

Summary
Biogas production is performed anaerobically by complex microbial communities with key species driving the process. Hence, analyses of their in situ activities are crucial to understand the process. In a previous study, metagenome sequencing and subsequent genome binning for different production‐scale biogas plants (BGPs) resulted in four genome bins of special interest, assigned to the phyla Thermotogae, Fusobacteria, Spirochaetes and Cloacimonetes, respectively, that were genetically analysed. In this study, metatranscriptome sequencing of the same BGP samples was conducted, enabling in situ transcriptional activity determination of these genome bins. For this, mapping of metatranscriptome reads on genome bin sequences was performed providing transcripts per million (TPM) values for each gene. This approach revealed an active sugar‐based metabolism of the Thermotogae and Spirochaetes bins and an active amino acid‐based metabolism of the Fusobacteria and Cloacimonetes bins. The data also hint at syntrophic associations of the four corresponding species with methanogenic Archaea.
Introduction
The availability of fossil fuels is limited while the demand for energy increases steadily in the private and industrial sector, due to factors like affluence and population growth (Malik et al., 2016). Additionally, the consumption of natural gas and especially petroleum and coal leads to large amounts of greenhouse gas (GHG) emissions, mainly CO2 and CH4, implicating climate change and global warming (Liao et al., 2016; Malik et al., 2016).
Fuels produced from renewable sources are increasingly important alternatives to provide environmental‐friendly energy (Weiland, 2010; Zhang et al., 2016). Biogas is one of these important alternatives, which is produced by anaerobic digestion (AD) and mostly consists of CH4 with smaller proportions of CO2 and other impurities (Ge et al., 2016). In industrial‐sized biogas plants (BGPs), biogas production and usage take place under controlled conditions as they are connected to combined heat and power (CHP) systems where biogas is combusted to provide electricity and heat. For biogas production, a wide variety of substrates, e.g. energy crops like maize and organic household, industrial, slaughterhouse and agricultural wastes, can be used mostly in mixtures (Weiland, 2010; Mao et al., 2015; Ge et al., 2016; Zhang et al., 2016). Aside from substrate input, main differences in the setup of biogas plants concern the process temperature, as they can be run at mesophilic (35–42°C) and thermophilic (45–60°C) conditions. While mesophilic biogas processes are more stable and feature lower energy demand, biomass turnover is faster and methane yield is higher in thermophilic BGPs (Weiss et al., 2008; Weiland, 2010; Mao et al., 2015; Ruile et al., 2015).
The anaerobic digestion of biomass into methane can formally be subdivided into four phases, namely hydrolysis, acidogenesis, acetogenesis and methanogenesis. Within these phases, specialized groups of Bacteria and Archaea are responsible for the degradation of their respective substrates and are sometimes closely linked by syntrophic interactions. Hydrolysis is the first step in which bacteria break down complex polymers, like carbohydrates, lipids and proteins, into mono‐ and oligomers that are subsequently fermented by acidogenic and acetogenic bacteria to volatile fatty acids, alcohols, acetate, H2 and CO2. The last step, in which acetate (acetoclastic) or CO2 and H2 (hydrogenotrophic) are converted into methane, is solely performed by methanogenic Archaea (Weiland, 2010; Mao et al., 2015; Campanaro et al., 2016). Within the last years, the complex biogas‐producing microbial communities have been studied with regard to their members and their respective functions, but are still not fully understood. Culture‐dependent approaches include isolation, culturing, phenotypic analyses and sequencing of single community members (e.g. Maus et al., 2016). However, the culturing approach is limited as not all Bacteria and Archaea can be cultured and do not necessarily represent dominant and therefore functionally important members of the community. Thus, culture‐independent approaches, like metagenome and metatranscriptome sequencing, are frequently used to access the communities’ functional potential and determine transcriptional activity (e.g. Zakrzewski et al., 2012; Eikmeyer et al., 2013; Kovács et al., 2013; Bremges et al., 2015; Stolze et al., 2015, 2016). However, it is important to determine in situ functions of single microorganisms within the fermenters to better understand the process and, in the long run, enable optimization of the biogas production process. Therefore, metagenome assembly and subsequent binning of assembled contigs into genome bins are used as an approach to access single genomes within the microbial community, circumventing the need of cultivation (Kunath et al., 2017; Sczyrba et al., 2017). In this approach, species genomes are reconstructed from metagenome data sets representing a microbial community, enabling the reconstruction of their metabolic potential and abundance determination by mapping back metagenome reads on the respective genome bins (Mande et al., 2012; Sharpton, 2014; Sangwan et al., 2016). Binning has previously been used on biogas communities from laboratory‐ and production‐scale biogas production reactors, resulting in reconstruction of unknown species (Campanaro et al., 2016; Stolze et al., 2016; Treu et al., 2016; Xia et al., 2016; Kougias et al., 2017). Still, a study on the actual role within the community and these species’ in situ metabolic transcriptional activity within their respective habitats is missing.
In this study, we determined the in situ transcriptional activity of four genome bins originating from deeply sequenced metagenomes obtained from mesophilic and thermophilic agricultural biogas systems using corresponding metatranscriptome data. The four genome bins, of which three are novel and uncharacterized, represent species of the bacterial phyla Thermotogae, Fusobacteria, Spirochaetes and Cloacimonetes (WWE1) respectively. They have been previously selected due to their taxonomic affiliation and genomically characterized (Stolze et al., 2016). Analyses on the four species represented by the genome bins gave insights into their actual transcriptional activities and showed their respective metabolism and role within their habitats.
Results and Discussion
Metatranscriptome sequencing and read mapping
In this study, the actual in situ transcriptional activity of the species represented by four distinct genome bins was analysed to determine their transcriptional profiles and with this their roles within the biogas production process. For this purpose, RNA was extracted simultaneously from the same samples as the metagenomic DNA was derived from and metatranscriptome sequencing was performed in duplicates. In total, 900 million reads (137 Gbp; Table 1) were generated for one mesophilic and one thermophilic BGP. For the evaluation of transcriptional activities, the metatranscriptome reads from the BGPs were mapped on selected genome bins, counted and normalized on gene length and data set size resulting in transcripts per million (TPM) values.
Table 1.
Metatranscriptome sequencing results
| Biogas plant sample | Technical replicate | No. of reads | No. of bases |
|---|---|---|---|
| Mesophilic | 1 | 261 433 302 | 39 214 995 300 |
| 2 | 258 702 414 | 38 805 362 100 | |
| Thermophilic | 1 | 161 677 326 | 24 251 598 900 |
| 2 | 233 914 040 | 35 087 106 000 | |
| Total | – | 915 727,082 | 137 359 062 300 |
To determine whether the postulated metabolic potentials of the four bins correlate with their transcriptomic activities and whether relevant genes show high transcriptional rates under in situ conditions, their TPM values were further analysed. Next to the general evaluation of the 25 most highly transcribed genes of the genome bins (see Table S1, S2, S3 and S4), analyses of the bins’ activity in carbohydrate degradation, fermentation pathways and syntrophic associations were performed in depth by determining TPMs for respective meaningful genes. To enable a direct examination of high, moderate or low transcriptional activity of these genes, their TPM values were assigned to categories, ranging from 1 (within the lowest 10%) up to 10 (within the top 10% transcripts). Table 2 lists TPM values and respective categories for genes encoding carbohydrate‐active enzymes, chosen by their relevance in anaerobic digestion according to Vanwonterghem et al. (2016). Table 3 shows those for enzymes involved in fermentation pathways. It was recently suggested that all four genome bins may be syntrophically associated with methanogenic Archaea (Stolze et al., 2016). Therefore, transcriptional activities of genes encoding enzymes potentially involved in syntrophy, according to Worm et al. (2014), were analysed regarding presence and transcriptional activity for each genome bin. The results are shown in Table 4. For better clarity, in all three Tables, only those genes are shown that were found in at least one of the genome bins (the complete tables are represented in the Tables S1, S2, S3, S4, S5, S6 and S7). In the following chapters, each genome bin is discussed regarding these analyses.
Table 2.
Glycosyl hydrolase (GH) families relevant for anaerobic digestion according to Vanwonterghem et al. (2016) and their respective transcript per million (TPM) values and transcriptional categories for each of the four analysed genome bins. The categories range from 0 (no transcription) and 1 (lowest 10% of transcripts) to 10 (top 10% transcripts). n.d.: not detected
| Enzymes | Glycoside | Thermotogae bin | Fusobacteria bin | Spirochaetes bin | Cloacimonetes bin | ||||
|---|---|---|---|---|---|---|---|---|---|
| Enzyme type | Hydrolase family | TPM | Category | TPM | Category | TPM | Category | TPM | Category |
| Endo – and exo‐1,4‐β‐D‐glucanase (cellulase) | GH5 | 3.639 | 3 | n.d. | – | n.d. | – | n.d. | – |
| Hemicellulose | GH16 | 4.340 | 3 | n.d. | – | n.d. | – | 0 | 0 |
| GH28 | 2.167 | 2 | n.d. | – | 0.023 | 2 | n.d. | – | |
| GH53 | 41.236 | 8 | n.d. | – | n.d. | – | n.d. | – | |
| GH115 | n.d. | – | n.d. | – | 0.132 | 6 | n.d. | – | |
| GH76 | n.d | – | n.d | – | 0.018 | 1 | n.d | – | |
| Starch and glycogen Hydrolase | GH13 | 3.958 | 3 | 1.574 | 6 | 0.129 | 6 | 0.007 | 2 |
| GH77 | n.d. | – | 9.716 | 10 | 0.102 | 6 | 0 | 0 | |
| GH57 | 35.201 | 8 | 3.788 | 8 | 0.165 | 7 | 0.021 | 5 | |
| Lysozyme, chitinase (cell wall degradation) | GH18 | 6.172 | 3 | n.d. | – | n.d. | – | 0.021 | 5 |
| GH23 | 20.029 | 6 | 0.655 | 3 | n.d. | – | 0.063 | 8 | |
| GH73 | n.d. | – | n.d | – | n.d | – | n.d | – | |
| Glycosidase (hydrolysis of single sugar residues from non–reducing ends) | GH1 | 0.822 | 1 | n.d. | – | 0.074 | 4 | n.d. | – |
| GH2 | 38.604 | 8 | n.d. | – | 0.107 | 6 | n.d. | – | |
| GH3 | 3.022 | 2 | n.d. | – | 0.142 | 6 | n.d. | – | |
| 0.069 | 4 | ||||||||
| GH4 | 22.249 | 6 | n.d. | – | 0.019 | 2 | n.d. | – | |
| 144.144 | 10 | ||||||||
| GH38 | n.d. | – | n.d. | – | n.d. | – | 0.004 | 1 | |
| GH51 | 3.355 | 3 | n.d | – | 0.113 | 6 | n.d | – | |
| Oligosaccharide phosphorylase | GH130 | n.d. | – | n.d. | – | n.d. | – | 0.002 | 1 |
Table 3.
Fermentation pathway proteins and their respective transcript per million (TPM) values and transcriptional categories for each of the four analysed genome bins. The categories range from 0 (no transcription) and 1 (lowest 10% of transcripts) to 10 (top 10% transcripts) n.d.: not detected
| Fermentation type | Fermentation pathway | Enzyme | Interpro number | Thermotogae bin | Fusobacteria bin | Spirochaetes bin | Cloacimonetes bin | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| TPM | Category | TPM | Category | TPM | Category | TPM | Category | ||||
| Propionic acid fermentation | Acrylyl‐CoA pathway | CoA‐transferase (EC 2.8.3.1) | IPR003702 | n.d. | – | n.d. | – | n.d. | – | n.d. | – |
| Lactoyl‐CoA dehydratase dehydratase | IPR010327 | n.d | – | n.d. | – | 0.065 | 3 | 0.009 | 2 | ||
| Acyl‐CoA dehydrogenase (E.C. 1.3.99.3) | IPR034179 | n.d. | – | 0.052 | 1 | n.d. | – | n.d. | – | ||
| IPR034180 | |||||||||||
| Methylmalonyl‐ CoA pathway | Pyruvate carboxylase (EC 6.4.1.1) | IPR005930 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | |
| Malate dehydrogenase (EC 1.1.1.37) | IPR001252 | 25.110 | 7 | 1.308 | 5 | n.d. | – | 0.035 | 7 | ||
| IPR023958 | |||||||||||
| IPR011275 | |||||||||||
| Fumarate hydratase (EC 4.2.1.2) | IPR018951 | 18.872 | 6 | 0.982 | 4 | 0.13 | 6 | 0.0104 | 3 | ||
| IPR011167 | |||||||||||
| Fumarate reductase (EC 1.3.5.4) | IPR005884 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| Succinyl‐CoA synthetase (EC 6.2.1.4; EC 6.2.1.5) | IPR034722 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| IPR005809 | |||||||||||
| IPR005810 | |||||||||||
| Methylmalonyl‐CoA mutase (5.4.99.2) | IPR004608 | n.d. | – | 0.021 | 1 | n.d. | – | n.d. | – | ||
| IPR024067 | |||||||||||
| Methylmalonyl‐CoA epimerase (EC 5.1.99.1) | IPR017515 | 38.108 | 8 | n.d. | – | n.d. | – | n.d. | – | ||
| Methylmalonyl‐CoA decarboxylase (EC 4.1.1.41) | – | 24.441 | 7 | n.d. | – | n.d. | – | n.d. | – | ||
| CoA‐transferase (EC 2.8.3.1) | IPR003702 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| Ethanol fermentation | Pyruvate dehydrogenase (EC 1.2.4.1) | IPR017597 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | |
| IPR027110 | |||||||||||
| Pyruvate decarboxylase (EC 4.1.1.1) | – | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| Alcohol dehydrogenase (EC 1.1.1.1) | IPR023921 | 26.691 | 7 | 1.563 | 6 | 0.337 | 9 | 0 | 0 | ||
| 0.3203 | 8 | ||||||||||
| 0.028 | 2 | ||||||||||
| Formic acid fermentation | 2,3‐Butanediol fermentation | Pyruvate formate‐lyase (EC 2.3.1.54) | IPR005949 | n.d. | – | n.d. | – | n.d. | – | n.d. | – |
| Formate Hydrogen Lyase (EC 1.2.1.2) | IPR006478 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| IPR033689 | |||||||||||
| Acetolactate synthase (EC 2.2.6.1) | IPR004789 | 57.686 | 9 | 2.784 | 8 | n.d. | – | n.d. | – | ||
| IPR012782 | 18.144 | 6 | 1.922 | 7 | |||||||
| IPR012846 | |||||||||||
| IPR019455 | |||||||||||
| Acetolactate decarboxylase (EC 4.1.1.5) | IPR005128 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| Butanediol dehydrogenase (EC 1.1.1.4) | – | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| Mixed‐acid fermentation | Pyruvate carboxylase (EC 6.4.1.1) | IPR005930 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | |
| Malate dehydrogenase (EC 1.1.1.37) | IPR001252 | 25.110 | 7 | 1.309 | 5 | n.d. | – | 0.035 | 7 | ||
| IPR023958 | |||||||||||
| IPR011275 | |||||||||||
| Fumarase (EC 4.2.1.2) | IPR018951 | 18.873 | 6 | n.d. | – | 0.130 | 6 | n.d. | – | ||
| IPR011167 | |||||||||||
| Fumarate reductase (EC 1.3.1.6) | IPR027477 | n.d. | – | n.d. | – | n.d. | – | 0.007 | 2 | ||
| Lactate dehydrogenase (EC 1.1.1.28) | – | 19.016 | 6 | n.d. | – | 0.169 | 7 | n.d. | – | ||
| 18.631 | 6 | 0.189 | 7 | ||||||||
| Phosphotransacetylase (EC 2.3.1.8) | IPR016475 | 1.765 | 2 | 3.875 | 8 | n.d. | – | 0.021 | 5 | ||
| IPR004614 | |||||||||||
| IPR002505 | |||||||||||
| IPR012147 | |||||||||||
| Acetate kinase (EC 2.7.2.1) | IPR000890 | 79.588 | 9 | 4.573 | 8 | 0.136 | 6 | 0.029 | 6 | ||
| IPR004372 | |||||||||||
| IPR023865 | |||||||||||
| Butyric acid fermentation | Thiolase (EC 2.3.1.9) | – | n.d. | – | n.d. | – | n.d. | – | n.d. | – | |
| 3‐hydroxybutyryl‐ CoA dehydrogenase(EC 1.1.1.157) | – | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| Crotonase (EC 4.2.1.150) | – | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| Butyryl‐CoA dehydrogenase (EC 1.3.8.1) | – | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| Phosphate butyryl transferase (EC 2.3.1.19) | IPR014079 | 1.765 | 2 | n.d. | – | n.d. | – | n.d. | – | ||
| Homoacetogenesis |
Butyrate kinase (2.7.2.7) Pyruvate: ferredoxin oxidoreductase (EC 1.2.7.1) |
IPR011245 | 3.494 | 3 | n.d. | – | n.d. | – | 0.029 | 6 | |
| – | 50.748 | 8 | 14.265 | 10 | 0.053 | 3 | 0.060 | 8 | |||
| 49.909 | 8 | 0.050 | 8 | ||||||||
| 11.622 | 5 | ||||||||||
| 61.543 | 9 | ||||||||||
| Phosphotransacetylase(EC 2.3.1.8) | IPR016475 | 1.765 | 2 | 3.874 | 8 | n.d. | – | 0.021 | |||
| IPR004614 | |||||||||||
| IPR002505 | |||||||||||
| IPR012147 | |||||||||||
| Acetate kinase (EC 2.7.2.1) | IPR000890 | 79.588 | 9 | 4.573 | 8 | 0.136 | 6 | 0.029 | 6 | ||
| IPR004372 | |||||||||||
| IPR023865 | |||||||||||
| Lactic acid Fermentation | Homolactic acid fermentation | Glucose‐6‐phosphate isomerase (EC 5.3.1.9) | IPR001672 | 22.717 | 6 | 5.772 | 9 | 0.520 | 9 | 0.011 | 3 |
| IPR010551 | |||||||||||
| IPR016758 | |||||||||||
| IPR018189 | |||||||||||
| IPR023096 | |||||||||||
| 6‐phospho‐fructokinase (EC 2.7.1.11) | IPR000023 | 74.683 | 9 | 7.921 | 9 | n.d. | – | 0.057 | 8 | ||
| IPR012003 | |||||||||||
| IPR012004 | |||||||||||
| IPR012828 | |||||||||||
| IPR015912 | |||||||||||
| IPR022953 | |||||||||||
| Fructose‐bisphosphate aldolase (EC 4.1.2.13) | IPR023014 | 152.525 | 10 | 15.580 | 10 | 1.015 | 10 | n.d. | – | ||
| IPR000741 | 0.111 | 5 | |||||||||
| IPR011289 | |||||||||||
| IPR029768 | |||||||||||
| Triosephosphate isomerase (5.3.1.1) | IPR000652 | 81.170 | 9 | n.d. | – | n.d. | – | n.d. | – | ||
| IPR020861 | |||||||||||
| IPR022891 | |||||||||||
| IPR022896 | |||||||||||
| Lactate dehydrogenase (EC 1.1.1.28) | – | 19.016 | 6 | n.d. | – | 0.169 | 7 | n.d. | – | ||
| 18.631 | 6 | 0.189 | 7 | ||||||||
| Heterolactic acid fermentation | Hexokinase (EC 2.7.1.1) | IPR001312 | 81.171 | 9 | n.d. | – | 0.115 | 5 | n.d. | – | |
| IPR019807 | |||||||||||
| IPR022672 | |||||||||||
| IPR022673 | |||||||||||
| Glucose‐6 phosphate dehydrogenase (EC 1.1.1.49) | IPR001282 | 18.460 | 6 | n.d. | – | 0.128 | 6 | n.d. | – | ||
| IPR019796 | |||||||||||
| IPR022674 | |||||||||||
| IPR022675 | |||||||||||
| 6‐phosphogluconolactonase (EC 3.1.1.31) | IPR022528 | n.d. | – | n.d. | – | 0.346 | 9 | n.d. | – | ||
| Phospho‐gluconate dehydrogenase (EC 1.1.1.44) | IPR006184 | 6.055 | 3 | n.d. | – | 0.095 | 5 | n.d. | – | ||
| IPR006114 | |||||||||||
| IPR006113 | |||||||||||
| IPR006183 | |||||||||||
| Ribulose‐phosphate 3‐epimerase (EC 5.1.3.1) | IPR000056 | 28.915 | 7 | 2.740 | 8 | 0.578 | 9 | 0.012 | 3 | ||
| IPR026019 | |||||||||||
| Xylulose‐5‐phosphate phosphoketolase (EC 4.1.2.9) | – | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| Acyl‐phosphatase (EC 3.6.1.7) | IPR001792 | 15.719 | 5 | n.d. | – | n.d. | – | 0.021 | 5 | ||
| IPR017968 | |||||||||||
| IPR020456 | |||||||||||
| IPR028627 | |||||||||||
| Acetate kinase (EC 2.7.2.1) | IPR000890 | 79.588 | 9 | 4.573 | 8 | 0.136 | 6 | 0.029 | 6 | ||
| IPR004372 | |||||||||||
| IPR023865 | |||||||||||
| Phosphotransacetylase (EC 2.3.1.8) | IPR016475 | 1.765 | 2 | 3.875 | 8 | n.d. | – | 0.028 | 6 | ||
| IPR004614 | |||||||||||
| IPR002505 | |||||||||||
| IPR012147 | |||||||||||
| Acetaldehyde dehydrogenase (EC 1.2.1.10) | IPR003361 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| IPR015426 | |||||||||||
| Alcohol dehydrogenase (EC 1.1.1.1) | IPR023921 | 26.691 | 7 | 1.563 | 6 | 0.337 | 9 | 0 | 0 | ||
| 0.303 | 8 | ||||||||||
| 0.028 | 2 | ||||||||||
Table 4.
Proteins possibly associated with syntrophy according to Worm et al. (2014) and their respective transcript per million (TPM) values and transcriptional categories for each of the four analysed genome bins. The categories range from 0 (no transcription) and 1 (lowest 10% of transcripts) to 10 (top 10% transcripts). n.d.: not detected
| Protein | Subunit | Interpro number | Thermotogae bin | Fusobacteria bin | Spirochaetes bin | Cloacimonetes bin | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| TPM | Category | TPM | Category | TPM | Category | TPM | Category | |||
| Capsule synthesis protein, CapA | – | IPR019079 | n.d. | – | n.d. | – | n.d. | – | 0.04 | 7 |
| Cell cycle, FtsW, RodA SpoVE | – | IPR018365 | n.d. | – | 0.42 | 2 | n.d. | – | n.d. | – |
| 0.64 | 3 | |||||||||
| 0.42 | 2 | |||||||||
| Ribonuclease P, conserved site | – | IPR020539 | n.d. | – | n.d. | – | n.d. | – | 0.12 | 9 |
| Cytoplasmic FDH | NUO 51 kDa | IPR019575 | n.d. | – | n.d. | – | 0.054 | 3 | n.d. | – |
| IPR001949 | 79.06 | 9 | 0.36 | 8 | 0.054 | 3 | 0.02 | 5 | ||
| 0.02 | 5 | |||||||||
| Extracytopl. FDH | Alpha | IPR006443 | 79.06 | 9 | 0.36 | 8 | n.d. | – | 0.02 | 5 |
| FeFe‐hydrogenase | Alpha | IPR004108 | n.d. | – | n.d. | – | 0.075 | 4 | n.d. | – |
| IPR009016 | 110.75 | 9 | 2.07 | 7 | 0.075 | 4 | 0.05 | 8 | ||
| 9.26 | 10 | |||||||||
| IPR003149 | 110.75 | 9 | 2.07 | 7 | 0.075 | 4 | 0.05 | 8 | ||
| 9.27 | 10 | |||||||||
| IPR013352 | 110.75 | 9 | 9.27 | 10 | 0.075 | 4 | 0.05 | 8 | ||
| NiFe‐hydrogenase | – | IPR001501 | 110.75 | 9 | 9.27 | 10 | n.d. | – | 0.05 | 8 |
| IPR018194 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| Rnf complex | RnfB | IPR007202 | n.d. | – | n.d. | – | n.d. | – | n.d. | – |
| IPR010207 | 798.79 | 10 | 2.07 | 7 | 0.25 | 8 | 0.02 | 5 | ||
| 2.68 | 8 | |||||||||
| RnfC | IPR026902 | 798.79 | 10 | 2.68 | 8 | 0.34 | 9 | 0.02 | 5 | |
| IPR010208 | 14.37 | 5 | 4.01 | 8 | 0.34 | 9 | 0.01 | 2 | ||
| RnfD | IPR004338 | 14.37 | 5 | 4.01 | 8 | 3.24 | 10 | 0.01 | 2 | |
| IPR011303 | 7.63 | 4 | 0.93 | 4 | 0.39 | 9 | 0.01 | 2 | ||
| RnfG | IPR007329 | 7.63 | 4 | 0.93 | 4 | n.d. | – | 0.01 | 2 | |
| Ech complex | EchA | IPR001750 | 1100.12 | 10 | 12.01 | 10 | n.d. | – | 0.01 | 2 |
| 162.44 | 10 | 0.01 | 2 | |||||||
| IPR001516 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| EchB | IPR001694 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | |
| EchC | IPR006137 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | |
| EchD | IPR001268 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | |
| IPR012179 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | ||
| EchE | IPR001135 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | |
| Etf Alpha | – | IPR014731 | n.d. | – | n.d. | – | 0.41 | 9 | n.d. | – |
| Etf Beta | – | IPR012255 | n.d. | – | n.d. | – | 1.19 | 10 | 0.03 | 6 |
| Bcd | – | IPR006089 | n.d. | – | n.d. | – | n.d. | – | 0.02 | 5 |
| – | IPR009075 | n.d. | – | n.d. | – | n.d. | – | 0.05 | 8 | |
| – | IPR006092 | n.d. | – | n.d. | – | n.d. | – | 0.05 | 8 | |
| – | IPR006091 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | |
| – | IPR013786 | n.d. | – | n.d. | – | n.d. | – | 0.05 | 8 | |
| – | IPR009100 | n.d. | – | n.d. | – | n.d. | – | 0.05 | 8 | |
| DUF224 | – | IPR003816 | n.d. | – | n.d. | – | n.d. | – | 0.05 | 8 |
| – | IPR004017 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | |
| – | IPR023234 | n.d. | – | n.d. | – | n.d. | – | n.d. | – | |
The transcriptional profile of the Thermotogae bin indicates a metabolism based on sugar fermentation
The previous taxonomic and genetic analyses of the Thermotogae genome bin showed that it represents a species closely related to the thermophilic bacterium Defluviitoga tunisiensis L3 (Maus et al., 2015, 2016), or may even indicate another strain of this species. It is presumably able to utilize a wide variety of mono‐, di‐ and polysaccharides and was predicted to produce acetate, hydrogen and carbon dioxide as end‐products (Stolze et al., 2016). Analysis of the 25 most highly transcribed genes within the Thermotogae bin showed that 22 of them are functionally classified and 17 encode proteins involved in mandatory processes like transcription, translation, fatty acid metabolism, iron storage, electron transport, protein and RNA folding. Three highly transcribed genes encode proteins associated with ATP‐binding cassette (ABC) transporters (see Table S1), known as importers for sugars and other solutes (Davidson et al., 2008), with two of them specifically annotated as maltose import system. In total, 223 transcripts encoding proteins involved in sugar utilization were found for the bin. Most of these are involved in either binding and import (ABC sugar transporters) or sugar utilization within the cell, e.g. via the glycolysis pathway. Table 2 shows that the Thermotogae bin encodes eleven glycoside hydrolase (GH) family proteins, all of them being transcribed featuring TPM values above the average (TPM categories ≥ 6). Selected sugar utilization genes of the genome bin and their transcriptional activity are indicated in Fig. 1. It appeared that especially the sugar transporter genes and those for glycolysis enzymes are highly transcribed. In general, these findings strongly suggest that this species actively degrades and utilizes a variety of carbohydrates, whose end‐products are further channelled into the glycolysis pathway.
Figure 1.
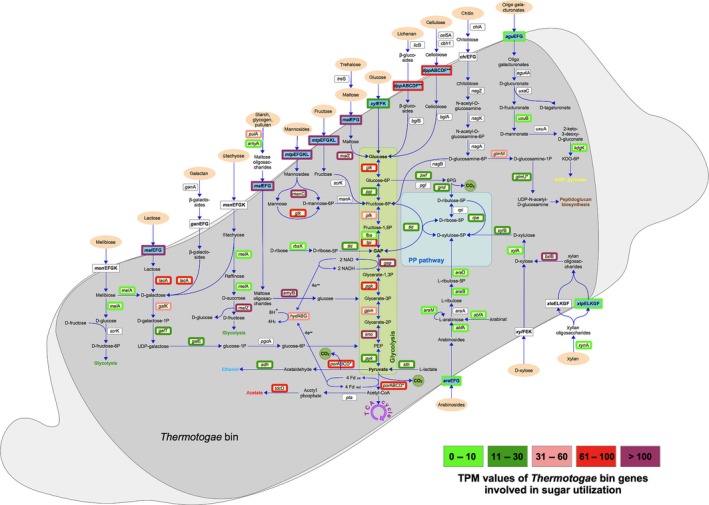
Metabolic reconstruction of sugar utilization pathways in the Thermotogae genome bin and transcript per million (TPM) values for genes encoding involved proteins. Figure modified according to Maus et al. (2016). Carbohydrates are labelled in light orange ovals, transporters in blue and corresponding genes in yellow rectangles. White rectangles represent genes lacking in the genome bin in comparison with its reference strain Defluviitoga tunisiensis L3. Frames indicate the category of TPM values for the respective gene, the five categories explained on the bottom right. The glycolysis and the pentose phosphate pathways are highlighted in green and blue, respectively. Abbreviations: CO 2, carbon dioxide; GAP, glyceraldehyde‐3‐phosphate; KDG‐6P, 2‐keto‐3‐deoxy‐d‐gluconase‐6‐phosphate.
Regarding the production of end‐products, Fig. 1 and Table 3 reflect the bin's activity in hydrogen, CO2, acetate and possibly lactate and ethanol production. Alternatively, lactate could be used for pyruvate production. Hydrogen and acetate production is known for D. tunisiensis L3 and was previously discussed as hint for a syntrophic lifestyle with aceticlastic or hydrogenotrophic Archaea (Maus et al., 2016; Stolze et al., 2016). This assumption is further supported by the presence and partially high transcriptional activities of genes encoding enzymes associated with syntrophy (Worm et al., 2014). The same applies for genes that are important for hydrogen production (see Table 4 and Fig. 1). For example, transcripts encoding the bifurcating FeFe‐hydrogenase (hydABG), pyruvate oxidoreductase (porABCD) and subunits of the Rnf complex were identified for the genome bin. The Rnf complex represents a membrane bound transporter that was shown to be able to conserve energy by coupling the oxidation of NADH to the simultaneous reduction of ferredoxin with ion transport (Biegel and Muller, 2010; Hess et al., 2013; Worm et al., 2014). The reduced ferredoxin may then be used as electron donor by a (bifurcating) hydrogenase to form H2 from protons (H+). The consumption of hydrogen from the producer would enable its formation despite the thermodynamically unfavourable nature of this process (Verhaart et al., 2010; Worm et al., 2014). Despite the lack of other putative syntrophy‐associated genes, the high TPM values for hydrogen production‐associated genes strongly indicate that the species represented by the Thermotogae bin is syntrophically associated with a partner consuming hydrogen, as it was previously described for other Thermotogae species (Balk et al., 2002; Johnson et al., 2006).
In summary, the in situ transcriptional profile of the Thermotogae genome bin shows partially high transcriptional activities regarding genes encoding proteins involved in (complex) sugar utilization, acetate, ethanol, CO2 and H2 production and those having predicted functions in a syntrophic association. The profile therefore reflects a possibly syntrophic and sugar‐based lifestyle of the corresponding species that in its thermophilic habitat occupies the role of a hydrolytic/acetogenic bacterium.
The transcriptional profile of the Fusobacteria bin indicates a motile species with a metabolism based on amino acid fermentation
Previous genetic analyses of the Fusobacteria genome bin suggested that the corresponding bacterium is an amino acid‐fermenting, acetogenic bacterium, possibly also syntrophically associated with methanogenic Archaea (Stolze et al., 2016). The species’ transcriptome analysis, based on metatranscriptome sequencing data from its mesophilic habitat, was supposed to uncover its in situ response to prevailing environmental conditions. Analyses of the genes with the 25 highest TPM values showed functional annotations for 15 genes, with six being involved in mandatory processes of translation, chromosome and RNA protection, reactive oxygen species (ROS) scavenging, fatty acid metabolism and cell division (see Table S2). Interestingly, eight transcripts encode flagellum‐associated proteins. In total, the bin encodes 46 proteins of this functional context and additional 73 proteins involved in chemotaxis (e.g. histidine kinases, Che proteins) (Bi and Lai, 2015; Micali and Endres, 2016), most of them being highly transcribed (category 8). Regarding the bin's metabolism, the top 25 list of the most highly transcribed genes did not provide any information, but Table 2 shows that the Fusobacteria bin features only three encoded GH families, however with transcriptional categories of 3, 6 and 10. Still, the low number of transcribed GH family genes and their predicted functional context rather suggest that the species represented by the bin does not utilize (complex) sugars. Previous analyses on the genetic content suggested that its metabolism is based on glutamate and lysine fermentation (Stolze et al., 2016). Transcriptional analyses showed that for glutamate utilization, some of the key enzymes of the hydroxyglutarate pathway have high (category 8), some others lower TPM values (categories 2 and 3). For lysine utilization, the pathway of d‐ and l‐lysine degradation to acetate is complete, with medium TPM values (category 6); both findings strongly indicate an active amino acid utilization.
The transcriptional data also clearly indicate ethanol, acetate, H2, CO2 and possibly lactate as end‐products of the species’ metabolism, as shown in Table 3. The key genes encoding alcohol dehydrogenase, lactate dehydrogenase and acetate kinase feature transcription categories of 6, 8 and 8 respectively. Table 3 also shows that hydrogen production is likely to occur as the pyruvate:ferredoxin oxidoreductase, converting pyruvate to acetyl‐CoA and CO2 and simultaneously reducing ferredoxin, is highly transcribed (categories 5, 8 and 9). Ferredoxin is, i.a., used for hydrogen production (Biegel and Muller, 2010; Hess et al., 2013) and features transcriptional categories of 10 (see Table S2). It can be used by a bifurcating FeFe‐hydrogenase to produce hydrogen. The genes encoding this enzyme are highly transcribed (categories 7 and 10) as shown in Table 4, which summarizes transcriptional activities of genes encoding proteins involved in syntrophy according to Worm et al. (2014). As the described reaction is thermodynamically unfavourable, it depends on a hydrogen‐consuming methanogenic archaeon (Sieber et al., 2012). The hypothesis of a possible syntrophic association is supported by other transcripts encoding putative syntrophy‐associated genes being highly transcribed by the species, and among them, subunits of the Rnf complex with transcriptional categories between 4 and 8 (Table 4).
However, these findings are not in line with the findings indicating a motile lifestyle, as it is known that the formation of mats or biofilms eventually results in motility loss (Alexandre, 2015). Flagella play important roles in the maintenance of the close physical contact between the syntrophic partners (McInerney et al., 2009; Krumholz et al., 2015), but this does not explain the high transcription rates of chemotaxis‐associated genes. However, the metatranscriptome‐based profile comprises the whole Fusobacteria bin‐represented population in situ. A subpopulation may be syntrophically associated, while other cells still were motile.
In summary, the transcriptional profile of the Fusobacteria genome bin, as deduced from metatranscriptome sequencing data, depicts a motile, acidogenic, mostly amino acid‐based metabolism with acetate, ethanol, CO2, H2 and probably lactate as fermentation end‐products.
The transcriptional profile of the Spirochaetes bin indicates a sugar fermentation‐based species
Previously, the Spirochaetes genome bin was analysed genetically and as deduced from its metabolic potential, the bin may constitute a syntrophic sugar‐fermenting bacterium producing acetate, CO2 and H2 (Stolze et al., 2016). Based on metatranscriptome sequencing data from the mesophilic BGP, the bin's activity and role within its habitat were analysed at the transcriptional level. Regarding the top 25 list of the most highly transcribed genes, 19 encoded gene products received functional annotations, with 15 of them being involved in mandatory processes of translation, transcription, fatty acid metabolism, protein folding and export and electron transfer (Table S3). Two of the annotated genes among the 25 most highly transcribed ones encode an ABC transporter substrate‐binding protein and a LacI family transcriptional regulator, both featuring transcriptional categories 10. ATP‐binding cassette (ABC) transporters are known as importers for sugars and also other solutes, while LacI family proteins function as transcription inhibitors for genes encoding proteins for lactose utilization (Davidson et al., 2008; Santillan and Mackey, 2008; Camas et al., 2010). In total, 290 genes (14% of all genes) encoding proteins involved in sugar import and utilization are present, all but six of them being transcribed. 48 genes encode (ABC) transporters directly associated with sugar import, mostly unspecific, some specific for lactose and the monosaccharides arabinose, rhamnose, ribose, fructose and xylose (categories 2 – 9). Additionally, Table 2 shows that the bin actively transcribed genes representing seven glycoside hydrolase families with transcriptional categories between 2 and 6.
Regarding the species’ fermentation end‐products, the transcriptional data strongly indicate the release of CO2, H2, acetate, ethanol and probably lactate. As shown in Table 3, the key genes for the production of the latter three compounds show high transcriptional categories. Additionally, high transcriptional categories of the encoded ferredoxin‐reducing and CO2‐producing pyruvate: ferredoxin oxidoreductase (see Table 3), ferredoxin (categories 4 and 9) and a bifurcating FeFe‐hydrogenase (category 10, Table 4) indicate hydrogen production from NADH and ferredoxin.
According to Worm et al. (2014), the bifurcating FeFe‐hydrogenase belongs to those enzymes possibly involved in syntrophy, as summarized in Table 4. Some other genes within this Table are actively transcribed by the analysed species, and among them, the majority of the Rnf complex subunit genes. They feature transcriptional categories between 8 and 10 and therefore belong to the most highly transcribed genes of this organism. It was proposed that the Rnf complex may play a crucial role in syntrophy (Worm et al., 2014), and its high transcription rate indicates a possible syntrophic interaction of the studied species at the time of RNA extraction. However, as only a small number of putative syntrophy‐associated genes are transcribed, this conclusion cannot be drawn with certainty, but in the context of hydrogen production and the seemingly acetogenic lifestyle, it is a likely assumption.
Summarizing, the metatranscriptome data mapping onto the Spirochaetes bin enabled in situ transcriptomic profiling of this so far unknown and uncharacterized species. Its metabolism seems to be mainly based on monosaccharides, but may also involve the degradation of some complex di‐ and polysaccharides. Transcriptional activities for associated genes also indicated acetate, ethanol, possibly lactate, CO2 and H2 as fermentation end‐products. A syntrophic association with methanogenic Archaea is presumed, due to the transcription of genes encoding proteins associated with syntrophy and hydrogen production, especially a bifurcating hydrogenase.
The transcriptional profile of the Cloacimonetes bin indicates an amino acid fermentation‐based species
In a previous study, the genome bin assigned to the phylum Cloacimonetes was analysed on the genetic level, concluding that it probably represents an amino acid‐fermenting, CO2 and H2‐producing bacterium, possibly syntrophically associated with methanogenic Archaea (Stolze et al., 2016). Mapping of metatranscriptome sequencing data from the mesophilic BGP on the Cloacimonetes genome bin was supposed to give insights into the in situ transcriptomic activity of the corresponding species and enable the uncovering of its response to prevailing environmental conditions.
Analyses of the 25 most highly transcribed genes, according to their TPM values, showed that 16 genes could be identified to encode proteins involved in mandatory bacterial functions like translation, chromosome structure maintenance, fatty acid metabolism and protein transport and protection (see Table S4). Interestingly, there are no other transcripts among them that encode proteins showing a certain response to the species’ environment or being involved in the genome bins’ postulated metabolism based on amino acids.
However, Table 2 shows that five encoded glycoside hydrolases feature only low transcription categories except for those two predicted to be involved in cell wall degradation. Generally, the bin lacks genes encoding proteins for the utilization of sugars; the only exception is glucose degradation via the glycolysis pathway whose enzymes are completely encoded and feature transcriptional categories between 2 and 9. However, the transcriptional data clearly show that fermentation of the amino acids glutamate, lysine, alanine, asparagine, aspartate, cysteine and proline is preferred by the bacterium. Genes encoding proteins involved in their conversion into pyruvate were identified featuring transcriptional activity categories between 5 and 9.
The end‐product of these pathways is most likely acetate, as all enzymes for its production are encoded and transcribed with categories between 5 and 8, while the other fermentation pathways are largely incomplete (Table 3). Interestingly, the studied species may have the potential to produce ethanol; however, no transcripts for the key enzyme, the alcohol dehydrogenase, were identified. This indicates that the Cloacimonetes species represents an acetogenic bacterium.
However, other end‐products seem to be CO2 and H2. Carbon dioxide is produced mainly via the conversion of pyruvate to acetyl‐CoA by the pyruvate:ferredoxin oxidoreductase (see Table 3), simultaneously reducing ferredoxin in the process. In addition to this, genes encoding the bifurcating FeFe‐hydrogenase using NADH and ferredoxin to produce hydrogen (Sieber et al., 2012) are actively transcribed and show high TPM values (see Table 4). Also, a second Fe‐only hydrogenase (transcriptional categories 8 to 10) and its assembly protein (categories 3–6) are transcribed by the Cloacimonetes genome bin. Transcripts encoding ferredoxins were also found, with categories 5 and 10 (not shown). These findings strongly indicate the production of hydrogen via this thermodynamically unfavourable reaction. Additionally, it indicates that this species is likely to be syntrophically associated with hydrogenotrophic (or aceticlastic) Archaea. This is also supported by partially high transcriptional activities of genes encoding proteins presumably associated with syntrophy (Table 4).
In summary, the metatranscriptome‐based profile of the Cloacimonetes bin showed that it actively ferments amino acids, producing acetate, H2 and CO2 in the process and is very likely associated with hydrogenotrophic and aceticlastic Archaea. The used methods of metatranscriptome sequencing and genome bin‐enabled transcriptional profiling therefore proved to be valuable tools for in situ characterization of unknown species and to deduce their role and importance within biogas‐producing communities.
Experimental procedures
Total microbial RNA extraction from three mesophilic and one thermophilic production‐scale biogas plants
Fermentation samples for whole‐microbial community RNA extraction were taken from three mesophilic and one thermophilic production‐scale biogas plants in Germany as described in Stolze et al. (2016). RNA extraction is based on acid phenol treatment followed by usage of the RNeasy Midi bacteria Kit (Qiagen, Hilden, Germany) and DNA digestion using DNase by Roche (Mannheim, Germany) and Qiagen. Whole‐community RNA was prepared from fermenter samples applying the protocol as follows: 4.4 g of fermenter sludge and 2.5 ml TE buffer (4°C) were mixed and applied on a nylon filter (40 μm nylon BD Biosciences, Heidelberg, Germany). Centrifugation at 400 g and 4°C for 2 min and filtrate mixing 1:1 (v/v) with acid phenol (4°C) followed. The mixture was added to 0.5 g glass beads (0.1 mm) in a 15 ml tube, vortexed for 4 min at the highest level, followed by centrifugation at 5000 g for 5 min. The upper phase was then mixed 1:4 (v/v) with RLT buffer (RNeasy Midi Kit, with 2‐Mercaptoethanol). Next steps followed the RNeasy Midi bacteria protocol (Qiagen) starting at step 5, with the upper phase/ethanol ratio being 1:2.8 (v/v) (without RLT). Finally, RNA was eluted in 150 μl RNase‐free water and remaining DNA was removed using DNase I by Roche and the RNeasy Mini kit by Qiagen following the manufacturer's instructions. Quality and quantity of the extracted RNA were evaluated using a Prokaryote RNA 600 Pico chip and the Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA).
For the mesophilic BGPs 1, 2 and 3, and the thermophilic BGP4, RNA extraction was performed in duplicates. cDNA library preparation for metatranscriptome sequencing and metagenome/metatranscriptome sequencing was performed at the DOE Joint Genome Institute (JGI) in Walnut Creek, CA, USA.
rRNA depletion, library preparation and high‐throughput metatranscriptome sequencing
For all eight whole RNA samples, rRNA depletion, cDNA library preparation and metatranscriptome sequencing were performed at the DOE JGI. Library preparation was performed following the TruSeq Stranded Total RNA sample preparation guide by Illumina (San Diego, CA, USA). Prior to library generation, rRNA depletion was performed using the Ribo‐Zero rRNA Removal Kit (Bacteria) (Epicentre, Chicago, IL, USA). For library construction, the TruSeq Stranded mRNA Sample Preparation Kit (Illumina; San Diego, CA, USA) was used. Depleted mRNA was fragmented and reverse‐transcribed using the Superscript II reverse transcriptase (Invitrogen, Waltham, MA, USA). After second‐strand synthesis, end‐repair, A‐tailing, adapter ligation of the double‐stranded cDNA and 10 cycles of PCR amplification followed.
Quantification of metatranscriptome libraries was performed using the next‐generation sequencing library qPCR kit (Kapa Biosystems, Wilmington, DE, USA) and the LightCycler 480 real‐time PCR instrument (Roche, Basel, Switzerland). Sequencing preparation was performed using the TruSeq paired‐end cluster kit (v3, Illumina; San Diego, CA, USA). Finally, metatranscriptome sequencing was performed on the HiSeq 2000 sequencer using the TruSeq SBS sequencing kits (v3) following the 2 × 150 indexed high‐output run instruction (both by Illumina).
Metatranscriptome sequence data processing
In order to determine the transcriptional profiles of the four genome bins, metatranscriptome data of the BGPs they derived from were used: BGP3 (Fusobacteria, Spirochaetes and Cloacimonetes (WWE1) bin), hereinafter referred to as the mesophilic BGP, and BGP4 (Thermotogae bin), hereinafter referred to as the thermophilic BGP. In total, 9001.8 million reads (137 269 Gbp; Table 1) were generated, the deepest sequencing of biogas metatranscriptomes so far. Kallisto (Bray et al., 2016; version 0.42.5) was used to quantify abundances of transcripts. The coding sequences predicted from contigs of the combined assembly of the four metagenome samples (see Stolze et al., 2016) were used as input to build the transcriptome index using kallisto index. FASTQ files from each metatranscriptome sample were ‘pseudoaligned’ using kallisto quant with default parameters. Transcripts per million (TPM) values for each gene were extracted from the resulting abundance file, for which the number of reads mapping on a gene is normalized on gene length and data set size.
Annotation of predicted genes of the metagenome assembly (see Stolze et al., 2016) was performed using InterProScan version 5.24‐63.0.
To predict genes encoding carbohydrate‐active enzymes, the carbohydrate‐active enzyme database (CAZy) annotation web server dbCAN (Yin et al., 2012) was used.
Conflict of interest
None declared.
Data sets
The datasets supporting the conclusions of this article are available in the short read archive (SRA, Metatranscriptome sequencing data): Metatranscriptome Data mesophilic BGP: https://www.ncbi.nlm.nih.gov/sra/?term=SRP096994, https://www.ncbi.nlm.nih.gov/sra/?term=SRP096993. Metatranscriptome Data thermophilic BGP: https://www.ncbi.nlm.nih.gov/sra/?term=SRP096995, https://www.ncbi.nlm.nih.gov/sra/?term=SRP096996.
Supporting information
Table S1. The 25 most highly transcribed genes of the Thermotogae bin, by Transcripts Per Million (TPM) values, their encoded proteins and functional contexts.
Table S2. The 25 most highly transcribed genes of the Fusobacteria bin, by Transcripts Per Million (TPM) values, their encoded proteins and functional contexts.
Table S3. The 25 most highly transcribed genes of the Spirochaetes bin, by Transcripts Per Million (TPM) values, their encoded proteins and functional contexts.
Table S4. The 25 most highly transcribed genes of the Cloacimonetes bin, by Transcripts Per Million (TPM) values, their encoded proteins and functional contexts.
Table S5. Unshortened table of glycosyl hydrolase (GH) families, their respective Transcript per Million (TPM) values and transcription categories between 0 and 10 for all four genome bins.
Table S6. Unshortened table of fermentation pathway proteins, their respective Transcript per Million (TPM) values and transcription categories between 0 and 10 for all four genome bins.
Table S7. Unshortened table of possibly syntrophy associated proteins, their respective Transcript per Million (TPM) values and transcription categories between 0 and 10 for all four genome bins.
Acknowledgements
The authors acknowledge the support for the article processing charge by the Deutsche Forschungsgemeinschaft, the Open Access Publication Fund of Bielefeld University, and sequencing work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, Contract No. DE‐AC02‐05CH11231. AB and IM acknowledge the CLIB Graduate Cluster Industrial Biotechnology, AB additionally the International DFG Research Training Group GRK 1906/1. ASch and YS acknowledge the support by the Bundesministerium für Ernährung und Landwirtschaft (BMEL) and the Fachagentur für Nachwachsende Rohstoffe (FNR), grant number 22006712, and ASch and IM the German Federal Ministry of Education and Research (BMBF) (Germany) and the Projektträger Jülich (PTJ), grant number 03SF0440C. AP acknowledges the support by the BMBF‐funded project ‘Bielefeld‐Gießen Center for Microbial Bioinformatics – BiGi (Grant number 031A533)’ within the German Network for Bioinformatics Infrastructure (de.NBI).
Microbial Biotechnology (2018) 11(4), 667–679
Funding Information
The authors acknowledge the support for the article processing charge by the Deutsche Forschungsgemeinschaft, the Open Access Publication Fund of Bielefeld University, and sequencing work conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, Contract No. DE‐AC02‐05CH11231. AB and IM acknowledge the CLIB Graduate Cluster Industrial Biotechnology, AB additionally the International DFG Research Training Group GRK 1906/1. ASch and YS acknowledge the support by the Bundesministerium für Ernährung und Landwirtschaft (BMEL) and the Fachagentur für Nachwachsende Rohstoffe (FNR), grant number 22006712, and ASch and IM the German Federal Ministry of Education and Research (BMBF) and the Projektträger Jülich (PtJ), grant number 03SF0440C. AP acknowledges the support by the BMBF‐funded project ‘Bielefeld‐Gießen Center for Microbial Bioinformatics – BiGi (Grant number 031A533)’ within the German Network for Bioinformatics Infrastructure (de.NBI).
References
- Alexandre, G. (2015) Chemotaxis control of transient cell aggregation. J Bacteriol 197: 3230–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balk, .M. , Weijma, J. and Stams, A.J. (2002) Thermotoga lettingae sp. nov., a novel thermophilic, methanol‐degrading bacterium isolated from a thermophilic anaerobic reactor. Int J Syst Evol Microbiol 52, 1361–1368. [DOI] [PubMed] [Google Scholar]
- Bi, S. , and Lai, L. (2015) Bacterial chemoreceptors and chemoeffectors. CMLS 72: 691–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegel, E. , and Muller, V. (2010) Bacterial Na+‐translocating ferredoxin:NAD+ oxidoreductase. Proc Natl Acad Sci USA 107: 18138–18142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, N.L. , Pimentel, H. , Melsted, P. , and Pachter, L. (2016) Near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol 34: 525–527. [DOI] [PubMed] [Google Scholar]
- Bremges, A. , Maus, I. , Belmann, P. , Eikmeyer, F. , Winkler, A. , Albersmeier, A. , et al (2015) Deeply sequenced metagenome and metatranscriptome of a biogas‐producing microbial community from an agricultural production‐scale biogas plant. GigaSci 4: 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camas, F.M. , Alm, E.J. , and Poyatos, J.F. (2010) Local gene regulation details a recognition code within the LacI transcriptional factor family. PLoS Comput Biol 6: e1000989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanaro, S. , Treu, L. , Kougias, P.G. , de Francisci, D. , Valle, G. , and Angelidaki, I. (2016) Metagenomic analysis and functional characterization of the biogas microbiome using high throughput shotgun sequencing and a novel binning strategy. Biotechnol Biofuels 9: 5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson, A.L. , Dassa, E. , Orelle, C. and Chen, J. (2008) Structure, function, and evolution of bacterial ATP‐binding cassette systems. MMBR 72, 317–364 table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikmeyer, F.G. , Rademacher, A. , Hanreich, A. , Hennig, M. , Jaenicke, S. , Maus, I. , et al (2013) Detailed analysis of metagenome datasets obtained from biogas‐producing microbial communities residing in biogas reactors does not indicate the presence of putative pathogenic microorganisms. Biotechnol Biofuels 6: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, X. , Xu, F. , and Li, Y. (2016) Solid‐state anaerobic digestion of lignocellulosic biomass: Recent progress and perspectives. Biores Technol 205: 239–249. [DOI] [PubMed] [Google Scholar]
- Hess, V. , Schuchmann, K. , and Muller, V. (2013) The ferredoxin:NAD+ oxidoreductase (Rnf) from the acetogen Acetobacterium woodii requires Na+ and is reversibly coupled to the membrane potential. J Biol Chem 288: 31496–31502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, M.R. , Conners, S.B. , Montero, C.I. , Chou, C.J. , Shockley, K.R. and Kelly, R.M. (2006) The Thermotoga maritima phenotype is impacted by syntrophic interaction with Methanococcus jannaschii in hyperthermophilic coculture. Appl Environ Microbiol 72, 811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kougias, P.G. , Campanaro, S. , Treu, L. , Zhu, X. , and Angelidaki, I. (2017) A novel archaeal species belonging to Methanoculleus genus identified via de‐novo assembly and metagenomic binning process in biogas reactors. Anaerobe 46: 23–32. [DOI] [PubMed] [Google Scholar]
- Kovács, E. , Wirth, R. , Maróti, G. , Bagi, Z. , Rákhely, G. , and Kovács, K.L. (2013) Biogas production from protein‐rich biomass: fed‐batch anaerobic fermentation of casein and of pig blood and associated changes in microbial community composition. PLoS ONE 8: e77265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumholz, L.R. , Bradstock, P. , Sheik, C.S. , Diao, Y. , Gazioglu, O. , Gorby, Y. , and McInerney, M.J. (2015) Syntrophic growth of Desulfovibrio alaskensis requires genes for H2 and formate metabolism as well as those for flagellum and biofilm formation. Appl Environ Microbiol 81: 2339–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunath, B.J. , Bremges, A. , Weimann, A. , McHardy, A.C. and Pope, P.B. (2017) Metagenomics and CAZyme discovery. Meth Mol Biol (Clifton, N.J.) 1588, 255–277. [DOI] [PubMed] [Google Scholar]
- Liao, J.C. , Mi, L. , Pontrelli, S. , and Luo, S. (2016) Fuelling the future: microbial engineering for the production of sustainable biofuels. Nat Rev Microbiol 14: 288–304. [DOI] [PubMed] [Google Scholar]
- Malik, A. , Lan, J. , and Lenzen, M. (2016) Trends in global greenhouse gas emissions from 1990 to 2010. Environ Sci Technol 50: 4722–4730. [DOI] [PubMed] [Google Scholar]
- Mande, S.S. , Mohammed, M.H. , and Ghosh, T.S. (2012) Classification of metagenomic sequences: methods and challenges. Brief Bioinform 13: 669–681. [DOI] [PubMed] [Google Scholar]
- Mao, C. , Feng, Y. , Wang, X. , and Ren, G. (2015) Review on research achievements of biogas from anaerobic digestion. Renew Sustain Energy Rev 45: 540–555. [Google Scholar]
- Maus, I. , Cibis, K.G. , Wibberg, D. , Winkler, A. , Stolze, Y. , König, H. , et al (2015) Complete genome sequence of the strain Defluviitoga tunisiensis L3, isolated from a thermophilic, production‐scale biogas plant. J Biotechnol 203: 17–18. [DOI] [PubMed] [Google Scholar]
- Maus, I. , Koeck, D.E. , Cibis, K.G. , Hahnke, S. , Kim, Y.S. , Langer, T. , et al (2016) Unraveling the microbiome of a thermophilic biogas plant by metagenome and metatranscriptome analysis complemented by characterization of bacterial and archaeal isolates. Biotechnol Biofuels 9: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney, M.J. , Sieber, J.R. , and Gunsalus, R.P. (2009) Syntrophy in anaerobic global carbon cycles. Curr Opin Biotechnol 20: 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micali, G. , and Endres, R.G. (2016) Bacterial chemotaxis: information processing, thermodynamics, and behavior. Curr Opin Microbiol 30: 8–15. [DOI] [PubMed] [Google Scholar]
- Ruile, S. , Schmitz, S. , Mönch‐Tegeder, M. , and Oechsner, H. (2015) Degradation efficiency of agricultural biogas plants–a full‐scale study. Biores Technol 178: 341–349. [DOI] [PubMed] [Google Scholar]
- Sangwan, N. , Xia, F. , and Gilbert, J.A. (2016) Recovering complete and draft population genomes from metagenome datasets. Microbiome 4: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santillan, M. , and Mackey, M.C. (2008) Quantitative approaches to the study of bistability in the lac operon of Escherichia coli . J R Soc 5(Suppl 1): S29–S39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczyrba, A. , Hofmann, P. , Belmann, P. , Koslicki, D. , Janssen, D. , Droege, J. , et al (2017) Critical Assessment of Metagenome Interpretation ‐ a benchmark of computational metagenomics software. Nat Methods 14, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpton, T.J. (2014) An introduction to the analysis of shotgun metagenomic data. Front Plant Sci 5: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber, J.R. , McInerney, M.J. , and Gunsalus, R.P. (2012) Genomic insights into syntrophy: the paradigm for anaerobic metabolic cooperation. Annu Rev Microbiol 66: 429–452. [DOI] [PubMed] [Google Scholar]
- Stolze, Y. , Zakrzewski, M. , Maus, I. , Eikmeyer, F. , Jaenicke, S. , Rottmann, N. , et al (2015) Comparative metagenomics of biogas‐producing microbial communities from production‐scale biogas plants operating under wet or dry fermentation conditions. Biotechnol Biofuels 8: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolze, Y. , Bremges, A. , Rumming, M. , Henke, C. , Maus, I. , Puhler, A. , et al (2016) Identification and genome reconstruction of abundant distinct taxa in microbiomes from one thermophilic and three mesophilic production‐scale biogas plants. Biotechnol Biofuels 9: 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treu, L. , Kougias, P.G. , Campanaro, S. , Bassani, I. , and Angelidaki, I. (2016) Deeper insight into the structure of the anaerobic digestion microbial community; the biogas microbiome database is expanded with 157 new genomes. Biores Technol 216: 260–266. [DOI] [PubMed] [Google Scholar]
- Vanwonterghem, I. , Jensen, P.D. , Rabaey, K. , and Tyson, G.W. (2016) Genome‐centric resolution of microbial diversity, metabolism and interactions in anaerobic digestion. Environ Microbiol 18: 3144–3158. [DOI] [PubMed] [Google Scholar]
- Verhaart, M.R.A. , Bielen, A.A.M. , van der Oost, J. , Stams, Alfons.J.M. , and Kengen, S.W.M. (2010) Hydrogen production by hyperthermophilic and extremely thermophilic bacteria and archaea: mechanisms for reductant disposal. Environ Technol 31: 993–1003. [DOI] [PubMed] [Google Scholar]
- Weiland, P. (2010) Biogas production: current state and perspectives. Appl Microbiol Biotechnol 85: 849–860. [DOI] [PubMed] [Google Scholar]
- Weiss, A. , Jérôme, V. , Freitag, R. , and Mayer, H.K. (2008) Diversity of the resident microbiota in a thermophilic municipal biogas plant. Appl Microbiol Biotechnol 81: 163–173. [DOI] [PubMed] [Google Scholar]
- Worm, P. , Koehorst, J.J. , Visser, M. , Sedano‐Nunez, V.T. , Schaap, P.J. , Plugge, C.M. , et al (2014) A genomic view on syntrophic versus non‐syntrophic lifestyle in anaerobic fatty acid degrading communities. Biochem Biophys Acta 1837: 2004–2016. [DOI] [PubMed] [Google Scholar]
- Xia, Y. , Wang, Y. , Wang, Y. , Chin, F.Y.L. , and Zhang, T. (2016) Cellular adhesiveness and cellulolytic capacity in Anaerolineae revealed by omics‐based genome interpretation. Biotechnol Biofuels 9: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin, Y. , Mao, X. , Yang, J. , Chen, X. , Mao, F. and Xu, Y. (2012) dbCAN: a web resource for automated carbohydrate‐active enzyme annotation. Nucleic Acids Res 40 , W445–W451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakrzewski, M. , Goesmann, A. , Jaenicke, S. , Jünemann, S. , Eikmeyer, F. , Szczepanowski, R. , et al (2012) Profiling of the metabolically active community from a production‐scale biogas plant by means of high‐throughput metatranscriptome sequencing. J Biotechnol 158: 248–258. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , O'Hara, I.M. , Mundree, S. , Gao, B. , Ball, A.S. , Zhu, N. , et al (2016) Biofuels from food processing wastes. Curr Opin Biotechnol 38: 97–105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The 25 most highly transcribed genes of the Thermotogae bin, by Transcripts Per Million (TPM) values, their encoded proteins and functional contexts.
Table S2. The 25 most highly transcribed genes of the Fusobacteria bin, by Transcripts Per Million (TPM) values, their encoded proteins and functional contexts.
Table S3. The 25 most highly transcribed genes of the Spirochaetes bin, by Transcripts Per Million (TPM) values, their encoded proteins and functional contexts.
Table S4. The 25 most highly transcribed genes of the Cloacimonetes bin, by Transcripts Per Million (TPM) values, their encoded proteins and functional contexts.
Table S5. Unshortened table of glycosyl hydrolase (GH) families, their respective Transcript per Million (TPM) values and transcription categories between 0 and 10 for all four genome bins.
Table S6. Unshortened table of fermentation pathway proteins, their respective Transcript per Million (TPM) values and transcription categories between 0 and 10 for all four genome bins.
Table S7. Unshortened table of possibly syntrophy associated proteins, their respective Transcript per Million (TPM) values and transcription categories between 0 and 10 for all four genome bins.