

Supplemental Digital Content is available in the text.
Keywords: angiotensin II, aorta, blood pressure, endothelial cells, vascular remodeling
Abstract
GTPCH (GTP cyclohydrolase 1, encoded by Gch1) is required for the synthesis of tetrahydrobiopterin; a critical regulator of endothelial NO synthase function. We have previously shown that mice with selective loss of Gch1 in endothelial cells have mild vascular dysfunction, but the consequences of endothelial cell tetrahydrobiopterin deficiency in vascular disease pathogenesis are unknown. We investigated the pathological consequence of Ang (angiotensin) II infusion in endothelial cell Gch1 deficient (Gch1fl/flTie2cre) mice. Ang II (0.4 mg/kg per day, delivered by osmotic minipump) caused a significant decrease in circulating tetrahydrobiopterin levels in Gch1fl/flTie2cre mice and a significant increase in the Nω-nitro-L-arginine methyl ester inhabitable production of H2O2 in the aorta. Chronic treatment with this subpressor dose of Ang II resulted in a significant increase in blood pressure only in Gch1fl/flTie2cre mice. This finding was mirrored with acute administration of Ang II, where increased sensitivity to Ang II was observed at both pressor and subpressor doses. Chronic Ang II infusion in Gch1fl/flTie2ce mice resulted in vascular dysfunction in resistance mesenteric arteries with an enhanced constrictor and decreased dilator response and medial hypertrophy. Altered vascular remodeling was also observed in the aorta with an increase in the incidence of abdominal aortic aneurysm formation in Gch1fl/flTie2ce mice. These findings indicate a specific requirement for endothelial cell tetrahydrobiopterin in modulating the hemodynamic and structural changes induced by Ang II, through modulation of blood pressure, structural changes in resistance vessels, and aneurysm formation in the aorta.
See Editorial Commentary, pp 61–62
Tetrahydrobiopterin (BH4) is a critical regulator of eNOS (endothelial NO synthase) function and eNOS-derived NO and reactive oxygen species (ROS) signaling in vascular physiology. Biosynthesis of BH4 is catalyzed by GTPCH (GTP cyclohydrolase 1, encoded by Gch1), the rate-limiting enzyme for de novo BH4 biosynthesis. We have previously shown that Gch1 expression is a key determinant of BH4 bioavailability, eNOS regulation, and NO generation.1,2 Evidence from experimental and clinical studies have demonstrated that reduced BH4 bioavailability is associated with the pathogenesis of endothelial dysfunction, a hallmark of vascular disease states. Reduced vascular BH4 levels and eNOS uncoupling have been observed in patients with endothelial dysfunction resulting from hypertension and diabetes mellitus.3,4 There is increasing evidence for the role of eNOS uncoupling in the development of pathologies, such as abdominal aortic aneurysms (AAA). Gao et al5 demonstrated that hph-1 (hyperphenylalaninemic) mice,6,7 which have moderate systemic BH4 deficiency, have increased aortic ROS generation, vascular hypertrophy, and an increase in the incidence of aortic aneurysms in response to Ang (angiotensin) II. However, global BH4 deficiency in the hph-1 mouse limits the interpretation of the specific role of endothelial cell BH4 in the pathogenesis of vascular disease. We recently reported that selective deficiency in endothelial cell BH4 biosynthesis, in an endothelial cell-specific Gch1 knockout (Gch1fl/flTie2cre) mouse, is sufficient to cause eNOS uncoupling, endothelial dysfunction, and mild hypertension, in healthy animals.8,9 However, it is unclear to what extent there is a specific requirement for endothelial cell BH4 in modulating susceptibility to structural vascular disease and whether selective endothelial cell BH4 deficiency is sufficient to drive pathological changes in the vascular wall.
Ang II causes vascular dysfunction, vascular smooth muscle cell (VSMC) proliferation and hypertrophy, and extracellular matrix degradation.10 Numerous studies have shown that Ang II–induced vascular hypertrophy and hypertension is mediated by ROS from VSMC and endothelial cells.11–14 Major sources of Ang II–derived ROS are the NADPH oxidases that cause eNOS uncoupling via oxidation of vascular BH4, leading to endothelial dysfunction, vascular hypertrophy, and hypertension.15,16 However, it is unclear whether increased endothelial cell-derived ROS from Ang II–induced eNOS uncoupling, because of selective endothelial cell BH4 deficiency, is alone sufficient to alter overall vascular function and the pathological response to Ang II stimulation, or whether these effects in endothelial cells are a consequence of disease processes throughout the vascular wall.
We sought to investigate the pathological effects of selective endothelial cell BH4 deficiency in the regulation of vascular homeostasis and blood pressure in response to Ang II, and the consequences of selective endothelial BH4 deficiency on vascular pathology, including resistance vessel remodeling and aortic aneurysm formation.
Methods
Most of the data that support the findings of this study are available within the article and in the online-only Data Supplement. The remaining supporting data are available from the corresponding author on reasonable request.
Conditional Gch1 Knockout Mice
We have generated a Gch1 conditional knockout (floxed) allele using Cre/loxP strategy as described previously.8 Gch1fl/fl animals were bred with Tie2cre transgenic mice to produce Gch1fl/flTie2cre mice on a C57BL/6J background, where Gch1 is deleted in endothelial cells, generating a mouse model of endothelial cell-specific BH4 deficiency. The Tie2cre transgene is active in the female germ line, so only male animals were used to establish breeding pairs to maintain conditional Gch1 deletion. Mice were genotyped according to the published protocol.8,17 Male Gch1fl/flTie2cre and their Gch1fl/fl littermates (wild-type [WT]) were used for all experiments at 12 to 22 weeks. All animal studies were conducted with ethical approval from the Local Ethical Review Committee and in accordance with the UK Home Office regulations (Guidance on the Operation of Animals, Scientific Procedures Act, 1986). Mice were housed in ventilated cages with a 12-hour light/dark cycle and controlled temperature (20°C–22°C) and fed normal chow and water ad libitum.
Statistical Analysis
Data are expressed as mean±SE of the means and analyzed using GraphPad Prism version 5.0 (San Diego, CA). Comparisons between WT and Gch1fl/flTie2cre were made by unpaired Student t test. Comparisons with concentration-response curves were compared by 2-way ANOVA for repeated measurements. When >2 independent variables were present a 2-way ANOVA with Tukey multiple comparisons test was used. A P value of <0.05 was considered statistically significant.
Results
Modulation of GTPCH/BH4 Biosynthesis by Ang II Infusion In Vivo
To investigate whether Gch1/GTPCH protein and BH4 biosynthesis are modulated by Ang II–induced hypertension, we infused WT (C56BL6) mice with either saline or a pressor dose of Ang II at 1 mg/kg per day by osmotic minipump for 28 days. Ang II infusion caused a marked increase in systolic blood pressure from 7 days, which was maintained throughout 28 days of infusion (Figure S1 in the online-only Data Supplement). During the 28 days of Ang II infusion, 1 out of 8 mice died because of aortic rupture. Among the survivors, 1 out of the 7 Ang II–infused mice had an AAA at harvest compared with no AAA in the saline group (Figure S1). After 28 days of Ang II infusion, aortic BH4 and total biopterins levels were significantly increased in Ang II–infused mice compared with saline-infused mice despite no significant alteration in GTPCH protein (Figure S1). In contrast to the aorta, there was no difference in BH4 content in plasma. In addition, we found no detectable iNOS (inducible NOS) protein in aortic tissues from Ang II–infused mice (Figure S1). Interestingly, we found a significant increase in aortic DHFR (dihydrofolate reductase) protein levels in Ang II–infused mice compared with saline-infused controls, which may indicate increased recycling of BH2 to BH4.
Ang II Infusion Exacerbates eNOS Uncoupling in Endothelial Cell BH4 Deficient Mice
After establishing the role of vascular Gch1/BH4 biosynthesis in hypertension in response to Ang II, we next determined whether an endothelial cell-specific reduction in BH4 plays a specific and causal role in disease pathogenesis, rather than being a consequence of the disease. We first validated the endothelial cell specificity of Gch1 deletion in Gch1fl/flTie2cre mice. Western blot analysis showed that GTPCH protein was markedly reduced in isolated primary endothelial cells from Gch1fl/flTie2cre mice (Figure S2A). Reduced GTPCH protein was accompanied by a reduction in BH4 levels and in the BH4/BH2 ratio in endothelial cells. The decrease in BH4 resulted in an increase in the L-NAME inhibitable superoxide and ROS production at baseline (Figure S2B and S2C), indicating eNOS uncoupling in Gch1fl/flTie2cre endothelial cells. In aortas, GTPCH protein and BH4 levels were significantly reduced in Gch1fl/flTie2cre mice compared with WT mice (Figure 1A and 1B). Removal of the endothelium and adventitia (ie, generating samples of vascular media) reduced GTPCH and BH4 levels in WT aortas, such that vascular GTPCH protein and BH4 levels were no longer different between WT and Gch1fl/flTie2cre mice (Figure 1B). Similarly, BH4 levels in adventitia were similar between the genotypes (Figure 1C). This result indicates that the endothelium is the principal site for de novo BH4 biosynthesis in mouse aorta. In addition, we found that both nNOS (neuronal NOS) and eNOS proteins were present in whole aortas in both genotypes (Figure 1A). However, neither nNOS or eNOS were detected in the medial layer from either genotypes, whereas the adventitia was the predominant source of nNOS in both genotypes. In contrast to eNOS and nNOS, there was no detectable iNOS protein in either whole aorta, media, or adventitia from either genotype (Figure 1A). These results indicate that loss of BH4 from the endothelial cell layer does not impact on medial BH4 content or on the presence of NOS isoforms in the aorta.
Figure 1.
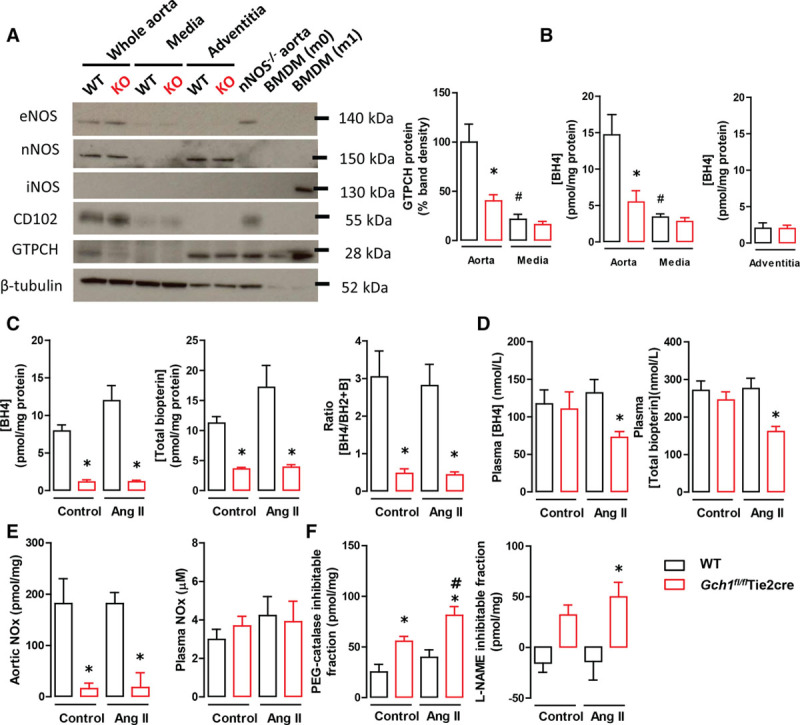
A, Representative immunoblots showing eNOS (endothelial NO synthase), nNOS (neuronal NOS), iNOS (inducible NOS), CD102 (endothelial cell marker), GTPCH (GTP cyclohydrolase I), and β-tubulin (loading control) proteins in whole aorta, media, and adventitia from wild-type (WT) and Gch1fl/flTie2cre knockout (KO) mice (left). Quantitative data, measured as percentage band density of β-tubulin, showing GTPCH protein in whole aorta and media from WT and Gch1fl/flTie2cre mice (right). B, HPLC analysis of biopterins in the whole aorta, media, and adventitia from WT and Gch1fl/flTie2cre mice (*P<0.05 comparing genotype; #P<0.05 comparing treatment; n=6 animals per group). C, Aortic tetrahydrobiopterin (BH4), total biopterins, and BH4/BH2 ratio. D, Plasma BH4 and total biopterins levels (*P<0.05 comparing genotype; n=5–6 animals per group). E, Aortic nitrite/nitrate (NOx) levels and plasma NOx levels, from WT and Gch1fl/flTie2cre mice infused with either saline or 0.4 mg/kg per day of Ang (angiotensin) II (*P<0.05 comparing genotype; n=5–6 animals per group). F, Aortic H2O2 production (expressed as the polyethylene glycol catalase inhibitable fraction) and aortic H2O2 production (expressed as the Nω-nitro-L-arginine methyl ester [L-NAME] inhibitable fraction) in WT and Gch1fl/flTie2cre mice infused with either saline or Ang II (*P<0.05 comparing genotype; #P<0.05 comparing treatment; n=6 animals per group).
Having demonstrated that endothelial cell BH4 deficiency leads to eNOS uncoupling in Gch1fl/flTie2cre mice, we next aimed to determine the effect of endothelial cell BH4 deficiency on the vascular response to Ang II in vivo. We infused WT and Gch1fl/flTie2cre mice with a subpressor dose (0.4 mg/kg per day) of Ang II by osmotic minipump for 5 days. In saline-infused mice, the levels of aortic BH4, total biopterins, and BH4/BH2 ratio were significantly reduced in Gch1fl/flTie2cre mice compared with WT controls (Figure 1C). In Ang II–infused mice, there was a trend toward increased vascular BH4 and total biopterin levels in WT mice, but this increase was attenuated in Gch1fl/flTie2cre mice (Figure 1C). There was no difference in circulating BH4 levels and total biopterins between groups in saline-treated mice (Figure 1D). However, Ang II infusion caused a significant reduction in circulating BH4 and total biopterin levels in Gch1fl/flTie2cre mice, whereas in WT mice circulating BH4 levels and total biopterins were unchanged after Ang II treatment, indicating that in pathological conditions, endothelial cells make a significant contribution to circulating BH4 levels. We next determined NO bioavailability in aortic tissues from Ang II–infused WT and Gch1fl/flTie2cre mice. In saline-infused mice, aortic nitrite/nitrate (NOx) accumulation was greatly decreased in Gch1fl/flTie2cre mice compared with WT control with no further reduction observed after Ang II infusion (Figure 1E). In plasma, there was no difference in circulating nitrate/nitrate between control mice and Ang II–infused mice in either WT or Gch1fl/flTie2cre mice (Figure 1E).
Because Ang II is known to increased aortic ROS generation, we next aimed to determine the impact of Ang II treatment on vascular ROS production in Gch1fl/flTie2cre mice. We observed that basal aortic H2O2 production (expressed as the polyethylene glycol catalase inhibitable fraction) was markedly increased in Gch1fl/flTie2cre mice compared with WT control and was further increased in Gch1fl/flTie2cre mice infused with Ang II. In contrast, no difference in aortic H2O2 production after Ang II infusion was observed in aortas from WT mice (Figure 1F). L-NAME markedly decreased aortic H2O2 production in Gch1fl/flTie2cre mice both at baseline and after Ang II infusion (Figure 1F), indicating that eNOS is the major source of aortic H2O2 production at baseline and after Ang II infusion. Taken together, these findings indicate that endothelial cell BH4 deficiency leads to exacerbated eNOS uncoupling and increased aortic H2O2 production in response to Ang II infusion.
Endothelial Cell BH4 Deficient Mice Have an Increased Pressor Response to Acute Administration of Ang II
To determine the functional importance of increased ROS production by eNOS in Gch1fl/flTie2cre mice, we measured the hemodynamic response to acute Ang II infusion. Using Millar catheters to measure invasive blood pressure changes, acute Ang II administration at a subpressor dose of 10 μg/kg, caused a significant increase in systolic blood pressure only in Gch1fl/flTie2cre mice (change from baseline, 7.0±1.2 versus 2.4±0.4 mm Hg; P<0.05). At pressor doses of Ang II, blood pressure was significantly augmented at both 50 and 100 μg/kg in Gch1fl/flTie2cre mice compared with WT controls (Figure 2A and 2B). Heart rate was markedly decreased in response to Ang II in both WT and Gch1fl/flTie2cre mice at both the 50 and 100 μg/kg dose of Ang II. However, a greater reduction in heart rate was observed in Gch1fl/flTie2cre mice compared with WT littermates (Figure 2B). These findings suggest that endothelial cell BH4 deficiency confers increased sensitivity to acute Ang II.
Figure 2.
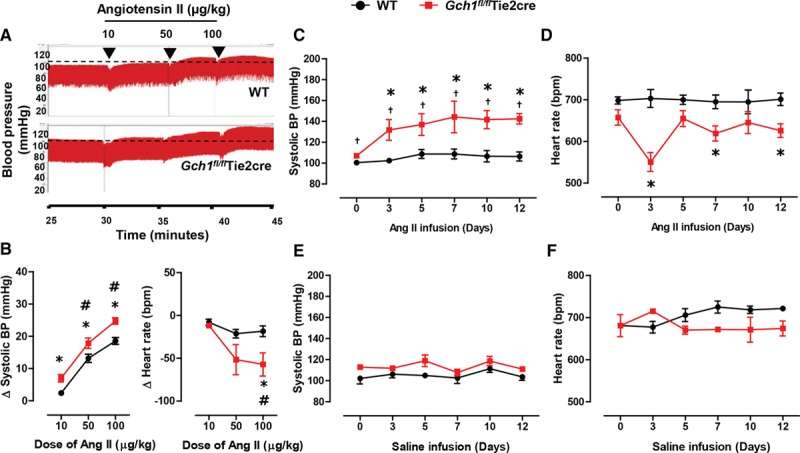
Hemodynamic response to acute and chronic Ang (angiotensin) II stimulation in Gch1fl/flTie2cre and wild-type (WT) mice. A, Representative continuous blood pressure (BP) traces, measured by Millar catheter, from WT and Gch1fl/flTie2cre mice before and after a serial intraperitoneal bolus doses of Ang II (10–100 μg/kg) with quantitative data for change (Δ) in B systolic BP and heart rate after Ang II administration. Ang II caused a significantly greater increase in systolic BP in Gch1fl/flTie2cre mice in a dose-dependent manner. Change in heart rate after Ang II administration, expressed in bpm (*P<0.05 comparing genotype; #P<0.05 comparing treatment; n=4–6 animals per group). C, Osmotic minipump containing Ang II (0.4 mg/kg per day) or saline was implanted in WT and Gch1fl/flTie2cre mice. Systolic BP and heart rate, measured by noninvasive tail cuff, in WT and Gch1fl/flTie2cre mice during 14 d of Ang II infusion. Systolic BP significantly increased in Gch1fl/flTie2cre mice after 3 d of Ang II infusion but was unaltered in WT mice (*P<0.05 comparing treatment; †P<0.05 comparing genotype; n=5–6 animals per group). D, Heart rate significantly reduced in Gch1fl/flTie2cre mice after Ang II infusion but unchanged in WT mice. E and F, Systolic BP and heart rate respectively in saline-infused WT and Gch1fl/flTie2cre mice (n=3 animals per group).
Increased Arterial Blood Pressure in Response to Chronic Ang II Infusion in Endothelial Cell BH4 Deficient Mice
We next aimed to establish whether the acute increase in blood pressure observed in Gch1fl/flTie2cre mice persisted during chronic Ang II infusion. Mice were infused with Ang II at a subpressor dose (0.4 mg/kg per day) for 14 days and blood pressure measured in conscious mice by the tail cuff. As expected, this subpressor dose of Ang II infusion did not have any effect on the systolic blood pressure of WT mice at any time point studied (Figure 2C). In contrast, the small increase in basal systolic blood pressure in Gch1fl/flTie2cre mice (108±1 versus 101±2 mm Hg; P<0.05;Figure 2C) was greatly increased in Gch1fl/flTie2cre mice as soon as 3 days after low-dose Ang II infusion (132±10 versus 102±2 mm Hg; P<0.05) and was maintained throughout the 14 day infusion. Basal heart rate was significantly reduced in Gch1fl/flTie2cre mice compared with WT littermates from day 3 onwards of Ang II infusion but remained unchanged in WT mice (551±23 versus 703±21 bpm; P<0.05; Figure 2D). Saline infusion had no significant effect on either systolic blood pressure or heart rates in either WT or Gch1fl/flTie2cre mice (Figure 2E and 2F).
Chronic Ang II Infusion Exacerbates Vascular Dysfunction in Resistance Mesenteric Arteries in Gch1fl/flTie2cre Mice
We next investigated the effect of eNOS uncoupling on vasomotor function in response to chronic Ang II infusion in isolated second-order resistance mesenteric arteries. In saline-treated mice, vasoconstrictions to the thromboxane A2 receptor mimetic, U46619, were significantly enhanced in Gch1fl/flTie2cre mesenteric arteries compared with WT controls. Ang II infusion (0.4 mg/kg per day for 28 days) caused a further increase in vasoconstrictions to U46619 in mesenteric arteries from Gch1fl/flTie2cre mice compared with saline-treated Gch1fl/flTie2cre mice (Figure 3A). In contrast, Ang II infusion had no significant effect on vasoconstriction to U46619 in WT mice. A significant increase in maximum vasoconstrictions to U46619 (1 µmol/L) was observed at baseline in saline-treated Gch1fl/flTie2cre mice and was further augmented after Ang II infusion (5.9±0.3 versus 9.7±0.9 mN; P<0.05) with no difference observed in WT mice infused with Ang II (3.7±0.4 versus 4.4±0.7 mN; Figure 3B).
Figure 3.
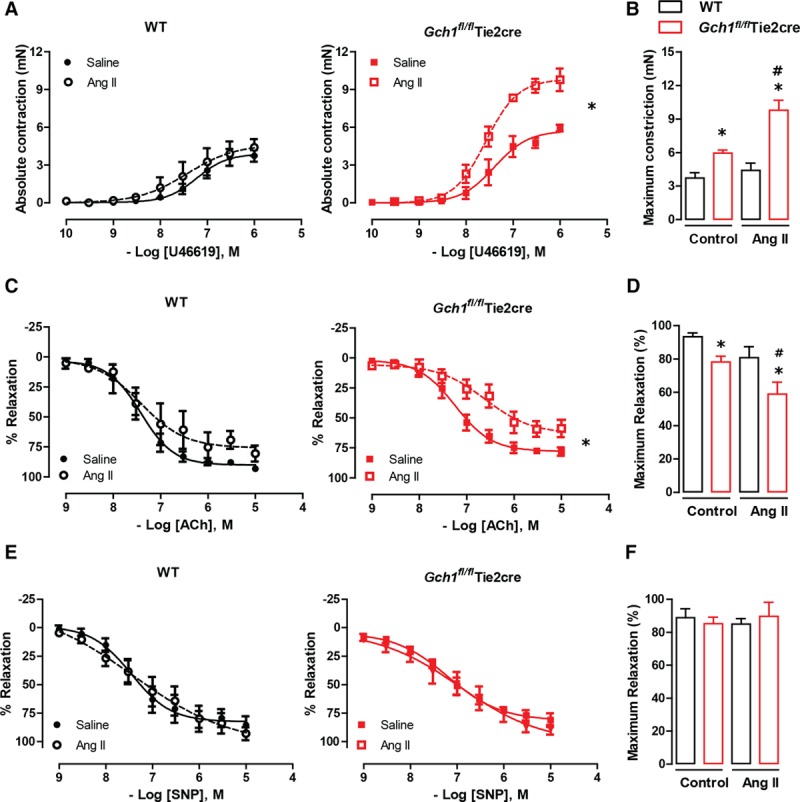
Impaired vasoconstrictions and endothelium-dependent vasodilatations in Gch1fl/flTie2cre resistance mesenteric arteries in response to Ang (angiotensin) II stimulation. Isometric tension study of second-order resistance mesenteric arteries from wild-type (WT) and Gch1fl/flTie2cre mice with either Ang II or saline infusion for 28 d. A, Vasoconstrictions in response to vasoconstrictor U46619 in mesenteric arteries. B, Maximum contraction (mN) in response to 1 µmol/L U46619. C, Endothelium-dependent vasodilatations in response to acetylcholine (ACh) in mesenteric arteries. D, Maximum relaxation (%) in response to 10 µmol/L ACh. E, Endothelium-independent vasodilatations in response to sodium nitroprusside (SNP) in mesenteric arteries. F, Maximum relaxation (%) in response to 10 µmol/L SNP (*P<0.05 comparing genotype; #P<0.05 comparing treatment; n=4–6 animals per group).
In saline-treated mice, a significant blunting of endothelial-dependent vasodilation was observed in mesenteric arteries from Gc11fl/flTie2cre mice (Figure 3C). This difference was even more pronounced after Ang II infusion, with a further impairment in endothelial-dependent relaxation observed in Gch1fl/flTie2cre mice. This was highlighted by the significant decrease in the maximal dilation response observed in mesenteric arteries from Gch1fl/flTie2cre mice after Ang II infusion (78.1±3.4% versus 58.9±7.2%; P<0.05; Figure 3D). In contrast, Ang II had no significant effect on endothelium-dependent vasodilatation in WT mice, with no significant difference observed in maximum relaxation after Ang II treatment (93.3±2.2% versus 80.7±6.6%; Figure 3C and 3D).
Furthermore, there were no difference in endothelium-independent vasodilatation between WT and Gch1fl/flTie2cre mice in either the saline or Ang II group, suggesting the impairment of vasodilatation to acetylcholine observed in Gch1fl/flTie2cre mesenteric arteries was not because of an alteration in vascular smooth muscle sensitivity to exogenous NO (Figure 3E and 3F).
eNOS Uncoupling Leads to Vascular Remodeling in Gch1fl/flTie2cre Resistance Mesenteric Arteries in Response to Ang II Infusion
Given the marked difference in constrictor response between the 2 groups after Ang II treatment and the role of vascular ROS production in vascular remodeling and hypertrophy, we next assessed medial changes in resistance arteries. Second-order mesenteric arteries from Gch1fl/flTie2cre mice and WT mice were perfusion fixed at 100 mm Hg after 28 days of saline or Ang II infusion. Ang II caused a marked increase in medial thickness and medial area in mesenteric arteries from Gch1fl/flTie2cre mice (Figure 4A through 4D). In contrast, no difference in either medial area of thickness was observed in mesenteric arteries from WT mice. In saline-infused mice, there was no significant difference in medial thickness and medial area between mesenteric arteries of WT and Gch1fl/flTie2cre mice (Figure 4A through 4D). Taken together, these results suggest that selective deficiency in endothelial cell BH4 biosynthesis leads to vascular dysfunction and medial remodeling in resistance arteries in response to chronic Ang II infusion.
Figure 4.
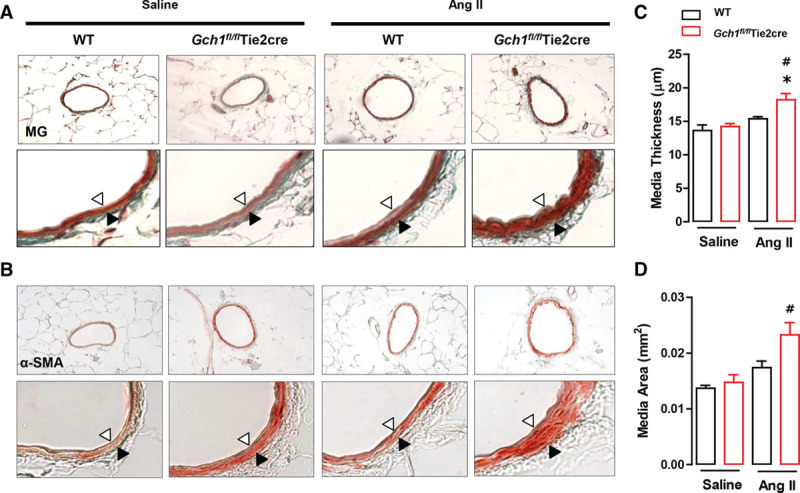
Exacerbated resistance mesenteric artery remodeling in Gch1fl/flTie2cre mice in response to Ang (angiotensin) II infusion. Vascular remodeling was analyzed in embed sections of the second-order branch of mesenteric arteries (perfusion fixed at 100 mm Hg) from wild-type (WT) and Gch1fl/flTie2cre mice with either Ang II or saline infusion (0.4 mg/kg/ per day, 28 d). Representative images are shown (A) Masson–Goldner (MG) staining and (B) α-smooth muscle actin (α-SMA) staining of mesenteric arteries. Vascular remodeling was evaluated by (C) media thickness and (D) media area. Opened arrow indicates internal elastic lamina and closed arrow indicates external elastic lamina. (*P<0.05 comparing genotype; #P<0.05 comparing treatment; n=4–6 animals per group).
Increased AAA Formation in Endothelial Cell BH4 Deficient Mice After Ang II Infusion
Having demonstrated increased vascular ROS and increased sensitivity in blood pressure and resistance artery function in response to Ang II, we next aimed to establish the consequence of endothelial cell BH4 deficiency on conduit artery remodeling. We infused WT and Gch1fl/flTie2cre mice with a subpressor dose (0.4 mg/kg per day) of Ang II by osmotic minipump for 28 days. The lumen diameter of abdominal aortas was assessed in mice before and at the end of 28 days of Ang II infusion, using ultrasound. There was no significant difference in the diameter of the abdominal aortas between WT and Gch1fl/flTie2cre mice at baseline (day 0; Figure 5A). However, after 28 days of Ang II infusion, the lumen diameter of the abdominal aorta in Gch1fl/flTie2cre mice was significantly increased compared with WT controls (Figure 5A and 5C). In saline-infused mice, there was no significant difference in the lumen diameter of the abdominal aortas between WT and Gch1fl/flTie2cre mice either at day 0 or day 28 (Figure 5B).
Figure 5.
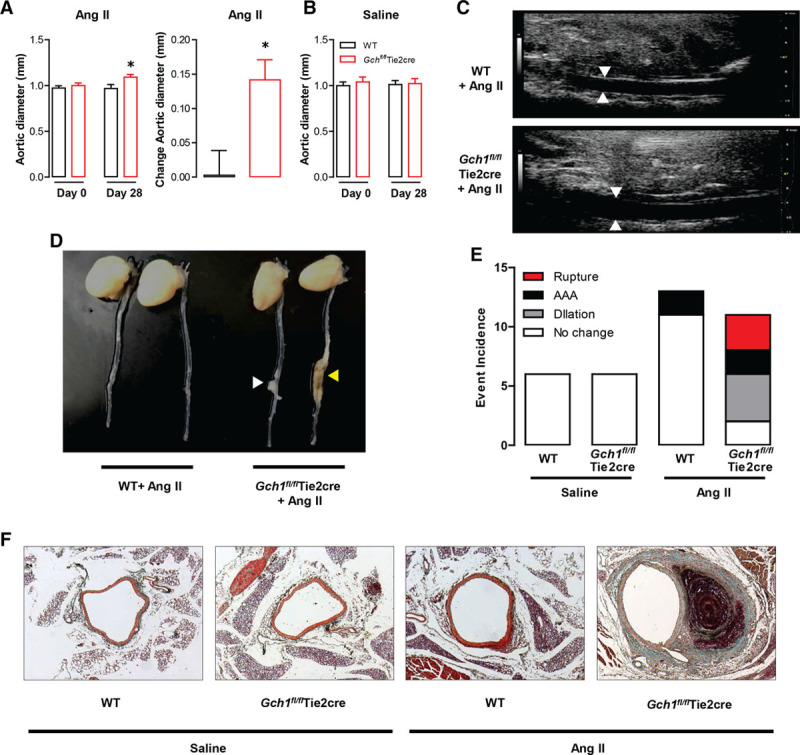
Ang (angiotensin) II–induced vascular pathology of abdominal aortic aneurysm (AAA) in Gch1fl/flTie2cre mice. Wild-type (WT) and Gch1fl/flTie2cre mice were treated with a subpressor dose (0.4 mg/kg per day) of Ang II by osmotic minipump for 28 d. The lumen diameter of abdominal aortas was assessed in mice before and at the end of 28 d of Ang II infusion by ultrasound. A, In Ang II–infused mice, there was no significant difference in the diameter of abdominal aortas between WT and Gch1fl/flTie2cre mice at day 0. After 28 d Ang II infusion, the lumen diameter of abdominal aorta of Gch1fl/flTie2cre mice was significantly increased compared with WT controls. Ang II infusion caused a greater increase in aortic lumen diameter (expressed as changed in lumen diameter) in Gch1fl/flTie2cre mice compared with WT mice (*P<0.05 comparing genotype; n=8–9 per group). B, In saline-infused mice, there was no significantly difference in the lumen diameter of abdominal aortas between WT and Gch1fl/flTie2cre mice either at day 0 or day 28 of saline infusion (n=5–6 per group). C, Representative of aortic lumen diameter by ultrasound after Ang II infusion in WT and Gch1fl/flTie2cre mice. D, Representative appearance of aortic dilation (opened arrow) and abdominal aortic aneurysm formation (yellow arrow). E, Event incidence of AAA pathology: dilation without AAA, AAA formation, and AAA rupture with sudden death in Ang II–infused Gch1fl/flTie2cre and WT mice. F, Representative images of Masson–Goldner staining of abdominal aortic sections from WT and Gch1fl/flTie2cre mice after saline or Ang II infusion for 28 d.
During 28 days of Ang II infusion, 3 out of 11 Gch1fl/flTie2cre mice died because of aortic rupture. In contrast, no WT mice died during the study period. Among the survivors, 75% (6 out of 8) of Ang II–infused Gch1fl/flTie2cre mice had classical features of AAA: aortic dilatation (50%; 4 out of 8 mice) and AAA formation (25%; 2 out of 8 mice; Figure 5D and 5E). In contrast, only 2 out of 13 WT mice showed the pathology of AAA formation. Importantly, none of the saline-infused WT or Gch1fl/flTie2cre mice died or developed any features of AAA pathology (Figure 5E). Histological sections of suprarenal aortas stained with Masson–Goldner demonstrated aortic dilatation and AAA formation in the suprarenal aorta after 28 days of Ang II infusion in Gch1fl/flTie2cre mice (Figure 5F).
As Tie2cre can lead to cre-mediated gene deletion in hematopoietic cells, we next assessed whether Ang II stimulation affects GTPCH/BH4 biosynthesis in macrophages. As expected, at baseline GTPCH protein was barely detectable in macrophages from Gch1fl/flTie2cre mice compared with WT controls (Figure S3A and S3B). This was accompanied by a marked reduction in BH4 and total biopterins in Gch1fl/flTie2cre macrophages compared with WT controls (Figure S3C). In addition, iNOS protein was undetectable in either WT or Gch1fl/flTie2cre macrophages. Ang II stimulation at 1 μmol\L for 16 hours did not alter GTPCH protein, BH4, or total biopterins levels in either WT or Gch1fl/flTie2cre macrophages compared with the control untreated group. Unlike lipopolysaccharide and IFNγ (interferon γ) stimulation, Ang II did not cause induction of iNOS protein either in WT or Gch1fl/flTie2cre macrophages.
Discussion
In this study, we used a mouse model of endothelial cell-targeted Gch1 deletion to test the requirement for endothelial cell BH4 in modulating the pathological vascular changes associated with Ang II stimulation. The major findings of this study are (1) loss of endothelial cell BH4 is sufficient to increase the sensitivity to Ang II–mediated hemodynamic responses; (2) Ang II infusion exacerbates eNOS uncoupling, increasing aortic eNOS-derived H2O2 production in Gch1fl/flTie2cre mice; (3) endothelial cell-specific Gch1 deficiency causes medial hypertrophy and dysfunction in resistance arteries in response to chronic Ang II infusion; and (4) chronic Ang II infusion leads to pathological vascular remodeling with a greatly increased incidence of AAA in Gch1fl/flTie2cre mice. Collectively, our studies demonstrate a specific role for endothelial cell BH4 in the pathogenesis of Ang II–mediated vascular hypertrophy and dysfunction, hypertension, and the development of AAA.
It is well-established that Ang II promotes hypertension through increased oxidative stress, vascular remodeling, and endothelial dysfunction.18,19 Increased levels of Ang II and angiotensin-converting enzyme, because of the DD genotype, are associated with hypertension and increased cardiovascular disease, respectively.20 Recently, the important role for vascular ROS signaling in the development of vascular remodeling and hypertension has been demonstrated in mice deficient in eNOS and NADPH oxidase.21,22 In our current study, we have also shown for the first time that endothelial cell BH4 deficiency and the associated changes in ROS and eNOS coupling are alone sufficient to increase the sensitivity of the acute pressor response to Ang II. We have shown that even low doses of Ang II, that are insufficient to increase blood pressure in wild-type mice, cause pressor responses in Gch1fl/flTie2cre mice. Furthermore, this increased sensitivity to Ang II, conferred by endothelial cell-specific BH4 deficiency, is sufficient to induce both functional and structural abnormalities in both resistance and conduit arteries.
To assess the physiological importance of endothelial cell BH4, we utilized a chronic model of Ang II infusion. It has been reported that chronic administration of a subpressor dose of Ang II mimics the development of human hypertension to a greater extent than the pressor dose.23 In this study, we, therefore, used the subpressor dose of Ang II (0.4 mg/kg per day) to investigate the pathological role of reduced endothelial cell Gch1 expression in the development of Ang II–induced hypertension. We found that a chronic subpressor infusion of Ang II resulted in a significant increase in blood pressure only in Gch1fl/flTie2cre mice. A growing body of evidence has suggested that ROS and NO play a critical role in the regulation of AT1R (angiotensin 1 receptor) signaling. Indeed, endogenous ROS increases AT1R density in rat cardiac fibroblasts,24 whereas NO decreases Ang II signaling by downregulating AT1R density through S-nitrosylation in the heart.24,25 It is possible that the alteration in NO/ROS signaling in Gch1fl/flTie2cre mice leads to increased Ang II sensitivity because of alteration in receptor density. In addition, it is possible that the increased baseline arterial ROS production observed in Gch1fl/flTie2cre mice primes the vasculature for an enhanced Ang II response. Indeed, it has been reported that vascular Nox expression, activity, and the associated ROS generation are increased in resistance arteries from human hypertensives, which is associated with increased sensitivity to Ang II stimulation.26,27 The results of this study indicate a critical role of endothelial cell BH4 in the sensitivity, and hence the resultant hemodynamic response to Ang II.
Hypertension has been shown to be associated with the impaired vascular function, particularly in resistance arteries, the key site for arterial blood pressure regulation. Previous studies have demonstrated eNOS uncoupling in resistance vessels from DOCA (deoxycorticosterone acetate)-salt and Ang II–induced hypertensive mice.16,28 However, what is unclear from these studies is whether eNOS uncoupling is causative in the resulting vascular dysfunction or was part of the disease process. In our study, we show for the first time that endothelial cell BH4 deficiency is alone sufficient to cause Ang II–mediated vascular dysfunction. We observed a further enhancement of the vasoconstrictor response and significantly impaired endothelium-dependent vasodilatation in resistance mesenteric arteries from Ang II–infused Gch1fl/flTie2cre mice. This is in contrast to wild-type mice, where no difference in the constrictor or dilator responses were observed with Ang II infusion. Previous studies have demonstrated that increased level of superoxide and H2O2 enhanced Ang II–stimulated redox signaling in resistance arteries from hypertensive patients.26 The combination of increased eNOS-derived ROS and reduced eNOS-derived NO production is likely to be the major contributor toward the development of vascular dysfunction, and hence hypertension observed in Ang II–treated Gch1fl/flTie2cre mice and contrasts with eNOS knockout mice where all functions of eNOS (ie, both NO and ROS generation) are deleted.
It has been demonstrated that vascular H2O2 plays a crucial role in smooth muscle growth and hypertrophy. Chronic Ang II infusion increases vascular superoxide and H2O2-derived NADPH oxidase, subsequently leading to vascular remodeling.12–14 In addition, treatment with either an Ang II receptor antagonist or an angiotensin-converting enzyme inhibitor reduced ROS generation and vascular hypertrophy in spontaneously hypertensive rats.29 Our studies now highlight the role for increased ROS generation specifically from endothelial cells, because of eNOS uncoupling, in the pathogenesis of Ang II–induced vascular remodeling. Ang II infusion at a subpressor dose caused a marked increase in mesenteric resistance artery medial thickness and area (typical feature of hypertrophic remodeling) in Gch1fl/flTie2cre mice, but no change in WT controls. It is possible that the increased medial area was in part because of increased pressor response to Ang II. However, it is unlikely that the increased blood pressure alone was responsible for the medial remodeling. Several in vivo studies have demonstrated that noradrenaline infusion, at concentrations producing a similar degree of hypertension to Ang II infusion, does not cause vascular hypertrophy.30 In endothelial cell-specific BH4 deficient mice, it is likely that increased endothelial cell ROS/H2O2 production, because of eNOS uncoupling, activates and primes the underlying VSMC, which in turn promote vascular hypertrophy in response to Ang II. Consistent with this notion, administration of polyethylene glycol catalase or overexpression of catalase in VSMC has been reported to reduce vascular H2O2 production and subsequently vascular hypertrophy in response to chronic Ang II stimulation.31
Important roles for eNOS uncoupling and BH4 deficiency in AAA formation have been recently reported in hph-1 mice, which have a moderate systemic BH4 deficiency because of a global reduction in Gch1 expression. Hph-1 mice treated with Ang II at 0.7 mg/kg per day, show an increased eNOS uncoupling and AAA formation.5 However, the hph-1 mouse has a global BH4 deficiency, affecting all cells and tissues, so is not possible to dissect out how BH4 deficiency mediates the increase in vascular pathology. Notably, in the present studies, Ang II infusion even at a subpressor dose (0.4 mg/kg per day) greatly increased the incidence of AAA in Gch1fl/flTie2cre mice, associated with eNOS uncoupling and increased aortic eNOS-derived H2O2 generation. Vascular H2O2 has been previously implicated in the pathology of AAA in various mouse models of AAA, including the Ang II, elastase, and CaCl2 model.16,32,33 Indeed, H2O2 has been reported to promote MMP-3 (matrix metalloproteinase-3), MMP-9, and MMP-12 expression, which are implicated in extracellular matrix degradation that is important in the pathogenesis of AAA.34 Administration of polyethylene glycol catalase or overexpression of catalase in VSMC has been reported to reduce the formation of AAA by enhancing VSMC survival and decreasing MMP activity.33 We have previously shown that increased endothelial cell superoxide by Nox-2 overexpression is sufficient to predispose mice to AAA formation.22 Taken together, these data strongly indicate that increased endothelial cell H2O2 production because of eNOS uncoupling is sufficient to increase AAA formation.
Ang II can promote inflammatory cell recruitment into the vascular wall and in particular to the abdominal aorta region.35 In the present study, we observed no difference in aortic protein levels of either iNOS or GTPCH between Ang II–treated mice and the saline controls after 28 days of Ang II infusion. These findings suggest that Ang II is unlikely to play a role in modulating aortic Gch1/BH4 biosynthesis or iNOS protein levels. In addition, Ang II stimulation of bone marrow-derived macrophages did not alter BH4 levels, GTPCH protein, or iNOS protein expression. Hence, the lack of Gch1 in myeloid cells is unlikely to be the cause of the increase in AAA formation in Gch1fl/flTie2cre mice; however, additional mechanistic studies would be required to confirm this.
Despite an increased aortic eNOS-derived H2O2 production in Gch1fl/flTie2ce mice, the levels of BH4, total biopterins, and BH4/BH2 ratio were unaffected by Ang II stimulation in Gch1fl/flTie2cre aortas. It is likely that other mechanisms, independent of endothelial cell BH4 levels, may contribute to further exacerbate eNOS uncoupling in response to Ang II stimulation. Indeed, several reports have demonstrated that Ang II can lead to eNOS uncoupling by eNOS-glutathionylation, independent of BH4 levels, in several experimental models of hypertension.36,37 In addition, it is possible that the increased blood pressure in response to Ang II may contribute to the development of AAA pathology in Gch1fl/flTie2cre mice. However, increased blood pressure is unlikely to be the cause of AAA formation in response to Ang II infusion5,22 because numerous studies have shown that blood pressure matching with noradrenaline does not induce AAA formation.22,38
A previous study reported that Ang II infusion at 0.7 mg/kg per day for 14 days caused a decrease in aortic BH4 levels because of a reduction in endothelial cell DHFR protein.5 However, consistent with our findings, Kossmann et al39 demonstrated that Ang II infusion at 1 mg/kg per day for 7 days increased aortic GTPCH protein and BH4 levels. The authors hypothesized that this increase was because of increased inflammatory cell influx as iNOS protein was detectable in the vascular wall after Ang II infusion. In contrast, we found no detectable iNOS protein in aortic tissues from Ang II-infused mice, suggesting that at the later time point inflammatory cells are either no longer abundant or are not expressing iNOS. Interestingly, we found a significant increase in aortic DHFR protein levels in Ang II–infused mice compared with saline-infused controls. This finding indicates that the increase in aortic BH4 and total biopterin content may be because of increased DHFR-mediated recycling of BH2 to BH4, rather than the presence of proinflammatory inflammatory cells. The increase in aortic BH4 in response to Ang II may represent a vascular protective mechanism. This result is consistent with the finding of an enhanced pathological response to Ang II in Gch1fl/flTie2cre mice, which are unable to upregulate aortic BH4 content in response to Ang II by either the de novo or salvage pathway. This increased aortic BH4 content may act as a protective measurement to compensate for the increase vascular oxidative stress in response to Ang II.
Interestingly, Ang II had no significant effect on plasma BH4 levels in WT mice, whereas plasma BH4 levels were greatly decreased in Ang II–infused Gch1fl/flTie2cre mice, indicating that under these pathological conditions endothelial cells make a significant contribution to circulating BH4 levels. Thus, circulating BH4 levels may be a useful indicator of AAA. Consistent with this finding, Gao et al5 demonstrated that decreased circulating BH4 is correlated with reduced vascular BH4 levels, which is associated with AAA formation in hph-1 mice. It would be interesting to determine if supplementation with BH4 could reverse the pathological changes observed in the current study. However, it has been challenging to achieve arterial BH4 supplementation because of the highly oxidative nature of BH4. Hence, alternative treatment strategies such as 5-methyl-tetrahydrofolate or folate, which have been shown to increase vascular BH4 levels in patients undergoing coronary artery bypass grafts, may represent a viable alternative.40
In summary, we describe for the first time a specific requirement for endothelial cell BH4 in modulating the hemodynamic and structural changes induced by Ang II, through modulation of blood pressure, structural changes in resistance vessels, and aneurysm formation in the aorta. This study demonstrates a specific role for endothelial cell BH4 deficiency/eNOS uncoupling in the pathogenesis of Ang II driven vascular disease.
Perspectives
Selective targeting endothelial cell Gch1 and BH4 biosynthesis under pathological conditions may provide a novel therapeutic target for the treatment of vascular disease.
Sources of Funding
This study was supported by a British Heart Foundation Program Grant (RG/12/5/29576), Chair award (CH/16/1/32013), Oxford British Heart Foundation Centre of Research Excellence (RE/13/1/30181), Wellcome Trust (090532/Z/09/Z), and the National Institute for Health Research Oxford Biomedical Research Centre.
Disclosures
None.
Supplementary Material
Footnotes
The online-only Data Supplement is available with this article at http://hyper.ahajournals.org/lookup/suppl/doi:10.1161/HYPERTENSIONAHA.118.11144/-/DC1.
Novelty and Significance
What Is New?
This is the first study to show that endothelial cell tetrahydrobiopterin deficiency is alone sufficient to cause pathophysiological changes in large and small vessels in response to the pathophysiological stimulus Ang (angiotensin) II.
What Is Relevant?
Targeting endothelial cell Gch1 and tetrahydrobiopterin biosynthesis under pathological conditions may provide a novel therapeutic target for the treatment of Ang II driven vascular disease.
Summary
Endothelial NO synthase uncoupling, because of selective deficiency in endothelial cell tetrahydrobiopterin biosynthesis, leads to vascular remodeling and vascular dysfunction in resistance vessels, hypertension, and an increased incidence of aneurysm formation in response to a subpressor does of Ang II. These findings indicate a specific requirement for endothelial cell tetrahydrobiopterin in modulating the hemodynamic and structural changes induced by Ang II, through modulation of blood pressure, structural changes in resistance vessels, and aneurysm formation in the aorta.
References
- 1.Vásquez-Vivar J, Martásek P, Whitsett J, Joseph J, Kalyanaraman B. The ratio between tetrahydrobiopterin and oxidized tetrahydrobiopterin analogues controls superoxide release from endothelial nitric oxide synthase: an EPR spin trapping study. Biochem J. 2002;362(pt 3):733–739. doi: 10.1042/0264-6021:3620733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crabtree MJ, Tatham AL, Al-Wakeel Y, Warrick N, Hale AB, Cai S, Channon KM, Alp NJ. Quantitative regulation of intracellular endothelial nitric-oxide synthase (eNOS) coupling by both tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status: insights from cells with tet-regulated GTP cyclohydrolase I expression. J Biol Chem. 2009;284:1136–1144. doi: 10.1074/jbc.M805403200. doi: 10.1074/jbc.M805403200. [DOI] [PubMed] [Google Scholar]
- 3.Heitzer T, Krohn K, Albers S, Meinertz T. Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with type II diabetes mellitus. Diabetologia. 2000;43:1435–1438. doi: 10.1007/s001250051551. doi: 10.1007/s001250051551. [DOI] [PubMed] [Google Scholar]
- 4.Higashi Y, Sasaki S, Nakagawa K, Fukuda Y, Matsuura H, Oshima T, Chayama K. Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am J Hypertens. 2002;15(4 pt 1):326–332. doi: 10.1016/s0895-7061(01)02317-2. [DOI] [PubMed] [Google Scholar]
- 5.Gao L, Siu KL, Chalupsky K, Nguyen A, Chen P, Weintraub NL, Galis Z, Cai H. Role of uncoupled endothelial nitric oxide synthase in abdominal aortic aneurysm formation: treatment with folic acid. Hypertension. 2012;59:158–166. doi: 10.1161/HYPERTENSIONAHA.111.181644. doi: 10.1161/HYPERTENSIONAHA.111.181644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cosentino F, Barker JE, Brand MP, Heales SJ, Werner ER, Tippins JR, West N, Channon KM, Volpe M, Lüscher TF. Reactive oxygen species mediate endothelium-dependent relaxations in tetrahydrobiopterin-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:496–502. doi: 10.1161/01.atv.21.4.496. [DOI] [PubMed] [Google Scholar]
- 7.Khoo JP, Zhao L, Alp NJ, Bendall JK, Nicoli T, Rockett K, Wilkins MR, Channon KM. A pivotal role for tetrahydrobiopterin in pulmonary hypertension. Circulation. 2005;111:2126–2133. doi: 10.1161/01.CIR.0000162470.26840.89. [DOI] [PubMed] [Google Scholar]
- 8.Chuaiphichai S, McNeill E, Douglas G, Crabtree MJ, Bendall JK, Hale AB, Alp NJ, Channon KM. Cell-autonomous role of endothelial GTP cyclohydrolase 1 and tetrahydrobiopterin in blood pressure regulation. Hypertension. 2014;64:530–540. doi: 10.1161/HYPERTENSIONAHA.114.03089. doi: 10.1161/HYPERTENSIONAHA.114.03089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chuaiphichai S, Crabtree MJ, Mcneill E, Hale AB, Trelfa L, Channon KM, Douglas G. A key role for tetrahydrobiopterin-dependent endothelial NOS regulation in resistance arteries: studies in endothelial cell tetrahydrobiopterin-deficient mice. Br J Pharmacol. 2017;174:657–671. doi: 10.1111/bph.13728. doi: 10.1111/bph.13728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stegbauer J, Coffman TM. New insights into angiotensin receptor actions: from blood pressure to aging. Curr Opin Nephrol Hypertens. 2011;20:84–88. doi: 10.1097/MNH.0b013e3283414d40. doi: 10.1097/MNH.0b013e3283414d40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bendall JK, Rinze R, Adlam D, Tatham AL, de Bono J, Wilson N, Volpi E, Channon KM. Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: studies in endothelial-targeted Nox2 transgenic mice. Circ Res. 2007;100:1016–1025. doi: 10.1161/01.RES.0000263381.83835.7b. [DOI] [PubMed] [Google Scholar]
- 12.Dikalova A, Clempus R, Lassègue B, Cheng G, McCoy J, Dikalov S, San Martin A, Lyle A, Weber DS, Weiss D, Taylor WR, Schmidt HH, Owens GK, Lambeth JD, Griendling KK. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–2676. doi: 10.1161/CIRCULATIONAHA.105.538934. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 13.Dikalova A, Sutcliffe D, Dikalov S, Lassegue B, Weber D, Rocic P, Cheng G, Lambeth D, Owens G, Griendling K. Nox1 overexpression in mice enhances Ang II-induced superoxide production and hypertension. Circulation. 2003;108:45–46. [Google Scholar]
- 14.Weber DS, Rocic P, Mellis AM, Laude K, Lyle AN, Harrison DG, Griendling KK. Angiotensin II-induced hypertrophy is potentiated in mice overexpressing p22phox in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2005;288:H37–H42. doi: 10.1152/ajpheart.00638.2004. doi: 10.1152/ajpheart.00638.2004. [DOI] [PubMed] [Google Scholar]
- 15.Oak JH, Cai H. Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes. 2007;56:118–126. doi: 10.2337/db06-0288. doi: 10.2337/db06-0288. [DOI] [PubMed] [Google Scholar]
- 16.Chalupsky K, Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 2005;102:9056–9061. doi: 10.1073/pnas.0409594102. doi: 10.1073/pnas.0409594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chuaiphichai S, Starr A, Nandi M, Channon KM, McNeill E. Endothelial cell tetrahydrobiopterin deficiency attenuates LPS-induced vascular dysfunction and hypotension. Vascul Pharmacol. 2016;77:69–79. doi: 10.1016/j.vph.2015.08.009. doi: 10.1016/j.vph.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Virdis A, Neves MF, Amiri F, Touyz RM, Schiffrin EL. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J Hypertens. 2004;22:535–542. doi: 10.1097/00004872-200403000-00016. [DOI] [PubMed] [Google Scholar]
- 19.Kimura S, Honda M, Tanabe M, Ono H. Noxious stimuli evoke a biphasic flexor reflex composed of A delta-fiber-mediated short-latency and C-fiber-mediated long-latency withdrawal movements in mice. J Pharmacol Sci. 2004;96:502–502. doi: 10.1254/jphs.95.94. [DOI] [PubMed] [Google Scholar]
- 20.Samani NJ, Thompson JR, O’Toole L, Channer K, Woods KL. A meta-analysis of the association of the deletion allele of the angiotensin-converting enzyme gene with myocardial infarction. Circulation. 1996;94:708–712. doi: 10.1161/01.cir.94.4.708. [DOI] [PubMed] [Google Scholar]
- 21.Thieme M, Sivritas SH, Mergia E, Potthoff SA, Yang G, Hering L, Grave K, Hoch H, Rump LC, Stegbauer J. Phosphodiesterase 5 inhibition ameliorates angiotensin II-dependent hypertension and renal vascular dysfunction. Am J Physiol Renal Physiol. 2017;312:F474–F481. doi: 10.1152/ajprenal.00376.2016. doi: 10.1152/ajprenal.00376.2016. [DOI] [PubMed] [Google Scholar]
- 22.Fan LM, Douglas G, Bendall JK, McNeill E, Crabtree MJ, Hale AB, Mai A, Li JM, McAteer MA, Schneider JE, Choudhury RP, Channon KM. Endothelial cell-specific reactive oxygen species production increases susceptibility to aortic dissection. Circulation. 2014;129:2661–2672. doi: 10.1161/CIRCULATIONAHA.113.005062. doi: 10.1161/CIRCULATIONAHA.113.005062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simon G, Abraham G, Cserep G. Pressor and subpressor angiotensin II administration. Two experimental models of hypertension. Am J Hypertens. 1995;8:645–650. doi: 10.1016/0895-7061(95)00047-S. doi: 10.1016/0895-7061(95)00047-S. [DOI] [PubMed] [Google Scholar]
- 24.Nishida M, Suda R, Nagamatsu Y, Tanabe S, Onohara N, Nakaya M, Kanaho Y, Shibata T, Uchida K, Sumimoto H, Sato Y, Kurose H. Pertussis toxin up-regulates angiotensin type 1 receptors through Toll-like receptor 4-mediated Rac activation. J Biol Chem. 2010;285:15268–15277. doi: 10.1074/jbc.M109.076232. doi: 10.1074/jbc.M109.076232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ichiki T, Usui M, Kato M, Funakoshi Y, Ito K, Egashira K, Takeshita A. Downregulation of angiotensin II type 1 receptor gene transcription by nitric oxide. Hypertension. 1998;31(1 pt 2):342–348. doi: 10.1161/01.hyp.31.1.342. [DOI] [PubMed] [Google Scholar]
- 26.Touyz RM, Yao G, Quinn MT, Pagano PJ, Schiffrin EL. p47phox associates with the cytoskeleton through cortactin in human vascular smooth muscle cells: role in NAD(P)H oxidase regulation by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25:512–518. doi: 10.1161/01.ATV.0000154141.66879.98. doi: 10.1161/01.ATV.0000154141.66879.98. [DOI] [PubMed] [Google Scholar]
- 27.Montezano AC, Dulak-Lis M, Tsiropoulou S, Harvey A, Briones AM, Touyz RM. Oxidative stress and human hypertension: vascular mechanisms, biomarkers, and novel therapies. Can J Cardiol. 2015;31:631–641. doi: 10.1016/j.cjca.2015.02.008. doi: 10.1016/j.cjca.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 28.Du YH, Guan YY, Alp NJ, Channon KM, Chen AF. Endothelium-specific GTP cyclohydrolase I overexpression attenuates blood pressure progression in salt-sensitive low-renin hypertension. Circulation. 2008;117:1045–1054. doi: 10.1161/CIRCULATIONAHA.107.748236. doi: 10.1161/CIRCULATIONAHA.107.748236. [DOI] [PubMed] [Google Scholar]
- 29.Yu H, Rakugi H, Higaki J, Morishita R, Mikami H, Ogihara T. The role of activated vascular angiotensin II generation in vascular hypertrophy in one-kidney, one clip hypertensive rats. J Hypertens. 1993;11:1347–1355. doi: 10.1097/00004872-199312000-00005. [DOI] [PubMed] [Google Scholar]
- 30.Black MJ, Bertram JF, Campbell JH, Campbell GR. Angiotensin II induces cardiovascular hypertrophy in perindopril-treated rats. J Hypertens. 1995;13:683–692. doi: 10.1097/00004872-199506000-00016. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Griendling KK, Dikalova A, Owens GK, Taylor WR. Vascular hypertrophy in angiotensin II-induced hypertension is mediated by vascular smooth muscle cell-derived H2O2. Hypertension. 2005;46:732–737. doi: 10.1161/01.HYP.0000182660.74266.6d. doi: 10.1161/01.HYP.0000182660.74266.6d. [DOI] [PubMed] [Google Scholar]
- 32.Grigoryants V, Hannawa KK, Pearce CG, Sinha I, Roelofs KJ, Ailawadi G, Deatrick KB, Woodrum DT, Cho BS, Henke PK, Stanley JC, Eagleton MJ, Upchurch GR. Tamoxifen up-regulates catalase production, inhibits vessel wall neutrophil infiltration, and attenuates development of experimental abdominal aortic aneurysms. J Vasc Surg. 2005;41:108–114. doi: 10.1016/j.jvs.2004.09.033. doi: 10.1016/j.jvs.2004.09.033. [DOI] [PubMed] [Google Scholar]
- 33.Parastatidis I, Weiss D, Joseph G, Taylor WR. Overexpression of catalase in vascular smooth muscle cells prevents the formation of abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2013;33:2389–2396. doi: 10.1161/ATVBAHA.113.302175. doi: 10.1161/ATVBAHA.113.302175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emeto TI, Moxon JV, Au M, Golledge J. Oxidative stress and abdominal aortic aneurysm: potential treatment targets. Clin Sci (Lond) 2016;130:301–315. doi: 10.1042/CS20150547. doi: 10.1042/CS20150547. [DOI] [PubMed] [Google Scholar]
- 35.McNeill E, Iqbal AJ, White GE, Patel J, Greaves DR, Channon KM. Hydrodynamic gene delivery of CC chemokine binding Fc fusion proteins to target acute vascular inflammation in vivo. Sci Rep. 2015;5:17404. doi: 10.1038/srep17404. doi: 10.1038/srep17404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Galougahi KK, Liu CC, Gentile C, Kok C, Nunez A, Garcia A, Fry NA, Davies MJ, Hawkins CL, Rasmussen HH, Figtree GA. Glutathionylation mediates angiotensin II-induced eNOS uncoupling, amplifying NADPH oxidase-dependent endothelial dysfunction. J Am Heart Assoc. 2014;3:e000731. doi: 10.1161/JAHA.113.000731. doi: 10.1161/JAHA.113.000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468:1115–1118. doi: 10.1038/nature09599. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ayabe N, Babaev VR, Tang Y, Tanizawa T, Fogo AB, Linton MF, Ichikawa I, Fazio S, Kon V. Transiently heightened angiotensin II has distinct effects on atherosclerosis and aneurysm formation in hyperlipidemic mice. Atherosclerosis. 2006;184:312–321. doi: 10.1016/j.atherosclerosis.2005.05.016. doi: 10.1016/j.atherosclerosis.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 39.Kossmann S, Hu H, Steven S, et al. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J Biol Chem. 2014;289:27540–27550. doi: 10.1074/jbc.M114.604231. doi: 10.1074/jbc.M114.604231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antoniades C, Shirodaria C, Warrick N, Cai S, de Bono J, Lee J, Leeson P, Neubauer S, Ratnatunga C, Pillai R, Refsum H, Channon KM. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation. 2006;114:1193–1201. doi: 10.1161/CIRCULATIONAHA.106.612325. doi: 10.1161/CIRCULATIONAHA.106.612325. [DOI] [PubMed] [Google Scholar]