

Abstract
Fast-scan cyclic voltammetry permits robust subsecond measurements of in vivo neurotransmitter dynamics, resulting in its established use in elucidating these species’ roles in the actions of behaving animals. However, the technique’s limitations, namely the need for digital background subtraction for analytical signal resolution, have restricted the information obtainable largely to that about phasic neurotransmitter release on the second-to-minute time scale. The study of basal levels of neurotransmitters and their dynamics requires a means of isolating the portion of the background current arising from neurotransmitter redox reactions. Previously, we reported on the use of a convolution-based method for prediction of the resistive-capacitive portion of the carbon-fiber microelectrode background signal, to improve the information content of background-subtracted data. Here we evaluated this approach for direct analytical signal isolation. First, protocol modifications (i.e., applied waveform and carbon-fiber type) were optimized to permit simplification of the interfering background current to components that are convolution-predictable. It was found that the use of holding potentials of at least 0.0 V, as well as the use of pitch-based carbon fibers, improved the agreement between convolution predictions and the observed background. Subsequently, it was shown that measurements of basal dopamine concentrations are possible with careful control of the electrode state. Successful use of this approach for measurement of in vivo basal dopamine levels is demonstrated, suggesting the approach may serve as a useful tool in expanding the capabilities of fast-scan cyclic voltammetry.
Fast-scan cyclic voltammetry (FSCV) at carbon-fiber microelectrodes is a powerful tool for in vivo measurements of electroactive neurotransmitters. Rapid scan rates enable high sensitivity measurements with subsecond time resolution while providing selectivity for neurotransmitters of interest (e.g., dopamine), which are often found at low concentrations relative to other electroactive extracellular species like ascorbic acid (AA) and 3,4-dihydroxyphenylacetic acid (DOPAC).1 However, rapid potential changes generate large electrode charging currents, mandating the use of digital background subtraction for analytical current resolution.2 Measurements are then limited to relative neurotransmitter changes on the time scale of background stability, restricting the use of the technique primarily to investigation of phasic neurotransmission. Information about absolute (i.e., basal) neurotransmitter concentrations, governed by phasic and tonic neuronal activity and believed to underlie important neurobiological phenomena,3−7 is lost in the background-subtraction step. Studies of basal concentrations have consequently largely been the domain of microdialysis.8−11 Microdialysis, coupled with an appropriate analysis technique, is readily capable of low- to subnanomolar detection of a variety of neurotransmitters, potentially simultaneously.12−14 As such, it has been able to provide a unique window into basal neurotransmitter levels, the information content encoded within them, and their correlation with behavior.15 The major traditional limitations reported on accessible information with microdialysis have stemmed from the relatively large size of microdialysis probes (typically a few hundred microns in diameter), coupled with the time needed to collect adequate sample volumes for robust analysis. These result in relatively low spatiotemporal resolution, hindering insight into fine details of localized and/or phasic neurotransmission, and also generate concerns about the effects of probe insertion on measurement fidelity.16 However, recent advancements, such as in the areas of probe fabrication (e.g., microfabrication),17,18 sampling methods (e.g., segmented flow and droplet microfluidics),19,20 and modulation of the immune response around the probe (e.g., delivery of the anti-inflammatory drug dexamethasone),21,22 have been reported to address these issues, effectively expanding the phenomena that may be accessible for study with microdialysis.23
Likewise, there has been a focus on altering the experimental protocols deployed with FSCV to access a wider array of neurochemical information and, in particular, generate data complementary to microdialysis concerning basal neurotransmitter concentrations. One reported approach has been the use of pharmacological agents with known effects, along with rapid drug administration techniques (i.e., microinfusion and intravenous administration), to manipulate neuronal release on an FSCV-compatible time scale.6 Additionally, multivariate data analysis (i.e., principal component regression) has been used to account for background artifacts appearing in the digitally subtracted data through incorporation of “background” voltammograms into the model, allowing for in vivo measurements of long-term dopamine concentration shifts following cocaine administration.24 Alternatively, modifications to measurement parameters have been explored. In a series of reports, Heien and colleagues have introduced fast-scan controlled adsorption voltammetry (FSCAV), a technique altering the waveform application frequency to modulate analyte adsorption behavior.25,26 Periods of rapid voltammetric scanning (100 Hz) are alternated with quiescent periods (∼10 s) at negative potentials to promote robust adsorptive preconcentration. Information from the initial scanning period allows determination of basal neurotransmitter concentrations, while analyte removal, and thus a blank background measurement, is achieved through continued rapid scanning prior to another preconcentration phase. The technique’s success for basal neurotransmitter measurement has been demonstrated in both brain slices and in vivo experiments.7,27,28
Recently, we reported on an alternative measurement protocol that relies on convolution to predict and remove nonfaradaic portions of the carbon-fiber FSCV background current.29 A small step placed immediately before each FSCV sweep is used to probe the electrode impedance and estimate its impulse response function through discrete differentiation. Convolution of this with the FSCV waveform allows prediction and digital subtraction of the nonfaradaic background signal. In the previous study, it was found that this approach only accounted for a portion of the total background current (i.e., that behaving like classical double-layer charging across the potential window), due mainly to the nonidealities introduced by a surface redox-active species (i.e., a quinone-like species). These moieties were observed to create a capacitive asymmetry across the potential window studied (between −0.6 and 1.3 V vs Ag/AgCl), apparently due to their redox state-dependent interactions with cations. Thus, the technique was explored for its potential to remove interferences in background-subtracted data introduced by ionic species affecting the predictable portion of the background in this potential range, and its success at removing these was demonstrated in vitro and in vivo.29−31 Here, we seek to explore further protocol changes to circumvent the nonidealities introduced by the quinone-like species and achieve more complete background removal. It is hypothesized that this will enable this technique to be used for direct resolution of the analytical current against this background to study basal neurotransmitter levels.
Here, we specifically explore modification of the waveform holding potential (to those positive of the commonly employed −0.4 V vs Ag/AgCl), as well as alternative carbon-fiber materials (i.e., pitch-based vs polyacrylonitrile-based), as a means to address these issues. While negative holding potentials promote adsorption of catecholamines, their use, combined with extensive oxidation of the electrode, exacerbates the current asymmetry across the potential window stemming from surface-active redox species.32 Thus, the use of holding potentials positive of the surface species’ redox potential is anticipated to mitigate these issues, albeit at the cost of sensitivity. Further, it will be shown that changing the carbon fiber used results in changes in the impedance characteristics that complement the convolution-based technique, though this results in a further decrease in sensitivity. However, there are other means of increasing sensitivity, namely the use of the higher scan rates and modulation of waveform application frequencies. It is shown that these strategies enable successful deployment of convolution-based background prediction to enable direct resolution of the analytical neurotransmitter current and access to information about the levels of, and changes in, basal neurotransmitter levels.
Experimental Section
Instrumentation and Software
T-650 (PAN-, or polyacrylonitrile-, based) and P-55 (pitch-based) type, cylindrical carbon-fiber microelectrodes (Thornel, Amoco Corporation, Greenville, SC; pulled in glass capillaries and cut to 50–100 μm exposed lengths) were used. Pulled electrodes were treated with epoxy as outlined in ref (29), as such treatment has been shown to result in beneficial changes to the electrical characteristics of the electrodes.31 Data was acquired in grounded Faraday cages, using a commercial interface (PCI-6052, 16 bit, National instruments, Austin TX) with a personal home computer and analyzed using locally constructed hardware and software (HDCV) written in LabVIEW (National Instruments, Austin, TX).33 Analog background subtraction (ABS) was implemented using the design described elsewhere.24 Of note, with the use of the convolution-based method, ABS current fed into the headstage was recorded separately and digitally added back to the measured data prior to convolution.
Electrochemical Experiments
Flow injection analysis experiments were performed using a syringe pump (Harvard Apparatus, Holliston, MA) operated at 0.8 mL/min using PEEK tubing (Sigma-Aldrich) connected to a pneumatically controlled six-port injection valve (Rheodyne, Rohnert Park, CA). All solutions were prepared in TRIS buffer (2.0 mM Na2SO4, 1.25 mM NaH2PO4·H2O, 140 mM NaCl, 3.25 KCl, 1.2 mM CaCl2·2H2O, 1.2 mM MgCl2·6H2O, and 15 mM Trizma HCl), adjusted to pH 7.4 with NaOH as necessary. Dopamine solutions were bubbled with nitrogen to prevent oxidative degradation. The tyramine fouling experiments follow the protocol described by Takmakov et al.34 However, the negative, not positive, potential limits were varied (−0.4 and 0.0 vs Ag/AgCl) during the recovery phase. Additionally, both waveforms (randomized order, n = 5 electrodes) were tested at each electrode, followed by conditioning and re-evaluation of the sensitivity before fouling.
The convolution-based approach used here is described in ref (21). Briefly, a waveform with a small amplitude pulse placed immediately prior to the FSCV sweep is used. The derivative of the step current (i.e., the system impulse response estimate) is convoluted with the applied waveform to generate a nonfaradaic current prediction, which is digitally subtracted.
In Vivo Measurements
Male Sprague–Dawley rats from Charles River (Wilmington, MA, USA) were pair-housed on a 12/12 h light/dark cycle. Animal procedures were approved by the UNC-Chapel Hill Institutional Animal Care and Use Committee (IACUC). For anesthetized experiments, animals (200–400 g) were administered urethane (1.5 mg/kg i.p.), and holes were drilled above the nucleus accumbens shell (AP + 1.7 mm, ML + 0.8 mm, DV −6.0 to −8.0 mm) and the contralateral hemisphere for lowering of the working and reference electrode, respectively. Additionally, a bipolar stimulating electrode (Plastics One, Roanoke, VA) was implanted at the ipsilateral ventral tegmental area (−2 mm, ML + 1.0 mm, DV −8.4 to −8.8 mm) to assist in positioning the working electrode.
Results and Discussion
Effect of Negative Holding Potentials on Background Currents
Waveforms with negative holding potentials are standard for FSCV catecholamine measurements, as they provide increased sensitivity by promoting adsorption.32 Here, the effect of using negative holding potentials on the observed background currents at carbon-fiber electrodes was evaluated.
Current at Negative Holding Potentials
Figure 1A shows backgrounds collected using increasingly positive holding potentials. As reported previously, the background decreases in both magnitude and complexity with more positive holding potentials.34,35 During FSCV waveform application, the electrode is held at the holding potential for the majority of the measurement window (typically <90%), although data is not typically collected during this time. However, current can often be observed throughout this period, which, after the return to the holding potential, decays to a steady state value proportional to that potential (Figure 1B). To understand its origin, slow-scan cyclic voltammetry (80 mV/s) was used (Figure 1C). The obtained voltammogram resembles that expected for the oxygen reduction reaction at a microelectrode, a two-step redox process that generates hydrogen peroxide at low overpotentials.36,37 Such a reaction was indeed suggested in the original report on the use of negative holding potentials and has been used recently to generate a microfabricated oxygen sensor.32,38 If one assumes the holding potential current originates from this reaction, Faraday’s law (n = It/Fz, z = 2 and I = 5 nA for −0.4 V) and the diffusion distance (x2 = 6Dt, DH202 = 1.8 × 10–5 cm2/s) suggest that the average concentration of peroxide around the electrode is above 1 μM after one holding period (∼92 ms).39 Further, collection of fast-scan voltammograms (400 V/s) using differing waveform application frequencies (0.5–30 Hz) indicates that this generated peroxide may be oxidized during the forward scan (Figure 1D). Using the 30 Hz waveform as the reference, differential CVs collected at lower frequencies have a peak that grows larger with increases in time at the holding potential (here, −0.4 V).
Figure 1.

Effects of negative holding potentials at carbon-fiber microelectrodes in TRIS buffer. (A) Fast-scan background voltammograms taken with different holding potentials (−0.5, −0.3, −0.1, and 0.0 V vs Ag/AgCl). (B) Amperometric current at various negative potentials over 5 min window at various holding potentials (0.0 to −0.8 V). (C) Slow scan voltammogram (80 mV/s, +0.7 to −1.7 V, forward scan shown only) showing significant redox current in the negative potential region. (D) Subtracted fast-scan voltammograms taken at various application frequencies (0.5–20 Hz), using the voltammograms taken at 30 Hz as the blank signal.
Electrode Surface Regeneration
It is known that the use of high positive potentials (>1.0 V) promotes etching of the carbon-fiber surface, which can be advantageous for maintaining sensitivity in the in vivo environment.34 However, the role of the negative holding potential in this process has not been characterized. To study this, a tyramine electrode fouling experiment, originally used to understand the positive potential limit effect, was carried out (Figure 2A). Carbon-fiber microelectrodes were fouled through electro-oxidation of tyramine, forming a surface film that decreases electrode capacitance and sensitivity. The negative holding potential’s effect on film removal and surface renewal was evaluated by application of a waveform with a positive potential limit known to promote etching (+1.3 V) and one of two negative holding potentials (−0.4 or 0.0 V). Successful removal was evaluated through dopamine sensitivity testing (−0.4 to 1.0 waveform). Both waveforms were tested at each electrode, using a randomized order and conditioning the electrode on a full waveform (−0.4 to 1.3 V) prior to fouling. The results are summarized in Figure 2B. The sensitivity was significantly lower after the use of the 0.0 V holding potential during the surface renewal phase than prior to fouling or after treatment. This suggests that a process occurring at negative potentials promotes this etching and surface renewal, driving the electrode surface evolution. While the specifics of this process are unknown, the generation of peroxide or interactions between the oxidized carbon surface and cations may underlie this phenomenon.40,41
Figure 2.
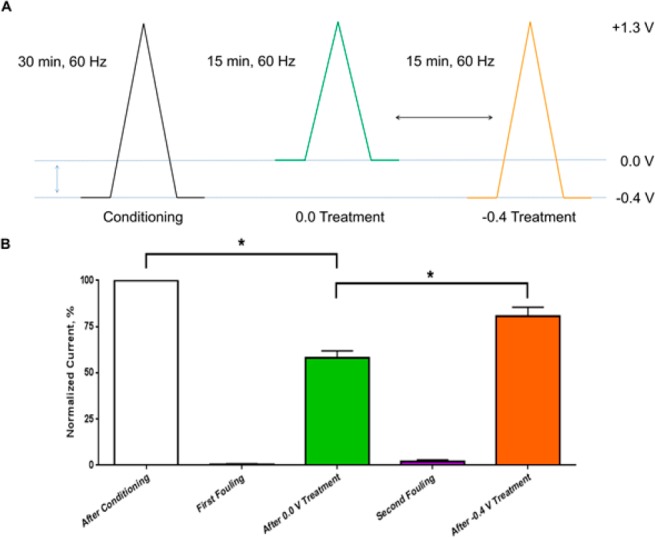
Sensitivity testing after tyramine fouling and treatment with waveforms with differing holding potentials. (A) Schematic of the experimental design. The arrow indicates that the order of waveform treatment (either 0.0 or −0.4 holding potential waveforms) was randomized. (B) Normalized peak currents observed for 1.0 μM of dopamine after each step in the experiment.
Convolution-Based Removal of Divalent Cation Interferences
Overall, these data suggest that some of the complexity and temporal evolution of carbon-fiber FSCV backgrounds stem from processes occurring at negative potentials. Further, as discussed in ref (29), the voltammetric waves (0.0 V and −0.3 V on the forward and backward scans in Figure 1A, respectively) originating from surface-bound, quinone-like species introduce capacitive nonidealities, increasing this complexity. In the context of convolution-based nonfaradaic current prediction, these effects are nonideal, as they introduce nonlinear features that cannot be modeled in this manner. However, it was hypothesized that the use of more positive holding potentials (≥0.0 V) would allow such issues to be avoided.
Previous deployment of the convolution-based approach with negative holding potentials (≤−0.4 V) found that only a subset of ionic signals was able to be successfully removed (i.e., those with traditional double-layer charging voltammograms). Other ions studied (i.e., the divalent cations Mg2+ and Ca2+) had more complex signals arising from interactions with surface quinone-like species, introducing nonlinearities that cannot be handled with the convolution-based method. For instance, Figure 3A shows the background-subtracted signal from a MgCl2-doped TRIS buffer using a holding potential of −0.5 V. Due to the capacitive asymmetry across the potential window, the use of the convolution-based method predicts an incorrectly large capacitive signal in the positive potential region. This results in strong artifacts in this region after prediction subtraction. However, avoidance of the potential region in which the quinone-like species undergoes its redox reaction avoids this issue, resulting in a considerably smaller ionic signal that can be successfully predicted with the convolution-based method (Figure 3B). This, in turn, permits dopamine signal resolution when flow injection analysis is performed for a mixture of MgCl2 and dopamine (Figure 3C).
Figure 3.

Removal of ionic artifacts seen during flow injection analysis of magnesium in TRIS buffer. (A-B) Raw (left) and convolution-treated (right) background-subtracted color plots using waveforms with −0.5 V (A) and 0.0 V (B) holding potentials. (C) Raw (left) and convolution-treated (right) background-subtracted color plots during flow injection analysis of a Mg2+-dopamine mixture.
Thus, the use of more positive holding potentials does indeed allow natural attenuation and convolution-based removal of a larger set of interferences. A similar attenuation was seen for signals caused by local pH changes (data not shown), which arise primarily through modulating the redox current of the quinone-like species. Overall, this is anticipated to allow for more complete isolation of neurotransmitter signals in unstable ionic environments. Additionally, longer measurements may be possible than with traditional FSCV protocols as background changes should be more successfully modeled and removed.
Optimization of Background Current for Prediction
Effect of Carbon Precursor
The use of more positive holding potentials should also make the background itself more amenable to convolution-based prediction. As noted in the previous report, considerable residual background current remained after prediction subtraction when using negative holding potentials, mandating background subtraction still be used for signal resolution. However, backgrounds observed with more positive holding potentials (representative background shown in Figure 1A, 0.0 V holding potential, purple line) resemble largely exponential charging curves, suggesting the convolution-based method may be able to more completely model it.
With the changes in the background seen at more positive holding potentials and attenuation of its redox current component, however, the fiber impedance characteristics become the dominant factor governing the background. Thus, two types of fibers (polyacrylonitrile- and pitch-based, referring to the carbon source) were evaluated to determine if one was preferable for use with the convolution-based approach. Traditionally, polyacrylonitrile (PAN)-based carbon fibers are used for FSCV, although pitch-based fibers have been explored. Of note, previous comparisons of the two found that the carbon precursor did not significantly affect the ability to detect neurotransmitters, although differences in electrochemical kinetics and sensitivity have been noted.42 PAN-based fibers, however, tend to have lower degrees of crystallinity, higher electrical resistivity, and lower densities (due to increased porosity).43 Thus, differences in impedance characteristics are expected. To explore this, apparent capacitances were determined from electrode backgrounds for two fibers (T-650, PAN; P-55, pitch) using cyclic voltammetry. The apparent capacitance was measured using the following equation
| 1 |
where C is the apparent capacitance, v is the scan rate (here, 400 V/s), and ip and in are the current amplitudes on the positive and negative sweeps, respectively. Figure 4A shows a representative set of apparent capacitances for the fibers at a range of times after capacitive charging (0.4 to 1.2 ms, corresponding to 0.16 to 0.48 V), along with a current trace during a step application for reference. In our previous report, we noted that the step current could not be adequately modeled by single-order exponential decay, suggesting time-varying impedance characteristics. Here, the PAN-based fiber (orange) shows a stronger time-dependence (increase of 7.6 μF/cm2 over this range) in the apparent capacitance relative to the pitch-based fiber (green, 1.6 μF/cm2). While the origin of these differences is uncertain, we hypothesize that the porosity of the PAN-based fibers plays a role, since porous carbons often exhibit multiple time constants due to these pores.44 Of note, this impedance variation occurs on a time scale that places information about it later in the step current (dashed line), where the signal-to-noise ratio is lower (and decreases with differentiation). The necessity of relying on this portion for capturing such changes in the impedance makes the use of the T-650 PAN-based fibers less useful than P-55 pitch-based fibers for convolution-based prediction.
Figure 4.

Evaluation of impedance characteristics and sensitivity of T-650 PAN-based and P-55 pitch-based carbon fibers (A) Apparent capacitance for T-650 PAN (orange) and P-55 pitch (green) fibers determined from a voltage sweep (400 V/s, 0.0 to 0.9 V vs Ag/AgCl) after capacitive charging (0.4–1.2 ms, corresponding to 0.16–0.48 V). A representative step current trace is shown on the same time scale for reference. (B) Peak current observed for dopamine oxidation (−0.4 to 1.3 V waveform) at both fiber types (n = 5 electrodes; error bars correspond to standard deviation).
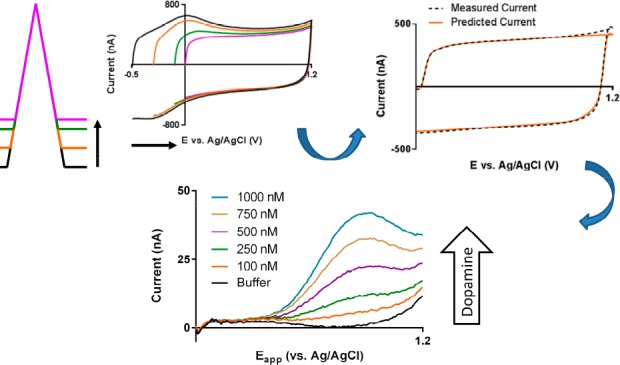
P-55 pitch-based fibers were used in the remaining experiments. To evaluate the analytical potential of these fibers, their sensitivity toward dopamine was determined and compared to the T-650 fibers (Figure 4B, 400 V/s, n = 5 electrodes). A decrease of approximately 60% (−0.4 to 1.3 V waveform; fiber type: slope ± S.E., T-650: 10.7 ± 0.3 nA/μM, P-55: 6.3 ± 0.3 nA/μM; ANCOVA comparison of slopes, F(1,6) = 71.26, p < 0.001) was observed. This sensitivity decline agrees with the previous comparison of these two fibers for another catecholamine, norepinephrine.42 The use of a more positive holding potential (0.0 vs −0.4 V) further decreased the sensitivity of the fibers to dopamine by approximately 25% at 400 V/s (data not shown). Despite this, the agreement between the convolution-based prediction and fiber background current is high under these latter conditions (Figure 5A). After subtraction (Figure 5B), only ∼2.5% of the total signal remains. In particular, the fit to the forward sweep (inset) at potentials below +0.8 V is robust, permitting the use of information in this region directly. Of note, above this voltage, a peak is seen, likely attributable to extraneous oxidation reactions.32 Additionally, slight mismatches between the prediction and measurement timings produce artifacts around the switching potentials.
Figure 5.

Prediction and removal of background currents using 0.0 V holding potential. (A) Measured (dashed black line) and predicted (orange line) from the convolution-based method. (B) Residual current after prediction subtraction, with a magnified inset. These data (average of 5 CVs, Bessel 4th order low-pass digital filter with 2 kHz cutoff) were collected with a 100 mV step placed 750 μs before a 400 V/s sweep in TRIS buffer at an unconditioned P-55 fiber.
Use of High Scan Rates
This lowered sensitivity resulting from these changes must be addressed for robust in vivo measurements. Traditional approaches for FSCV sensitivity enhancement include longer periods for adsorptive analyte preconcentration (e.g., lower waveform application frequencies) or higher scan rates.45−47 The latter approach does not sacrifice temporal resolution and is compatible with the convolution-based procedure. With higher scan rates, the high frequency impedance dominates the observed behavior. This leads to greater overlap of the sweep frequency components with the highest signal-to-noise region of the impulse response (Figure S-2A). As the focus here is high temporal resolution, scan rate modulation was the preferred means of sensitivity enhancement and is characterized here, with application frequency serving as a useful adjunct for further increases when necessary (see below).
As expected for adsorbed species, the sensitivity increased linearly with scan rate (Figure 6A).48 Positive shifts in the dopamine peak potential were also observed, attributable to slow electron transfer kinetics.1,47 There was also proportional background current amplification, mandating the use of analog background subtraction (ABS) to avoid saturation of the A/D converter used.24 With ABS, maximum scan rates of 4000–6000 V/s were attainable, the limit being determined by individual electrode impedances. This proved sufficient for in vitro convolution-based resolution of a dopamine signal. Figure 6B shows an example for dopamine solutions (TRIS buffer; 5000 V/s at 10 Hz) at an unconditioned P-55 fiber. It was first verified that the background could be predicted in a blank solution (black line). Addition of dopamine resulted in the appearance of a signal at approximately 0.9 V (orange line), which increased in a linear manner with subsequent additions.
Figure 6.

Use of high scan rates for dopamine detection. (A) Peak current observed for 1 μM dopamine oxidation as a function of scan rate. (B) Prediction-subtracted background voltammograms (5000 V/s, 10 Hz, forward sweep only) taken in TRIS buffer (black) and after sequential additions of dopamine (total concentrations of 100, 250, 500, 750, and 1000 nM).
Electrode Oxidation State
An established method for increasing sensitivity has been electrochemical conditioning (i.e., intentional surface oxide introduction). However, it was found that extended electrode oxidation can result in the unexpected appearance of a secondary background peak near the dopamine oxidation potential. Supplementary Figure S-2 shows an example of this peak seen during in vitro analysis of an extensively oxidized electrode in TRIS buffer alone after removal from the brain. It was found that the peak amplitude varied linearly with scan rate (data not shown). This suggests that this may be due to a surface redox-active functional group, which, due to its small contribution at lower scan rates (∼20 nA at 400 V/s), was not previously apparent. Fortuitously, the peak did not appear significantly responsive to local ionic changes (data not shown) and appears not to introduce nonidealities like those associated with the surface quinone-like species. Thus, given stability in this background signal, background subtraction can be used to resolve changes in basal levels in dopamine over long periods (not shown). However, the presence of this peak does prevent direct determination of the basal concentrations of dopamine. For this, the electrode state must be carefully controlled to prevent significant formation of the underlying species.
Selectivity
An important concern when conducting basal level measurements is the selectivity of such measurements over common interfering agents. Of particular concern are ascorbic acid (AA) and 3,4-dihydroxyphenylacetic acid (DOPAC), which are found at orders of magnitude larger concentrations in vivo than the low nanomolar levels expected for dopamine.1 To evaluate this aspect of the measurements, the sensitivity for these two interfering agents at the dopamine oxidation potential was determined at unoxidized electrodes (10 Hz, 0.0–1.2 V vs Ag/AgCl waveform, 5000 V/s; Supplementary Table S-1). Minimal responses were seen for either interfering agent, which is attributed to the slow electron transfer kinetics at unoxidized electrodes and the high scan rate used.32
In Vivo Measurement of Dopamine
Measurement of Basal Concentrations in Anesthetized Rats
First, the potential of the technique for measuring basal concentrations of dopamine was evaluated. For this experiment, an unconditioned electrode was first positioned in the cortex (−1.5 mm DV) and allowed to stabilize with the step-sweep convolution waveform. Measurements collected here (Figure 7A, black) were used to verify the lack of significant background contributions from the electrode. Subsequently, the electrode was lowered into the nucleus accumbens, using release evoked by electrical stimulation of the ventral tegmental area (VTA) for optimum placement, and allowed to stabilize.
Figure 7.

In vivo measurement of basal dopamine levels in an anesthetized rat. (A) Convolution-prediction background voltammograms (5000 V/s scan rate, forward sweep only) in the cortex (−1.5 mm DV; 10 Hz waveform application frequency) and the nucleus accumbens (NAc; −7.0 mm DV; 10, 5, 2, or 1 Hz waveform application frequency, shown in green, orange, and purple, respectively). (B) Comparison of background voltammogram with convolution prediction-subtracted voltammogram (black, 1 Hz) and digital background-subtracted, electrically evoked voltammogram.
After stabilization, an upward deflection of the signal was observed around the dopamine oxidation potential (Figure 7A, green) in the convolution-treated background voltammograms. However, given the low sensitivity of the electrode (likely exacerbated by in vivo fouling), robust measurements could not be made of the basal dopamine concentration under the initial experimental conditions (5000 V/s, 0.0 V holding potential, 10 Hz). To increase the sensitivity, the waveform application frequency was lowered, which resulted in an increase in the signal (Figure 7A, orange, purple, and brown for 5, 2, and 1 Hz, respectively). Of note, the voltammograms obtained compared favorably to those obtained by electrical stimulation and digital background subtraction in the same region (Figure 7B).
Basal dopamine concentration measurements (n = 3 rats) were then made using the 1 Hz waveform application frequency. For this, the current at the peak potential, after the subtraction of the convolution prediction from the total voltammogram, was used to generate the estimates (i.e., the current at the peak potential from the convolution estimate was used as the “zero” estimate). Postexperiment in vitro calibration was used to generate specific calibration factors for each electrode (representative calibration curve shown in Supplementary Figure S-3), and the estimates were obtained by averaging 30 voltammograms (30 s time bin) after electrode stabilization (representative trace shown in Supplementary Figure S-4). Of note, the electrode response to AA (200 μM) and DOPAC (20 μM) was tested to ensure no significant contribution to the current at the dopamine peak potential was observed. Additionally, delivery of raclopride (2 mg/kg, i.p., a D2 dopamine receptor antagonist) resulted in a 180 ± 20% (mean ± S.E.M.) increase in the signal at dopamine oxidation potential after 30 min.
These experiments generated an estimate of basal dopamine levels of 41 ± 13 nM (mean ± S.E.M.) in the rodent nucleus accumbens. This is slightly higher than estimates in the striatum reported by Gonan and Buda using slow electrochemical recordings (25 nM)49 in anesthetized (with chloral hydrate) rats and by Owesson-White et al. from FSCV paired with pharmacological manipulation (20–30 nM) in awake, freely moving Sprague–Dawley rats.6 All of these results are higher than those that tend to be reported using microdialysis. For example, Shou et al., using microdialysis coupled with online electrokinetic chromatography, reported a value of 18 nM in anesthetized (with ketamine/domitor) Sprague–Dawley rats,12 while Oslund et al. reported significantly lower values throughout the striatum (0.83, 0.73, 1.46, and 0.96 nM for the nucleus accumbens core, nucleus accumbens shell, dorsolateral striatum, and dorsomedial striatum, respectively) in awake, freely moving Long-Evans rats.15 While the origin of the discrepancies is unclear, the effect of probe size and damage, sensor placement/natural variability in the brain, the effects of anesthetic drugs, and the animal model used may play a role. Regardless, this experiment demonstrates another means of generating estimates of basal extracellular dopamine levels, expanding the approaches available to attempt to obtain their precise values. It is worth noting, however, that the lower sensitivity of the method, as compared to a technique like FSCAV, leads to less robust estimates of the extracellular dopamine concentration, albeit at higher temporal resolution.
Conclusions
High scan rates, holding potentials at or positive of 0.0 V vs Ag/AgCl, and alternative carbon-fiber materials facilitate the use of the convolution procedure for prediction and removal of the majority of the background current. With this background removed, information about basal levels of neurotransmitters, as demonstrated here for dopamine, can be accessed. However, as highlighted, measurement of absolute levels of dopamine requires careful control of the electrode state, typically requiring conditions that lead to low sensitivity. Further study is needed to optimize the electrode pretreatment and experimental waveform limits to maximize sensitivity without generating interfering signals. The instrumentation deployed here placed an upper limit on the achievable scan rate, and it is anticipated that further sensitivity increases could be gained with higher scan rates. Additionally, the work presented here exclusively focuses on the use of the positive sweep for measuring dopamine levels. However, greater confidence in signal assignment and quantification can be gained through measurement of the reductive wave, which requires addition of a negative potential excursion on the negative sweep. Preliminary studies suggest that the use of lower scan rates for the negative sweep may be a useful tool in this approach, allowing for robust measurement of the reductive wave within moderate potential limits (i.e., preventing severe reductive peak shifting). Finally, while not the focus here, it should be noted that the strategies employed here appear to be beneficial in signal isolation for traditional background-subtracted FSCV measurements, where the requirement for impedance ideality can be relaxed. Initial work suggests that this may be a promising route for lengthening the time that neurotransmitter fluctuations can be measured.
Acknowledgments
This research was supported by grants from NIH to R.M.W. (DA010900 and DA032530). This paper is dedicated to the Department of Chemistry at the University of North Carolina at Chapel Hill, in acknowledgment of its 200th anniversary, 1818–2018.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b04682.
Waveforms of varying scan rates in the time and frequency domain; convolution prediction-subtracted voltammogram showing peak following extensive electrode oxidation; representative post in vivo dopamine calibration curve; representative in vivo current-vs-time trace used for basal level estimates; in vitro sensitivity for dopamine, AA, and DOPAC (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Baur J. E.; Kristensen E. W.; May L. J.; Wiedemann D. J.; Wightman R. M. Anal. Chem. 1988, 60, 1268–1272. 10.1021/ac00164a006. [DOI] [PubMed] [Google Scholar]
- Howell J. O.; Kuhr W. G.; Ensman R. E.; Wightman R. M. J. Electroanal. Chem. Interfacial Electrochem. 1986, 209, 77–90. 10.1016/0022-0728(86)80187-5. [DOI] [Google Scholar]
- Berke J. D.; Hyman S. E. Neuron 2000, 25, 515–532. 10.1016/S0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- Schultz W. Annu. Rev. Neurosci. 2007, 30, 259–288. 10.1146/annurev.neuro.28.061604.135722. [DOI] [PubMed] [Google Scholar]
- Dreyer J. K.; Herrik K. F.; Berg R. W.; Hounsgaard J. D. J. Neurosci. 2010, 30, 14273–14283. 10.1523/JNEUROSCI.1894-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owesson-White C. A.; Roitman M. F.; Sombers L. A.; Belle A. M.; Keithley R. B.; Peele J. L.; Carelli R. M.; Wightman R. M. J. Neurochem. 2012, 121, 252–262. 10.1111/j.1471-4159.2012.07677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atcherley C. W.; Wood K. M.; Parent K. L.; Hashemi P.; Heien M. L. Chem. Commun. (Cambridge, U. K.) 2015, 51, 2235–2238. 10.1039/C4CC06165A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. D.; Justice J. B. J. Neurosci. Methods 1994, 54, 75–82. 10.1016/0165-0270(94)90161-9. [DOI] [PubMed] [Google Scholar]
- Newton A. P.; Justice J. B. Jr. Anal. Chem. 1994, 66, 1468–1472. 10.1021/ac00081a018. [DOI] [PubMed] [Google Scholar]
- Strecker R. E.; Sharp T.; Brundin P.; Zetterstrom T.; Ungerstedt U.; Bjorklund A. Neuroscience 1987, 22, 169–178. 10.1016/0306-4522(87)90207-7. [DOI] [PubMed] [Google Scholar]
- Sharp T.; Zetterstrom T.; Ljungberg T.; Ungerstedt U. Brain Res. 1987, 401, 322–330. 10.1016/0006-8993(87)91416-8. [DOI] [PubMed] [Google Scholar]
- Shou M.; Ferrario C. R.; Schultz K. N.; Robinson T. E.; Kennedy R. T. Anal. Chem. 2006, 78, 6717–6725. 10.1021/ac0608218. [DOI] [PubMed] [Google Scholar]
- Ngo K. T.; Varner E. L.; Michael A. C.; Weber S. G. ACS Chem. Neurosci. 2017, 8, 329–338. 10.1021/acschemneuro.6b00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Jaquins-Gerstl A.; Nesbitt K. M.; Rutan S. C.; Michael A. C.; Weber S. G. Anal. Chem. 2013, 85, 9889–9897. 10.1021/ac4023605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostlund S. B.; Wassum K. M.; Murphy N. P.; Balleine B. W.; Maidment N. T. J. Neurosci. 2011, 31, 200–207. 10.1523/JNEUROSCI.4759-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaquins-Gerstl A.; Michael A. C. Analyst 2015, 140, 3696–3708. 10.1039/C4AN02065K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. H.; Ngernsutivorakul T.; Mabrouk O. S.; Wong J. M.; Dugan C. E.; Pappas S. S.; Yoon H. J.; Kennedy R. T. Anal. Chem. 2016, 88, 1230–1237. 10.1021/acs.analchem.5b03541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngernsutivorakul T.; White T. S.; Kennedy R. T. ChemPhysChem 2018, 19, 1128–1142. 10.1002/cphc.201701180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Slaney T.; Mabrouk O.; Kennedy R. T. J. Neurosci. Methods 2010, 190, 39–48. 10.1016/j.jneumeth.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Hershey N. D.; Mabrouk O. S.; Kennedy R. T. Anal. Bioanal. Chem. 2011, 400, 2013–2023. 10.1007/s00216-011-4956-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varner E. L.; Leong C. L.; Jaquins-Gerstl A.; Nesbitt K. M.; Boutelle M. G.; Michael A. C. ACS Chem. Neurosci. 2017, 8, 1779–1788. 10.1021/acschemneuro.7b00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbitt K. M.; Varner E. L.; Jaquins-Gerstl A.; Michael A. C. ACS Chem. Neurosci. 2015, 6, 163–173. 10.1021/cn500257x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy R. T. Curr. Opin. Chem. Biol. 2013, 17, 860–867. 10.1016/j.cbpa.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans A.; Keithley R. B.; Kita J. M.; Sombers L. A.; Wightman R. M. Anal. Chem. 2008, 80, 4040–4048. 10.1021/ac800108j. [DOI] [PubMed] [Google Scholar]
- Atcherley C. W.; Laude N. D.; Parent K. L.; Heien M. L. Langmuir 2013, 29, 14885–14892. 10.1021/la402686s. [DOI] [PubMed] [Google Scholar]
- Atcherley C. W.; Laude N. D.; Monroe E. B.; Wood K. M.; Hashemi P.; Heien M. L. ACS Chem. Neurosci. 2015, 6, 1509–1516. 10.1021/cn500020s. [DOI] [PubMed] [Google Scholar]
- Abdalla A.; Atcherley C. W.; Pathirathna P.; Samaranayake S.; Qiang B.; Pena E.; Morgan S. L.; Heien M. L.; Hashemi P. Anal. Chem. 2017, 89, 9703–9711. 10.1021/acs.analchem.7b01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell M. H.; Atcherley C. W.; Heien M. L.; Lipski J. ACS Chem. Neurosci. 2015, 6, 1802–1812. 10.1021/acschemneuro.5b00120. [DOI] [PubMed] [Google Scholar]
- Johnson J. A.; Hobbs C. N.; Wightman R. M. Anal. Chem. 2017, 89, 6166–6174. 10.1021/acs.analchem.7b01005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs C. N.; Holzberg G.; Min A. S.; Wightman R. M. ACS Chem. Neurosci. 2017, 8, 2512–2521. 10.1021/acschemneuro.7b00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs C. N.; Johnson J. A.; Verber M. D.; Mark Wightman R. Analyst 2017, 142, 2912–2920. 10.1039/C7AN00508C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heien M. L. A. V.; Phillips P. E. M.; Stuber G. D.; Seipel A. T.; Wightman R. M. Analyst 2003, 128, 1413–1419. 10.1039/b307024g. [DOI] [PubMed] [Google Scholar]
- Bucher E. S.; Brooks K.; Verber M. D.; Keithley R. B.; Owesson-White C.; Carroll S.; Takmakov P.; McKinney C. J.; Wightman R. M. Anal. Chem. 2013, 85, 10344–10353. 10.1021/ac402263x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takmakov P.; Zachek M. K.; Keithley R. B.; Bucher E. S.; McCarty G. S.; Wightman R. M. Anal. Chem. 2010, 82, 9892–9900. 10.1021/ac102399n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh Y.; Park C.; Kim D. H.; Shin H.; Kang Y. M.; DeWaele M.; Lee J.; Min H. K.; Blaha C. D.; Bennet K. E.; Kim I. Y.; Lee K. H.; Jang D. P. Anal. Chem. 2016, 88, 10962–10970. 10.1021/acs.analchem.6b02605. [DOI] [PubMed] [Google Scholar]
- Sosna M.; Denuault G.; Pascal R. W.; Prien R. D.; Mowlem M. Sens. Actuators, B 2007, 123, 344–351. 10.1016/j.snb.2006.08.033. [DOI] [Google Scholar]
- Qu D. Y.; Tao Y. Z.; Guo L. P.; Xie Z. Z.; Tu W. N.; Tang H. L. Int. J. Electrochem Sci. 2015, 10, 3363–3371. [Google Scholar]
- Dengler A. K.; Wightman R. M.; McCarty G. S. Anal. Chem. 2015, 87, 10556–10564. 10.1021/acs.analchem.5b02866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanstroebiezen S. A. M.; Everaerts F. M.; Janssen L. J. J.; Tacken R. A. Anal. Chim. Acta 1993, 273, 553–560. 10.1016/0003-2670(93)80202-V. [DOI] [Google Scholar]
- Dikin D. A.; Stankovich S.; Zimney E. J.; Piner R. D.; Dommett G. H. B.; Evmenenko G.; Nguyen S. T.; Ruoff R. S. Nature 2007, 448, 457–460. 10.1038/nature06016. [DOI] [PubMed] [Google Scholar]
- Stankovich S.; Dikin D. A.; Dommett G. H. B.; Kohlhaas K. M.; Zimney E. J.; Stach E. A.; Piner R. D.; Nguyen S. T.; Ruoff R. S. Nature 2006, 442, 282–286. 10.1038/nature04969. [DOI] [PubMed] [Google Scholar]
- Huffman M. L.; Venton B. J. Electroanalysis 2008, 20, 2422–2428. 10.1002/elan.200804343. [DOI] [Google Scholar]
- Liu Y. D.; Kumar S. Polym. Rev. 2012, 52, 234–258. 10.1080/15583724.2012.705410. [DOI] [Google Scholar]
- Signorelli R.; Ku D. C.; Kassakian J. G.; Schindall J. E. Proc. IEEE 2009, 97, 1837–1847. 10.1109/JPROC.2009.2030240. [DOI] [Google Scholar]
- Bath B. D.; Martin H. B.; Wightman R. M.; Anderson M. R. Langmuir 2001, 17, 7032–7039. 10.1021/la0106844. [DOI] [Google Scholar]
- Bath B. D.; Michael D. J.; Trafton B. J.; Joseph J. D.; Runnels P. L.; Wightman R. M. Anal. Chem. 2000, 72, 5994–6002. 10.1021/ac000849y. [DOI] [PubMed] [Google Scholar]
- Keithley R. B.; Takmakov P.; Bucher E. S.; Belle A. M.; Owesson-White C. A.; Park J.; Wightman R. M. Anal. Chem. 2011, 83, 3563–3571. 10.1021/ac200143v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard A. J.; Faulkner L. R.. Electrochemical Methods: Fundamentals and Applications, 2nd ed.; John Wiley & Sons, Inc.: New York, NY, 2001. [Google Scholar]
- Gonon F. G.; Buda M. J. Neuroscience 1985, 14, 765–774. 10.1016/0306-4522(85)90141-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.