

Abstract
Exo–endocytotic turnover of synaptic vesicles (SVs) at synapses between hippocampal neurons in culture was examined by electron microscopy (EM). We carried out photoconversion (PC) of the fluorescent endocytotic marker FM 1-43 by using 3,3′-diaminobenzidine to convert the dye signal into an electron-dense product. Electron-dense products were located almost exclusively in SVs, whose densities were bimodally distributed in two sharply demarcated populations, PC-positive (PC+) and PC-negative (PC−). The median densities of these populations did not vary with the proportion of vesicles stained within a presynaptic terminal (bouton). The proportion of PC+ SVs remained constant across consecutive thin sections of single boutons, but varied greatly from one bouton to another, indicating marked heterogeneity in exo-endocytotic activity. Our experiments indicated that only a minority of SVs were stained in most boutons after stimuli known to cause complete turnover of the functional vesicular pool. A direct spatial correlation was found between FM 1-43 fluorescent spots seen with light microscopy and PC+ boutons by EM. The correlation was clearer in isolated boutons than in clusters of boutons. Photoconversion in combination with FM dyes allows clarification of important aspects of vesicular traffic in central nervous system nerve terminals.
Synaptic vesicle (SV) turnover in presynaptic terminals of central nervous system (CNS) neurons is critical for information transfer within the brain. Continued progress in understanding exocytosis and endocytosis at the level of single vesicles depends critically on physiological studies using light microscopy and ultrastructural analysis using electron microscopy (EM). FM 1-43 and structurally related styryl dyes offer a way to tie such approaches together. These molecules undergo a large increase in quantum yield when taken up into membranes. Activity-dependent sequestration of the dyes in presynaptic vesicles and release on subsequent exocytosis have been monitored as increases and decreases in nerve terminal fluorescence (1–4). Key properties of FM 1-43 as a fluorescent probe were demonstrated by Betz and colleagues (1, 5), working at the frog neuromuscular junction (NMJ), the same preparation used by Heuser and Reese (6) in their pioneering studies of vesicle turnover. Betz and colleagues went on to perform photoconversion of FM 1-43 and FM 2-10 by using photolysis of the dyes in the presence of 3,3′-diaminobenzidine (DAB) to cause an electron-dense reaction product in synaptic vesicles (7, 8). At CNS synapses, like the NMJ, FM 1-43 has been widely used as an optical marker for studies of vesicle dynamics by the light microscope (2, 9, 10). Photoconversion of FM 1-43 has also provided valuable information about the number of vesicles in the readily releasable pool (11).
This paper describes our efforts to use FM 1-43 photoconversion to strengthen links between light microscopic and EM approaches to vesicular properties. Working with rat hippocampal neurons in culture, we modified existing experimental procedures to improve ultrastructural preservation while minimizing photoconversion because of background fluorescence. Electron-dense products were essentially confined to the lumen of synaptic vesicles, and large differences in staining intensity of photoconverted and nonphotoconverted vesicles allowed us to unambiguously identify vesicles that had taken up dye. Terminals containing photoconverted vesicles corresponded closely to FM 1-43 fluorescent spots visualized under the light microscope. After stimulation known to cause the functional vesicular pool to turn over completely, only a minority of vesicles were stained.
Materials and Methods
Preparation of Cultured Neurons.
Hippocampal neurons in culture were prepared from 1- to 4-day-old Sprague–Dawley rats as described (4), and used between 14 and 43 days in vitro (div).
Staining with FM Dyes.
Neurons were loaded with 15 μM FM 1-43 by either field stimulation or high K+ application. With field stimulation, neurons were exposed to FM 1-43 during and up to 60 s after stimulation with silver chloride electrodes (2–120 s, 10 or 20 Hz; see figure legends for details). After loading, neurons were washed with low Ca2+ solution for 5–10 min. With high K+ application, neurons were exposed to 70 mM K+ solution for 90 s in the presence of FM 1-43, washed in low Ca2+ solution for 10–15 min, challenged with 70 mM K+ solution without FM 1-43 for 120 s (destaining), restained with FM 1-43 by using the same condition, and washed again with low Ca2+ solution for 10–15 min.
Photoconversion.
After FM staining, neurons were fixed with 2% glutaraldehyde in 100 mM sodium phosphate buffer (PB) for 20 min, and washed with 100 mM glycine (in PB) for 1 h. They were washed further in 100 mM ammonium chloride (in distilled water) for 5 min. After brief rinsing in PB, neurons were incubated in DAB (1 mg/ml in PB) for 10–20 min. Fluorescence excitation light was then continuously applied for 10–20 min in DAB solution. Neurons were washed in ice-cold PB, and processed for EM. Only nonsomatic regions were selected for analysis. This procedure differs from published protocols (7, 11) in that it uses glutaraldehyde as the main fixative to optimize preservation of ultrastructure under the harsh conditions of photoconversion, as well as subsequent washing with glycine and ammonium chloride to reduce the autofluorescence of glutaraldehyde and unwanted background photoconversion. Our protocol§ has been adopted in modified form in other studies (8, 12, 13).
Imaging.
Imaging was accomplished by either of the following two methods.
Confocal microscopy.
Fluorescence images were acquired by averaging four frames obtained at a spatial sampling of 160 nm per pixel and a dwell time of 2 μs per pixel through an Olympus 40× 1.3 numerical aperture objective by using a modified Bio-Rad MRC 500 confocal laser scanning unit coupled to a Zeiss IM-35 inverted microscope. The optical section thickness was ≈0.5 μm. Fluorescence images were acquired by using a total power of ≈15 μW of 488-nm light (measured at the back aperture) slightly overfilling the aperture. Photoconversion was accomplished with ≈150 μW of power with higher spatial sampling during the scan (40 nm per pixel).
Wide-field.
Images were acquired by using a video camera (SIT-66X, DAGE-MTI, Michigan City, IN) mounted on an inverted phase-contrast microscope (Diaphot 200, Nikon, Tokyo) equipped with epifluorescence system (100-W mercury bulb) and a Nikon 40× air objective (numerical aperture, 0.75). Eight-bit images were acquired through an image-acquisition board (IMAQ PCI-1408, National Instruments, Austin, TX). A standard fluorescein filter set was used (47°/40-nm exciter, 500-nm dichroic and 515-nm long-pass filter). Bright-field images were monitored every 10 s by averaging 10 frames. Shutter control, image acquisition, and analysis were done by using a program written by N.H. in LABVIEW (National Instruments).
Bright-field images were expressed in an 8-bit gray scale (white-255, black-0; Fig. 1F). Change in absorbance of photoconversion products (ΔA) was calculated by using Beer's law with modification.
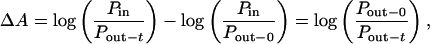 |
where Pin is power incident on the sample and Pout-t is power leaving the sample at time t. Pout-0 represents baseline power leaving the sample. We assumed constant shape of boutons and no interference among reaction products (14). To reduce effects of stray light (partition error), region of interest (ROI) was made small (3 × 3 to 4 × 4 pixels, 0.6 × 0.6 to 0.8 × 0.8 μm2) in comparison to bouton size.
Figure 1.

Photoconversion of FM 1-43 in cultured hippocampal neurons. (A) Phase-contrast image. Region shown by a yellow rectangle was repeatedly imaged in B–E. ✻, Acellular background (see F and G). (B) Fluorescence image after exposure to 15 μM FM 1-43 in 70 mM K+ solution for 90 s, followed by washout. Arrowheads, four puncta further analyzed in F and G (not marked). ∗∗, An FM 1-43-negative dendrite (cellular background; see F and G). (C–E) Time course of photoconversion in DAB solution. Elapsed time is shown after start of continuous fluorescence excitation. (F) Absolute bright-field density plotted against time for the 6 ROIs. Upper four lines correspond to ROIs with FM 1-43 fluorescence (arrowheads in B). (G) Data in F transformed into absorbance by using Beer's law (see Materials and Methods). For EM studies, photoconversion reactions were ended within the interval between the broken lines. (Inset) Slopes of change in absorbance. (H) Representative image of a photoconverted bouton, previously subjected to field stimulation (20 Hz for 60 s). Dye exposure during stimulation and an additional 60 s of rest. Arrow points to a PC+ structure that was docked at the active zone. Neurons in A–G and H were 16 and 21 div, respectively.
EM.
After photoconversion, neurons were processed for EM by using a Pelco 3450 microwave oven (Ted Pella, Redding, CA). Neurons were washed with 100 mM cacodylate buffer containing 150 mM sucrose for 5 min, postfixed with 1% osmium tetroxide in 100 mM cacodylate buffer containing 0.8% potassium ferricyanide (15) for 5 min on ice, and then given a 10-s pulse in the microwave. Neurons were then washed in distilled water for 5 min, stained en bloc with 2% aqueous uranyl acetate for 20 min, and given another 10-s pulse in the microwave. They were dehydrated in an ascending ethanol series (50%, 70%, 95%, 100% three times) in the microwave for 10 s each. Neurons were infiltrated in microwave in 1:2, 1:1, 2:1 (100% resin/100% ethanol) for 5 min each, and in 100% resin for 5 min by using EMbed 812 resin (Electron Microscopy Sciences, Fort Washington, PA). After overnight polymerization at 60°C, glass coverslips were removed by using hydrofluoric acid (16). Thin sections (50–70 nm) were cut parallel to the coverslip surface, in serial fashion, and mounted on Formvar-coated slot grids. Grids were poststained with 2% aqueous uranyl acetate and Sato's lead citrate (17) and examined on Philips 410, CM12, or JEOL 200CXII at 60-kV accelerating voltage.
Electron micrographs were digitally scanned with an 8-bit scale (white-0, black-255). For density analyses, brightness was adjusted by using PHOTOSHOP (Adobe Systems, San Jose, CA), such that intensities for cytoplasmic membrane and cytoplasm had similar median values (100–140 and 50–80). Luminal density (Fig. 2) was measured by averaging density over 3–5 pixels (2.8–4.7 nm) at the SV center. SV diameter was measured as the diameter of a circle fitted to the outer rim of SV membrane.
Figure 2.

Density profiles of SVs. (A) Overlay of line density profiles from 8 small clear SVs classified as PC−. No photoconversion was performed. (B) Overlay of line density profiles from 14 small structures classified as PC+. Centers of the lumen (A) and peaks of positive structures (B) were arbitrarily aligned at 40 nm on distance scale. (C) Histogram of luminal density for PC− (open bars) and PC+ SVs (filled bars) from a single photoconverted bouton (Upper) and a negative control (illuminated without prior exposure to FM 1-43, Lower, open bars). (D) Cumulative histograms of luminal density from 5 boutons. Dotted line represents the data from C. Continuous lines represent other boutons taken from the same electron micrograph. (E) Difference in median densities of PC− and PC+ SVs (Δ median density) plotted against proportion of PC+ SVs (%PC+ SVs). Data were obtained from specimens with field stimulation (FS, ●) or with high K+ stimulation (○). Linear regression line was drawn for all boutons (r = −0.54, P > 0.1, n = 8). (F) Distributions of SV diameters.
Solutions.
Tyrode solution contained 124 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 30 mM glucose, and 25 mM Hepes (pH, 7.4). Low Ca2+ solution was prepared with the following modification: 0.2 mM CaCl2, 5 mM MgCl2, 0.001 mM tetrodotoxin. High K+ solution (70 mM) was prepared by replacing 65 mM NaCl in Tyrode solution with equimolar KCl. Kynurenic acid (1 mM) or 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μM) and dl-2-amino-5-phosphonovaleric acid (AP5, 100 μM) were added to solutions.
Chemicals.
Other than FM 1-43 (Molecular Probes) and glutaraldehyde (Polysciences, Warrington, PA), chemicals were purchased from Sigma.
Results
Time Course of Photoconversion.
Fig. 1 illustrates the time course of photoconversion in cultured hippocampal neurons. A rectangular region containing a dendrite (Fig. 1A) is shown with FM 1-43 imaging in Fig. 1B. A bright-field image of the same dendrite did not show any dark regions (Fig. 1C). As neurons were progressively exposed to fluorescent light, FM 1-43 fluorescent puncta gradually faded (data not shown) and became darker as DAB reaction product formed (Fig. 1D). These boutons became intensely dark as photoconversion proceeded for 15 min (Fig. 1E). Cellular background devoid of FM 1-43 puncta showed only slight photoconversion (Figs. 1 B–E, ∗∗).
For quantitative analyses, we measured bright-field density in multiple ROIs (Fig. 1F). Density remained unchanged during the first 5 min, but after this latent period, all regions with FM 1-43 fluorescence (Fig. 1B, arrowheads) darkened. The latent period varied between 5–10 min in other neurons. We used Beer's law to determine changes in absorbance (Fig. 1G). The absorbance in ROIs at fluorescent puncta increased to a much greater degree than at nonfluorescent cellular (∗∗, Fig. 1 B, F, and G) or acellular regions (∗, Fig. 1 A , F, and G), although these eventually darkened as well. Typically, the rate of change of absorbance leveled off at a maximal slope (Fig. 1G Inset).
Further analysis was carried out to determine the relationship between initial FM 1-43 fluorescence and overall change in absorbance, for both fluorescent puncta and background regions. The correlation coefficients ranged between 0.55 and 0.93 with a mean of 0.78 ± 0.07 (n = 6, all with P < 0.01), demonstrating the consistency of photoconversion reaction. It was consistently found that fluorescence mapped to photoconverted boutons. However, some of the photoconversion product did not correspond to fluorescence at the light microscopic level, and was ascribed to mitochondrial darkening that was not related to vesicular recycling.
In this example, neurites became morphologically distorted beyond 15 min of illumination. At the EM level, boutons were so electron dense that ultrastructural detail was obscured (data not shown). In most experiments, we stopped the illumination when an identifiable reaction product appeared at FM 1-43 puncta (window between broken lines, Fig. 1G) but before there was appreciable photoconversion in cellular background.
A representative electron micrograph is shown in Fig. 1H. An isolated bouton contained numerous small clear SVs and made a synapse with a dendritic spine. Distributed among the SVs were small dark structures, some of which were docked at a single active zone (arrow). Because the dark structures were not found in negative controls (photoconverted in the absence of FM 1-43), we conclude that they represent SVs that were positively stained by photoconversion.
Analysis of Density Profiles of Single Vesicles.
We first gained quantitative information on single SVs by analyzing density profiles along a line (Fig. 2). In control experiments without illumination, the majority of identifiable structures were small clear SV. The density profile was characterized by a valley flanked by two peaks, corresponding to vesicular lumen and membrane (Fig. 2A). Similar analyses were performed in photoconverted boutons. A small PC+ structure showed a central peak whose density was far greater than that seen at any point along the profile of nonilluminated SVs (Fig. 2B). The majority of positive structures were identified as SVs. First, small PC+ structures were often the most numerous organelles in presynaptic terminals (Figs. 1H and 4 E–G), as were SVs in negative control. Second, PC+ structures were similar in size to SVs in negative controls (see below). Third, other organelles, such as mitochondria, large dense-core vesicles, and endosomes, were found at a frequency of <3 per section, not numerous enough to account for the PC+ profiles.
Figure 4.

Correspondence between FM 1-43 fluorescence, photoconversion, and EM. (A) Nomarski differential interference contrast microscopy image of a neuron (43 div). Boxed region was enlarged in B and C. (B) Confocal fluorescence image of boxed region in A. Field stimulation for 40 s at 20 Hz and additional dye exposure for 30 s after stimulus, followed by wash. A rectangle encompasses two fluorescent puncta. (C) Bright-field image of B after photoconversion. (E–G) EM of three thin sections obtained from the region. Numbers 1 and 2 indicate presynaptic structures identified as fluorescent puncta in D. The more fluorescent structure (1) contained more PC+ vesicles. Note that mitochondrion (m) was not photoconverted. Scale bar in C applies to B and C. Scale bar in G applies to E–G.
Unequivocal Distinctions Between PC− and PC+ Vesicles.
A basic question is how clear a distinction can be made between SVs that have undergone photoconversion and those that have not. Accordingly, we carried out an analysis of line density profiles of all of the vesicles in a section from a large bouton (n = 139). The number of distinct peaks was used to classify SVs as PC− (two peaks, Fig. 2A) or PC+ (one peak, Fig. 2B). Fig. 2C plots the electron density at the SV center for PC− SVs (open bars, n = 69) and PC+ SVs (filled bars, n = 70) from a photoconverted sample (Upper). The respective distributions showed virtually no overlap, and had median densities of 110 and 224, a factor of 2 apart. The median density of the PC− SVs (110) agreed well with that of sham photoconverted vesicles (112), determined in a sample that was illuminated without previous exposure to FM 1-43 (Lower, open bars, n = 115).
Pooled data from five boutons, presented as cumulative histograms (Fig. 2D), show several key features. There was always a clear separation between PC− and PC+ SVs, as indicated by the leveling off at intermediate densities. The median densities of each of the PC− and PC+ peaks were very similar across boutons. Thus, like the shape of the profile, the relative density of its central region would have been sufficient to allow unambiguous distinctions. On the other hand, the relative sizes of negative and positive peaks varied widely from one bouton to the next, ranging from a ratio of 1:2 to 2:1 (≈30% to ≈60%). Because this variability was found even in the same electron micrograph, it must be attributed to a genuine bouton-to-bouton variability in the likelihood of FM 1-43 uptake. There was no correlation between the proportion of PC+ SVs and the difference in the median densities of negative and positive SVs (Fig. 2E, r = −0.54, P > 0.1, n = 8). Additional features of these results support the idea that photoconversion is an all-or-none process at the level of individual SVs (see Discussion).
Because SV diameter determines volume, and, in turn, influences neurotransmitter content (18), it would be important to know whether SVs that underwent exo–endocytosis showed any deviation in size from that of the SV population as a whole. The median diameter of PC+ SVs was 37.2 nm, slightly smaller than that of PC− SVs (39.5 nm; Fig. 2F) or SVs in a negative control in which no treatment was performed (39.9 nm). Although statistically significant (P < 0.01 by Kolmogorov–Smirnov test), the discrepancy was far smaller than the standard deviation of any of distributions (8.1, 10.0, and 8.2 nm respectively). PC− SVs in photoconverted samples were no different from SVs in nonilluminated controls (P > 0.1).
Interbouton Variability Greatly Exceeds Intrabouton Variability.
The proportion of PC+ SVs varied greatly among different boutons. For example, a variability ranging from 1% to ≈50% is illustrated in Fig. 3A. How much of this variability arose from uncertainty in the process of photoconversion as opposed to genuine biological variability? We addressed this question by examining the percentage of PC+ SVs in consecutive sections through individual boutons. If the main source of variability were the stochastic nature of photoconversion, operating at the single vesicle level, one would expect similar variability among multiple sections of a given bouton as among sections of different boutons. Fig. 3A plots data from a set of 27 boutons from one coverslip, each represented by a string of connected symbols. Intrabouton variation was small, as quantified by values of the coefficient of variation (CV) for the proportion of PC+ SVs. The median CV value for all of the individual boutons in the set was 21.0%; similar CVs were found for the three cases where the most consecutive sections (7) were obtained (15.5%, 17.6%, and 19.8%). In contrast, interbouton variability across the entire set of boutons was much greater, with an overall CV of 75.4%. These results suggested that the photoconversion process was reliable and that bouton-to-bouton differences largely reflected a genuine physiological variability.
Figure 3.

Uniformity of photoconversion within individual boutons, and high variability among different boutons. (A Left) Plots of proportion of PC+ SVs in each section of a bouton. Each symbol represents data from an individual bouton (n = 27). (Right) Filled circles and vertical bars show intrabouton variability, seen in the mean and SD for 3 cases where a bouton was sampled with seven distinct sections (7-section boutons). Filled triangle and vertical bars show interbouton variability, expressed as mean and SD of the mean values for all boutons. (B) Comparison of interbouton variability with different stimulation. Mean and SD are from three specimens. FS, field stimulation (dye exposure during stimulation at 10 Hz for 120 s followed by an additional 60 s of dye exposure, 24 div). High K+, high K+ application for 90 s (17 and 16 div). Number of boutons examined is indicated.
Interbouton variability was compared among different stimulation protocols. One coverslip of neurons was loaded with FM 1-43 by field stimulation (FS in Fig. 3B; n = 124 boutons), and two others by high K+ (High K+ in Fig. 3B; n = 92 and 159). Median values of the proportion of PC+ SV ranged from 9.6% to 17.8%. Once again, the CV values were large, ranging between 76.6% and 102.6%.
Correspondence Between FM 1-43 Fluorescence and Photoconversion EM.
We turned next to how closely FM 1-43 fluorescence corresponds to photoconverted vesicles seen with EM. A boxed region of a neuronal field (Fig. 4A) was enlarged and shown as a fluorescence image in Fig. 4B. After photoconversion, the same field was viewed with bright-field optics (Fig. 4C). A precise correspondence between FM 1-43 puncta and photoconversion products was found (Fig. 4 B and C). A rectangular box in Fig. 4B is enlarged in Fig. 4D, which highlights two puncta of different sizes (labeled 1 and 2). The same structures were visible at the EM level (Fig. 4 E–G). In addition to spatial correspondence, there was also a good correlation between the intensity of fluorescence and the density of photoconverted product (r = 0.99, P < 0.01, n = 5). Another notable feature of this sample was that the two puncta were actually fused together as a single large bouton, albeit with two distinguishable clusters of synaptic vesicles (Fig. 4 E–G).
Complexity of Hippocampal Neurons in Culture.
A precise fluorescence-photoconversion correlation was obtained at nonbranching points as shown above. However, as illustrated by Fig. 5, the correlation was less stringent at branching points or multineuritic intersections where synaptic boutons were clustered (ref. 19; and D. Baranes, personal communication). FM 1-43 fluorescence (Fig. 5A) and PC+ structures (Fig. 5B) corresponded reliably in most boutons (arrows in Fig. 5 A and B), but less reliably in other cases. A possible cause for imperfect correlation is that not all of the boutons were in the same plane of the thin section.
Figure 5.

Analysis of a bouton cluster. (A) Confocal FM 1-43 fluorescence image of neurons (34 div). Field stimulation for 2 s at 10 Hz, followed by 60 s of additional dye exposure. White arrows point to FM 1-43 fluorescent puncta. (B) Corresponding electron micrograph of the same field in A. Mitochondria (m) were photoconverted positively in this sample. ∗ In B denotes a bouton observed in multiple thin sections (1–8). (1–8) Selected serial sections. Arrows in 2 indicate two active zones, visible in panels 1–4. Upper active zone was further seen in 5 and 6. Arrowheads in 4 indicate PC+ SVs. Scale bar under 8 applies to 1–8.
A nearly complete set of serial sections is shown for a bouton in Fig. 5 (marked by ✻), illustrating the structural complexity sometimes observed. This terminal appeared to contain two independent active zones (Fig. 5, panel 2, arrows). PC+ SVs (e.g., panel 4, arrowheads) were widely distributed throughout the overall vesicle pool, suggesting that functional SVs were shared between both active zones.
Discussion
When we used photoconversion with FM 1-43 (7, 8, 11–13), we obtained robust labeling of presynaptic boutons at synapses between cultured hippocampal neurons. When the photoconversion was monitored with online absorbance measurements and illumination was terminated at an appropriately early point, the DAB reaction product was not found in the cytoplasm, but was largely restricted to small clear SVs identified on the basis of their round profile, external diameter and abundance. Excellent correspondence in position and intensity was observed between puncta of FM dye fluorescence visualized by light microscopy and foci of photoconversion product detected by EM. Thus, photoconversion proved to be a reliable method for marking functional synapses or individual vesicles at active synapses.
Key Properties of Vesicular Photoconversion.
Several characteristics of the photoconversion procedure set out here supported its usefulness for quantitative analysis of vesicular trafficking. The uniformity of the photoconversion reaction was reflected by the relative consistency in the proportion of PC+ vesicles found in multiple sections through the same nerve terminals (Fig. 3A). This supported the use of data from single sections as an index of the behavior of the bouton as a whole. Photoconversion of individual SVs appeared to be independent of the presence or absence of photoconverted product in neighboring SVs. The adequacy of stimulation methods was verified by concordance between data obtained with electrical stimulation and with high K+ application, each applied for long enough to evoke multiple cycles of exo–endocytosis (20, 21). Although K+-rich solutions produce a continuous depolarization and considerably greater presynaptic Ca2+ entry than stimulus trains (J. Pyle and E. Piedras-Rentería, personal communication), both procedures evoked similar effects on FM dye uptake detected by photoconversion (Fig. 3B), consistent with near-maximal stimulation of vesicular turnover in both cases.
Our experiments supported the idea that the photoconversion reaction is an abrupt process at the level of single vesicles, as previously suggested by Betz and colleagues at the neuromuscular junction (7). In our study of hippocampal boutons, the electron densities of individual PC+ SVs were narrowly distributed, with hardly more spread than that found for the distribution of PC− SVs (Fig. 2C). This argues against the slow, graded formation of electron-dense products, which would be expected to produce a much greater dispersion. A stepwise, saturating photoconversion in individual vesicles would be expected if the reactive oxygen species arising from fluorescent excitation of FM dye triggered the nucleation and/or assembly of DAB (22), probably in a nonlinear manner. Under light microscopy, the absorbance of presynaptic boutons increased in an apparently smooth fashion (Fig. 1G), as predicted if increments at the level of individual vesicles were too small to be clearly resolved as single steps.
Interbouton Heterogeneity.
Interbouton comparisons in thin sections revealed a large variability in exo–endocytotic activity from one bouton to the next (Fig. 3). This result was in line with the wide variability in FM 1-43 fluorescent intensity at presynaptic terminals in hippocampal cultures, previously studied by light microscopy (9, 23).
Most of our experiments relied on monitoring of the progression of photoconversion by light microscopy, seen as darkening of dye-stained boutons. We deliberately stopped the illumination near the point when the rate of change of absorbance was maximal, before the signal leveled off. This ensured that extravesicular photoconversion was negligible, but it left open the possibility that some fraction of the FM 1-43-containing vesicles were not photoconverted. Thus, the number of PC+ vesicles should be proportional to, but not necessarily identical to, the number that took up dye. Some information about the actual percentage of vesicles undergoing exo–endocytotic turnover can be derived from experiments in which illumination was more aggressive, enough to cause ≈100% photoconversion in a few of the nerve terminals. Under such conditions, most boutons contained a substantial percentage of unstained vesicles, supporting the general idea that a significant fraction of vesicles may be spared from turnover even under strong stimulation (24).
Conclusion
Photoconversion of FM 1-43 provides a reliable way to link light and electron microscopic studies of nerve terminals of central neurons. It could readily be applied in conjunction with fluorescence imaging or electrical recordings to allow physiological studies to be followed up with microanatomical analysis. Our results illustrate the usefulness of the approach to investigations at the level of single nerve terminals or even individual vesicles. Future applications might include studies of diverse aspects of synapses such as the localization of readily releasable and recycling pools within a bouton.
Acknowledgments
We thank Dr. W. J. Betz for help in the initial stage of this research, Drs. Mark Ellisman, John Heuser, Thomas S. Reese, Harald Reuter, and Noam Ziv for comments, and Frances Thomas, Nafisa Ghori, and Dr. Thomas S. Reese for use of the EM facility. This work was supported by grants from the National Institutes of Health (to T.A.R., S.J.S., and R.W.T.), the Silvio Conte National Institute of Mental Health Center for Neuroscience Research (S.J.S. and R.W.T.), the Mathers Charitable Trust (to S.J.S. and R.W.T.), and the McKnight Foundation (to R.W.T.). T.A.R. is an Alfred P. Sloan Research Fellow.
Abbreviations
- EM
electron microscopy
- PC+
photoconversion-positive
- PC−
photoconversion-negative
- SV
synaptic vesicle
- div
days in vitro
- CNS
central nervous system
- DAB
3,3′-diaminobenzidine
- ROI
region of interest
Footnotes
Harata, N., Buchanan, J. & Tsien, R. W. (1998) Soc. Neurosci. Abstr. 24, 77.
References
- 1.Betz W J, Bewick G S. Science. 1992;255:200–203. doi: 10.1126/science.1553547. [DOI] [PubMed] [Google Scholar]
- 2.Murthy V N, Stevens C F. Nat Neurosci. 1999;2:503–507. doi: 10.1038/9149. [DOI] [PubMed] [Google Scholar]
- 3.Lagnado L, Gomis A, Job C. Neuron. 1996;17:957–967. doi: 10.1016/s0896-6273(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 4.Ryan T A, Reuter H, Wendland B, Schweizer F E, Tsien R W, Smith S J. Neuron. 1993;11:713–724. doi: 10.1016/0896-6273(93)90081-2. [DOI] [PubMed] [Google Scholar]
- 5.Betz W J, Mao F, Bewick G S. J Neurosci. 1992;12:363–375. doi: 10.1523/JNEUROSCI.12-02-00363.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heuser J E, Reese T S. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henkel A W, Lübke J, Betz W J. Proc Natl Acad Sci USA. 1996;93:1918–1923. doi: 10.1073/pnas.93.5.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richards D A, Guatimosim C, Betz W J. Neuron. 2000;27:551–559. doi: 10.1016/s0896-6273(00)00065-9. [DOI] [PubMed] [Google Scholar]
- 9.Ryan T A, Reuter H, Smith S J. Nature (London) 1997;388:478–482. doi: 10.1038/41335. [DOI] [PubMed] [Google Scholar]
- 10.Klingauf J, Kavalali E T, Tsien R W. Nature (London) 1998;394:581–585. doi: 10.1038/29079. [DOI] [PubMed] [Google Scholar]
- 11.Schikorski T, Stevens C F. Nat Neurosci. 2001;4:391–395. doi: 10.1038/86042. [DOI] [PubMed] [Google Scholar]
- 12.Teng H, Cole J C, Roberts R L, Wilkinson R S. J Neurosci. 1999;19:4855–4866. doi: 10.1523/JNEUROSCI.19-12-04855.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teng H, Wilkinson R S. J Neurosci. 2000;20:7986–7993. doi: 10.1523/JNEUROSCI.20-21-07986.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strobel H A. Chemical Instrumentation: A Systematic Approach. Menlo Park, CA: Addison-Wesley; 1973. [Google Scholar]
- 15.McDonald K. J Ultrastruct Res. 1984;86:107–118. doi: 10.1016/s0022-5320(84)80051-9. [DOI] [PubMed] [Google Scholar]
- 16.Buchanan J, Sun Y-A, Poo M-M. J Neurosci. 1989;9:1540–1554. doi: 10.1523/JNEUROSCI.09-05-01540.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato T. J Electron Microsc. 1968;17:158–159. [PubMed] [Google Scholar]
- 18.Bruns D, Riedel D, Klingauf J, Jahn R. Neuron. 2000;28:205–220. doi: 10.1016/s0896-6273(00)00097-0. [DOI] [PubMed] [Google Scholar]
- 19.Kavalali E T, Klingauf J, Tsien R W. Proc Natl Acad Sci USA. 1999;96:12893–12900. doi: 10.1073/pnas.96.22.12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryan T A, Smith S J. Neuron. 1995;14:983–989. doi: 10.1016/0896-6273(95)90336-4. [DOI] [PubMed] [Google Scholar]
- 21.Liu G, Tsien R W. Nature (London) 1995;375:404–408. doi: 10.1038/375404a0. [DOI] [PubMed] [Google Scholar]
- 22.Deerinck T J, Martone M E, Lev-Ram V, Green D P L, Tsien R Y, Spector D L, Huang S, Ellisman M H. J Cell Biol. 1994;126:901–910. doi: 10.1083/jcb.126.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Murthy V N, Sejnowski T J, Stevens C F. Neuron. 1997;18:599–612. doi: 10.1016/s0896-6273(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 24.Kuromi H, Kidokoro Y. Neuron. 1998;20:917–925. doi: 10.1016/s0896-6273(00)80473-0. [DOI] [PubMed] [Google Scholar]