells were incubated with macrophages with LPS in the absence or presence of 50 μM indomethacin, and the extent of cell killing was measured after 3 days of co-cultivation. Indeed, indomethacin sensitized the resistant tumor cells to macrophage cytotoxicity (Fig. 2C). Further studies showed that when the macrophage-sensitive A9 cells were incubated with macrophages that have been pre-incubated with the resistant NIH3T3 or M109 cells, the cell killing was reduced by 75% (Fig. 2D). This reduction in cell killing could be reversed by treating the co-cultures with indomethacin (Fig. 2D), further emphasizing the central role of PGE2 in mediating the macrophage inhibitory effect. This is further manifested by the suppression of A9 cell killing when PGE2 was added to the co-culture of A9 and macrophages (Fig. 2E). Maximum inhibition was obtained at 1–10 nM PGE2 (47–51% inhibition; Fig. 2E).
Table 2.
Production of PGE2 by NIH3T3 and M109 cells. The PGE2 concentrations in the conditioned medium (CM) of the indicated cell lines was determined by radioimmunoassay as described in the Section 2.7. Conditioned media were collected 24 h after seeding 2 × 105 cells in 1.5 ml DMEM with 5% FCS in a 24-well culture plate.
| Cell lines | PGE2 conc in CM |
|---|---|
| NIH3T3 | 502pg/ml (1.4nM) |
| M109 | 397 pg/ml (1.1 nM) |
| A9 | n.d. |
| MEF | 185 pg/ml (0.5 nM) |
MEF: mouse embryonic fibroblasts. n.d.: non detectable.
Fig. 2.
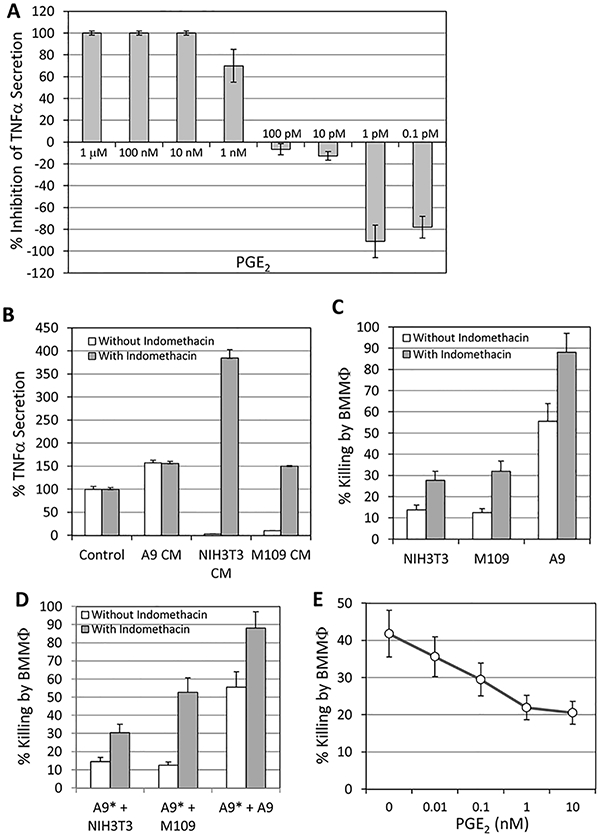
(A) Effect of PGE2 on TNFα secretion by LPS-stimulated macrophages. Hundred thousand macrophages that have been seeded in each well of a 96-flat bottomed tissue culture plate, were exposed to 1 μg/ml LPS in the absence or presence of various concentrations of PGE2 as indicated. The amount of TNFα secreted was analyzed 24h later. 1 nM corresponds to 352.4ng/ml PGE2. p<0.05 for 1 nM-1 μM and 0.1–1 pM PGE2 versus control medium. (B) Indomethacin treatment of N1H3T3 and M109 cells abolished the macrophage inhibitory effect of conditioned medium. Hundred thousand macrophages were incubated in control medium or conditioned media from untreated or indomethacin (50 μM, 24h)-treated M109, N1H3T3 and A9 cells in the presence of 1 μg/ml LPS, and the amount of TNFα secreted was determined 24h later. The data are presented as relative TNFα secretion by macrophages under each treatment condition in comparison to that of control medium. p<0.05 for N1H3T3 CM and M109 CM in the absence of indomethacin versus control and A9 CM; and p<0.05 for N1H3T3 CM and M109 CM in the presence versus in the absence of indomethacin. (C) Indomethacin sensitized the resistant tumor cells to macrophage cytotoxicity. N1H3T3, M109 and A9 cells were incubated with macrophages in the presence of 1 μg/ml LPS with or without 50 μM indomethacin for 3 days, and the extent of tumor cell killing determined. p < 0.05 for cells in the presence versus in the absence of indomethacin. (D) The presence of resistant cells prevented macrophage cytotoxicity on sensitive cells that could be reversed by indomethacin. Five thousand [3 H]-thymidine-labeled A9* cells were addedto 1 × 105 macrophagesthat have been pre-incubated with 5 × 103 unlabeled N1H3T3, M109 or A9 cells for 24 h. The extent of A9 cell killingwas determined after 3 days co-incubation. p<0.05 forkilling of A9 cells inthe presenceversus inthe absence of indomethacin. (E) PGE2 suppressed macrophage-mediated cytotoxicity of A9 cells. PGE2 was added at 10pM, 100pM, 1 nM and 10 nM to the co-culture of A9 cells and macrophages in the presence of 1 μg/ml LPS, and the extent of cell killing determined 3 days later. p<0.05 forcell killing in the presence versus in the absence of PGE2.
4. Discussion
Our pioneering study, performed in the late 1980’s, is fully relevant today in light of the current recognition that PGE2 is a key player in the carcinogenesis of colon cancer and several other cancer cell types [16–19], along with the introduction of aspirin in the clinics for the prevention of colon cancer [20,21]. During the years, it has been repeatedly shown that PGE2 suppresses diverse macrophage functions [22–26] and may even promote the shift towards the M2 phenotype [27–30] as well as the appearance of myeloid-derived suppressor cells with tumor-promoting function [31]. Other studies have demonstrated PGE2 production by various cancer cells [27,32,33,34–36], besides being produced by the macrophages themselves [37,38]. Our study combines these two issues showing that tumor-secreted PGE2 protects the tumor cells from macrophage cytotoxicity, a tumor immune escape mechanism that can be overcome by the drug indomethacin. Indomethacin and aspirin belong to the same group of non-steroid anti-inflammatory drugs (NSAIDs), and their benefits as adjuvants in cancer therapy seem thus to be of a dual nature. Namely, by preventing PGE2 production in the tumor cells, these drugs may directly inhibit PGE2-dependent tumor cell growth, and, simultaneously, increase macrophage cytotoxicity towards the tumor cells (Fig. 3). Both mechanisms contribute to the reduction of the tumor cell mass. Our study suggests a mechanistic explanation for the anti-tumor effects observed for indomethacin in various tumor models (e.g., [39–41]).
Fig. 3.
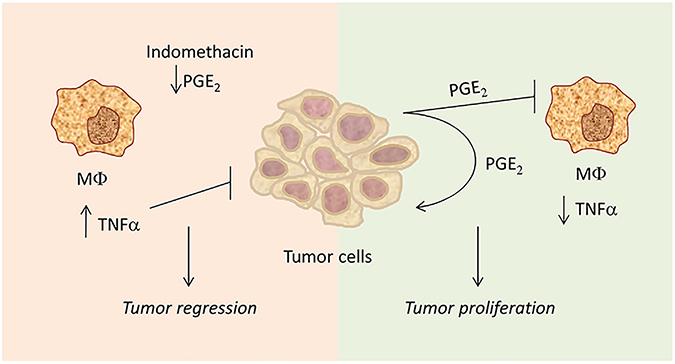
The Tumor-PGE2-Macrophage Cross-Talk. Tumor cells produce PGE2 that is sometimes necessary for tumor autonomous cell growth, and at the same time suppresses macrophage activation required for anti-tumor function. Interruption of this cross talk by indomethacin, restores the tumoricidal macrophage function that contributes to limit tumor growth.
The dose-dependent effects of PGE2 observed by us on macrophage secretion of TNFα has also been observed by another research group [42]. That group observed that the differential effects of PGE2 are mediated by changes in the intracellular cAMP/cGMP ratio [42]. A dose-dependent effect of PGE2 on macrophage adhesion and migration has also recently been documented [43]. An interesting fact is that reducing the PGE2 level by indomethacin may not only prevent the inhibitory effect of PGE2 on macrophages, but may even lead to such low levels that the tumoricidal effect of macrophages is enhanced. Indomethacin even increased the tumoricidal effect of activated macrophages on A9 cells that barely produce PGE2 (Fig. 2C). This might be due to inhibition of PGE2 production by the macrophages themselves during co-cultivation, where the negative feedback mechanism triggered upon macrophage activation is interrupted.
Of note, we could only reach up to 50% inhibition of A9 cell killing by macrophages when adding PGE2, even though the higher concentration (10 nM) completely blocked TNFα secretion. This might be due to the induction of another tumoricidal factor, such as the nitric oxide radical by PGE2 [44]. Nitric oxide has been shown to be involved in macrophage killing of L929 fibrosarcoma cells [3]. This scenario might also explain how PGE2 can induce tumoricidal activity of resident macrophages on L929 cells in the absence of any other activation signal [45]. Thus, PGE2 may support some anti-tumor macrophage activities despite abolishing TNFα production. This duality of PGE2 action might be important for maintaining essential macrophage functions under conditions where excessive immune responses are suppressed.
Altogether, our study sheds new light on the tumor cellmacrophage interrelationship, where macrophage tumoricidal activity can be regained by preventing excessive PGE2 production using the NSAID drug indomethacin. Tumor-secreted PGE2 likely acts in concert with other immune suppressive factors such as TGFβ [9], based on the observation that indomethacin couldn’t reverse macrophage killing of A9 cells when co-cultured in the presence of resistant transformed cells to a level similar to that observed in their absence (Fig. 2D). Our data would therefore suggest a potential use of indomethacin as an adjuvant agent in cancer immunotherapy [28]that ought to be combined, for instance, with a TGFβ inhibitor.
Acknowledgments
We are in depth grateful to Prof. David Wallach for doing the TNFα binding assay on our cell lines. This work was supported by the Concern Foundation of Los Angeles Society of Research Associates of the Lautenberg Center. HTJ is supported by the NIH, National Heart, Lung, and Blood InstituteR00HL109133add space and R01HL128411.
Abbreviations
- COX
cyclooxygenase
- PGE2
prostaglandin E2
- M Φ
macrophages
- NSAID
non-steroid anti-inflammatory drug
- TNF
tumor necrosis factor α
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Kawata K, Iwai A, Muramatsu D, Aoki S, Uchiyama H, Okabe M, Hayakawa S, Takaoka A, Miyazaki T, Stimulation of macrophages with the beta-glucan produced by aureobasidiu pullulans promotes the secretion of tumor necrosis factor-related apoptosis inducing ligand (TRAlL), PLoS One 10 (2015)e0124809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Long KB, Beatty GL, Harnessing the antitumor potential of macrophages for cancer immunotherapy, Oncoimmunology 2 (2013) e26860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nascimento FR, Gomes EA, Russo M, Lepique AP, Interferon regulatory factor (IRF)-1 is a master regulator of the cross talk between macrophages and L929 fibrosarcoma cells for nitric oxide dependent tumoricidal activity, PLoS One 10(2015)e0117782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sionov RV, Gallily R, Engulfment and intracellular killing of F9 teratocarcinoma cells by non-activated murine macrophages, Int. Immunol 2 (1990)291–301. [DOI] [PubMed] [Google Scholar]
- [5].Ruffell B, Coussens LM, Macrophages and therapeutic resistance in cancer, CancerCell 27 (2015) 462–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mosser DM, Edwards JP, Exploring the full spectrum of macrophage activation, Nat. Rev. Immunol 8 (2008) 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Caplan S, Gallily R, Barenholz Y, Characterization and purification of a mycoplasma membrane-derived macrophage-activating factor, Cancer Immunol. Immunother 39 (1994) 27–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sionov RV, Fridlender ZG, Granot Z, The multifaceted roles neutrophils play in the tumor microenvironment, Cancer Microenviron. 8 (2015) 125–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P, The polarization of immune cells in the tumour environmen by TGFbeta, Nat. Rev. Immunol 10(2010)554–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Galdiero MR, Bonavita E, Barajon I, Garlanda C, Mantovani A, Jaillon S, Tumor associated macrophages and neutrophils in cancer, Immunobiology (2013). [DOI] [PubMed] [Google Scholar]
- [11].Mantovani A, Allavena P, The interaction of anticancertherapies with tumor-associated macrophages, J. Exp. Med 212 (2015) 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sade-Feldman M, Kanterman J, Ish-Shalom E, Elnekave M, Horwitz E, Baniyash M, Tumor necrosis factor-alpha blocks differentiation and enhances suppressive activity of immature myeloid cells during chronic inflammation, Immunity 38(2013) 541–554. [DOI] [PubMed] [Google Scholar]
- [13].Tatano Y, T Shimizu, Tomioka H, Unique macrophages different from M1/M2 macrophages inhibit T cell mitogenesis while upregulating Th17 polarization, Sci. Rep 4 (2014) 4146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Holtmann H, Wallach D, Down regulation of the receptors fortumornecrosis factor by interleukin 1 and 4 beta-phorbol-12-myristate-13-acetate, J. Immunol 139(1987) 1161–1167. [PubMed] [Google Scholar]
- [15].Kunkel SL, Wiggins RC, Chensue SW, Larrick J, Regulation of macrophage tumor necrosis factor production by prostaglandin E2, Biochem. Biophys. Res. Commun 137 (1986) 404–410. [DOI] [PubMed] [Google Scholar]
- [16].Liu B, Qu L, Yan S, Cyclooxygenase-2 promotes tumorgrowth and suppresses tumor immunity, CancerCell. Int 15 (2015) 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang D, Fu L Sun H, Guo L, DuBois RN, Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice, Gastroenterology 149 (2015) 1884–1895, e1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pan Y, Jiang Y, Tan L, Ravoori MK, Gagea M, Kundra V, Fischer SM, Yang P, Deletion of cyclooxygenase-2 inhibits K-ras-induced lung carcinogenesis, Oncotarget 6 (2015) 38816–38826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].O’Callaghan G, Houston A, P rostaglandin E2 and the EP receptors in malignancy: possible therapeutic targets? Br.J. Pharmacol 172 (2015) 5239–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Garcia-Albeniz X, Chan AT, Aspirin forthe prevention of colorectal cancer, Best Pract. Res. Clin. Gastroenterol 25 (2011)461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nelson N, Ontrial: evidence from using aspirin to prevent cancer, J. Natl. CancerInst 107(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kim JG, Hahn YS, IFN-gamma inhibits the suppressive effects of PGE2 on the production of tumor necrosis factor-alpha by mouse macrophages, Immunol. Invest 29 (2000) 257–269. [DOI] [PubMed] [Google Scholar]
- [23].Qian X,Zhang J,Liu J, Tumor-secreted PGE2 inhibits CCL5 production in activated macrophages through cAMP/PKA signaling pathway,J. Biol. Chem 286(2011)2111–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Konopski Z, Seljelid R, Eskeland T, Cytokines and PGE2 modulate the phagocytic function of the beta-glucan receptor in macrophages, Scand. J. Immunol 37 (1993) 587–592. [DOI] [PubMed] [Google Scholar]
- [25].Strassmann G, Patil-Koota V, Finkelman F, Fong M, Kambayashi T, Evidence forthe involvement of interleukin 10in the differential deactivation of murine peritoneal macrophages by prostaglandin E2,J. Exp. Med 180 (1994) 2365–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sokolowska M, Chen LY, Liu Y, Martinez-Anton A, Qi HY, Logun C, Alsaaty S, Park YH, Kastner DL,Chae JJ,Shelhamer JH, Prostaglandin E2 inhibits NLRP3 inflammasome activation through EP4 receptor and intracellular cyclic AMP in human macrophages, J. Immunol 194 (2015) 5472–5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Heusinkveld M, de Vos van Steenwijk PJ, Goedemans R, Ramwadhdoebe TH, Gorter A, Welters MJ, van Hall T, van der Burg SH, M2 macrophages induced by prostaglandin E2 and IL-6 from cervical carcinoma are switched to activated M1 macrophages by CD4+ Th1 cells, J. Immunol 187(2011)1157–1165. [DOI] [PubMed] [Google Scholar]
- [28].Liu L, Ge D, Ma L, Mei J, Liu S, Zhang Q, Ren F, Liao H, Pu Q, Wang T, You Z, Interleukin-17 and prostaglandin E2 are involved in formation of an M2 macrophage-dominant microenvironment in lung cancer,J. Thorac. Oncol 71091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M, CREB pathway links PGE2 signaling with macrophage polarization, Proc. Natl. Acad. Sci. U. S. A 112(2015) 15642–15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhang Q, Cai DJ, Li B, Ovarian cancer stem-like cells elicit the polarization of M2 macrophages, Mol. Med. Rep 11 (2015) 4685–4693. [DOI] [PubMed] [Google Scholar]
- [31].Chang J, Vacher J, Yao B, Fan X, Zhang B, Harris RC, Zhang MZ, Prostaglandin E receptor 4 (EP4) promotes colonic tumorigenesis, Oncotarget 6(2015)33500–33511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sasaki Y,Nakatani Y, Hara S, Role of microsomal prostaglandin E synthase-1 (mPGES-1)-derived prostaglandin E2 in colon carcinogenesis, Prostag. Other Lipid Mediat 121 (2015) 42–45. [DOI] [PubMed] [Google Scholar]
- [33].Half E, Tang XM, Gwyn K, Sahin A, Wathen K, Sinicrope FA, Cyclooxygenase-2 expression in human breast cancers and adjacent ductal carcinoma in situ, Cancer Res 62 (2002) 1676–1681. [PubMed] [Google Scholar]
- [34].Abrahao AC, Castilho RM, Squarize CH, Molinolo AA, dos Santos-Pinto D Jr., Gutkind JS, A role forCOX2-derived PGE2 and PGE2-receptorsubtypes in head and neck squamous carcinoma cell proliferation, Oral Oncol 46 (2010) 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rundhaug JE, Simper MS, Surh I, Fischer SM, The role of the EP receptors for prostaglandin E2 in skin and skin cancer, Cancer Metastasis Rev 30 (2011) 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kaminska K, Szczylik C, Lian F, Czarnecka AM, The role of prostaglandin E2 in renal cell cancer development: future implications for prognosis and therapy, Future Oncol 10 (2014) 2177–2187. [DOI] [PubMed] [Google Scholar]
- [37].Kuroda E, Yamashita U, Mechanisms of enhanced macrophage-mediated prostaglandin E2 production and its suppressive role in Th1 activation in Th2-dominant BALB/c mice, J. Immunol 170 (2003) 757–764. [DOI] [PubMed] [Google Scholar]
- [38].Endo Y, Blinova K, Romantseva T, Golding H, Zaitseva M, Differences in PGE2 production between primary human monocytes and differentiated macrophages: role of IL-1betaand TRIF/IRF3, PLoS One 9 (2014) e98517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Moon CM,Kwon JH,Kim JS, Oh SH, Jin Lee K, Park JJ, Pil Hong S,Cheon JH, Kim TI, Kim WH, Nonsteroidal anti-inflammatory drugs suppress cancer stem cells via inhibiting PTGS2 (cyclooxygenase 2) and NOTCH/HES1 and activating PPARG in colorectal cancer, Int.J. Cancer 134 (2014) 519–529. [DOI] [PubMed] [Google Scholar]
- [40].Santander S, Cebrian C, Esquivias P, Conde B, Esteva F, Jimenez P, Ortego J, Lanas A, Piazuelo E, Cyclooxygenase inhibitors decrease the growth and induce regression of human esophageal adenocarcinoma xenografts in nude mice, Int. J. Oncol 40 (2012) 527–534. [DOI] [PubMed] [Google Scholar]
- [41].Connolly EM,Harmey JH, O’Grady T, Foley D, Roche-Nagle G, Kay E, Bouchier-Hayes DJ, Cyclo-oxygenase inhibition reduces tumour growth and metastasis in an orthotopic model of breast cancer, Br.J. Cancer 87 (2002) 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Renz H,Gong JH, Schmidt A, Nain M, Gemsa D, Release of tumor necrosis factor-alpha from macrophages. Enhancement and suppression are dose-dependently regulated by prostaglandin E2 and cyclic nucleotides,J. Immunol 141 (1988) 2388–2393. [PubMed] [Google Scholar]
- [43].Osma-Garcia IC, Punzon C, Fresno M, Diaz-Munoz MD, Dose-dependent effects of prostaglandin E2 in macrophage adhesion and migration, Eur.J. Immunol (2015). [DOI] [PubMed] [Google Scholar]
- [44].Milano S, Arcoleo F, Dieli M, D’Agostino R, D’Agostino P, De Nucci G, Cillari E, Prostaglandin E2 regulates inducible nitric oxide synthase in the murine macrophage cell line J774, Prostaglandins 49(1995) 105–115. [DOI] [PubMed] [Google Scholar]
- [45].Snider ME, Fertel RH, Zwilling BS, Prostaglandin regulation of macrophage function: effect of endogenous and exogenous prostaglandins, Cell. Immunol 74(1982) 234–242. [DOI] [PubMed] [Google Scholar]