

Abstract
Effector proteins play crucial roles in plant-parasite interactions by suppressing plant defenses and hijacking plant physiological responses to facilitate parasite invasion and propagation. Although effector proteins have been characterized in many microbial plant pathogens, their nature and role in adaptation to host plants are largely unknown in insect herbivores. Aphids rely on salivary effector proteins injected into the host plants to promote phloem sap uptake. Therefore, gaining insight into the repertoire and evolution of aphid effectors is key to unveiling the mechanisms responsible for aphid virulence and host plant specialization. With this aim in mind, we assembled catalogues of putative effectors in the legume specialist aphid, Acyrthosiphon pisum, using transcriptomics and proteomics approaches. We identified 3,603 candidate effector genes predicted to be expressed in A. pisum salivary glands (SGs), and 740 of which displayed up-regulated expression in SGs in comparison to the alimentary tract. A search for orthologs in 17 arthropod genomes revealed that SG-up-regulated effector candidates of A. pisum are enriched in aphid-specific genes and tend to evolve faster compared with the whole gene set. We also found that a large fraction of proteins detected in the A. pisum saliva belonged to three gene families, of which certain members show evidence consistent with positive selection. Overall, this comprehensive analysis suggests that the large repertoire of effector candidates in A. pisum constitutes a source of novelties promoting plant adaptation to legumes.
Keywords: Acyrthosiphon pisum, salivary proteins, host adaptation, positive selection, pest evolution, plant defenses
Introduction
Insects comprise the most diverse group of metazoans, and evidence indicates that the evolution of herbivory has played a fundamental role in promoting their species richness and diversification (Wiens et al. 2015). Almost half of the currently known insect species feed on plants (Wu and Baldwin 2010), and herbivorous insect groups exhibit faster rates of diversification compared with nonherbivorous species (Wiens et al. 2015). A central hypothesis accounting for higher species richness in herbivorous insects proposes an evolutionary interaction between plant defense mechanisms and plant exploitation strategies of insects (Janz 2011). Furthermore, continuous interactions between host plants and herbivorous insects are predicted to make herbivore generalism difficult and constrain a given insect species to one or a few host species (Forister et al. 2015). Since plants provide not only food resources, but also habitats and mating sites to many herbivorous insects, plant specialization may induce divergent selection in insect populations at a range of traits that can lead to reproductive isolation and speciation (Peccoud et al. 2010; Mullen and Shaw 2014). Attempting to unveil the basic mechanisms of insect herbivory provides opportunities to understand the evolutionary and mechanistic basis of plant specialization by herbivorous insects, in particular, and the diversification of metazoan life, in general.
Aphids (Insecta: Aphidomorpha) are pests of wild and cultivated plants that directly reduce plant nutrients by ingesting phloem sap and indirectly cause diseases by transmitting plant pathogens (Blackman and Eastop 2000; Harris and Maramorosch 2014). Aphids are also excellent subjects for host specialization studies. Their clade is composed of approximately 5,000 species (Blackman and Eastop 2000), and most are considered to be plant specialists (Peccoud et al. 2010). Aphid mouthparts are modified into a rostrum or beak with the mandibles and maxillae forming needle-like stylets. Aphids secrete gelling saliva during the early stages of feeding to form a feeding sheath surrounding the stylets, and then secrete watery saliva into various plant cells (Moreno et al. 2011). Saliva contains effectors that modulate physiological responses to herbivory and permit feeding (Rodriguez et al. 2017). These effectors are likely exposed to natural selection, in particular by plant surveillance systems and defense mechanisms (Will et al. 2013). Interference with plant defenses through various mechanisms has been demonstrated for several effectors secreted by microbial plant pathogens, which ultimately promotes persistence and even spread of these pathogens (Varden et al. 2017). A subset of effectors, the so-called avirulence proteins, are detected by plant surveillance systems and trigger strong immunity in specific plants, determining the incompatibility (Bent and Mackey 2007).
Effector genes are diverse, making prediction of effector functions often difficult from the amino acid sequences. As a result, relatively few salivary effectors from aphids have characterized interactions with active host defense responses. In planta expression of salivary effectors C002, Mp1, and Mp2 from the green peach aphid, Myzus persicae, increases M. persicae fecundity on the host plants Arabidopsis thaliana and Nicotiana benthamiana, whereas expression of orthologous genes from another aphid species (the pea aphid, Acyrthosiphon pisum) in these plants has no effect on M. persicae growth (Pitino and Hogenhout 2013). This observation supports specialization of orthologous effectors to distinct plant species during aphid divergence. Some salivary proteins that are known to contribute to aphid plant exploitation are expressed in salivary glands (Wang et al. 2015a, 2015b) and a few showed sites under positive selection (Pitino and Hogenhout 2013; Thorpe et al. 2016). However, a global and comprehensive evolutionary analysis of aphid salivary genes has yet to be reported.
A catalogue of putative salivary effectors was created for A. pisum upon completion of the genome sequence (The International Aphid Genomics Consortium 2010). Combined transcriptomics and proteomics produced a catalog of 324 secreted proteins (Carolan et al. 2011). A small number of effectors predicted in this catalogue were functionally characterized and have been shown to be involved in plant-aphid interactions (Mutti et al. 2006, 2008; Wang et al. 2015a, 2015b). Significant developments in RNA-seq technology and high-resolution mass-spectrometry (MS) provide new impetus to revise the salivary gene catalogue and to define a new and expanded set of candidate salivary effector genes for further analyses. In addition, the genome sequences of two specialist aphids, the Russian wheat aphid, Diuraphis noxia (Nicholson et al. 2015) and the soybean aphid, Aphis glycines (Wenger et al. 2017), together with that of the generalist green peach aphid, M. persicae, (Mathers et al. 2017) have been recently published. These data offer the opportunity to better understand the evolutionary dynamics of salivary effector candidates, in particular, their suspected role in the adaptation of aphid lineages to their host plants (Pitino and Hogenhout 2013; Rodriguez et al. 2017). The critical effectors that drive aphid–plant interactions may display peculiar gene expression and evolutionary patterns, which we search by combining transcriptomics and proteomics in A. pisum and by conducting a comparative analysis of aphid genomes.
Materials and Methods
Aphids, Plants, and Growth Conditions
Acyrthosiphon pisum lineage LSR1 (used for whole-genome sequencing; The International Aphid Genomics Consortium 2010) was maintained in a growth chamber at 18 °C with a 16 h day/8 h night photoperiod on broad bean, Vicia faba (Castel cultivar), at low density to avoid the production of winged individuals. Vicia faba was grown in a growth chamber at 18 °C with a 16 h day/8 h night photoperiod for 10 days before installation of the aphids.
RNA Sequencing
To prepare RNA samples from aphid salivary glands and alimentary tracts, 9-day-old individuals reared at a density of 10–15 aphids per V. faba plant were rapidly dissected with fine forceps in saline solution. The dissected organs were soaked in RNA later (QIAGEN) immediately after dissection to avoid RNA degradation. The dissected tissues were pooled in several batches, and RNA was extracted by NucleoSpin RNA XS (Macherey-Nagel) and quantified. On average, RNA samples from 200 pairs of salivary glands or 20 alimentary tracts that were dissected on the same day were pooled for one replicate of RNA-seq experiment. Four replicates were prepared by 4 days of dissection with 5–6 persons.
rRNA depletion, single stranded-RNA library preparation, multiplexing, and sequencing were performed by Genewiz (New Jersey, USA). Sequencing was performed on the Illumina HiSeq2500 platform, in a 2 × 125 bp paired-end (PE) configuration in High Output mode (V4 chemistry). Each sample was sequenced on four different flowcell lanes to avoid lane effect. In total, 471,933,074 reads were obtained for eight samples. Raw data is available in NCBI Sequence Read Archive (https://trace.ncbi.nlm.nih.gov/Traces/sra/; last accessed May 22, 2018) with reference number SRP14110.
Mapping and Differential Expression Analysis
Gene expression of A. pisum salivary glands and alimentary tracts was analyzed using the Acyr_2.0 (GCF_000142985.2) reference genome assembly and the NCBI Acyrthosiphon pisum Annotation Release 102, both available at ftp://ftp.ncbi.nlm.nih.gov/genomes, last accessed May 22, 2018. The paired-end libraries were mapped on the reference genome using STAR v2.5.2 (Dobin et al. 2013) with the following parameters: outFilterMultimapNmax = 5, outFilterMismatchNmax = 3, alignIntronMin = 10, alignIntronMax = 50,000, alignMatesGapMax = 50,000. Fragment counts per genes were estimated by Subread featureCounts (Liao et al. 2014) using default parameters. Differential expression analysis between SGs and AT was then conducted following the workflow proposed by Law et al. (2016). The raw fragment counts were converted to counts per million (CPM) using the edgeR (Robinson et al. 2010) R-implemented package (R-Core Team 2017). Expressed genes were filtered based on a CPM > 1 in at least three libraries among the eight analyzed libraries and CPMs were normalized by the edgeR TMM method for Normalization Factor calculation (Robinson and Oshlack 2010). The mean–variance relationship of the log-CPM was estimated by the voom function (Law et al. 2014) and incorporated in the empirical Bayes analysis from the limma package (Ritchie et al. 2015) to fit linear models and compare SG vs. AT tissues. Validation of the described differential expression analysis is presented in supplementary fig. S1, Supplementary Material online. In addition, normalized fragments per kilobase million (FPKM) were calculated with edgeR on the four salivary glands samples and the four alimentary tracts samples separately to obtain whole transcriptomes of each organ. Genes with FPKM > 0.5 in at least three libraries per tissue were considered as expressed.
Saliva and Salivary Gland Proteomics
Saliva and Salivary Gland Collection
LSR1 aphids of mixed ages were reared on V. faba and approximately 2,000 aphids were installed on 12 perspex rings (radius 4.5 cm, height 5 cm), each containing 5 ml of a chemically defined diet formulation AP3 (Febvay et al. 1988) held between two stretched sheets of ParafilmTM. The aphids were reared at 18 °C with 16 h day/8 h night photoperiod and the diets were collected and replaced every 24 h. New V. faba-reared aphids were added to the diets to maintain aphid numbers. The daily collected diets were pooled and stored at −80 °C for later use. Three independent replicates were produced by pooling the collected diet from three daily collections (approximately 150 ml). Pooled diets were concentrated at 4 °C in a Vivacell 250 Pressure Concentrator using a 5,000 molecular weight cut-off (MWCO) polyethersulfone (PES) membrane. The volume of concentrate was further reduced by centrifugation at 3,400 × g in a Vivaspin 6 with a 5000 MWCO. Proteins from this final concentrate (300 µl) were purified using a 2D Clean-up Kit (GE HealthCare) following the manufacturer’s instructions and the resulting protein pellet was suspended in 6 M urea, 2 M thiourea, 0.1 M Tris–HCl, pH 8.0, and quantified using the Qubit™ protein quantification system (Invitrogen). Ten micrograms were removed from each sample for protein digestion.
Adult LSR1 aphids (14–16-days old, reared on V. faba) were dissected in ice-cold saline and dissected SGs were immediately transferred to 60 µl PBS supplemented with Roche cOmpleteTM protease inhibitor cocktail (PIC; final EDTA concentration: 0.2 mM). Thirty pairs of SGs were pooled per replicate and homogenized with a disposable pestle. Sixty microliter of 12 M urea, 4 M thiourea, and PIC was added and samples were homogenized further, centrifuged at 9,000 × g for 5 min to pellet cellular debris and the supernatant was removed and quantified. One hundred microgram of protein was removed and purified using the 2D Clean-up Kit. The solubilized protein lysates were resuspended in 6 M urea, 2 M thiourea, 0.1 M Tris–HCl, pH 8.0, and requantified. Twenty micrograms were removed from each sample for protein digestion.
Sample Preparation for Mass Spectrometry
For mass spectrometry, three independent biological replicates were analyzed. Fifty-mM ammonium bicarbonate was added to each sample, and proteins were reduced with 0.5 M dithiothreitol (DTT) at 56 °C for 20 min. The proteins were then alkylated with 0.55 M iodoacetamide (IAA) at room temperature for 15 min, in the dark. One microliter of a 1% w/v solution of Protease Max Surfactant Trypsin Enhancer (Promega) and 0.5 µg of Sequence Grade Trypsin (Promega) was added to obtain a protein: trypsin ratio of 40:1 and 80:1 for saliva and salivary glands, respectively. The protein/trypsin mixture was incubated at 37 °C for 18 h. Digestion was terminated by adding 1 µl of 100% trifluoroacetic acid (Sigma Aldrich) and incubation at room temperature for 5 min. Samples were centrifuged for 10 min at 13,000 × g and purified for mass spectrometry using either the ZipTip pipette procedure (Millipore) for saliva and C18 Spin Columns (Pierce) for salivary glands, following the manufacturer’s instructions. The eluted peptides were dried using a SpeedyVac concentrator (Thermo Scientific Savant DNA120) and resuspended in 2% v/v acetonitrile and 0.05% v/v trifluoroacetic acid (TFA). Samples were sonicated for 5 min to aid peptide resuspension followed by centrifugation for 5 min at 13,000 × g. The supernatant was removed and used for mass spectrometry.
Mass Spectrometry
One microgram of each digested sample was loaded onto a QExactive (ThermoFisher Scientific) high-resolution accurate mass spectrometer connected to a Dionex Ultimate 3000 (RSLCnano) chromatography system. The peptides were separated by a 2–40% gradient of acetonitrile on a Biobasic C18 PicofritTM column (100 mm length, 75 mm ID), using a 120-min reverse-phase gradient at a flow rate of 250 nl min−1 with a runtime of 50 and 130 min for saliva and salivary glands, respectively. All data were acquired with the mass spectrometer operating in automatic data dependent switching mode. A full MS scan at 70,000 resolution and a range of 400–1,600 m/z was followed by an MS/MS scan at resolution 17,500 and a range of 200–2,000 m/z, selecting the 15 most intense ions prior to MS/MS.
Protein identification of MS/MS data was performed using MaxQuant v1.5.6.5 (www.maxquant.org; last accessed May 22, 2018) following the general procedures and settings outlined in Hubner et al. (2010). The Andromeda search algorithm (Cox et al. 2011) implemented in the MaxQuant software was used to correlate MS/MS data against the protein reference sequence set of A. pisum obtained from the NCBI (27,984 entries, May 2016) and a contaminant sequence set provided by MaxQuant. The following search parameters were used: first search peptide tolerance of 20 ppm, second search peptide tolerance 4.5 ppm with cysteine carbamidomethylation as a fixed modification and N-acetylation of protein and oxidation of methionine as variable modifications and a maximum of two missed cleavage sites allowed. False Discovery Rates (FDR) were set to 1% for both peptides and proteins and the FDR was estimated following searches against a target-decoy database. Peptides with minimum length of seven amino acids were considered for identification.
Data Analysis
Perseus v.1.5.5.3 (www.maxquant.org, last accessed May 22, 2018) was used for data analysis, processing and visualization. The data matrix was first filtered for the removal of contaminants and peptides identified by site. Label Free Quantitation (LFQ) intensity values were log2 transformed and proteins not found in all three replicates in at least one group were omitted from the analysis. A data-imputation step was conducted to replace missing values with values that simulate signals of low abundant proteins chosen randomly from a distribution specified by a downshift of 1.8 times the mean standard deviation (SD) of all measured values and a width of 0.3 times this SD.
Gene Ontology and Secretion Prediction
The GO annotation was performed on the whole A. pisum proteome available on NCBI using Blast2GO v2.5.0 (Götz et al. 2008). Associations were realized using a blastp [BLAST+ v2.5.0 (Camacho et al. 2009)] search against the nonredundant protein database (release 2017-2-4) with the following parameters: e-value = 1e−8, max_target_seqs = 20, soft_making = false, and Interproscan v5.13.52.0 (Jones et al. 2014). Assigned GO terms for genes of interest groups were categorized by molecular function (MF), biological process (BP) and cellular component (CC) and GO enrichment was investigated using hypergeometric tests in R. The P-values were adjusted for multiple comparisons using Bonferroni correction. The whole A. pisum proteome was analyzed with SignalP v3.0 and v4.1 (Dyrløv Bendtsen et al. 2004; Petersen et al. 2011) and SecretomeP v2.0 (Bendtsen et al. 2004) to characterize the presence of signal peptide or nonclassical secretion signal, respectively. Then, membrane inserted domains were predicted using TMHMM v2.0 (Krogh et al. 2001) for transmembrane domains and PredGPI (Pierleoni et al. 2008) for GPI anchors. Finally, the results were combined to define the list of A. pisum secreted proteins that have secretion signals and no membrane insertion domains.
Orthology Analysis
To determine groups of orthologs among the 17 genomes (supplementary table S1, Supplementary Material online), we first kept the longest protein isoforms from each species with an home-made perl script and then ran the OrthoDB_soft_1.6 (Kriventseva et al. 2015) using standard parameters. To establish the species phylogeny, 478 groups of conserved single-copy orthologs were extracted and their protein sequences aligned with MAFFT (Katoh et al. 2005). Alignments were concatenated and used to generate a maximum likelihood tree with RAxML (Stamatakis 2006) with default parameters and 1,000 replicates, considering Tetranychus urticae as an outgroup. The phylogeny was then used to establish the level of orthology of each A. pisum gene. The enrichments of orthologous categories among data set were analyzed using hypergeometric test implemented in R.
Evolution Analyses of Salivary Genes
Single Copy Genes
Ortholog groups that were represented by a single gene in the A. pisum genome and in at least one of the other Aphididae genomes were extracted. If alternative transcripts of a gene were present, the longest coding sequence (CDS) for each species was used. Pairs of orthologous CDS (one CDS per species: A. pisum/A. glycines, A. pisum/D. noxia, A. pisum/M. persicae) were aligned using MAFFT (Katoh et al. 2005) within the Hot algorithm from GUIDANCE2 (Sela et al. 2015) to produce reliable codon-based alignments without poorly aligned regions. From these codon alignments, pairwise dN/dS values were calculated with PAML v4.8 using the YN00 program (Yang and Nielsen 2000; Yang 2007). We removed ortholog pairs with dS > 2 or dN > 0.5 to avoid mutational saturation, as well as pairs with dS = 0. dN/dS of different gene categories within each species pairs were compared using a Kruskal–Wallis test, and Nemenyi-Tests for multiple comparisons were realized with the R package PMCMR (Pohlert 2014).
Orthologs of characterized salivary effectors were searched in all Aphididae sequences available in Genbank database and in 454-sequenced contigs of Rhopalosiphum padi and Schizaphis graminum assembled for this study (see supplementary material and methods S1, Supplementary Material online), in order to compute more accurate evolutionary rates and to test selection models. For this task, reciprocal blast searches were run with blastn (BLAST+ v2.5.0) using A. pisum, M. persicae, A. glycines and D. noxia CDS sequences against the different databases (e-value < 10−10). Then, for each obtained ortholog group, a codon-based alignments was generated with PRANK (Löytynoja and Goldman 2005) and cleaned from poorly aligned regions using GUIDANCE2, specifying a minimum alignment quality threshold of 0.93. The cleaned alignment was used to generate a maximum likelihood phylogenetic tree with RAxML and both alignment and tree served to estimate dN/dS with codeml [implemented in PAML v4.8 (Yang 2007)] under different models (Yang et al. 2000; Yang and Nielsen 2000). The null models (M0 = one ratio, M1 = neutral, M7 = β) were compared with alternative models (M2 = selection, M8 = β + ω) using the likelihood ratio test (LRT), which compares twice the difference in log likelihood to a χ2 distribution. Finally, if selection models were found more likely, the Bayes Empirical Bayes (BEB) procedure was used to compute the posterior probability of evolution under positive selection for each site (Yang et al. 2005).
Gene Families
Families containing genes expressed in A. pisum salivary glands and their orthologs in arthropod species were extracted from OrthoDB groups. Then, we examined if the two salivary gene catalogs contain more members of multigene families than expected by chance. For each catalog, a significant effect is demonstrated if the number of genes that belong to multigene families that are represented by two or more copies in the catalogue lies above the 95% confidence interval (CI) of the expectation. 95% CI was computed by randomly sampling the number of genes contained in a catalog (740 and 3,603 genes) from the list of 18,603 genes and counting the number of gene-family members (that belong to a multigene family represented by two more copies in the sample) in this random sample. This step was repeated 10,000 times.
To test for positive selection on certain sites and branches (selection acting on foreground branch compared with background branches) of the three selected gene families (Cysteine-Rich Protein, Angiotensin-Converting-Enzyme-like and Aminopeptidase-N families), a cleaned alignment and a maximum-likelihood tree were generated as described above, and were fed to the codeml branch-site (BS) model (Yang and Nielsen 2002). The BS model classified the sites in four categories: class 0 where sites were under negative selection (ω0 < 1) on both foreground and background branches, class 1 where sites evolved under neutral evolution (ω1 = 1) on both foreground and background branches, class 2a where the sites were under positive selection (ω2 ≥ 1) on the foreground branch and under negative selection (ω0 < 1) on background branches, and class 2b where the sites evolved under positive selection (ω2 ≥ 1) on the foreground branch and under neutral evolution (ω1 = 1) on background branches. The null model in which the foreground branch may have different proportions of sites under neutral evolution (dN/dS of 1), and the alternative model, in which the foreground branch may have sites under positive selection, were applied to all branches of each gene family. The likelihood of the selection model was computed as described in previous section and P-values were corrected for multiple testing comparisons as described by Anisimova and Yang (2007). In case the selection model was retained, the posterior probability of particular sites evolving under positive selection was computed.
All phylogenetic trees shown in figures were designed in iTOL (Letunic and Bork 2007).
Results
Generation of the A. pisum Salivary Effector Candidate Gene Sets
We conducted transcriptome analyses of A. pisum salivary glands (SGs) to generate two catalogues of salivary effector candidates: one that considers up-regulation of their expression in SGs and another that does not. This approach assumes that the vast majority of salivary proteins secreted into plants are expressed in aphid SGs (Mutti et al. 2008; Carolan et al. 2011; Naessens et al. 2015) and that most salivary effectors are highly expressed in SGs compared with other organs (Wang et al. 2015a, 2015b). Up-regulation of expression in SGs was determined in comparison to expression levels in the alimentary tract (AT), which we chose due to ease of isolation and RNA extraction. RNA-seq by Illumina technology was conducted on dissected SG and AT tissues of A. pisum. In SGs, 12,040 genes passed the cut-off value for gene expression (fig. 1A) and encode proteins that may or may not be secreted in saliva. A protein secretion prediction pipeline was applied to the global gene set of A. pisum (N = 18,601) using SignalP v3 or v4.1 (Dyrløv Bendtsen et al. 2004; Petersen et al. 2011), as well as SecretomeP (Bendtsen et al. 2004), which predicts noncanonical secretion signals. Genes encoding transmembrane domains [predicted by TMHMM 2.0 (Krogh et al. 2001)] or GPI-anchors [predicted with PredGPI (Pierleoni et al. 2008)] were considered as not secreted, leaving 3,603 encoded proteins predicted to be secreted. These constituted the candidate SG-expressed effector set (fig. 1A and supplementary table S2, Supplementary Material online).
Fig. 1.

—Pipelines used to establish sets of candidate Acyrthosiphon pisum salivary effector genes expressed and up-regulated in salivary glands (A). Pipeline used to identify proteins from saliva injected in artificial diet and composition of the secreted proteins (B).
To create the candidate SG-up-regulated (or SG-up) effector set, the expression levels of individual RNAs were compared between SGs and AT. After filtering and normalization steps, 12,378 protein coding genes from SGs and AT were retained. Among these genes, 1,989 genes were up-regulated in SG tissues, of which 740 genes were predicted to encode secreted proteins. These constituted the candidate SG-up effector set (fig. 1A and supplementary table S2, Supplementary Material online). All were found in the 3,603 SG-expressed effector set except one (gene LOC100574271), which was expressed at too low level to pass the criterion used to define the SG-expressed effector set (FPKM ≥ 0.5) although it passed the criterion used to define the SG-up effector set (CPM > 1).
Identification of Aphid Proteins in Artificial Diets and Salivary Glands
Proteins from artificial diets fed upon by aphids were analyzed by proteomics-based mass spectrometry (MS). Fifty-one proteins were supported by more than one peptide detected by MS in at least two out of the three replicates or by one peptide in all three replicates of saliva samples from artificial diets (fig. 1B and supplementary table S3, Supplementary Material online). Of these, 35 proteins (68.62%) were included in the SG-up effector set, two belonged to the SG-expressed effector set, and 14 were not predicted as secreted proteins and were encoded by genes up-regulated in SGs (11 genes) or expressed in SGs (three genes) (fig. 1B and supplementary table S2, Supplementary Material online). Five out of the 11 SG-up-regulated genes seemed to be truncated in the genome sequence and were predicted to be not secreted.
In all, 1,837 proteins (supported by more than one peptide in at least two of the three replicates or by one peptide in all the three replicates) encoded by 1,809 unique genes were detected directly from dissected SGs (supplementary table S3, Supplementary Material online), among which 205 proteins belonged to the SG-up effector set and 447 proteins to the SG-expressed effector set. Signal intensity of proteins in the SGs significantly correlated (Pearson’s r = 0.5446, P < 0.001) with the transcription level of corresponding genes in the organ although such correlation was not observed for the proteins detected in the artificial diets (supplementary fig. S2, Supplementary Material online).
Aphididae Specific Genes Are Enriched in the SG-Up-Regulated Effector Set
To assess the phylogenetic distribution of genes constituting the SG-up and SG-expressed effector sets, an analysis of orthology was conducted between A. pisum and 16 other arthropods whose genome sequences were available, and which cover a wide range of divergence from A. pisum (fig. 2A). Relative to the whole set of genes annotated in the A. pisum genome, the SG-expressed effector set was significantly enriched in highly conserved genes found across the arthropod or insect clades (hypergeometric test, P < 0.01). The set was also slightly enriched in genes found only in Aphidomorpha, whereas the proportion of A. pisum-specific genes was significantly reduced (hypergeometric test, P < 0.001). In contrast, in the SG-up effector set, the proportion of genes found only in Aphididae was significantly higher than in the whole gene set, and highly conserved genes found across arthropods were less frequent (hypergeometric test, P < 0.001) (fig. 2B). In our comparison, this pattern was specific to the SG-up effector set as the genes expressed or up-regulated in ATs showed significant enrichment of highly conserved genes found across arthropod and insect clades (supplementary fig. S3A, Supplementary Material online).
Fig. 2.

—Orthology profile of Acyrthosiphon pisum SG effector candidate gene sets (478 single-copy ortholog groups). (A) Phylogeny of the 17 arthropod species analyzed to determine ortholog groups. The colored squares indicate the levels of orthology, as indicated on the bottom right-hand legend. (B) Proportions of the different orthology levels among genes of the salivary effector sets and the A. pisum genome. Stars indicate the significance of differences in the proportion of genes presenting a given orthology level between a given effector set and the whole gene set (hypergeometric test): *P < 0.05, ***P < 0.001.
Evolutionary rates vary among of A. pisum Salivary Candidate Effectors
The enrichment of Aphididae-specific genes in the SG-up effector set may be associated with rapid molecular evolution related to aphid-specific gene functions. This hypothesis was tested for the SG-up effector set by estimating the ratio of nonsynonymous to synonymous substitutions (dN/dS or ω) in three categories: SG-up effector set, SG-expressed effector set and the remaining genes (fig. 3). Only single-copy gene pairs of orthologs between A. pisum and the three other Aphididae species were included in the orthology search, and pairwise evolutionary rates were analyzed (A. pisum/A. glycines, A. pisum/D. noxia, A. pisum/M. persicae). On average, genes of SG-up effector set showed higher dN/dS ratios than the genes of other sets (fig. 3). The same analysis comparing AT-up, AT-expressed and remaining genes revealed that the observed overall higher dN/dS ratio was specific to the genes of SG-up effector set (supplementary fig. S3B, Supplementary Material online).
Fig. 3.
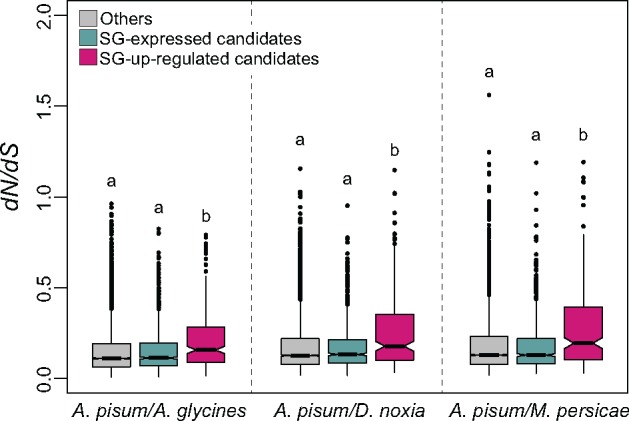
—Pairwise evolutionary rates (dN/dS) of single-copy Aphididae genes orthologous to Acyrthosiphon pisum effector candidates. Here, SG-expressed effector set does not contain SG-up effector set. For the three species pairs indicated on the X axis 5297, 5379, and 5562 single copy ortholog pairs were retained, respectively. Letters above boxes denote significant differences determined by multiple Kruskal–Wallis test within each species pair (A. pisum/Aphis glycines: H = 33.944, 2 d.f., P < 0.001; A. pisum/Diuraphis noxia: H = 51.17, 2 d.f., P < 0.001; A. pisum/Myzus persicae: H = 39.373, 2 d.f., P < 0.001).
The 11 single-copy salivary effector genes that were previously functionally characterized and shown to be involved in plant-aphid interactions were examined (table 1). All these single copy salivary effectors or their A. pisum orthologs were found in the SG-up effector set with the exception of Mif1 (Naessens et al. 2015), which was not up-regulated in SGs and not predicted to be secreted in our study (table 1). Most of these genes were highly expressed, and genes C002, Mp1, Ap25, Me23, Me10, Mp55, Shp, and Mp2 were Aphididae- or Aphidomorpha-specific.
Table 1.
Aphid Single-Copy Salivary Effector Genes Characterized for their Role in the Interaction with Host Plants
| Gene | Acyrthosiphon pisum Gene | Effector set | Expression Ranka | Orthology Level | ωb dN/dS | Aphid Phenotype | References* |
|---|---|---|---|---|---|---|---|
| Mp1 | LOC100165393 | SG-up | 19/740 | Aphididae | 0.64653 | Expression promotes fecundity | [3, 4, 6, 10, 15] |
| ACYPI006346 | |||||||
| C002 | LOC100167863 | SG-up | 12/740 | Aphididae | 0.62307 | Expression promotes fecundity | [1, 2, 3, 4, 6, 13] |
| ACYPI008617 | Silencing reduces survival/fecundity | ||||||
| Ap25 | LOC100169287 | SG-up | 57/740 | Aphididae | 0.56219 | Expression promotes fecundity | [14] |
| ACYPI009919 | |||||||
| Me23 | LOC100161198 | SG-up | 109/740 | Aphididae | 0.53025 | Expression promotes fecundity | [5] |
| ACYPI002439 | |||||||
| Me10 | LOC100167427 | SG-up | 5/740 | Aphididae | 0.51787 | Expression promotes fecundity | [5] |
| ACYPI008224 | |||||||
| Shp | LOC100169243 | SG-up | 1/740 | Aphidomorpha | 0.48863 | Silencing reduces survival/fecundity | [8, 12] |
| ACYPI009881 | |||||||
| Mp55 | LOC100569515 | SG-up | 18/740 | Aphididae | 0.42972 | Expression promotes fecundity | [7] |
| ACYPI33755 | Silencing reduces survival/fecundity | ||||||
| Mp2 | LOC100160479 | SG-up | 107/740 | Aphididae | 0.40948 | Expression promotes fecundity | [6] |
| ACYPI001774 | Silencing reduces survival/fecundity | ||||||
| Mp10 | LOC100145855 | SG-up | 303/740 | Insect | 0.12212 | Expression reduces fecundity | [3] |
| ACYPI000097 | |||||||
| Mif1 | LOC100161225 | None | 4700/12040 | Arthropod | 0.09454 | Silencing reduces fecundity | [9] |
| ACYPI002465 | |||||||
| Armet | LOC100167188 | SG-up | 122/740 | Arthropod | 0.06298 | Silencing reduces survival | [11] |
| ACYPI008001 |
Expression ranks show the rank of effector SG expression level (based on Log[FPKM]) within the SG-up effector set except for Mif1 which does not belong to any effector sets (global SG expression scale).
ω values represent the evolutionary rates within closely related Aphididae species.
1Mutti et al. 2006; 2Mutti et al. 2008; 3Bos et al. 2010; 4Pitino et al. 2011; 5Atamian et al. 2013; 6Pitino and Hogenhout 2013; 7Elzinga et al. 2014; 8Abdellatef et al. 2015; 9Naessens et al. 2015; 10Pan et al. 2015; 11Wang et al. 2015a; 12Will and Vilcinskas 2015; 13Zhang et al. 2015; 14Guy et al. 2016; 15Rodriguez et al. 2017.
To examine the evolutionary rates of these 11 effectors, we retrieved the orthologs from SG transcripts of two additional aphid species, Schizaphis graminum and Rhopalosiphum padi (supplementary material and methods S1 and table S4, Supplementary Material online). In addition, orthologs from other aphids were retrieved from public databases (supplementary table S4, Supplementary Material online) and used to estimate dN/dS ratios under different site-models of selection. The global dN/dS for each gene showed a range from 0.06298 (Armet) to 0.64653 (Mp1) (table 1 and supplementary table S5, Supplementary Material online). Differences were found between insect- or arthropod-conserved effector genes like Armet, Mif1 and Mp10, which seem to have evolved under strong negative selection (dN/dS < 0.12), and the aphid-specific, fast-evolving effector genes (dN/dS > 0.40, in the top 5% of values obtained from pairwise comparisons). Despite these faster evolutionary rates, the examination of null and alternative models of codon substitutions [M1 vs. M2 and M7 vs. M8] revealed signatures of positive selection only for genes Mp1 and Me10 (supplementary table S5, Supplementary Material online). In Mp1 and Me10, sites detected as under positive selection were mainly found in the region coding for the mature protein injected into the host plant (supplementary fig. S4, Supplementary Material online).
Episodic positive selection occurred in salivary expanded gene families in the A. pisum Lineage
Evolutionary novelties are usually brought by gene duplication followed by diversification in functions (Conant and Wolfe 2008; Kondrashov 2012). Interestingly, duplicated genes are more frequently observed in SG-up and SG-expressed effector sets than expected. Indeed, the observed number of genes that are represented by two or more members of gene family in the two catalogs always lies above the 95% confidence interval (CI) (SG-up: 80 genes, 95% CI = [23, 55] and SG-expressed: 654 genes, 95% CI = [442, 540]). Three multigene families in particular, a cystein-rich protein family (CRP), the Angiotensin-Converting-Enzyme-like family (ACE) and the Aminopeptidase-N (apN) family, represented 46.9% of the proteins detected in A. pisum saliva (supplementary table S6, Supplementary Material online). Gene ontology analysis showed that the ACE and apN gene families belong to the metallopeptidase family and are involved in proteolysis, two functions for which the SG-up effector set was enriched (supplementary fig. S5, Supplementary Material online).
The Cysteine-Rich Protein (CRP) Family
The CRP Aphididae specific gene family was originally described as a family of 12 genes in A. pisum (Guo et al. 2014). Here, 15 gene members were identified from genomic data, and encode proteins of <200 amino acids with 14 highly conserved cysteine residues. The family is expanded in A. pisum with 15 copies compared with the other aphid species D. noxia, A. glycines and M. persicae, which have 6, 4, and 1 copies, respectively. Among the 15 A. pisum gene copies, three were not detected in either SG or AT tissues by RNAseq or protein mass spectrometry. Eleven genes were up-regulated in SGs, nine encode proteins that were predicted to be secreted (supplementary table S6, Supplementary Material online), two of which (CRP-12 and 14) were found in saliva. The branch-site (BS) model of codon substitutions (Yang and Nielsen 2002) detected positive selection in five branches of the Aphididae CRP family tree (fig. 4A and B): the branch leading to D. noxia gene CRP-5 (branch #1), the ancestral branches #2, #4, and those leading to A. pisum CRP-2 (branch #3) and CRP-12 (branch #5). The Bayes Empirical Bayes (BEB) posterior probability (Yang et al. 2005) revealed sites under positive selection (BEB above 0.75) in each aforementioned branch (supplementary fig. S6A, Supplementary Material online), which are scattered across the protein (supplementary fig. S6B, Supplementary Material online). The functional importance of these sites is unknown, due to the absence of known domains within the proteins. Although positive selection seems to occur in some A. pisum CRP copies, no site under positive selection was detected in CRP-13, the most highly expressed A. pisum CRP.
Fig. 4.

—Evolutionary analysis of the Aphididae CRP gene family. (A) Codon-based maximum-likelihood tree used to compute dN/dS under the branch-site (BS) model of codon substitution. Numbers below branches indicate bootstrap support (percentage of the 1,000 replicates) when >40. DNOX, MPER, AGLY, and APIS indicate sequences from Diuraphis noxia, Myzus persicae, Aphis glycines, and Acyrthosiphon pisum, respectively. Branches where positive selection affected certain sites are in bold. (B) Estimated evolutionary parameters for these branches (numbered as on the tree). ω0, ω1, and ω2 indicate the average dN/dS ratio for sites assigned to class 0 (ω < 1, negative selection), class 1 (ω = 1, neutral evolution) and class 2 (ω > 1, positive selection), respectively, in the branch, and n.c. indicates insufficient dS to compute ω. p0, p2a, and p2b show proportions of sites in classes 0, 2a, and 2b, respectively, for each branch (Yang and Nielsen 2002).
The Angiotensin-Converting-Enzyme-like (ACE) Gene Family
In contrast to the Aphididae-specific CRP gene family, the ACE gene family is found among arthropods (fig. 5 and supplementary table S6, Supplementary Material online). A search for the ortholog groups yielded ten members in the A. pisum genome [three were previously identified by (Wang et al. 2015b)], which are distributed in four clades along with orthologs from other insect species (fig. 5 and supplementary table S6, Supplementary Material online). Seven of these genes were up-regulated in A. pisum SG tissue, including three members (ACE1, ACE2, and ACE5) of clade 4 encoding predicted secreted ACEs. ACE1, 5 and 10 were detected in saliva, although the latter lacks an encoded signal peptide, indicating errors in gene annotation or secretion prediction (supplementary table S6, Supplementary Material online). It is notable that the other aphid genomes comprise just one ACE of clade 4 (fig. 6A). All clade-4 ACE proteins except ACE10 are determined as functional peptidases, based on the presence of the M2 peptidase HEXXH domain (IPR001548) (fig. 6A). Positive selection was inferred in three branches of this clade (fig. 6B and C): the ancestral branch (#1) of A. pisum ACE 1 and ACE5, the A. pisum ACE1 gene (#2), and the ancestral branch #3. Sites detected to evolve under positive selection are scattered across the protein (supplementary fig. S7A and B, Supplementary Material online).
Fig. 5.

—Protein-based maximum likelihood phylogeny of the ACE gene family among insect species, with bootstrap support indicated next to branches (percentage of the 1,000 replicates) when >40. Species abbreviations: AGAM, Anopheles gambiae; AGLY, Aphis glycines; AMEL, Apis mellifera; APIS, Acyrthosiphon pisum; BMOR, Bombyx mori; CLEC, Cimex lectularius; DMEL, Drosophila melanogaster; DNOX, Diuraphis noxia; DVIT, Daktulosphaira vitifoliae; MDES, Mayetiola destructor; MPER, Myzus persicae; MSEX, Manduca sexta; NLUG, Nilaparvata lugens; NVIT, Nasonia vitripennis; RPRO, Rhodnius prolixus; TCAS, Tribolium castaneum; TURT, Tetranychus urticae.
Fig. 6.

—Evolutionary analysis of clade 4 of the Aphididae ACE gene family. (A) Protein structure of the family members with protease active sites shown as blue lines. (B) Codon-based maximum likelihood tree used to compute dN/dS under the branch-site (BS) model of codon substitution. Numbers below branches indicate bootstrap support (percentage of the 1,000 replicates) when >40. DNOX, MPER, AGLY, and APIS indicate sequences from Diuraphis noxia, Myzus persicae, Aphis glycines and Acyrthosiphon pisum, respectively. Branches where positive selection was detected at specific sites are shown in bold. (C) Estimated evolutionary parameters for these branches (numbered as on the tree). See fig. 4B for the definition of terms used.
The Aminopeptidase-N (apN) Gene Family
The aminopeptidase-N (apN) gene family was the most represented in A. pisum saliva proteome with 18 apN proteins detected, accounting for 36.7% of the 51 saliva proteins identified. Another apN protein not detected here was identified in saliva by Carolan et al. (2009) (supplementary table S6, Supplementary Material online). Forty-seven apN genes were identified in the A. pisum genome, 27 of them belonged to ortholog groups present among insects, and the remaining 20 did not show orthologs in other species (supplementary table S6, Supplementary Material online). All 20 A. pisum-specific apN genes were up-regulated in SG tissue, ten encode proteins with predicted secretion, four of which were detected in saliva. Four of the ten proteins not predicted as secreted were also detected in saliva, indicating incorrect gene annotation and/or secretion prediction. The 27 apN genes with orthologs in other species were clustered in eight distinct clades (fig. 7). The A. pisum genes from clades 1, 2, 3, 4, and 7 were up-regulated in SGs with the exception of only three members. Genes from clades 5, 6, and 8 were not up-regulated in SGs. Apart from the one A. pisum gene that belongs to clade 1, all the other SG-up candidate effector genes were found in clade 4 (fig. 7).
Fig. 7.

—Protein-based maximum likelihood phylogeny of the apN gene family among insect species, with bootstrap support indicated next to branches (percentage of the 1,000 replicates) when >40. Species abbreviations are the same as on fig. 5.
Clade 4 is remarkable for the presence of 14 closely related gene copies in A. pisum and only a few copies from other insect species, including aphids (fig. 7). Eleven A. pisum copies were SG-up-regulated and detected in saliva, but two encode proteins that were not predicted as secreted (fig. 7). Genes of this clade generally encode proteins with a signal peptide followed by a M1 peptidase domain (IPR014782) and an ERAP1-C domain (IPR024571). However, some have lost one of the two domains, and the vast majority (apart from D. noxia apN-3, A. pisum apN-1 and A. pisum apN-5) possesses the HEXXH active site ensuring the peptidase function (fig. 8A).
Fig. 8.

—Evolutionary analysis of the Aphididae clade 4 of the apN gene family. (A) Protein structure of the family members. (B) Codon-based maximum likelihood tree used to compute dN/dS under the branch-site (BS) model of codon substitution. Numbers below branches indicate bootstrap support (percentage of the 1,000 replicates) when >40. DNOX, MPER, AGLY, and APIS indicate sequences from Diuraphis noxia, Myzus persicae, Aphis glycines, and Acyrthosiphon pisum, respectively. The branches where positive selection was detected at certain sites are in bold. (C) Estimated parameters of these branches (numbered as on the tree). See fig. 4B for the definition of terms used.
Traces of positive selection were detected by the branch-site model in seven branches of apN clade 4 (fig. 8B and C): the branch leading to D. noxia gene apN-1 (branch #1), the ancestral branches #3, #5, #7, and those leading to A. pisum apN-3 (#2), apN-8 (#4), and apN-12 (#6) genes. Along these branches, sites under positive selection (with BEB > 0.75) were mainly located in the M1 peptidase and ERAP1-C domains, and in the uncharacterized part between the signal peptide sequence and M1 peptidase domain for branches #1, #2, and #3 (supplementary figs. S8 and S9, Supplementary Material online).
Discussion
New Salivary Candidate Effector Gene Sets
As evidence accumulates that aphid salivary effectors play important roles in plant-aphid interactions (Elzinga and Jander 2013), a comprehensive identification of aphid salivary genes and analysis of their evolutionary patterns are essential to shed light on the process of aphid adaptation to specific host plants. Here, we have assembled new catalogues of candidate A. pisum salivary effector genes based primarily on comparative transcriptional analysis of SG and AT genes. We identified 3,603 genes expressed in SGs and potentially secreted in saliva. A subset of 740 genes have their expression up-regulated in SGs compared with AT. These sets combined are substantially larger than the previously established candidate effector catalogue (Carolan et al. 2011), reflecting the greater sensitivity of Illumina's RNA-seq compared with EST sequencing. The salivary gland secretome established for A. pisum by Carolan et al. (2011) contains 324 genes, of which 95 are found in our SG-up effector set and 93 others are contained in the SG-expressed effector set. The remaining 120 encompass 111 SG-expressed genes encoding proteins considered as not secreted in our analysis, and 9 genes that were absent from our source transcriptome. Finally, 16 genes did not exist in the updated gene annotation of the A. pisum genome used here (NCBI Acyrthosiphon pisum Annotation Release 102). The aphid line used here (LSR1) was collected from Medicago sativa (The International Aphid Genomics Consortium 2010) and feeds well on the universal host plant of A. pisum, Vicia faba (our unpublished data). We used V. faba to rear the aphids for RNA-seq experiment because of the ease of producing large quantity of synchronized aphids on this plant. It was also because previous study revealed that A. pisum reared on two suitable legume plants show very similar expression patterns of salivary effector candidates (Eyres et al. 2016). There is a possibility that our catalogues missed the salivary genes that were expressed exclusively when the aphid was feeding on M. sativa, but we assume that such genes are rare.
Comparing expression levels between SGs and AT was fundamental in restricting the list of candidate effectors, which may be refined by sequencing RNAs from other tissues. Nonetheless, salivary genes not up-regulated in SGs should not be ignored. Some may indeed encode salivary effectors like Mif1, which suppresses induction of plant defense responses against aphids (Naessens et al. 2015). In addition, some effector proteins could be produced in tissues other than SGs and injected into plants. Proteins can move from the hemocoel of aphids to SGs to be secreted through saliva. However, only the chaperonin GroEL, which is produced by the aphid obligate endosymbiont in bacteriocytes, is known to be injected into plant tissue through this pathway (Chaudhary et al. 2014).
As 46 proteins out of the 51 (90%) detected in saliva through proteomics were encoded by SG-up genes, most of A. pisum salivary proteins seem encoded by genes whose expression is up-regulated in SGs. The lack of significant correlation between the signal intensities of the proteins secreted in saliva and their gene expression level in SGs may result from insufficient statistical power and/or regulation of salivary protein secretion. Plant cues may be required to trigger certain aphid responses, including protein secretion (Powell et al. 2006), and this may explain why some proteins encoded by highly transcribed salivary genes were not detected in the artificial diet. For example, C002 is reported to be secreted into the plant and required for aphid feeding (Mutti et al. 2008), but was not detected by our saliva proteomics despite its high transcription level. The comparison of transcriptomic and proteomic results revealed limits in the secretion prediction pipeline as 14 proteins detected in saliva were not predicted to be secreted. Five of them may be miss-annotated, but the rest might have been secreted by pathways that were not considered for the secretion prediction.
Fast Evolutionary Rates of Candidate Effectors Genes
SG-up genes showed lower degree of gene conservation than SG-expressed genes. Indeed, 50% of the A. pisum SG-up effector set had either no ortholog in other genomes or only in Aphidomorpha and Aphididae, whereas 64.2% of the SG-expressed effector set had orthologs found among arthropods. Furthermore, single-copy SG-up genes presented higher dN/dS ratio than SG-expressed genes on average. This indicates that the evolution of SG-up genes tends to be less constrained (relaxed selection) or accelerated (positive selection). We did not observe such contrast in between AT-up and AT-expressed gene sets, highlighting the peculiar evolutionary history of genes constituting the SG-up effector set.
Interestingly, the analysis of the functionally characterized single-copy genes revealed different selective regimes. The more conserved effectors (e.g., Mif1, Armet, and Mp10) with low dN/dS ratio (≤0.12) could be involved in fundamental functions required for plant feeding, for example, prevention of phloem clogging, repression/manipulation of general plant defenses, such that orthologous proteins of similar sequences may be effective on numerous host species (Bos et al. 2010; Furch et al. 2015; Naessens et al. 2015). In contrast, 11 effectors with dN/dS ratios above 0.40 have been characterized as modulators of plant-aphid interactions in functional studies, and their non-synonymous divergence may reflect adaptation to specific host plants. Some of them (C002, Mp1 and Mp2) were indeed shown to act in a species-specific manner to promote aphid performances on host plants (Pitino and Hogenhout 2013). Moreover, sites detected as under positive selection in Mp1 (Pitino and Hogenhout 2013; this study) and Me10 salivary effector genes may be involved in adaptation of their respective aphid lineages.
Although we cannot globally evaluate the relative contributions of relaxed and positive selection regimes on faster gene evolution, several evidences underline the importance of positive selection in shaping the evolution of some candidate effectors identified in our study. It would be unintuitive to hypothesize that SG-up candidate effector genes are more prone to relaxed selection than SG-expressed ones, especially when considering that 90% of the proteins detected in saliva in this study were encoded by SG-up genes. These SG-up effector candidates are more likely secreted by the aphid and thus exposed to selective pressures exerted by the plant surveillance and defense systems. Moreover, we did find traces of positive selection acting on certain sites and branches of SG-up candidate effector genes. Based on these observations and previous functional studies, we argue that the evolutionary history of some salivary effectors has been conditioned by the specialization of SG tissue during establishment of aphid-plant interactions.
Salivary Gene Family Expansions
In addition to single-copy genes, evolutionary histories of three multigenic families were examined. Remarkably, these three gene families encoded nearly half of the proteins detected in saliva and showed gene expansion in the A. pisum lineage. Irrespective of their specificity to aphid lineages (CRP family) or conservation among insects (ACE and apN), these families show the highest number of members in A. pisum, with 15, 10, and 47 genes, respectively. Gene duplication and diversification therefore appear to have largely contributed to the battery of A. pisum salivary proteins. Accordingly, SG-up and SG-expressed effector sets were enriched in duplicated genes. Aphididae-specific duplications were also present, but in lower numbers, in the D. noxia genome for CRP and apN families, as well as in A. glycines for the CRP. Interestingly, clade 4 of the apN family, which contains a majority of candidate A. pisum SG-up effector genes, did not comprise any A. glycines members. Further analyses may indicate whether the loss happened in an ancestor of the Aphidini subtribe or only in A. glycines (clade 4 is present in Aphidomorpha) or resulted from technical artifacts. Positive selection was detected on specific duplicated copies in D. noxia, but a tissue specific expression analysis or saliva proteomics is required to check whether they are SG specialized potential effectors.
Effector genes are often aggregated in large clusters in the genome of plant pathogens (e.g., CRN and RXLR effectors of the oomycete Phytophthora infestans, Haas et al. 2009). These clusters are thought to result from non-allelic homologous recombination and tandem duplications (facilitated in repeat-rich genomic regions), generally associated with rapid birth and death evolution (Jiang et al. 2008; Haas et al. 2009). Effector gene clusters have also been observed in a few herbivore insects, including the gall midge Mayetiola destructor, which shows a massive expansion of effectors organized in clusters (Zhao et al. 2015). Although some members of each of the three A. pisum gene families are clustered on the same genomic scaffolds (supplementary table S6, Supplementary Material online), we were not able to identify clear gene clusters resulting from tandem duplication events, possibly due to assembly errors in the pea aphid reference genome (Jaquiéry et al. 2018).
The functions of the CRP, ACE and apN gene families in insects, including aphids, are largely unknown. The Aphididae-specific CRP gene family was first described by Guo et al. (2014), who demonstrated that CRP-13 expression in A. pisum was induced by feeding on plant in comparison to feeding on an artificial diet. However, silencing of CRP-13 did not affect aphid survival on host plant. Proteins with numerous cysteine residues, like those of the CRP family, were previously characterized for their antifungal activities in different insect species (Banzet et al. 2002). Interestingly, some small secreted cysteine-rich proteins act as effectors in the interaction between the Asian soybean rust fungus, Phakopsora pachyrhizi, and its host plant (Qi et al. 2016). One of them was shown to suppress plant immunity by interacting with a soybean transcription factor essential for negative regulation of immunity. Thus, the various cysteine-rich proteins appear to be active in different systems including plant-pathogen interactions, so the aphid-specific CRP family may play a role in aphid nutrition once injected into the host plant.
The A. pisum ACE1 and ACE2 genes were previously reported to contribute to aphid growth on plants, but not on artificial diet (Wang et al. 2015b). As the encoded proteins are predicted to have protease functions, their involvement in cleavage of certain plant defense proteins or signaling components is speculated (Wang et al. 2015b). Aminopeptidases have been found in aphid guts and reported as involved in digestion (Rahbé et al. 1995; Cristofoletti et al. 2003) or in virus binding (Linz et al. 2015), but little is known about their function in aphid saliva. Furch et al. (2015) showed that A. pisum saliva was able to degrade a major phloem protein 1 (PP1) in vitro. Because PP1 is involved in protein deposition on sieve plates after severe metabolic disturbance (Gaupels et al. 2008), its degradation by aphid saliva suggests that proteases in aphid saliva degrade PP1 to prevent sieve-element occlusion triggered by aphid feeding (Furch et al. 2015). In all cases, protease function relies on peptide processing activity ensured by the M2 peptidase domain. Despite the detection of positive selection in several A. pisum M1 and M2 peptidase genes, the active site HEXXH is conserved in most of the members, indicating a functional cleaving activity. Some copies may have lost active sites or domains, particularly in the A. pisum apN family members that were too divergent to be clustered, reflecting possible functional changes after high diversification.
Gene family expansion was previously described in A. pisum, particularly in cathepsin B gut proteases (Rispe et al. 2007), chemoreceptors (Smadja et al. 2009), small RNAs machinery (Jaubert-Possamai et al. 2010), amino acid transporters (Price et al. 2011) and cuticular proteins (The International Aphid Genomics Consortium 2010). In some cases, fast evolution and positive selection occurred on recently duplicated genes (Rispe et al. 2007; Smadja et al. 2009). The expansion of salivary gene families reported here in A. pisum and subsequent putative positive selection on some gene copies highlight the importance of gene duplication in adaptation in the A. pisum ancestry. Notably, A. pisum constitutes a complex that comes from the recent adaptive radiation of least 15 biotypes (races or cryptic species), each specialized to one or few related plant species within the Fabaceae (legume) family (Peccoud et al. 2009a, 2009b; Nouhaud et al. 2014; Peccoud et al. 2015). Population genomics analyses on A. pisum biotypes have highlighted salivary and chemosensory genes as likely contributing to host-specific adaptation in the A. pisum complex (Jaquiéry et al. 2012; Smadja et al. 2012; Nouhaud et al. 2014; Duvaux et al. 2015; Eyres et al. 2017). We may relate these results to the existence of many SG-up effector candidates that have undergone duplications and episodic positive selection in the A. pisum lineage. A large repertoire of effector genes might have increased the chance of adaptive mutations selected during biotype formation, reflecting evolutionary patterns seen in other parasites’ effectors (Stergiopoulos et al. 2012; Zhao et al. 2015).
Strikingly, we found only one member of each CRP and apN (clade 4) family in the genome of M. persicae, consistent with the overall lower rate of gene expansion compared with the A. pisum lineage (Mathers et al. 2017). M. persicae is perhaps the most generalist aphid known, being able to colonize hundreds of plant species across 40 families (Blackman and Eastop 2000). Monitoring of gene expression during host switches of M. persicae laboratory lineages have suggested that acclimation to different plant species was enabled by a rapid transcriptional plasticity of duplicated genes (Mathers et al. 2017). By contrast, very little changes in gene expression have been measured in A. pisum lineages when shifted from one suitable legume plant to another (Eyres et al. 2016). Therefore, while large gene families, including salivary effectors, may be key in the rapid diversification of specialized A. pisum on various plant species, transcriptional plasticity may enable ecological generalism in M. persicae. These hypotheses require further investigation. In particular, comparative genomics using more species of the M. persicae and A. pisum lineages in combination with functional analyses of candidate effectors, would help to precisely date the origins of genetic changes and to establish more robust correlations between these changes and ecological shifts.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
This work is part of H.B. PhD thesis funded by Plant Health and Environment division of INRA and Région Bretagne. This work was funded by Agence Nationale de la Recherche (ANR) Bugspit (ANR-13-JSV7-0012-01) to A.S. and ANR Speciaphid (ANR-11-BSV7-005-01) to J.C.S., Campus France Ulysses (907424D) to A.S. and J.C. and IOS-1258028 from the National Science Foundation and Kansas State University to F.W. and J.O. The Q-Exactive quantitative mass spectrometer was funded under the SFI Research Infrastructure Call 2012, Grant Number 12/RI/2346 (3) to Sean Doyle. We are grateful to the Phylloxera Genomic Initiative (and especially François Delmotte, Claude Rispe and Denis Tagu) for providing Daktulosphaira vitifoliae genome sequences. Funding for Daktulosphaira vitifoliae clone Pcf genomic sequencing was provided by INRA (AIP Bioressources) and BGI Biotech in the frame of i5k initiative. Parts of the D. vitifoliae transcriptomic resources were obtained within the 1KITE projects (Bernhard Misof, Bonn, Germany). We thank John Reese (Kansas State University) for providing R. padi and S. graminum aphid samples. We thank Gaëtan Denis, Jean-François Le Gallic, Frédérique Mahéo, and Sylvie Tanguy for technical support. We are grateful to Claude Rispe for critical reading of the M.S.
Literature Cited
- Abdellatef E, et al. , 2015. Silencing the expression of the salivary sheath protein causes transgenerational feeding suppression in the aphid Sitobion avenae. Plant Biotechnol J. 136:849–857. [DOI] [PubMed] [Google Scholar]
- Anisimova M, Yang Z.. 2007. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol Biol Evol. 245:1219–1228. [DOI] [PubMed] [Google Scholar]
- Atamian HS, et al. , 2013. In planta expression or delivery of potato aphid Macrosiphum euphorbiae effectors Me10 and Me23 enhances aphid fecundity. Mol Plant Microbe Interact. 261:67–74. [DOI] [PubMed] [Google Scholar]
- Banzet N, et al. , 2002. Expression of insect cystein-rich antifungal peptides in transgenic tobacco enhances resistance to a fungal disease. Plant Sci. 1626:995–1006. [Google Scholar]
- Bendtsen JD, Jensen LJ, Blom N, von Heijne G, Brunak S.. 2004. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel. 174:349–356. [DOI] [PubMed] [Google Scholar]
- Bent AF, Mackey D.. 2007. Elicitors, effectors, and R genes: the new paradigm and a lifetime supply of questions. Annu Rev Phytopathol. 45:399–436. [DOI] [PubMed] [Google Scholar]
- Blackman RL, Eastop VF.. 2000. Aphids on the world’s crops: an identification and information guide. United Kingdom: John Wiley & Sons Ltd. [Google Scholar]
- Bos JIB, et al. , 2010. A functional genomics approach identifies candidate effectors from the aphid species Myzus persicae (green peach aphid). PLoS Genet. 611:e1001216.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, et al. , 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carolan JC, et al. , 2011. Predicted effector molecules in the salivary secretome of the pea aphid (Acyrthosiphon pisum): a dual transcriptomic/proteomic approach. J Proteome Res. 104:1505–1518. [DOI] [PubMed] [Google Scholar]
- Carolan JC, Fitzroy CIJ, Ashton PD, Douglas AE, Wilkinson TL.. 2009. The secreted salivary proteome of the pea aphid Acyrthosiphon pisum characterised by mass spectrometry. Proteomics 99:2457–2467. [DOI] [PubMed] [Google Scholar]
- Chaudhary R, Atamian HS, Shen Z, Briggs SP, Kaloshian I.. 2014. GroEL from the endosymbiont Buchnera aphidicola betrays the aphid by triggering plant defense. Proc Natl Acad Sci U S A. 11124:8919–8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant GC, Wolfe KH.. 2008. Turning a hobby into a job: how duplicated genes find new functions. Nat Rev Genet. 912:938–950. [DOI] [PubMed] [Google Scholar]
- Cox J, et al. , 2011. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 104:1794–1805. [DOI] [PubMed] [Google Scholar]
- Cristofoletti PT, Ribeiro AF, Deraison C, Rahbé Y, Terra WR.. 2003. Midgut adaptation and digestive enzyme distribution in a phloem feeding insect, the pea aphid Acyrthosiphon pisum. J Insect Physiol. 491:11–24. [DOI] [PubMed] [Google Scholar]
- Dobin A, et al. , 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 291:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvaux L, et al. , 2015. Dynamics of copy number variation in host races of the pea aphid. Mol Biol Evol. 321:63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyrløv Bendtsen J, Nielsen H, von Heijne G, Brunak S.. 2004. Improved prediction of signal peptides: signalP 3.0. J Mol Biol. 3404:783–795. [DOI] [PubMed] [Google Scholar]
- Elzinga DA, Jander G.. 2013. The role of protein effectors in plant–aphid interactions. Curr opin Plant Biol. 164:451–456. [DOI] [PubMed] [Google Scholar]
- Elzinga DA, De Vos M, Jander G.. 2014. Suppression of plant defenses by a Myzus persicae (green peach aphid) salivary effector protein. Mol Plant Microbe Interact. 277:747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyres I, et al. , 2017. Targeted re-sequencing confirms the importance of chemosensory genes in aphid host race differentiation. Mol Ecol. 261:43–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyres I, et al. , 2016. Differential gene expression according to race and host plant in the pea aphid. Mol. Ecol 2517:4197–4215. [DOI] [PubMed] [Google Scholar]
- Febvay G, Delobel B, Rahbé Y.. 1988. Influence of the amino acid balance on the improvement of an artificial diet for a biotype of Acyrthosiphon pisum (Homoptera: Aphididae). Can J Zool. 6611:2449–2453. [Google Scholar]
- Forister ML, et al. , 2015. The global distribution of diet breadth in insect herbivores. Proc Natl Acad Sci U S A. 1122:442–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furch ACU, van Bel AJ, Will T.. 2015. Aphid salivary proteases are capable of degrading sieve-tube proteins. J Exp Bot. 662:533–539. [DOI] [PubMed] [Google Scholar]
- Gaupels F, et al. , 2008. Nitric oxide generation in Vicia faba phloem cells reveals them to be sensitive detectors as well as possible systemic transducers of stress signals. New Phytol. 1783:634–646. [DOI] [PubMed] [Google Scholar]
- Götz S, et al. , 2008. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 3610:3420–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo K, et al. , 2014. Characterization of an aphid-specific, cysteine-rich protein enriched in salivary glands. Biophys Chem. 189:25–32. [DOI] [PubMed] [Google Scholar]
- Guy E, et al. , 2016. Optimization of agroinfiltration in Pisum sativum provides a new tool for studying the salivary protein functions in the pea aphid complex. Front Plant Sci. 7:1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas BJ, et al. , 2009. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 4617262:393–398. [DOI] [PubMed] [Google Scholar]
- Harris KF, Maramorosch K.. 2014. Aphids as virus vectors. Amsterdam: Elsevier. [Google Scholar]
- Hubner NC, et al. , 2010. Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J Cell Biol. 1894:739–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz N. 2011. Ehrlich and Raven Revisited: mechanisms underlying codiversification of plants and enemies. Annu Rev Ecol Evol Syst. 421:71–89. [Google Scholar]
- Jaquiéry J, et al. , 2018. Disentangling the causes for faster-X evolution in aphids. Genome Biol Evol. 10:507–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaquiéry J, et al. , 2012. Genome scans reveal candidate regions involved in the adaptation to host plant in the pea aphid complex. Mol Ecol. 2121:5251–5264. [DOI] [PubMed] [Google Scholar]
- Jaubert-Possamai S, et al. , 2010. Expansion of the miRNA pathway in the hemipteran insect Acyrthosiphon pisum. Mol Biol Evol. 275:979–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang RHY, Tripathy S, Govers F, Tyler BM.. 2008. RXLR effector reservoir in two Phytophthora species is dominated by a single rapidly evolving superfamily with more than 700 members. Proc Natl Acad Sci U S A. 10512:4874–4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, et al. , 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 309:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Kuma K, Toh H, Miyata T.. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 332:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov FA. 2012. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc Biol Sci 2791749:5048–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriventseva EV, et al. , 2015. OrthoDB v8: update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res. 43(D1):D250–D256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer ELL.. 2001. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol. 3053:567–580. [DOI] [PubMed] [Google Scholar]
- Law CW, Alhamdoosh M, Su S, Smyth GK, Ritchie ME.. 2016. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Research 5:1408.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK.. 2014. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 152:R29.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P.. 2007. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 231:127–128. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W.. 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 307:923–930. [DOI] [PubMed] [Google Scholar]
- Linz LB, Liu S, Chougule NP, Bonning BC.. 2015. In vitro evidence supports membrane alanyl aminopeptidase N as a receptor for a plant virus in the pea aphid vector. J Virol. 8922:11203–11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löytynoja A, Goldman N.. 2005. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 10230:10557–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathers TC, et al. , 2017. Rapid transcriptional plasticity of duplicated gene clusters enables a clonally reproducing aphid to colonise diverse plant species. Genome Biol. 18:27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno A, et al. , 2011. Aphids secrete watery saliva into plant tissues from the onset of stylet penetration. Entomol Exp Appl. 1392:145–153. [Google Scholar]
- Mullen SP, Shaw KL.. 2014. Insect speciation rules: unifying concepts in speciation research. Annu Rev Entomol. 591:339–361. [DOI] [PubMed] [Google Scholar]
- Mutti NS, et al. , 2008. A protein from the salivary glands of the pea aphid, Acyrthosiphon pisum, is essential in feeding on a host plant. Proc Natl Acad Sci U S A. 10529:9965–9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutti NS, Park Y, Reese JC, Reeck GR.. 2006. RNAi Knockdown of a salivary transcript leading to lethality in the pea aphid, Acyrthosiphon pisum. J Insect Sci. 6:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naessens E, et al. , 2015. A secreted MIF cytokine enables aphid feeding and represses plant immune responses. Curr Biol. 2514:1898–1903. [DOI] [PubMed] [Google Scholar]
- Nicholson SJ, et al. , 2015. The genome of Diuraphis noxia, a global aphid pest of small grains. BMC Genomics 16:429.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouhaud P, et al. , 2014. Genomic regions repeatedly involved in divergence among plant-specialized pea aphid biotypes. J Evol Biol. 279:2013–2020. [DOI] [PubMed] [Google Scholar]
- Pan Y, Zhu J, Luo L, Kang L, Cui F.. 2015. High expression of a unique aphid protein in the salivary glands of Acyrthosiphon pisum. Physiol Mol Plant Pathol. 92:175–180. [Google Scholar]
- Peccoud J, Mahéo F, de la Huerta M, Laurence C, Simon J-C.. 2015. Genetic characterisation of new host-specialised biotypes and novel associations with bacterial symbionts in the pea aphid complex. Insect Conserv Divers. 85:484–492. [Google Scholar]
- Peccoud J, et al. , 2010. Evolutionary history of aphid-plant associations and their role in aphid diversification. C R Biol. 333(6–7):474–487. [DOI] [PubMed] [Google Scholar]
- Peccoud J, Ollivier A, Plantegenest M, Simon J-C.. 2009. A continuum of genetic divergence from sympatric host races to species in the pea aphid complex. Proc Natl Acad Sci U S A. 10618:7495–7500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peccoud J, Simon J-C, McLaughlin HJ, Moran NA.. 2009. Post-Pleistocene radiation of the pea aphid complex revealed by rapidly evolving endosymbionts. Proc Natl Acad Sci U S A. 10638:16315–16320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H.. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 810:785.. [DOI] [PubMed] [Google Scholar]
- Pierleoni A, Martelli PL, Casadio R.. 2008. PredGPI: a GPI-anchor predictor. BMC Bioinformatics 9:392.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitino M, Coleman AD, Maffei ME, Ridout CJ, Hogenhout SA.. 2011. Silencing of aphid genes by dsRNA feeding from plants. PLoS One 610:e25709.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitino M, Hogenhout SA.. 2013. Aphid protein effectors promote aphid colonization in a plant species-specific manner. Mol Plant Microbe Interact. 261:130–139. [DOI] [PubMed] [Google Scholar]
- Pohlert T. 2014. The Pairwise Multiple Comparison of Mean Ranks Package (PMCMR). R package. http://CRAN.R-project.org/package=PMCMR, last accessed May 22, 2018.
- Powell G, Tosh CR, Hardie J.. 2006. Host plant selection by aphids: behavioral, evolutionary, and applied perspectives. Annu Rev Entomol. 51:309–330. [DOI] [PubMed] [Google Scholar]
- Price DRG, Duncan RP, Shigenobu S, Wilson ACC.. 2011. Genome expansion and differential expression of amino acid transporters at the aphid/Buchnera symbiotic interface. Mol Biol Evol. 2811:3113–3126. [DOI] [PubMed] [Google Scholar]
- Qi M, et al. , 2016. A small cysteine-rich protein from the Asian soybean rust fungus, Phakopsora pachyrhizi, suppresses plant immunity. PLoS Pathog. 129:e1005827.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahbé Y, Sauvion N, Febvay G, Peumans WJ, Gatehouse AMR.. 1995. Toxicity of lectins and processing of ingested proteins in the pea aphid Acyrthosiphon pisum. Entomol Exp Appl. 762:143–155. [Google Scholar]
- R-Core Team. 2017. R: A Language and Environment for Statistical Computing. https://www.R-project.org/, last accessed June 12, 2018.
- Rispe C, et al. , 2007. Large gene family expansion and variable selective pressures for cathepsin B in aphids. Mol Biol Evol. 251:5–17. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, et al. , 2015. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 437:e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK.. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 261:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Oshlack A.. 2010. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 113:R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez PA, Escudero-Martinez C, Bos JIB.. 2017. an aphid effector targets trafficking protein VPS52 in a host-specific manner to promote virulence. Plant Physiol. 1733:1892–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela I, Ashkenazy H, Katoh K, Pupko T.. 2015. GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 43(W1):W7–W14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smadja C, Shi P, Butlin RK, Robertson HM.. 2009. Large gene family expansions and adaptive evolution for odorant and gustatory receptors in the pea aphid, Acyrthosiphon pisum. Mol Biol Evol. 269:2073–2086. [DOI] [PubMed] [Google Scholar]
- Smadja CM, et al. , 2012. Large-scale candidate gene scan reveals the role of chemoreceptor genes in host plant specialization and speciation in the pea aphid. Evolution 669:2723–2738. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2221:2688–2690. [DOI] [PubMed] [Google Scholar]
- Stergiopoulos I, et al. , 2012. In silico characterization and molecular evolutionary analysis of a novel superfamily of fungal effector proteins. Mol Biol Evol. 2911:3371–3384., [DOI] [PubMed] [Google Scholar]
- The International Aphid Genomics Consortium 2010. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 82:e1000313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe P, Cock PJ, Bos J.. 2016. Comparative transcriptomics and proteomics of three different aphid species identifies core and diverse effector sets. BMC Genomics 171:172.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varden FA, De la Concepcion JC, Maidment JH, Banfield MJ.. 2017. Taking the stage: effectors in the spotlight. Curr Opin Plant Biol. 38:25–33. [DOI] [PubMed] [Google Scholar]
- Wang W, et al. , 2015. Armet is an effector protein mediating aphid–plant interactions. FASEB J. 295:2032–2045. [DOI] [PubMed] [Google Scholar]
- Wang W, et al. , 2015. Angiotensin-converting enzymes modulate aphid–plant interactions. Sci Rep. 51: 8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger JA, et al. , 2017. Whole genome sequence of the soybean aphid, Aphis glycines. Insect Biochem Mol Biol. doi: 10.1016/j.ibmb.2017.01.005. [DOI] [PubMed] [Google Scholar]
- Wiens JJ, Lapoint RT, Whiteman NK.. 2015. Herbivory increases diversification across insect clades. Nat Commun. 61: 8370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will T, Vilcinskas A.. 2015. The structural sheath protein of aphids is required for phloem feeding. Insect Biochem Mol Biol. 57:34–40. [DOI] [PubMed] [Google Scholar]
- Will T, Furch ACU, Zimmermann MR.. 2013. How phloem-feeding insects face the challenge of phloem-located defenses. Front Plant Sci. 4:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Baldwin IT.. 2010. New insights into plant responses to the attack from insect herbivores. Annu Rev Genet. 44:1–24. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 248:1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R.. 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol. 171:32–43. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R.. 2002. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 196:908–917. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen A-MK.. 2000. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wong WSW, Nielsen R.. 2005. Bayes Empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 224:1107–1118. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Fan J, Sun J, Chen J.. 2015. Cloning and RNA interference analysis of the salivary protein C002 gene in Schizaphis graminum. J Integr Agric. 144:698–705. [Google Scholar]
- Zhao C, et al. , 2015. A massive expansion of effector genes underlies gall-formation in the wheat pest Mayetiola destructor. Curr Biol. 255:613–620. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.