

Summary
This study examined whether the abundance and expression of microbial 16S rRNA genes were associated with elemental concentrations and substrate conversion biokinetics in 20 full‐scale anaerobic digesters, including seven municipal sewage sludge (SS) digesters and 13 industrial codigesters. SS digester contents had higher methane production rates from acetate, propionate and phenyl acetate compared to industrial codigesters. SS digesters and industrial codigesters were distinctly clustered based on their elemental concentrations, with higher concentrations of NH 3‐N, Cl, K and Na observed in codigesters. Amplicon sequencing of 16S rRNA genes and reverse‐transcribed 16S rRNA revealed divergent grouping of microbial communities between mesophilic SS digesters, mesophilic codigesters and thermophilic digesters. Higher intradigester distances between Archaea 16S rRNA and rRNA gene profiles were observed in mesophilic codigesters, which also had the lowest acetate utilization biokinetics. Constrained ordination showed that microbial rRNA and rRNA gene profiles were significantly associated with maximum methane production rates from acetate, propionate, oleate and phenyl acetate, as well as concentrations of NH 3‐N, Fe, S, Mo and Ni. A co‐occurrence network of rRNA gene expression confirmed the three main clusters of anaerobic digester communities based on active populations. Syntrophic and methanogenic taxa were highly represented within the subnetworks, indicating that obligate energy‐sharing partnerships play critical roles in stabilizing the digester microbiome. Overall, these results provide new evidence showing that different feed substrates associate with different micronutrient compositions in anaerobic digesters, which in turn may influence microbial abundance, activity and function.
Introduction
Anaerobic digestion is a microbial process that produces renewable methane from various organic waste streams and is expanding globally due to a growing interest in sustainable energy and nutrient recovery (McCarty et al., 2011; Batstone and Virdis, 2014). Efficient and stable conversion of organic matter into methane relies on a series of interdependent relationships between fermenters, acidogens, acetogenic syntrophs and methanogenic archaea (Stams, 1994; Briones and Raskin, 2003). Due to the large microbial biodiversity present in anaerobic digesters (Nelson et al., 2011; Sundberg et al., 2013) and the large portion of uncultured species in such systems (Li et al., 2009), culture‐independent molecular tools have been critical in advancing our understanding of microbial communities within anaerobic digesters (Rivière et al., 2009; Werner et al., 2011; Vanwonterghem et al., 2014, 2016; De Vrieze et al., 2015, 2016a; Hao et al., 2016). Despite these advancements, anaerobic digesters are still typically operated based on empirical knowledge of process parameters, owing to a general lack of understanding regarding relationships between microbial community structure and digester function in terms of process biokinetics (Carballa et al., 2015). While some recent studies have found correlations between specific population abundances and substrate conversion biokinetics (Venkiteshwaran et al., 2017; Ziels et al., 2017), these studies focused on the presence of DNA, which may provide little information about the active portion of the microbial community. Discrepancies have been observed between DNA‐based abundance and RNA‐ or protein‐based expression of methanogen populations in anaerobic digesters (Zakrzewski et al., 2012), which could be attributed to their high activity but low biomass yields (Smith et al., 2015) or conversely due to changes in their RNA/protein expression in response to environmental perturbations (De Vrieze et al., 2016b). For instance, changes in mRNA expression of the methyl‐coenzyme M reductase gene (mcrA) involved in methanogenesis were observed along with decreased methane production rates at high ammonia loadings, whereas DNA levels of the mcrA gene remained stable (Zhang et al., 2014a). While changes in the expression of ribosomal RNA (rRNA) may not always reflect changes in microbial activity over short temporal scales (e.g. 1–3 days) (Flärdh et al., 1992; Wagner et al., 1995), rRNA gene expression levels have been related to microbial activity and growth rates within bioreactors under steady‐state conditions (Poulsen et al., 1993). Comparing the rRNA (RNA) and rRNA gene (DNA) profiles of an anaerobic digester microbial community may highlight active populations more so than DNA‐only based approaches, due to the shorter half‐life of rRNA and constant turnover of digester contents by the solids retention time.
Many factors can impact the digester efficiency as well as the microbial community structure, such as temperature (De Vrieze et al., 2015; Westerholm et al., 2015), the hydraulic and solids retention times (Lee et al., 2011; Hao et al., 2016), salinity (De Vrieze et al., 2016b), free ammonia (Westerholm et al., 2015), feeding regime (Conklin et al., 2006; De Vrieze et al., 2013; Mulat et al., 2016; Ziels et al., 2017) and micronutrients (Karlsson et al., 2012). Among these important parameters, macro‐ and micronutrient levels are gaining attention because they are necessary for many biological and enzyme‐mediated processes in methanogenic communities, and they can be controlled via supplementation (Demirel and Scherer, 2011). A vast body of research has examined the effect of nutrient supplementation on the performance of the anaerobic digestion process (Worm et al., 2009; Banks et al., 2012; Karlsson et al., 2012; Williams et al., 2013; Schmidt et al., 2014; Zhang et al., 2015; Moestedt et al., 2016), highlighting that many anaerobic digesters may have suboptimal organic matter conversion and methane production due to deficiencies in one or multiple essential elements.
Our previous study of 21 full‐scale anaerobic digesters in Sweden demonstrated that different feed substrate types were associated with different methanogenic pathways and community structure (Sundberg et al., 2013). We suggested that differences in the elemental compositions of the feed substrates may be influential to establishing different microbial activities and functional performance of the digesters. However, there have been no studies examining the association of macro‐ and micronutrient levels with process biokinetics and the activities and abundance of microbial populations in full‐scale anaerobic digestion systems.
The objective of this research was to elucidate relationships between the activity of microbial populations and elemental concentrations and biokinetics within a diverse set of 20 full‐scale anaerobic digesters, including municipal sewage sludge (SS) digesters and industrial codigesters (Table 1). The digester Bacteria and Archaea community profiles were examined through amplicon sequencing of 16S rRNA genes and reverse‐transcribed 16S rRNA, and the concentrations of 21 biologically relevant elements were measured in the digester contents. Additionally, the biokinetics of methane production from acetate, propionate, phenyl acetate and oleate were assessed in batch assays with the full‐scale digester contents to infer their capacity to degrade critical intermediates in the methanogenic food chain. We examined potential correlations between 16S rRNA and rRNA gene profiles and elemental concentrations as well as biokinetics to elucidate whether different substrate types create distinct ecological conditions that drive differences in microbial activity and function within the full‐scale digesters.
Table 1.
Operational parameters of the reactors including feed substrate type, temperature, organic loading rate (OLR), free ammonia and hydraulic retention time (HRT)
| Reactor | Temp. (°C) | Substrate (% volume fraction of total substrate) | pH | Free Ammonia (mg NH3‐N l−1) | OLR (kg VS m−3 day−1) | HRT (days) |
|---|---|---|---|---|---|---|
| CD1 | 38 | Wheat stillage (47%), grain (37%) and water (14%) | 7.5 | 204 | 3.3 | 55 |
| CD10 | 38 | OFMSW (77%), fat (12%) and silage (11%) | 7.7 | 162 | 2.2 | 28 |
| CD11ap | 38 | SHW in majority | 8.0 | 558 | 3.7 | 55 |
| CD11 bp | 38 | SHW in majority | 8.0 | 541 | 3.7 | 55 |
| CD2T | 53 | OFMSW (62%), dry fodder (13%) and SHW (9%) | 7.9 | 424 | 2.4 | 25 |
| CD3T | 52 | SHW (45%), manure (cow) (29%) and whey (14%)a | 7.9 | 751 | 3.9 | 20 |
| CD4ap | 37 | SHW (45%), manure (pig and cow) (32%) and OFMSW (20%)a | 7.7 | 178 | 3.4 | 25 |
| CD6ap | 38 | Manure (pig and cow) (67%) and SHW/OFMSW (32%)b | 7.9 | 289 | 3.6 | 26 |
| CD6 bp | 38 | Manure (pig and cow) (67%) and SHW/OFMSW (32%)b | 7.9 | 261 | 3.6 | 26 |
| CD7a | 37 | OFMSW (57%), FIW (21%) and SHW (12%)2)c | 7.6 | 123 | 2.5 | 35 |
| CD7b | 37 | OFMSW (57%), FIW (21%) and SHW (12%)2)c | 7.3 | 49 | 3.6 | 24 |
| CD8Tap | 53 | OFMSW (95%) and fat (5%) | 7.7 | 176 | 3.2 | 20 |
| CD9T | 55 | OFMSW (85%) and SHW (15%) | 8.2 | 803 | 1.8 | ‐ |
| SS1as | 37 | FIW (58%) and PS/WAS (42%) | 7.1 | 13 | 3 | 39 |
| SS1bs | 37 | Sludge from SS1as | 7.3 | 20 | 1.1 | 39 |
| SS2T | 52 | PS/WAS | 7.5 | 112 | 2.5 | 12 |
| SS3a | 37 | PS | 7.1 | 16 | 2.7 | 14 |
| SS3b | 37 | PS (54%) and WAS (46%) | 7.2 | 26 | 2.4 | 10 |
| SS4ap | 37 | WAS (55%), PS (42%), sludge from external source (3%) | 7.6 | 39 | 2.4 | 18 |
| SS5 | 38 | PS | 7.3 | 26 | – | 50 |
FIW, food industry waste; OFMSW, organic fraction municipal solid waste; PS, primary sewage sludge; SHW, slaughterhouse waste; WAS, waste‐activated sludge.
a. 2–6% fat is included in the remaining substrate.
b. 1–1.5% iron‐rich sludge is included in the remaining substrate.
c. 8% pig manure is included in the remaining substrate.
Results and discussion
Elemental composition in full‐scale anaerobic digesters
We assessed the concentrations of 21 elements within 20 full‐scale anaerobic digesters (Table 1) with concurrent analysis of biokinetics and microbial rRNA and rRNA gene profiles. NMDS analysis based on Bray–Curtis distances of elemental concentrations in the digestate samples demonstrated that the SS digesters were grouped separately from codigesters (Fig. S1). The dissimilarity in elemental profiles in codigesters versus SS digesters was confirmed by ANOSIM (R ANOSIM = 0.75, P < 0.001). The SS digesters had higher Cu, Al, Ti, W, Ni and Fe concentrations (Fig. 1A) than codigesters. The higher Cu concentration in SS digesters may be related to leaching from drinking water and wastewater collection pipes (Boulay and Edwards, 2001; Sörme and Lagerkvist, 2002), while higher Al and Fe levels could be due to their common supplementation for phosphate removal prior to anaerobic digestion at wastewater treatment plants (Wilfert et al., 2015). Trace amounts of Ni present in chemical stocks used for phosphate removal have been shown to cause elevated Ni levels in sewage sludge (Sörme and Lagerkvist, 2002). Codigesters typically had higher concentrations of NH3‐N, Cl, K and Na (Fig. 1A). Many of the codigesters received slaughterhouse waste and food waste (Table 1), which are protein‐rich substrates that could lead to high digester free ammonia levels (Palatsi et al., 2011). Within the overall cluster of codigester elemental profiles, CD1, CD11ap and CD11 bp were closely grouped (Fig. S1). These codigesters are operated by a private company and are supplemented with a proprietary nutrient additive containing mainly Fe, Co and Ni (data not shown). Overall, these results demonstrate that municipal sludge digesters and codigesters have distinct elemental compositions, which may be attributed to differences in influent feedstocks and/or differences in chemical supplementation (e.g. salts for P removal).
Figure 1.
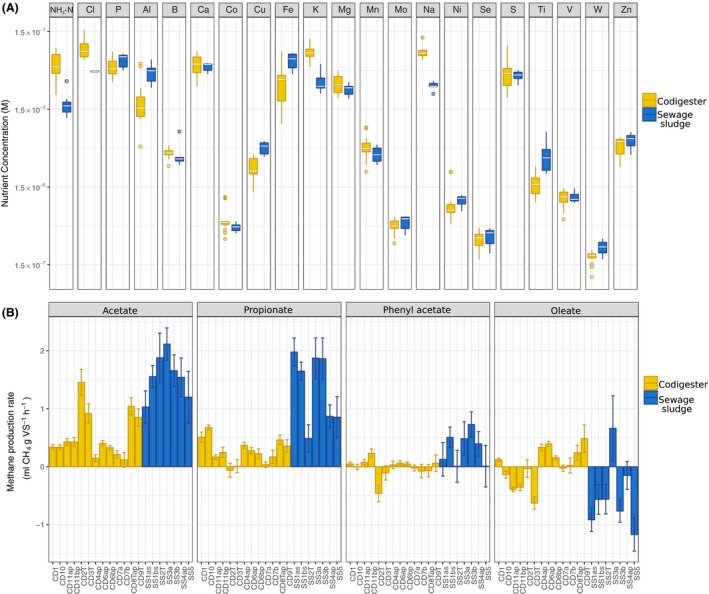
A. Box plot illustrating average concentrations of the measured elements in the codigesters and sludge digesters.
B. Maximum methane production rates (R m) from batch assays with acetate, propionate, phenyl acetate and oleate with all digester contents determined via fitting Gompertz models to methane production curves. R m values refer to standard temperature and pressure. Error bars indicate the standard errors for R m using the nonlinear regression analysis.
Degradation kinetics of key substrates by the full‐scale digester contents
Batch assays were conducted over 280 h with digestate sampled from the 20 full‐scale digesters to measure their degradation kinetics for acetate, propionate, phenyl acetate and oleate (Fig. 1B). The purpose of the batch incubations was to capture the kinetics of substrate degradation by the microbial communities, not to monitor the time until total substrate degradation. Background VFA levels in the full‐scale digester contents were below 20 mM (Table S1) and, thus, likely did not impact maximum observed rates in the batch assays. Maximum methane production rates (R m) from acetate and propionate were higher with contents from the SS digesters compared to the codigesters (P < 0.01, Welch's t‐test) (Fig. 1B). Yet, acetate conversion in the codigesters was faster under thermophilic conditions than under mesophilic conditions (P < 0.01, Welch's t‐test) (Fig. 1B). Maximum methane production rates in the batch assays supplemented with phenyl acetate were lower than in the unsupplemented control assays for several codigesters (Table S2), resulting in negative net substrate conversion rates (Fig. 1B). However, the rates of phenyl acetate degradation were slightly positive in the SS reactors, except for SS1as, SS2T and SS5. Phenyl acetate was previously shown to stimulate methanogenic rates in anaerobic digesters fed with swine manure, but had inhibitory effects in digesters fed with kitchen food waste (Hecht and Griehl, 2009). Thus, the methane production rate from phenyl acetate could depend on the microbial community structure, as the anaerobic degradation of phenyl acetate was hypothesized to rely on syntrophic interactions (Glissmann et al., 2005). Oleate addition resulted in lower methane production rates than in unsupplemented controls for assays with SS digester contents, except for SS3a, while five codigesters had slightly positive net oleate conversion rates (Fig. 1B). NMDS analysis showed that the biokinetic profiles of the SS digesters and the codigesters were distinctly clustered (Fig. S1), and the significance of the clustering was confirmed by ANOSIM (R ANOSIM = 0.8, P < 0.001). Overall, these results demonstrate that the biokinetic characteristics of full‐scale digester communities were distinct among codigesters and SS digesters, with SS digesters having higher acetate, propionate and phenyl acetate conversion rates and lower oleate conversion rates in comparison with codigesters.
Microbial community structure and activity in full‐scale digesters
The microbial community structure and activity of the full‐scale digester contents were assessed with parallel amplicon sequencing of both 16S rRNA and rRNA genes. Principal components analysis (PCA) of OTU read counts in the rRNA and rRNA gene pools of the 20 full‐scale digesters showed that Bacteria and Archaea community structures were clustered into three main groups: thermophilic digesters, mesophilic SS digesters and mesophilic codigesters (Fig. 2). This finding confirms previous observations that temperature and substrate type are major factors that influence the microbial community structure in anaerobic digestion (Sundberg et al., 2013; Zhang et al., 2014b; De Vrieze et al., 2015). However, this work is the first to demonstrate such clustering based on active communities via reverse‐transcribed 16S rRNA profiles of full‐scale anaerobic digesters.
Figure 2.
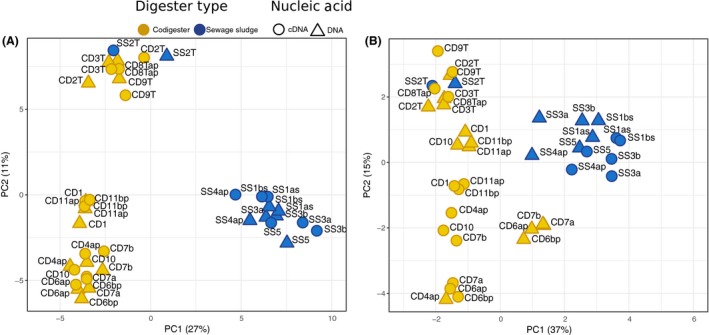
Principal components analysis (PCA) plots of (A) Bacteria and (B) Archaea OTU read counts after variance stabilizing transformation with DESeq2 (Love et al., 2014). Points are shown for both rRNA gene (DNA) and rRNA (RNA) samples for each digester. Values in parentheses represent the fraction of the total variance explained by each PC axis.
The distances between intradigester Bacteria community DNA and RNA profiles were smaller than for Archaea (Fig. 2A), meaning that the bacterial DNA profiles were more reflective of their activity (RNA). The largest intradigester DNA and RNA distances were observed for Archaea community profiles within the mesophilic codigester samples, including CD1, CD4ap, CD6ap, CD6 bp, CD7a, CD7b, CD10, CD11ap and CD11 bp (Fig. 2B). Notably, these codigesters had the lowest acetate conversion rates of all digesters tested (Fig. 1A). Large deviations between archaeal RNA and DNA gene profiles for 16S rRNA were previously associated with stressful conditions during anaerobic digestion caused by high salinity (De Vrieze et al., 2016b) and high LCFA levels (Amha et al., 2017). If larger archaeal 16S rRNA and rRNA gene distances are indicative of stressful conditions, this would suggest that the full‐scale codigesters had higher stress levels than SS digesters. This supports the hypothesis that rRNA signatures may be representative of deterministic processes under stable digester conditions, while under periods of process perturbation, the rRNA signatures may reflect stochastic processes (De Vrieze et al., 2016b). The Archaea communities in the mesophilic codigesters in this study may have had divergent rRNA and rRNA gene profiles due to stress induced from high ammonia levels (Table 1), or possibly from variable feed substrate compositions during codigestion operations. For instance, De Francisci et al. (2015) showed that a changing feed composition during codigestion impacted the microbial community structure based on rRNA genes. Yet, to the best of our knowledge, the impact of changing feed substrate composition on 16S rRNA gene expression has not been elucidated. Further time‐series analysis is needed to verify whether Archaea rRNA and rRNA gene distances can be predictive of lower digester biokinetics in full‐scale systems, and whether a changing feed composition during codigestion impacts Archaea 16S rRNA gene expression.
The taxonomic profiles of the Bacteria communities also differed between the SS digesters and codigesters, corresponding to the observed OTU clustering patterns between the digester groups. Over 85% of bacterial reads were attributed to 12 phyla (Fig. S2). The SS digesters had higher fractions of Syntrophaceae in both the rRNA and rRNA gene fractions in comparison with the codigesters (Fig. 3A). The presence of Syntrophaceae in the SS digesters was mostly attributed to the propionate‐oxidizing genus of Smithella (Fig. S3). The Candidatus Cloacamonas genus was also detected in higher abundance and activity levels in the SS digesters relative to the codigesters (Fig. S3). This genus includes potential amino acid‐ and propionate‐degrading syntrophs (Pelletier et al., 2008; Sieber et al., 2012) and could have been enriched in the SS digesters due to hydrolysis of cellular proteins in the feed primary and waste‐activated sludge (PS+WAS). Conversely, the Syntrophomonadaceae family typically had higher rRNA and rRNA gene sequence fractions in the codigesters relative to the SS digesters, except for the thermophilic SS2T digester (Fig. 3A). Syntrophomonadaceae sequences consisted mostly of the Syntrophomonas genus (Fig. S3), which includes short‐ and long‐chain fatty acid β‐oxidizing species (Sousa et al., 2009). Syntrophomonas has also been purported to belong to the core community within anaerobic digesters (De Vrieze et al., 2016a; Treu et al., 2016), which is corroborated by its presence in 95% of the digester rRNA samples in this study (19 of 20). Thermophilic codigesters generally had higher bacterial rRNA and rRNA gene sequence fractions of the family Thermotogaceae, which comprised over 80% of the rRNA reads in CD9T (Fig. 3A) and was mainly comprised of the genus Defluviitoga (Fig. S3). Defluviitoga was also the dominant genus in the rRNA and rRNA gene profiles in CD1, comprising 38% and 27% of bacterial reads respectively (Fig. S3). Genome reconstruction of a Defluviitoga spp. indicated it is a hydrolytic fermenting organism (Maus et al., 2016a). Defluviitoga spp. have been detected at high levels in other thermophilic digesters (Maus et al., 2016b) indicating its potential significance to methane production at high temperatures. Notably, genera harbouring known syntrophic acetate‐oxidizing (SAO) bacteria of Tepidanaerobacter and Syntrophaceticus (Westerholm et al., 2010, 2011) were detected at higher rRNA and rRNA gene fractions in the codigesters compared to SS digesters, again with the exception of the thermophilic SS2T digester (Fig. S3). The presence and activity of SAO in codigesters but not SS digesters were likely attributed to the higher level of ammonia–nitrogen in the codigesters (Fig. 1A), which is considered a driving factor for SAO in anaerobic digestion (Schnürer and Nordberg, 2008). This result also aligns with the potentially higher stress in the mesophilic codigester Archaea populations, as indicated by their large intradigester rRNA and rRNA gene distances (Fig. 2B). The SAO genus Tepidanaerobacter often had higher rRNA fractions relative to its rRNA gene fraction, indicating that it might have been highly active but slow growing.
Figure 3.
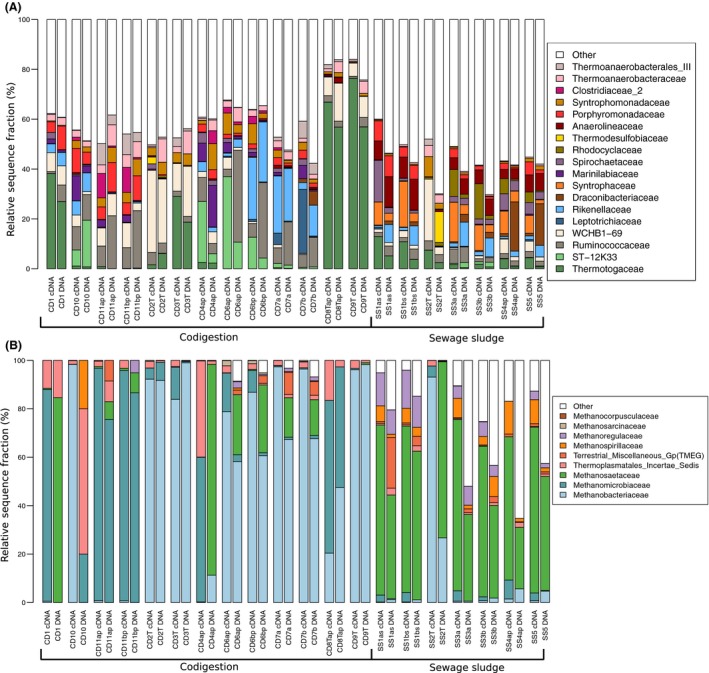
Relative sequence fractions of (A) the 18 most abundant Bacteria families and (B) the 10 most abundant Archaea families, in the 16S rRNA and rRNA gene profiles of all full‐scale digesters. rRNA fractions are denoted ‘cDNA’, and rRNA gene fractions are denoted ‘DNA’. Read fractions not taxonomically classified within the most abundant families are included as ‘other’. WCHB1‐69 and ST‐12K33 are uncultured groups under the order Sphingobacteriales.
The presence of SAO bacteria in codigesters corresponded with an absence of aceticlastic methanogens from their rRNA sequence pools (Fig. 3B). Methanosaetaceae was the primary aceticlastic methanogen family detected at high abundance in the Archaea rRNA and rRNA gene sequences of the mesophilic SS digesters, whereas Methanosaetaceae was only detected at a maximum 3% read fraction in the codigester rRNA samples (Fig. 3B). Methanosaeta is an obligate aceticlast with a higher affinity for acetate than the other aceticlastic genus of Methanosarcina (Conklin et al., 2006). While we have previously reported the dominance of Methanosaeta rRNA genes in digesters treating municipal sludge (Sundberg et al., 2013), the high rRNA gene expression of Methanosaeta in SS digesters found in this study (Fig. S3) shows that aceticlastic methanogenesis is likely the main acetate utilization pathway in such systems. The observation that Methanosaeta rRNA gene expression was low in thermophilic SS digesters and codigesters, even though it was present based on rRNA genes, suggests that rRNA profiling may accurately capture stochastic processes in methanogenic communities due to suboptimal growth conditions, as was suggested by De Vrieze et al. (2016b) during high salinity exposure. The hydrogenotrophic genus of Methanospirillum was also detected in the mesophilic SS digester Archaea rRNA and rRNA gene libraries, but was not detected in the rRNA sequences of the codigesters nor the thermophilic SS2T, suggesting that it was inactive in those systems (Fig. 3B, S3). In contrast, the codigester rRNA libraries were dominated by the hydrogenotrophic families of Methanomicrobiaceae and Methanobacteriaceae (Fig. 3B), which were mostly comprised of the genera Methanoculleus and Methanothermobacter, respectively (Fig. S3). Methanoculleus was often dominant in the mesophilic codigesters, whereas Methanothermobacter was prevalent in thermophilic codigesters and SS2T. Both Methanoculleus and Methanothermobacter are known partners of SAO bacteria at high free ammonia levels (Schnürer et al., 1999; Hattori et al., 2000; Manzoor et al., 2016). Often the Methanoculleus rRNA read fraction was larger than its rRNA gene fraction (Fig. S3), suggesting that members of this genus were important active methanogens in these codigesters despite their low DNA abundance. The lack of aceticlastic Methanosaeta in the rRNA libraries of thermophilic digesters and codigesters and the corresponding activity of hydrogenotrophic Methanoculleus and Methanothermobacter provide further evidence to suggest that SAO is the dominant acetate utilization pathway in those systems. However, further work is needed using isotopically labelled substrates (Schnürer et al., 1999) to confirm the dominant methanogenic pathway across the full‐scale digesters.
Association of microbial community structure and activity with nutrients and biokinetics in full‐scale digesters
We investigated whether differences in observed biokinetics and nutrient levels were linked to the variation in the abundance (rRNA genes) and activity (rRNA) profiles of microbial populations in the full‐scale digesters. Redundancy analysis (RDA) and permutational model selection showed that five elements—free ammonia (NH3‐N), Ni, S, Mo and Fe—could significantly (P < 0.05) explain the variance in the microbial rRNA gene and rRNA profiles (Fig. 4A). The codigester rRNA and rRNA gene profiles generally associated more closely with higher NH3‐N concentrations than the SS digesters based on RDA (Fig. 4A), which is consistent with the higher measured NH3‐N in these systems (Table 1). Free ammonia nitrogen is a well‐known growth inhibitor of certain microbial populations in AD, especially aceticlastic methanogens (Schnürer and Nordberg, 2008), and has previously been associated with rRNA gene profiles of anaerobic digesters (De Vrieze et al., 2015). Notably, the rRNA profile of the thermophilic sludge digester, SS2T, was more closely associated with higher NH3‐N concentrations than its corresponding rRNA gene profile. This discrepancy could be caused by the ‘seeding’ of background microbial DNA through influent sewage sludge, whereas the active community in SS2T was shaped by the selective pressure induced by high temperature and free ammonia (higher free ammonia with higher temperature). S levels were associated with the RNA and DNA profiles of the CD11ap and CD11 bp codigesters, which received slaughterhouse waste that could have contributed to high S levels through protein hydrolysis. Ni and Mo were also associated with the RNA and DNA profiles of the CD11ap and CD11 bp codigesters, which received a proprietary blend of trace elements. Ni and Mo have previously been shown to impact rRNA gene fingerprints of Bacteria and Archaea communities in digester systems (Feng et al., 2010) and are both known to have stimulating effects on methanogenic (Schönheit et al., 1979) and syntrophic activities (Plugge et al., 2009; Worm et al., 2011a,b). Ni is a requirement for factor F 430 involved in methanogenesis (Diekert et al., 1981), and Mo is a cofactor of formate dehydrogenase enzymes (Hille, 2002) that enable energy conservation in syntrophs and methanogens (Stams and Plugge, 2009). Ni and Fe are both critical for hydrogenase enzymes used by methanogens (Thauer et al., 2010) and syntrophs (Li et al., 2011; Worm et al., 2011a,b). In this case, Fe was more associated with the RNA and DNA profiles of SS digesters rather than codigesters (Fig. 4A). Fe was previously shown to influence the rRNA gene profiles of digester microbial communities by modulating the Fe:S ratio, which can reduce the concentration of toxic H2S (Shakeri Yekta et al., 2017). Our findings indicate that the differences in elemental composition among the full‐scale biogas digesters were not only associated with variations in the microbial community rRNA gene abundance, but were also significantly associated with microbial activity based on rRNA gene expression profiles. These results offer additional evidence to support the notion that the activities of digester communities may be adjusted through trace element supplementation (Feng et al., 2010; Banks et al., 2012; Karlsson et al., 2012).
Figure 4.
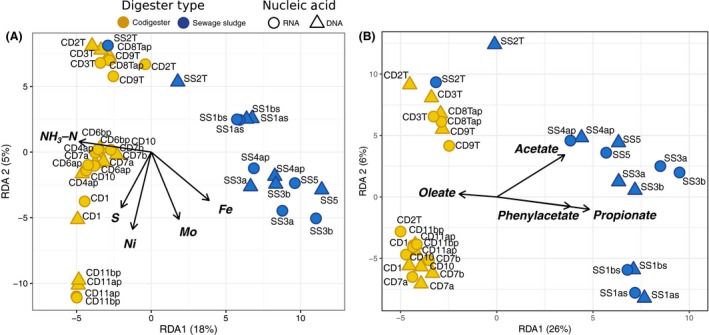
Redundancy analysis (RDA) of OTU read counts in the total microbial community (Bacteria + Archaea) rRNA gene (DNA) and rRNA (RNA) libraries after variance stabilizing transformation with DESeq2 (Love et al., 2014), constrained by significant environmental variables from (A) elemental molar concentrations, and (B) maximum methane production rates (R m) in batch assays with different substrates. Values in parentheses represent the proportion of total variance explained by each RDA axis. The RDA models were significant at the P < 0.001 level, and the explanatory variables were significant at the P < 0.05 level.
We also investigated whether measured maximum rates of methane production (R m) from acetate, propionate, phenyl acetate and oleate by the digester contents were associated with microbial population abundance and activity. The R m from all substrates could significantly (P < 0.05) explain the variance in the total microbial rRNA gene and rRNA profiles (Fig. 4B). The higher propionate oxidation rates observed in the SS digesters (Fig. 1B) may have been attributed to the higher activity of syntrophic propionate‐oxidizing Smithella in those systems compared to the codigesters (Fig. S3). The higher oleic acid conversion rates of the codigesters (Fig. 1B) corresponded with higher rRNA gene and rRNA fractions of LCFA‐degrading Syntrophomonas (Fig. S3). Higher concentrations of Syntrophomonas have previously been correlated with higher degradation rates of LCFA in anaerobic digestion (Ziels et al., 2015, 2016, 2017). Recently, 16S rRNA gene expression of Syntrophomonas was correlated with methane production from LCFA‐rich waste (Amha et al., 2017). The presence and activity of SAO bacteria in the codigesters were associated with lower maximum rates of acetate consumption (Fig. 1B), which supports previous findings that the biokinetics of SAO may be slower than aceticlastic methanogenesis (Schnürer et al., 1999). Overall, the apparent grouping of the full‐scale digester rRNA and rRNA gene profiles based on digester type (Fig. 2) corresponded with different biokinetics of the digester contents, indicating an important relationship between microbial activity levels and digester function. Thus, while AD‐communities may be considered functionally redundant based on substrate type and temperature (Sundberg et al., 2013; De Vrieze et al., 2015), this redundancy does not necessarily implicate equivalent physiologies in terms of the digester biokinetic capacities (i.e. the different community types had different biokinetics).
Bacteria and Archaea diversity in full‐scale anaerobic digesters
We assessed whether the levels of diversity in the Bacteria and Archaea rRNA and rRNA gene libraries were different between the codigesters and municipal SS digesters. Diversity was measured with OTU read counts based on Hill numbers (q D) at various levels (q = 0, 1, 2), which provides information on both the richness (q = 0) and evenness (q > 0) of the community (Jost, 2006). All levels of Hill numbers were statistically lower (P < 0.05, Welch's t‐test) for both Bacteria and Archaea rRNA and rRNA genes in the codigesters relative to the SS digesters (Fig. S4). Lower Hill numbers with codigestion have been observed in prior works (Ziels et al., 2017) and may be caused by the enrichment of specialized microorganisms on influent substrates with certain organic matter compositions. Higher diversity in the SS digesters could also have been attributed to immigration of populations from the PS+WAS feed source, as was shown to occur in other biological wastewater treatment processes (Wells et al., 2014). There were no significant differences within intragroup diversity (i.e. within codigesters and municipal sludge digesters per se) for the rRNA and rRNA gene libraries at all Hill numbers (q = 0, 1, 2; P > 0.1, t‐test), indicating that the diversity in DNA libraries generally reflected the diversity of the active communities (RNA). An exception to this general trend was the thermophilic SS2T digester, which had lower diversity in its rRNA library relative to its rRNA gene library at all levels of Hill numbers libraries for both Bacteria and Archaea (Fig. S4). Similarly, the thermophilic codigesters, CD2T, CD3T and CD9T, had lower Bacteria rRNA diversity relative to rRNA genes at all Hill numbers. Thermophilic digesters (both in SS and CD) had significantly lower diversity at all Hill numbers in rRNA profiles than mesophilic digesters (P < 0.005, Welch's t‐test). This lower rRNA diversity under thermophilic conditions could be caused by a greater selective growth pressure leading to a community with fewer active members, as was suggested in prior works based on rRNA genes (Levén et al., 2007; Pervin et al., 2013). Further research is thus warranted to monitor the ratio of diversity in rRNA to rRNA genes during periods of process instability to determine its utility as a potential monitoring indicator for microbial stress in AD.
Co‐occurrence of microbial rRNA signatures in full‐scale digesters
A total of 2339 statistically significant (P < 0.001) pairs (i.e. positive edges) were detected among 174 OTUs (i.e. nodes) based on correlation analysis of their 16S rRNA gene expression profiles (Fig. 5). Three main subclusters were detected by dissecting the overall co‐occurrence network (Table S4). Inspecting the phylogenetic distribution of the taxa within each subcluster revealed that their arrangement corresponded to the major groupings observed by PCA (Fig. 2) reflecting the three main digester types (i.e. mesophilic codigesters, mesophilic SS digesters and thermophilic digesters). This was further supported by the inclusion of nodes for digester temperature and NH3‐N within one subcluster, a node for Ni within a second subcluster and a node for R m‐propionate, R m‐phenylacetate and R m‐acetate in the third subcluster (Fig. 5).
Figure 5.
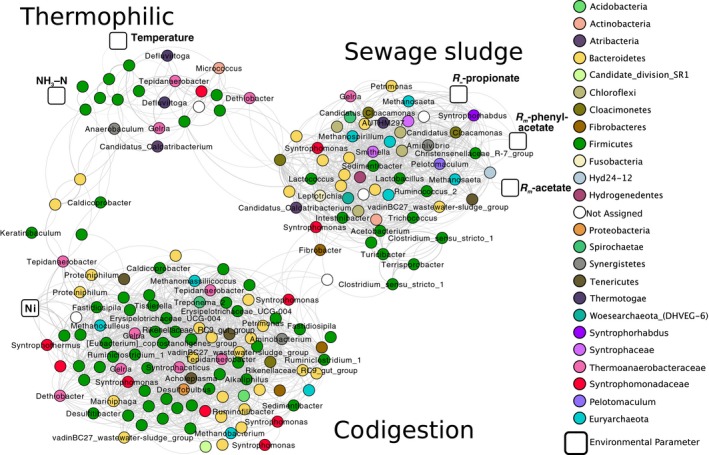
Co‐occurrence network based on correlation of OTU read counts in 16S rRNA gene expression profiles across the 20 full‐scale anaerobic digesters. Nodes are coloured by taxonomy with labelled genera names when possible, and edges are shown as grey lines. The thickness of the edge is proportional to the r value of the correlation (from 0.8 to 0.99). Syntrophic bacteria groups and archaeal groups are depicted in pinks/reds and turquoise respectively. Boxed nodes represent environmental parameters.
The subcluster associated with R m‐propionate, R m‐phenyl acetate and R m‐acetate contained OTUs belonging to Methanosaeta, Methanospirillum, Smithella, Syntrophaceae, Candidatus Cloacamonas, Pelotomaculum and Syntrophorhabdus, which were representative of rRNA profiles in the SS digesters (Fig. S3). Notably, the co‐occurrence of potential propionate‐oxidizing OTUs belonging to Pelotomaculum, Candidatus Cloacimonas and Smithella (Liu et al., 1999; Imachi et al., 2002) and aromatics‐degrading Syntrophorhabdus (Qiu et al., 2008) and Syntrophaceae could explain the higher propionate and phenyl acetate degradation kinetics observed with SS digester contents (Fig. 1B). The ability of Candidatus Cloacimonas acidaminovorans to syntrophically degrade propionate was hypothesized based on in‐silico genome analysis (Pelletier et al., 2008). The first neighbours of the R m‐propionate node included a Methanosaeta‐OTU, a Candidatus Cloacimonas‐OTU, a Syntrophorhabdus‐OTU and a Syntrophaceae‐OTU, suggesting their potential involvement in syntrophic propionate degradation (Schink, 1997). OTUs assigned to the uncultured archaeal groups of Woesearchaeota and the Terrestrial Miscellaneous Euryarchaeotic Group (TMEG) also shared edges with the R m‐propionate node. It was inferred that members of Woesearchaeota may have fermentative and symbiotic lifestyles based on genome reconstruction from metagenomes (Castelle et al., 2015), but their role in anaerobic digesters is unknown and warrants further investigations based on these findings that their activity correlates with higher substrate turnover rates.
The second subcluster was associated with Ni levels and contained OTUs within Methanoculleus, Syntrophomonadaceae, Tepidanaerobacter, Syntrophaceticus and Gelria, which all had significantly higher rRNA gene expression levels in the municipal codigesters in comparison with SS digesters (P < 0.05, Welch's t‐test; Fig. S3). The first neighbours of the Ni node included several potential SAO OTUs, including Thermoanaerobacteraceae and Tepidanaerobacter. SAO activity relies on a Ni‐containing CO‐dehydrogenase (Müller et al., 2013), and Ni addition was previously shown to enhance acetate degradation in SAO‐dominated systems (Karlsson et al., 2012). These results suggest that the activity of SAO is positively associated with Ni levels, but the mechanism for this association needs further confirmation by enzymatic and transcriptomic‐based research.
The third cluster was associated with higher NH3‐N and temperature, which were characteristic of the thermophilic digesters (Table 1). The OTU sharing an edge with the NH3‐N node belonged to the uncultured group of Firmicutes OPB54. In a recent study, Firmicutes OPB54‐assigned OTUs increased in abundance concurrently with a shift towards SAO in high‐ammonia digesters (Müller et al., 2016). Further directed studies are needed to examine the ecophysiology of Firmicutes OPB54 in high ammonia digesters, as it was previously suggested that SAO can be conducted by diverse uncultured bacterial lineages of which little genetic information is known (Werner et al., 2014). The first neighbours of the temperature node included OTUs assigned to Defluviitoga and Tepidanaerobacter (Fig. 5), which agrees with the higher rRNA gene expression of those genera in the thermophilic digesters (Fig. S3). The thermophilic subcluster had the lowest edge count of all subclusters (Table S4). Similarly, edge counts were negatively correlated to temperature in co‐occurrence networks based on rRNA profiles in anaerobic digesters (Lin et al., 2017), which could potentially be explained by a lower diversity at higher temperatures (Fig. S4) limiting the number of species interactions. However, richness and evenness were not correlated with positive edge percentages in networks across various environments (Faust et al., 2015), and thus, the lower edge count in the thermophilic subcluster could be attributed to other community interactions.
Notably, syntrophic and methanogenic taxa were prevalent throughout the overall network, indicating that such obligate energy‐sharing partnerships play critical roles in stabilizing the digester microbiome. An exception was the lack of a methanogenic taxon in the thermophilic subcluster. Metabolic interdependencies, such as syntrophy, were identified as a major driver of species co‐occurrence across a diverse set of microbial ecosystems (Zelezniak et al., 2015). The high representation of syntrophs and methanogens in the co‐occurrence network may also be attributed to their metabolic specialization (Sieber et al., 2012), suggesting that their ecological niches are not easily filled by other taxa (Werner et al., 2011). Yet, recent work has shown high strain variability within digester syntrophic communities under different feeding strategies (Ziels et al., 2017, 2018), indicating that selective forces may still govern the syntrophic community structure in different digester environments.
Overall, co‐occurrence analysis of 16S rRNA gene expression profiles revealed three distinct subclusters of active microbes within different digester types, which were linked to differences in their biokinetics and elemental compositions. While these results are indicative of alternative stability states or high functional redundancy (Shade et al., 2012) for digester microbiomes based on different environmental conditions, the co‐occurrence data alone are purely observational and do not provide a mechanistic understanding of the types of interactions between species. However, these findings warrant further longitudinal studies with planned perturbations and/or physiological investigations using metagenomics and metatranscriptomics to elucidate the nature of interactions among microbes based on varying digester environments. For instance, such experiments should be focused to elucidate the role of uncultured archaeal groups in SS digesters, as well as to illuminate the nature of interactions between micro‐ and macronutrients and uncultivated SAO taxa to develop solutions for improved digester capacity and methane production efficiency.
Experimental procedures
Sample sources and analysis of nutrient compositions
Digestate samples were collected from the effluent of 20 full‐scale anaerobic digesters for parallel biokinetic profiling, elemental analysis and sequencing of 16S rRNA and rRNA genes. The operational parameters of each digester including influent substrate composition, temperature, organic loading rate (OLR) and hydraulic retention time (HRT) are presented in Table 1.
Samples were analysed for elemental contents including NH4‐N, Cl, P, Al, B, Ca, Co, Cu, Fe, K, Mg, Mn, Mo, Na, Ni, Se, S, Ti, V, W and Zn by Eurofin Environment Testing Sweden AB (Sweden). Elemental analyses were carried out per the Swedish Standard Method SS028150‐2, except for NH4‐N and Cl, which were determined per the Standard Methods 4500‐ammonia and 4500‐chloride. The measured elemental concentrations and analytical uncertainties are presented in the (Table S3). Free ammonia levels were calculated based on measured levels of NH4‐N, pH and temperature for each digester.
Batch assays for substrate conversion biokinetics
Degradation biokinetics of acetate, propionate, phenyl acetate and oleic acid by the digester contents were analysed in batch assays according to Karlsson et al. (2012). In short, 15 ml of digestate samples, corresponding to 0.12–0.68 g VS, was inoculated in 330 ml gas‐tight bottles containing 135 ml of growth medium at 37°C. A detailed description of composition of the growth medium is found in Karlsson et al. (1999). The headspace was purged with a gas mixture of 80% nitrogen and 20% carbon dioxide. Acetate, propionate, phenyl acetate and oleic acid were separately added to triplicate bottles with final concentrations of 15, 15, 5 and 5 mM respectively. Triplicate control assays with only digestate were used to determine the methane production from the background organic matter of the samples. Volumetric biogas production was determined by measuring the gas pressure in the bottles using a digital pressure meter (Testo 3123; Testo, Sparta, NJ, USA) at 37°C. Gas samples (1 ml) were also collected from the bottles at nine occasions during 240 h incubation for methane concentration measurements using GC‐FID (HP 5880A; Hewlett Packard, Palo Alto, California, USA). Total solid and VS contents of the samples were determined per the Swedish Standard Method (SS‐028113).
The maximum methane production rates (R m) (ml CH4 g VS−1 h−1) in batch assays were calculated by fitting a Gompertz model to the accumulated methane production (M) curve versus incubation time (t):
Parameters P and λ are model parameters termed as methane production potential (ml CH4) and lag phase (h) respectively. Methane volumes refer to standard temperature and pressure. Model fitting was conducted in R version 3.3. Fitted R m values are presented in Table S2, and example profiles of the model fitting are shown in Fig. S5.
DNA and RNA extraction, 16S rRNA amplicon sequencing and statistics
A detailed protocol for sample processing for DNA and RNA extraction, reverse transcription to complimentary DNA (cDNA) and PCR amplification is provided in the Supplemental Methods (found in Supporting Information). Briefly, PCR amplification of DNA and cDNA was conducted according to Sundberg et al. (2013) with primers 515F and a modified 915R (Casamayor et al., 2002). The sequence library was prepared according to Ziels et al. (2015). The amplicon libraries were sequenced on an Illumina MiSeq in 2 × 250 bp mode at the University of Copenhagen Molecular Microbial Ecology Lab. Raw sequence reads have been submitted to the NCBI SRA database under BioProject PRJNA402061.
16S rRNA and rRNA gene reads were quality filtered and trimmed and denoised into exact sequences using DADA2 (Callahan et al., 2016). Forward and reverse reads were initially truncated at 240 bp and then trimmed by the first 28 and 35 bp, respectively, to remove primers and adapters. A maximum expected error of 2 was used to remove low‐quality reads. After denoising and merging the forward and reverse reads, sequences were clustered into OTUs using UPARSE (Edgar, 2013) based on a 99% similarity threshold, resulting in 4725 OTUs. Representative sequences of each OTU were taxonomically classified against the SILVA SSU Ref NR dataset (v.123) using the RDP classifier (Wang et al., 2007). Denoising and error‐correcting sequences with DADA2 before OTU clustering allowed for a smaller clustering radius to be used without generating spurious or erroneous OTUs (Callahan et al., 2016), thus, enabling a fine‐scale analysis of community structure. A detailed description of read processing and statistical analysis is given in the Supplementary Information.
OTU relative abundance values were calculated with DESeq2 (Love et al., 2014) normalized sequence counts, which account for the different sequencing depths among samples. Diversity was presented using Hill diversity numbers (q D) of orders q = 0, 1 and 2 (Jost, 2006) using OTU read counts. Diversity of order 0 (i.e. 0 D) is equal to species richness, 1 D is equal to exp(Shannon entropy), and 2 D is equivalent to 1/(Simpson concentration) (Jost, 2006).
Principal components analysis (PCA) was conducted using OTU read counts after variance stabilizing (VS) transformation with DESeq2 (Love et al., 2014). Redundancy analysis (RDA) was used to summarize linear relationships between OTU read counts (VS‐transformed) and maximum methane production rates (R m) and elemental concentrations in the digesters. For the elemental dataset, multicollinearity was identified using Pearson correlation, and variables with r values above 0.7 were removed prior to RDA. Model selection was conducted using forward–backward selection with the ordistep function within the vegan package v.2.4.2 (Oksanen et al., 2007) for R version 3.3. Significance of the RDA solution and the selected exploratory variables were assessed through permutational ANOVA in vegan. Differences in biokinetic and nutrient profiles between the digester types were assessed using analysis of similarities (ANOSIM) based on Bray–Curtis distances with the anosim function within the vegan package in R.
A co‐occurrence matrix was constructed by calculating correlations between OTU rRNA read counts based on an ensemble approach of Spearman, Pearson and Kendall correlations using CoNet v.1.1.1 (Faust and Raes, 2016). A statistically significant correlation was defined by an r value >0.75 and P < 0.01 after multiple‐test correction with the Bonferroni procedure (Faust and Raes, 2016). Only OTUs that had >5 read counts in seven digester samples were included in the correlation analysis to reduce spurious correlations (Berry and Widder, 2014), and only positive associations were included in the subsequent network. Correlations between OTUs and environmental parameters were also considered in the network analysis, including NH3‐N, Fe, Ni, Mo, temperature and R m values of acetate, propionate, oleate and phenyl acetate. Only NH3‐N, Ni, temperature, R m‐phenyl acetate, R m‐acetate and R m‐propionate had significant correlations (r > 0.75, P < 0.01). The network was visualized and analysed for topological features in Cytoscape v. 3.5.1 (Shannon et al., 2003). Distinct clusters within the network were detected using the OH‐PIN algorithm within CytoCluster.
Conflict of interest
The authors declare no conflict of interests.
Supporting information
Fig. S1. (A) NMDS plot based on Bray‐Curtis distances of the element concentrations in the 20 digesters.
Fig. S2. Relative sequence fractions of: (A) the 12 most abundant Bacteria phyla in the 16S rRNA and rRNA gene profiles of all full‐scale digesters; (B) the 4 most abundant Archaea phyla in the 16S rRNA and rRNA gene profiles of all full‐scale digesters.
Fig. S3. Relative sequence fractions of: (A) the 25 most abundant Bacteria genera in the 16S rRNA and rRNA gene profiles of all full‐scale digesters; (B) the 15 most abundant Archaea genera in the 16S rRNA and rRNA gene profiles of all full‐scale digesters.
Fig. S4. Hill diversity numbers (q D) in 16S rRNA and rDNA profiles for Bacteria at orders of (A) q = 0.
Fig. S5. Example plots of fitting the Gompertz model (dashed lines) to cumulative methane production in the batch kinetics assays inoculated with sludge samples from SS1bs (A) and CD1 (B) reactors.
Table S1. Volatile fatty acid profiles of sludge sampled from full‐scale digesters.
Table S2. Values of the maximum methane production rate (Rm) based on fitting a Gompertz model to cumulative methane production over time in batch assays with the various digester sludges spiked with acetate, propionate, oleate, and phenyl acetate, as well as un‐amended controls.
Table S3. Elemental concentrations (M) and uncertainties for the 20 full‐scale anaerobic digester biomass samples.
Table S4. Network topology parameters estimated in Cytoscape v. 3.5.1.
Acknowledgements
This research was funded by the Swedish Energy Agency and the Biogas Research Centre at Linköping University, Sweden. R.M.Z received support from NSF grant [DGE‐1256082].
Microbial Biotechnology (2018) 11(4), 694–709
Funding Information
This research was funded by the Swedish Energy Agency and the Biogas Research Centre at Linköping University, Sweden. R.M.Z received support from NSF grant [DGE‐1256082].
References
- Amha, Y.M. , Sinha, P. , Lagman, J. , Gregori, M. , and Smith, A.L. (2017) Elucidating microbial community adaptation to anaerobic co‐digestion of fats, oils, and grease and food waste. Water Res 123: 277–289. [DOI] [PubMed] [Google Scholar]
- Banks, C.J. , Zhang, Y. , Jiang, Y. , and Heaven, S. (2012) Trace element requirements for stable food waste digestion at elevated ammonia concentrations. Bioresour Technol 104: 127–135. [DOI] [PubMed] [Google Scholar]
- Batstone, D.J. , and Virdis, B. (2014) The role of anaerobic digestion in the emerging energy economy. Curr Opin Biotechnol 27: 142–149. [DOI] [PubMed] [Google Scholar]
- Berry, D. and Widder, S. (2014) Deciphering microbial interactions and detecting keystone species with co‐occurrence networks. Front Microbiol 5, https://doi.org/10.3389/fmicb.2014.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay, N. , and Edwards, M. (2001) Role of temperature, chlorine, and organic matter in copper corrosion by‐product release in soft water. Water Res 35: 683–690. [DOI] [PubMed] [Google Scholar]
- Briones, A. , and Raskin, L. (2003) Diversity and dynamics of microbial communities in engineered environments and their implications for process stability. Curr Opin Biotechnol 14: 270–276. [DOI] [PubMed] [Google Scholar]
- Callahan, B.J. , McMurdie, P.J. , Rosen, M.J. , Han, A.W. , Johnson, A.J.A. , and Holmes, S.P. (2016) DADA2: high‐resolution sample inference from Illumina amplicon data. Nat Methods 13: 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballa, M. , Regueiro, L. , and Lema, J.M. (2015) Microbial management of anaerobic digestion: exploiting the microbiome‐functionality nexus. Curr Opin Biotechnol 33: 103–111. [DOI] [PubMed] [Google Scholar]
- Casamayor, E.O. , Massana, R. , Benlloch, S. , Øvreås, L. , Díez, B. , Goddard, V.J. , et al (2002) Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ Microbiol 4: 338–348. [DOI] [PubMed] [Google Scholar]
- Castelle, C.J. , Wrighton, K.C. , Thomas, B.C. , Hug, L.A. , Brown, C.T. , Wilkins, M.J. , et al (2015) Genomic expansion of domain Archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol 25: 690–701. [DOI] [PubMed] [Google Scholar]
- Conklin, A. , Stensel, H.D. , and Ferguson, J. (2006) Growth kinetics and competition between methanosarcina and methanosaeta in mesophilic anaerobic digestion. Water Environ Res 78: 486–496. [DOI] [PubMed] [Google Scholar]
- De Francisci, D. , Kougias, P.G. , Treu, L. , Campanaro, S. , and Angelidaki, I. (2015) Microbial diversity and dynamicity of biogas reactors due to radical changes of feedstock composition. Bioresour Technol 176: 56–64. [DOI] [PubMed] [Google Scholar]
- De Vrieze, J. , Verstraete, W. , and Boon, N. (2013) Repeated pulse feeding induces functional stability in anaerobic digestion. Microb Biotechnol 6: 414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vrieze, J. , Saunders, A.M. , He, Y. , Fang, J. , Nielsen, P.H. , Verstraete, W. , and Boon, N. (2015) Ammonia and temperature determine potential clustering in the anaerobic digestion microbiome. Water Res 75: 312–323. [DOI] [PubMed] [Google Scholar]
- De Vrieze, J. , Raport, L. , Roume, H. , Vilchez‐Vargas, R. , Jáuregui, R. , Pieper, D.H. , and Boon, N. (2016a) The full‐scale anaerobic digestion microbiome is represented by specific marker populations. Water Res 104: 101–110. [DOI] [PubMed] [Google Scholar]
- De Vrieze, J. , Regueiro, L. , Props, R. , Vilchez‐Vargas, R. , Jáuregui, R. , Pieper, D.H. , et al (2016b) Presence does not imply activity: DNA and RNA patterns differ in response to salt perturbation in anaerobic digestion. Biotechnol Biofuels 9: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirel, B. , and Scherer, P. (2011) Trace element requirements of agricultural biogas digesters during biological conversion of renewable biomass to methane. Biomass Bioenerg 35: 992–998. [Google Scholar]
- Diekert, G. , Konheiser, U. , Piechulla, K. , and Thauer, R.K. (1981) Nickel requirement and factor F430 content of methanogenic bacteria. J Bacteriol 148: 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10: 996–998. [DOI] [PubMed] [Google Scholar]
- Faust, K. and Raes, J. (2016) CoNet app: inference of biological association networks using Cytoscape. F1000Research 5, 1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust, K. , Lima‐Mendez, G. , Lerat, J.‐S. , Sathirapongsasuti, J.F. , Knight, R. , Huttenhower, C. , et al (2015) Cross‐biome comparison of microbial association networks. Front Microbiol 6, https://doi.org/10.3389/fmicb.2015.01200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, X.M. , Karlsson, A. , Svensson, B.H. , and Bertilsson, S. (2010) Impact of trace element addition on biogas production from food industrial waste – linking process to microbial communities. FEMS Microbiol Ecol 74: 226–240. [DOI] [PubMed] [Google Scholar]
- Flärdh, K. , Cohen, P.S. , and Kjelleberg, S. (1992) Ribosomes exist in large excess over the apparent demand for protein synthesis during carbon starvation in marine Vibrio sp. strain CCUG 15956. J Bacteriol 174: 6780–6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glissmann, K. , Hammer, E. , and Conrad, R. (2005) Production of aromatic compounds during methanogenic degradation of straw in rice field soil. FEMS Microbiol Ecol 52: 43–48. [DOI] [PubMed] [Google Scholar]
- Hao, L. , Bize, A. , Conteau, D. , Chapleur, O. , Courtois, S. , Kroff, P. , et al (2016) New insights into the key microbial phylotypes of anaerobic sludge digesters under different operational conditions. Water Res 102: 158–169. [DOI] [PubMed] [Google Scholar]
- Hattori, S. , Kamagata, Y. , Hanada, S. , and Shoun, H. (2000) Thermacetogenium phaeum gen. nov., sp. nov., a strictly anaerobic, thermophilic, syntrophic acetate‐oxidizing bacterium. Int J Syst Evol Microbiol 50: 1601–1609. [DOI] [PubMed] [Google Scholar]
- Hecht, C. , and Griehl, C. (2009) Investigation of the accumulation of aromatic compounds during biogas production from kitchen waste. Bioresour Technol 100: 654–658. [DOI] [PubMed] [Google Scholar]
- Hille, R. (2002) Molybdenum and tungsten in biology. Trends Biochem Sci 27: 360–367. [DOI] [PubMed] [Google Scholar]
- Imachi, H. , Sekiguchi, Y. , Kamagata, Y. , Hanada, S. , Ohashi, A. , and Harada, H. (2002) Pelotomaculum thermopropionicum gen. nov., sp. nov., an anaerobic, thermophilic, syntrophic propionate‐oxidizing bacterium. Int J Syst Evol Microbiol 52: 1729–1735. [DOI] [PubMed] [Google Scholar]
- Jost, L. (2006) Entropy and diversity. Oikos 113: 363–375. [Google Scholar]
- Karlsson, A. , Ejlertsson, J. , Nezirevic, D. , and Svensson, B.H. (1999) Degradation of phenol under meso‐ and thermophilic, anaerobic conditions. Anaerobe 5: 25–35. [DOI] [PubMed] [Google Scholar]
- Karlsson, A. , Einarsson, P. , Schnürer, A. , Sundberg, C. , Ejlertsson, J. , and Svensson, B.H. (2012) Impact of trace element addition on degradation efficiency of volatile fatty acids, oleic acid and phenyl acetate and on microbial populations in a biogas digester. J Biosci Bioeng 114: 446–452. [DOI] [PubMed] [Google Scholar]
- Lee, I.‐S. , Parameswaran, P. , and Rittmann, B.E. (2011) Effects of solids retention time on methanogenesis in anaerobic digestion of thickened mixed sludge. Bioresour Technol 102: 10266–10272. [DOI] [PubMed] [Google Scholar]
- Levén, L. , Eriksson, A.R.B. , and Schnürer, A. (2007) Effect of process temperature on bacterial and archaeal communities in two methanogenic bioreactors treating organic household waste. FEMS Microbiol Ecol 59: 683–693. [DOI] [PubMed] [Google Scholar]
- Li, T. , Mazéas, L. , Sghir, A. , Leblon, G. , and Bouchez, T. (2009) Insights into networks of functional microbes catalysing methanization of cellulose under mesophilic conditions. Environ Microbiol 11: 889–904. [DOI] [PubMed] [Google Scholar]
- Li, X. , McInerney, M.J. , Stahl, D.A. , and Krumholz, L.R. (2011) Metabolism of H2 by Desulfovibrio alaskensis G20 during syntrophic growth on lactate. Microbiology 157: 2912–2921. [DOI] [PubMed] [Google Scholar]
- Lin, Q. , De Vrieze, J. , Li, C. , Li, J. , Li, J. , Yao, M. , et al (2017) Temperature regulates deterministic processes and the succession of microbial interactions in anaerobic digestion process. Water Res 123, 134–143. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Balkwill, D.L. , Aldrich, H.C. , Drake, G.R. , and Boone, D.R. (1999) Characterization of the anaerobic propionate‐degrading syntrophs Smithella propionica gen. nov., sp. nov. and Syntrophobacter wolinii . Int J Syst Evol Microbiol 49: 545–556. [DOI] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. , and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoor, S. , Schnürer, A. , Bongcam‐Rudloff, E. , and Müller, B. (2016) Complete genome sequence of Methanoculleus bourgensis strain MAB1, the syntrophic partner of mesophilic acetate‐oxidising bacteria (SAOB). Stand Genomic Sci 11: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maus, I. , Cibis, K.G. , Bremges, A. , Stolze, Y. , Wibberg, D. , Tomazetto, G. , et al (2016a) Genomic characterization of Defluviitoga tunisiensis L3, a key hydrolytic bacterium in a thermophilic biogas plant and its abundance as determined by metagenome fragment recruitment. J Biotechnol 232: 50–60. [DOI] [PubMed] [Google Scholar]
- Maus, I. , Koeck, D.E. , Cibis, K.G. , Hahnke, S. , Kim, Y.S. , Langer, T. , et al (2016b) Unraveling the microbiome of a thermophilic biogas plant by metagenome and metatranscriptome analysis complemented by characterization of bacterial and archaeal isolates. Biotechnol Biofuels 9: 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty, P.L. , Bae, J. , and Kim, J. (2011) Domestic wastewater treatment as a net energy producer‐can this be achieved? Environ Sci Technol 45: 7100–7106. [DOI] [PubMed] [Google Scholar]
- Moestedt, J. , Nordell, E. , Shakeri Yekta, S. , Lundgren, J. , Martí, M. , Sundberg, C. , et al (2016) Effects of trace element addition on process stability during anaerobic co‐digestion of OFMSW and slaughterhouse waste. Waste Manag 47: 11–20. [DOI] [PubMed] [Google Scholar]
- Mulat, D.G. , Jacobi, H.F. , Feilberg, A. , Adamsen, A.P.S. , Richnow, H.‐H. , and Nikolausz, M. (2016) Changing feeding regimes to demonstrate flexible biogas production: effects on process performance, microbial community structure, and methanogenesis pathways. Appl Environ Microbiol 82: 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, B. , Sun, L. , and Schnürer, A. (2013) First insights into the syntrophic acetate‐oxidizing bacteria – a genetic study. Microbiol Open 2: 35–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller, B. , Sun, L. , Westerholm, M. , and Schnürer, A. (2016) Bacterial community composition and fhs profiles of low‐ and high‐ammonia biogas digesters reveal novel syntrophic acetate‐oxidising bacteria. Biotechnol Biofuels 9: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, M.C. , Morrison, M. , and Yu, Z. (2011) A meta‐analysis of the microbial diversity observed in anaerobic digesters. Bioresour Technol 102: 3730–3739. [DOI] [PubMed] [Google Scholar]
- Oksanen, J. , Kindt, R. , Legendre, P. , O'Hara, B. , Stevens, M.H.H. , Oksanen, M.J. , and Suggests, M. (2007) The vegan package. Community Ecol Package 10: 631–637. [Google Scholar]
- Palatsi, J. , Viñas, M. , Guivernau, M. , Fernandez, B. , and Flotats, X. (2011) Anaerobic digestion of slaughterhouse waste: main process limitations and microbial community interactions. Bioresour Technol 102: 2219–2227. [DOI] [PubMed] [Google Scholar]
- Pelletier, E. , Kreimeyer, A. , Bocs, S. , Rouy, Z. , Gyapay, G. , Chouari, R. , et al (2008) “Candidatus Cloacamonas Acidaminovorans”: genome sequence reconstruction provides a first glimpse of a new bacterial division. J Bacteriol 190: 2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervin, H.M. , Dennis, P.G. , Lim, H.J. , Tyson, G.W. , Batstone, D.J. , and Bond, P.L. (2013) Drivers of microbial community composition in mesophilic and thermophilic temperature‐phased anaerobic digestion pre‐treatment reactors. Water Res 47: 7098–7108. [DOI] [PubMed] [Google Scholar]
- Plugge, C.M. , Jiang, B. , de Bok, F.A.M. , Tsai, C. , and Stams, A.J.M. (2009) Effect of tungsten and molybdenum on growth of a syntrophic coculture of Syntrophobacter fumaroxidans and Methanospirillum hungatei . Arch Microbiol 191: 55–61. [DOI] [PubMed] [Google Scholar]
- Poulsen, L.K. , Ballard, G. , and Stahl, D.A. (1993) Use of rRNA fluorescence in situ hybridization for measuring the activity of single cells in young and established biofilms. Appl Environ Microbiol 59: 1354–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, Y.‐L. , Hanada, S. , Ohashi, A. , Harada, H. , Kamagata, Y. , and Sekiguchi, Y. (2008) Syntrophorhabdus aromaticivorans gen. nov., sp. nov., the first cultured anaerobe capable of degrading phenol to acetate in obligate syntrophic associations with a hydrogenotrophic methanogen. Appl Environ Microbiol 74: 2051–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivière, D. , Desvignes, V. , Pelletier, E. , Chaussonnerie, S. , Guermazi, S. , Weissenbach, J. , et al (2009) Towards the definition of a core of microorganisms involved in anaerobic digestion of sludge. ISME J 3: 700–714. [DOI] [PubMed] [Google Scholar]
- Schink, B. (1997) Energetics of syntrophic cooperation in methanogenic degradation. Microbiol Mol Biol Rev 61: 262–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, T. , Nelles, M. , Scholwin, F. , and Pröter, J. (2014) Trace element supplementation in the biogas production from wheat stillage – Optimization of metal dosing. Bioresour Technol 168: 80–85. [DOI] [PubMed] [Google Scholar]
- Schnürer, A. , and Nordberg, Å. (2008) Ammonia, a selective agent for methane production by syntrophic acetate oxidation at mesophilic temperature. Water Sci Technol 57: 735–740. [DOI] [PubMed] [Google Scholar]
- Schnürer, A. , Zellner, G. , and Svensson, B.H. (1999) Mesophilic syntrophic acetate oxidation during methane formation in biogas reactors. FEMS Microbiol Ecol 29: 249–261. [Google Scholar]
- Schönheit, P. , Moll, J. , and Thauer, R.K. (1979) Nickel, cobalt, and molybdenum requirement for growth of Methanobacterium thermoautotrophicum. Arch Microbiol 123: 105–107. [DOI] [PubMed] [Google Scholar]
- Shade, A. , Peter, H. , Allison, S.D. , Baho, D.L. , Berga, M. , Bürgmann, H. , et al (2012) Fundamentals of microbial community resistance and resilience. Front Microbiol 3, https://doi.org/10.3389/fmicb.2012.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakeri Yekta, S. , Ziels, R.M. , Björn, A. , Skyllberg, U. , Ejlertsson, J. , Karlsson, A. , et al (2017) Importance of sulfide interaction with iron as regulator of the microbial community in biogas reactors and its effect on methanogenesis, volatile fatty acids turnover, and syntrophic long‐chain fatty acids degradation. J Biosci Bioeng 123: 597–605. [DOI] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N.S. , Wang, J.T. , Ramage, D. , et al (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sieber, J.R. , McInerney, M.J. , and Gunsalus, R.P. (2012) Genomic insights into syntrophy: the paradigm for anaerobic metabolic cooperation. Annu Rev Microbiol 66: 429–452. [DOI] [PubMed] [Google Scholar]
- Smith, A.L. , Skerlos, S.J. , and Raskin, L. (2015) Membrane biofilm development improves COD removal in anaerobic membrane bioreactor wastewater treatment. Microb Biotechnol 8: 883–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sörme, L. , and Lagerkvist, R. (2002) Sources of heavy metals in urban wastewater in Stockholm. Sci Total Environ 298: 131–145. [DOI] [PubMed] [Google Scholar]
- Sousa, D.Z. , Smidt, H. , Alves, M.M. , and Stams, A.J.M. (2009) Ecophysiology of syntrophic communities that degrade saturated and unsaturated long‐chain fatty acids. FEMS Microbiol Ecol 68: 257–272. [DOI] [PubMed] [Google Scholar]
- Stams, A.J.M. (1994) Metabolic interactions between anaerobic bacteria in methanogenic environments. Antonie Van Leeuwenhoek 66: 271–294. [DOI] [PubMed] [Google Scholar]
- Stams, A.J.M. , and Plugge, C.M. (2009) Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat Rev Microbiol 7: 568–577. [DOI] [PubMed] [Google Scholar]
- Sundberg, C. , Al‐Soud, W.A. , Larsson, M. , Alm, E. , Yekta, S.S. , Svensson, B.H. , et al (2013) 454 pyrosequencing analyses of bacterial and archaeal richness in 21 full‐scale biogas digesters. FEMS Microbiol Ecol 85: 612–626. [DOI] [PubMed] [Google Scholar]
- Thauer, R.K. , Kaster, A.‐K. , Goenrich, M. , Schick, M. , Hiromoto, T. , and Shima, S. (2010) Hydrogenases from methanogenic archaea, nickel, a novel cofactor, and H2 storage. Annu Rev Biochem 79: 507–536. [DOI] [PubMed] [Google Scholar]
- Treu, L. , Kougias, P.G. , Campanaro, S. , Bassani, I. , and Angelidaki, I. (2016) Deeper insight into the structure of the anaerobic digestion microbial community; the biogas microbiome database is expanded with 157 new genomes. Bioresour Technol 216: 260–266. [DOI] [PubMed] [Google Scholar]
- Vanwonterghem, I. , Jensen, P.D. , Ho, D.P. , Batstone, D.J. , and Tyson, G.W. (2014) Linking microbial community structure, interactions and function in anaerobic digesters using new molecular techniques. Curr Opin Biotechnol 27: 55–64. [DOI] [PubMed] [Google Scholar]
- Vanwonterghem, I. , Jensen, P.D. , Rabaey, K. , and Tyson, G.W. (2016) Genome‐centric resolution of microbial diversity, metabolism and interactions in anaerobic digestion. Environ Microbiol 18: 3144–3158. [DOI] [PubMed] [Google Scholar]
- Venkiteshwaran, K. , Milferstedt, K. , Hamelin, J. , Fujimoto, M. , Johnson, M. , and Zitomer, D.H. (2017) Correlating methane production to microbiota in anaerobic digesters fed synthetic wastewater. Water Res 110: 161–169. [DOI] [PubMed] [Google Scholar]
- Wagner, M. , Rath, G. , Amann, R. , Koops, H.‐P. , and Schleifer, K.‐H. (1995) In situ identification of ammonia‐oxidizing bacteria. Syst Appl Microbiol 18: 251–264. [Google Scholar]
- Wang, Q. , Garrity, G.M. , Tiedje, J.M. , and Cole, J.R. (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73: 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells, G.F. , Wu, C.H. , Piceno, Y.M. , Eggleston, B. , Brodie, E.L. , DeSantis, T.Z. , et al (2014) Microbial biogeography across a full‐scale wastewater treatment plant transect: evidence for immigration between coupled processes. Appl Microbiol Biotechnol 98: 4723–4736. [DOI] [PubMed] [Google Scholar]
- Werner, J.J. , Knights, D. , Garcia, M.L. , Scalfone, N.B. , Smith, S. , Yarasheski, K. , et al (2011) Bacterial community structures are unique and resilient in full‐scale bioenergy systems. Proc Natl Acad Sci 108: 4158–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner, J.J. , Garcia, M.L. , Perkins, S.D. , Yarasheski, K.E. , Smith, S.R. , Muegge, B.D. , et al (2014) Microbial community dynamics and stability during an ammonia‐induced shift to syntrophic acetate oxidation. Appl Environ Microbiol 80: 3375–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerholm, M. , Roos, S. , and Schnürer, A. (2010) Syntrophaceticus schinkii gen. nov., sp. nov., an anaerobic, syntrophic acetate‐oxidizing bacterium isolated from a mesophilic anaerobic filter. FEMS Microbiol Lett 309: 100–104. [DOI] [PubMed] [Google Scholar]
- Westerholm, M. , Roos, S. , and Schnürer, A. (2011) Tepidanaerobacter acetatoxydans sp. nov., an anaerobic, syntrophic acetate‐oxidizing bacterium isolated from two ammonium‐enriched mesophilic methanogenic processes. Syst Appl Microbiol 34: 260–266. [DOI] [PubMed] [Google Scholar]
- Westerholm, M. , Müller, B. , Isaksson, S. , and Schnürer, A. (2015) Trace element and temperature effects on microbial communities and links to biogas digester performance at high ammonia levels. Biotechnol Biofuels 8: 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfert, P. , Kumar, P.S. , Korving, L. , Witkamp, G.‐J. , and van Loosdrecht, M.C.M. (2015) The relevance of phosphorus and iron chemistry to the recovery of phosphorus from wastewater: a review. Environ Sci Technol 49: 9400–9414. [DOI] [PubMed] [Google Scholar]
- Williams, J. , Williams, H. , Dinsdale, R. , Guwy, A. , and Esteves, S. (2013) Monitoring methanogenic population dynamics in a full‐scale anaerobic digester to facilitate operational management. Bioresour Technol 140: 234–242. [DOI] [PubMed] [Google Scholar]
- Worm, P. , Fermoso, F.G. , Lens, P.N.L. , and Plugge, C.M. (2009) Decreased activity of a propionate degrading community in a UASB reactor fed with synthetic medium without molybdenum, tungsten and selenium. Enzyme Microb Technol 45: 139–145. [Google Scholar]
- Worm, P. , Fermoso, F.G. , Stams, A.J.M. , Lens, P.N.L. , and Plugge, C.M. (2011a) Transcription of fdh and hyd in Syntrophobacter spp. and Methanospirillum spp. as a diagnostic tool for monitoring anaerobic sludge deprived of molybdenum, tungsten and selenium. Environ Microbiol 13: 1228–1235. [DOI] [PubMed] [Google Scholar]
- Worm, P. , Stams, A.J.M. , Cheng, X. , and Plugge, C.M. (2011b) Growth‐ and substrate‐dependent transcription of formate dehydrogenase and hydrogenase coding genes in Syntrophobacter fumaroxidans and Methanospirillum hungatei. Microbiology 157: 280–289. [DOI] [PubMed] [Google Scholar]
- Zakrzewski, M. , Goesmann, A. , Jaenicke, S. , Jünemann, S. , Eikmeyer, F. , Szczepanowski, R. , et al (2012) Profiling of the metabolically active community from a production‐scale biogas plant by means of high‐throughput metatranscriptome sequencing. J Biotechnol 158: 248–258. [DOI] [PubMed] [Google Scholar]
- Zelezniak, A. , Andrejev, S. , Ponomarova, O. , Mende, D.R. , Bork, P. , and Patil, K.R. (2015) Metabolic dependencies drive species co‐occurrence in diverse microbial communities. Proc Natl Acad Sci 112: 6449–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, C. , Yuan, Q. , and Lu, Y. (2014a) Inhibitory effects of ammonia on methanogen mcrA transcripts in anaerobic digester sludge. FEMS Microbiol Ecol 87: 368–377. [DOI] [PubMed] [Google Scholar]
- Zhang, W. , Werner, J.J. , Agler, M.T. , and Angenent, L.T. (2014b) Substrate type drives variation in reactor microbiomes of anaerobic digesters. Bioresour Technol 151: 397–401. [DOI] [PubMed] [Google Scholar]
- Zhang, W. , Wu, S. , Guo, J. , Zhou, J. , and Dong, R. (2015) Performance and kinetic evaluation of semi‐continuously fed anaerobic digesters treating food waste: role of trace elements. Bioresour Technol 178: 297–305. [DOI] [PubMed] [Google Scholar]
- Ziels, R.M. , Beck, D.A.C. , Martí, M. , Gough, H.L. , Stensel, H.D. and Svensson, B.H. (2015) Monitoring the dynamics of syntrophic β‐oxidizing bacteria during anaerobic degradation of oleic acid by quantitative PCR. FEMS Microbiol Ecol 91, https://doi.org/10.1093/femsec/fiv028. [DOI] [PubMed] [Google Scholar]
- Ziels, R.M. , Karlsson, A. , Beck, D.A.C. , Ejlertsson, J. , Yekta, S.S. , Bjorn, A. , et al (2016) Microbial community adaptation influences long‐chain fatty acid conversion during anaerobic codigestion of fats, oils, and grease with municipal sludge. Water Res 103: 372–382. [DOI] [PubMed] [Google Scholar]
- Ziels, R.M. , Beck, D.A.C. , and Stensel, H.D. (2017) Long‐chain fatty acid feeding frequency in anaerobic codigestion impacts syntrophic community structure and biokinetics. Water Res 117: 218–229. [DOI] [PubMed] [Google Scholar]
- Ziels, R.M. , Sousa, D.Z. , Stensel, H.D. , and Beck, D.A. (2018) DNA‐SIP based genome‐centric metagenomics identifies key long‐chain fatty acid‐degrading populations in anaerobic digesters with different feeding frequencies. ISME J 12: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. (A) NMDS plot based on Bray‐Curtis distances of the element concentrations in the 20 digesters.
Fig. S2. Relative sequence fractions of: (A) the 12 most abundant Bacteria phyla in the 16S rRNA and rRNA gene profiles of all full‐scale digesters; (B) the 4 most abundant Archaea phyla in the 16S rRNA and rRNA gene profiles of all full‐scale digesters.
Fig. S3. Relative sequence fractions of: (A) the 25 most abundant Bacteria genera in the 16S rRNA and rRNA gene profiles of all full‐scale digesters; (B) the 15 most abundant Archaea genera in the 16S rRNA and rRNA gene profiles of all full‐scale digesters.
Fig. S4. Hill diversity numbers (q D) in 16S rRNA and rDNA profiles for Bacteria at orders of (A) q = 0.
Fig. S5. Example plots of fitting the Gompertz model (dashed lines) to cumulative methane production in the batch kinetics assays inoculated with sludge samples from SS1bs (A) and CD1 (B) reactors.
Table S1. Volatile fatty acid profiles of sludge sampled from full‐scale digesters.
Table S2. Values of the maximum methane production rate (Rm) based on fitting a Gompertz model to cumulative methane production over time in batch assays with the various digester sludges spiked with acetate, propionate, oleate, and phenyl acetate, as well as un‐amended controls.
Table S3. Elemental concentrations (M) and uncertainties for the 20 full‐scale anaerobic digester biomass samples.
Table S4. Network topology parameters estimated in Cytoscape v. 3.5.1.