

Abstract
Purpose
GI stromal tumors (GISTs) are commonly associated with somatic mutations in KIT and PDGFRA. However, a subset arises from mutations in NF1, most commonly associated with neurofibromatosis type 1. We define the anatomic distribution of NF1 alterations in GIST.
Methods
We describe the demographic/clinicopathologic features of 177 patients from two institutions whose GISTs underwent next-generation sequencing of ≥ 315 cancer-related genes.
Results
We initially identified six (9.7%) of 62 GISTs with NF1 genomic alterations from the first cohort. Of these six patients, five (83.3%) had unifocal tumors at the duodenal-jejunal flexure (DJF). Two additional patients with DJF GISTs had non-NF1 (KIT and BRAF) genomic alterations. After excluding one DJF GIST with an NF1 single nucleotide polymorphism, four (57.1%) of seven sequenced DJF tumors demonstrated deleterious NF1 alterations, whereas only one (1.8%) of 55 sequenced non-DJF GISTs had a deleterious NF1 somatic mutation (P < .001). One patient with DJF GIST had a germline NF1 variant that was associated with incomplete penetrance of clinical neurofibromatosis type 1 features along with a somatic NF1 mutation. Of the five DJF GISTs with any NF1 alteration, three (60%) had KIT mutations, and three (60%) had Notch pathway mutations (NOTCH2, MAML2, CDC73). We validated these findings in a second cohort of 115 GISTs, where two (40%) of five unifocal NF1-mutated GISTs arose at the DJF, and one of these also had a Notch pathway mutation (EP300).
Conclusion
Broad genomic profiling of adult GISTs has revealed that NF1 alterations are enriched in DJF GISTs. These tumors also may harbor concurrent activating KIT and/or inactivating Notch pathway mutations. In some cases, germline NF1 genetic testing may be appropriate for patients with DJF GISTs.
INTRODUCTION
GI stromal tumor (GIST) represents the most common type of sarcoma in the GI tract, with an annual incidence of 6.8 per million people in the United States.1 GISTs arise from the interstitial cell of Cajal lineage within the enteric nervous system.2 Approximately 70% to 80% of all sporadic GISTs have activating genomic alterations in KIT, whereas 5% to 10% have activating genomic alterations in PDGFRA.3,4 Of the remaining GISTs, 10% to 15% have activation of the Ras pathway (K/H/N-RAS, BRAF, NF1); 2% arise from mutations or deficiencies in the succinate dehydrogenase (SDH) subunits (A, B, C, or D); and a subset occurs as a result of kinase fusions (ETV6-NTRK3, FGFR1-TACC1, or FGFR1-HOOK3) or additional mutations in genes, including ARID1A and ARID1B.3-9
Within sporadic and hereditary GISTs driven by Ras pathway alterations, a subset possesses NF1 inactivating germline mutations that are associated with neurofibromatosis type 1 (NF-1 [ie, von Recklinghausen disease]).10,11 NF-1 is an autosomal-dominant disorder with variable penetrance: Patients with NF-1 often present in childhood or adolescence12 and have a 34-fold increased risk of developing GIST.13 At the molecular level, the NF1 gene encodes neurofibromin, which functions as a GTPase-activating protein that negatively regulates Ras family GTPases (ie, KRAS, NRAS, HRAS) by increasing the catalytic conversion of the active form (RAS-GTP) to the inactive form (RAS-GDP).14 Pathogenic NF1 variants disrupt the normal function of neurofibromin and result in constitutive RAS activation.15 This activation increases downstream signaling through BRAF/CRAF-mediated activation of the mitogen-activated protein kinase pathway and hence, facilitates tumor initiation and progression.16 Until recently, NF1 mutations in GIST were believed to be primarily of germline origin, associated with clinical NF-1, and combined with a second somatic mutation in the tumor. However, Belinsky et al17 reported the first case of somatic inactivation of NF1 in a patient with GIST without germline NF1 mutation or clinical NF-1. Thus, our understanding of NF1 in GIST continues to evolve.
Approximately 30% of all GISTs occur in the small intestine,1 which measures approximately 5 to 6 m in length.18 Most small bowel GISTs are associated with somatic KIT mutations, whereas a subset is associated with NF1 mutations.19 The small bowel is divided into three anatomically, histologically, and functionally distinct segments, namely the duodenum, jejunum, and ileum.18 Although some studies distinguish duodenal GISTs from other small bowel GISTs, most combine them and do not characterize the biology of tumors on the basis of these distinct locations. Moreover, the exact transition from jejunum to ileum is somewhat arbitrary, but the duodenal-jejunal flexure (DJF), also known as the ligament of Treitz, represents a clear anatomic site that marks the transition from duodenum to jejunum. To date, no study of GISTs has specifically characterized tumors that arise from the DJF. We investigated the biology of GISTs at the DJF and report on an association with somatic and germline genomic alterations in NF1. Most published case series have focused primarily on GIST in patients with known clinical NF-1. The current study, however, started with presumably sporadic GISTs, which led to the unexpected finding that somatic NF1 mutations in GISTs are more prevalent than previously suspected and that they are enriched in tumors that arise from the DJF.
METHODS
Primary Study Population
Patient demographic and tumor clinicopathologic data were retrospectively collected in an unbiased fashion from every patient with pathologically confirmed GIST seen within the University of California, San Diego (UCSD), health system from January 1, 2000, to April 30, 2017, under a UCSD institutional review board–approved protocol. Data collected were age, sex, race, ethnicity, primary GIST site, tumor size, TNM disease stage, and pathologic characteristics. Available operating notes or imaging reports were reviewed for these 165 patients to distinguish the primary site of origin of any small bowel GIST as duodenal, DJF, jejunal, or ileal.
Comprehensive Genomic Profiling
Of the 165 patients with GIST in the cohort, 62 underwent next-generation sequencing (NGS) of coding regions of cancer-related genes. NGS data from 61 of these patients were prospectively collected continuously as part of our institutional standard that began on May 1, 2014. One additional DJF GIST specimen from 2011 was retrospectively included in the NGS cohort (UCSD patient 1). Of note, 17 tumors in the NGS cohort, including two DJF GISTs (UCSD patients 3 and 5), were included in a prior study of pooled international data that aimed to identify novel deleterious genomic alterations in GISTs that lacked canonical driver mutations.7 GIST tumor specimens were either submitted to Foundation Medicine (a Clinical Laboratory Improvement Act–certified and College of American Pathologists– and New York State–accredited laboratory) or sequenced internally by the UCSD Clinical Genomics Laboratory. DNA was extracted from formalin-fixed paraffin-embedded (FFPE) sections that contained a minimum of 20% tumor tissue and were used for comprehensive genomic profiling with hybridization-captured, adaptor ligation–based libraries. The FoundationOne assay is an NGS-based genomic test that sequences coding regions of cancer-related genes. The number of genes in the FoundationOne panel has evolved over time as new data on cancer-related genes have been published and currently includes 315 cancer-related genes plus select introns from 28 genes often rearranged or altered in solid tumor cancers. However, all versions of the assay simultaneously analyze the extracted DNA for base substitutions, short insertions and deletions, amplifications and homozygous deletions, and gene rearrangements with ≥ 99% sensitivity.20 Specimens submitted to the UCSD Clinical Genomics Laboratory underwent NGS of the exons of 397 genes involved in pathways that control growth or differentiation and are known to be frequently mutated in solid tumors. The UCSD gene list includes all genes and select introns in the FoundationOne panel in addition to 82 genes identified from the COSMIC (Catalog of Somatic Mutations in Cancer) database as being mutated in > 3% of breast, lung, colon, prostate, skin, or CNS tumors (Data Supplement). Coverage depth ranged from 283× to 830× across all sequenced tumor samples from both the FoundationOne and the UCSD assays.
Nontumor Tissue Sequencing
Patients found to have NF1 mutations in their tumor tissue were also tested for germline NF1 mutation. Four samples tested by using DNA extracted from FFPE sections of adjacent normal tissue or peripheral blood underwent NGS cancer-related mutation panel testing by the UCSD Clinical Genomics Laboratory as just described. One additional sample was tested by using commercially available targeted sequencing of the NF1 gene from peripheral blood by ARUP Laboratories (Salt Lake City, UT).
Secondary Study Population
A confirmatory cohort of > 1,000 patients with pathologically confirmed GISTs from Memorial Sloan Kettering Cancer Center (MSKCC) with retrospectively collected patient demographic and tumor clinicopathologic data also were analyzed under an institutional review board–approved protocol. Data included age, sex, primary GIST site, tumor size, and pathologic characteristics. One hundred fifteen patients with GISTs had NGS results available in the MSK-IMPACT (Integrated Mutation Profiling of Actionable Cancer Targets) platform, which characterized frozen or FFPE tumor specimens for somatic DNA mutations, copy number alterations, and select rearrangements of 341 cancer-associated genes.21
Statistical Analysis
All statistical analyses were performed with Stata 9.0 software (StataCorp, College Station, TX). Comparisons between groups were performed by using the two-sample test of proportions. Statistical significance was accepted at the 5% level.
RESULTS
NF1 Genomic Alterations in GIST Occur in the Absence of Clinical NF-1
From the UCSD institutional cohort of 165 consecutive adult patients (> 18 years old) with pathologically confirmed GISTs, 62 underwent NGS for coding regions of at least 315 cancer-related genes. We identified six (9.7%) of these 62 GISTs with NF1 genomic alterations. None of these patients had previously known clinical manifestations of NF-1 (eg, café au lait spots; axillary/inguinal freckling; dermal neurofibromas; Lisch nodules; iris hamartomas; nervous system tumors, including malignant peripheral nerve sheath tumors [MPNSTs], pancreatic neuroendocrine tumors, pheochromocytomas, or other sarcomas]).10 Therefore, the application of expanded NGS panels to characterize the mutational profile of GISTs identified an unappreciated subset of patients with NF1 genomic alterations in their tumors but no clinical evidence of NF-1.
NF1 Genomic Alterations in GIST Frequently Occur at the DJF
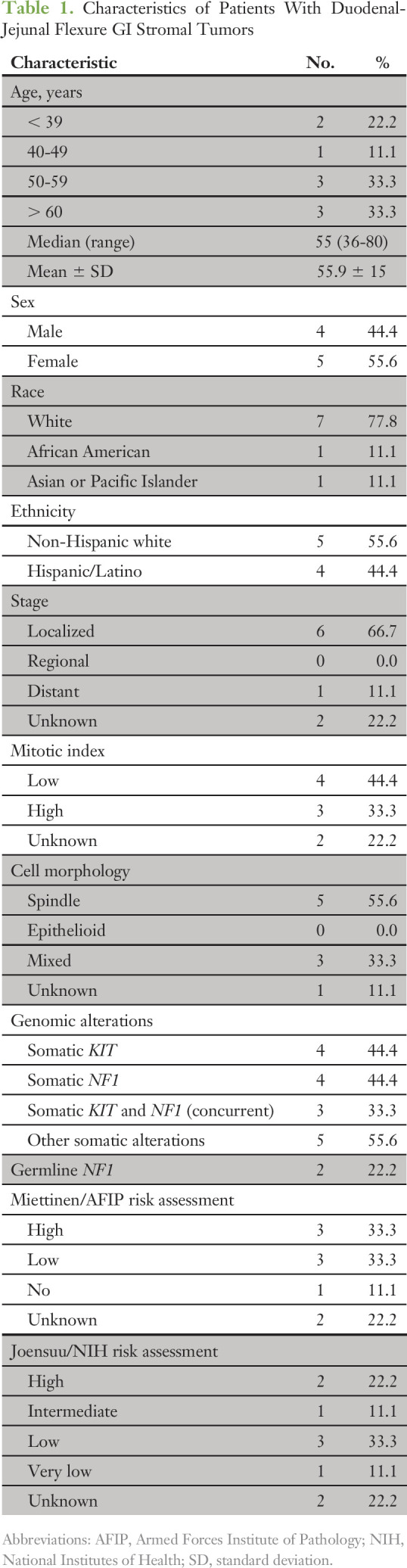
Of the six patients with GIST for whom we identified NF1 genomic alterations, five (83.3%) had unifocal tumors at the DJF, and one (16.7%) had a unifocal tumor in the stomach (Fig 1A). In the remaining cohort of 103 patients without available NGS data, two additional patients had GISTs at the DJF. Finally, in the 56 patients with GIST and NGS data that indicated non-NF1 mutations, two GISTs arose at the DJF. The demographic, clinicopathologic, and genomic characteristics of these nine patients with DJF tumors are listed in Table 1 (n = 7 NGS GISTs) and the Data Supplement (n = 2 non-NGS GISTs). Taken together, within the entire cohort of 165 patients, DJF GISTs (n = 9) represented an infrequent tumor site (5.5%). Given the infrequency of NF1 genomic alterations in GIST, the DJF appears to be an over-represented focus of NF1 mutant tumors.
Fig 1.
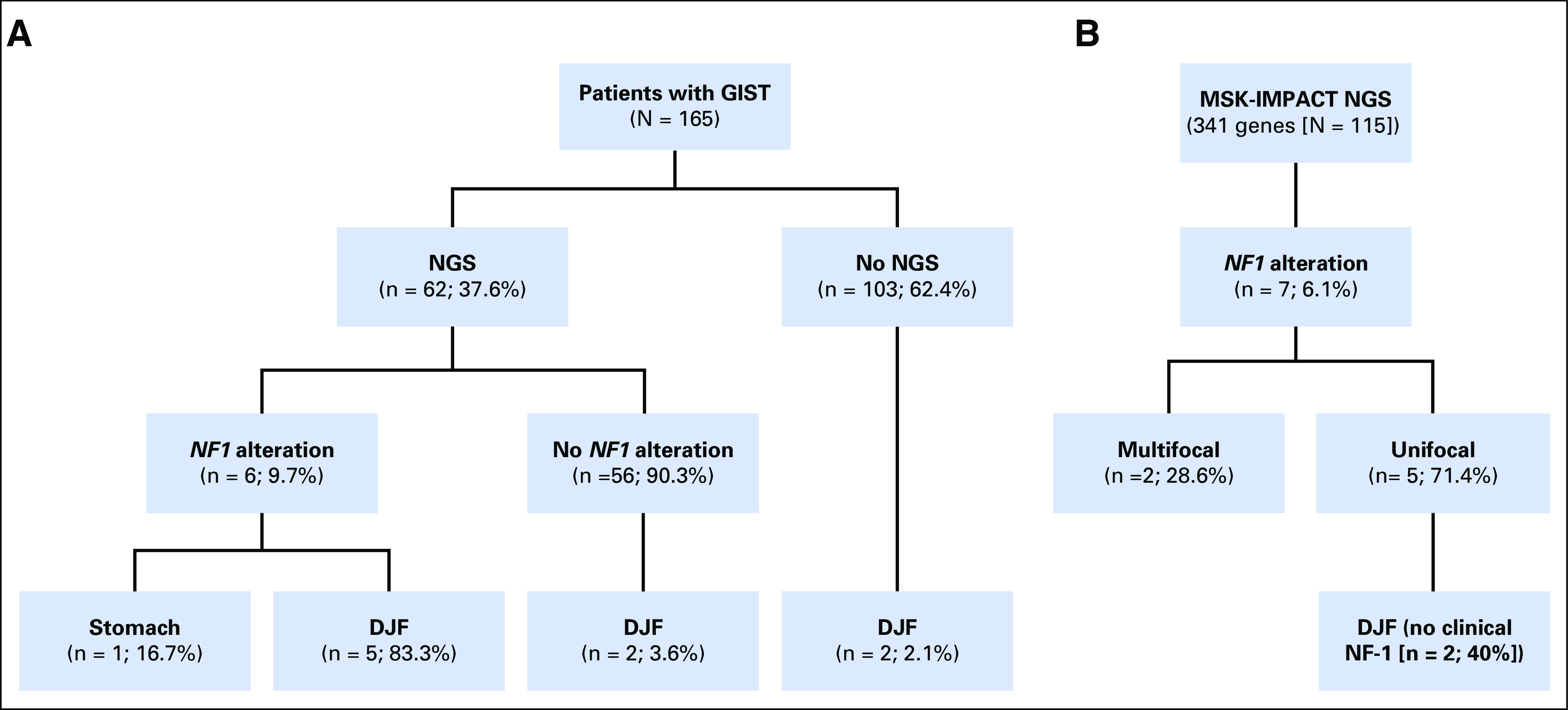
Schematic of patients with NF1-altered GI stromal tumors (GISTs). (A) From a total of 165 patients (University of California, San Diego [UCSD]) with pathologically confirmed GISTs, 62 had available next generation sequencing (NGS). Of these 62 patients, six had NF1 genomic alterations. (Seventeen tumors in the UCSD NGS cohort, including two duodenal-jejunal flexure [DJF] GISTs, were previously included in an earlier study of pooled international data that aimed to identify novel deleterious genomic alterations in GISTs that lack canonical driver mutations.7) (B) From a validation cohort (Memorial Sloan Kettering Cancer Center) of 115 patients with pathologically confirmed GISTs and available NGS, seven had NF1 genomic alterations. Five of these tumors were unifocal, and from two of these five, unifocal NF1-mutated GISTs arose from the DJF. MSK-IMPACT, Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets.
Table 1.
Characteristics of Patients With Duodenal-Jejunal Flexure GI Stromal Tumors
NF1 Genomic Alterations in GIST Are Primarily Somatic but Can Herald Mild Neurofibromatosis
NF1 genomic alterations for each patient are listed in Table 2. Of the 62 patients with GISTs and NGS data, four (57.1%) of seven DJF GISTs had known deleterious NF1 genomic alterations, but only one (1.8%) of 55 tumors outside the DJF had a deleterious NF1 mutation (P < .001). Although none of the patients had known prior clinical evidence of NF-1, we next obtained nontumor DNA from blood or nontumor tissue to determine whether any of these patients had germline NF1 mutations. UCSD patient 2 (Table 2) had two NF1 variants noted on his tumor profile; one was classified as a mutation and the other as a variant of unknown significance, which had been previously established as a pathogenic low-penetrance germline NF1 mutation seen in patients with mild NF-1 manifestations.22 The patient underwent genetic counseling and confirmatory germline testing; his c.2970_2972delAAT (M992del) mutation was confirmed to be germline in origin. A follow-up dermatology examination revealed no evidence of neurofibromas, but several café au lait spots and mild axillary freckling were identified. This finding is consistent with the mild phenotype/low penetrance reported in other patients with this germline NF1 mutation.22
Table 2.
Clinicopathologic Data of Seven Patients With Duodenal-Jejunal Flexure GI Stromal Tumors in the First Cohort With Next-Generation Sequencing

NF1 Genomic Alterations in GISTs Are Associated With Other Cancer-Related Mutations
At the genomic level, the UCSD cohort NGS data generally parallel the known distribution of driver mutations in GISTs (Fig 2). Of the 65 driver mutations identified in the 62 GISTs, oncogenic KIT (63%), PDGFRA (11%), and SDH subunit (11%) mutations were most frequent, whereas BRAF and KRAS mutations were less common. Moreover, NF1 mutations (six of 65) represented 9% of total genomic alterations. The loss of the NF1 tumor suppressor gene often leads to additional chromosomal alterations, but second-hit mutations and/or loss of heterozygosity are necessary to promote tumorigenesis.23 Therefore, we analyzed a cohort of five patients with NF1 mutant DJF GISTs for additional cancer-related gene mutations. Within these GISTs, three (60%) also had oncogenic KIT mutations (exons 9 or 11). Moreover, three (60%) had inactivating Notch pathway mutations (NOTCH2, MAML2, CDC73). In comparison, only two (3.6%) of the 55 non-DJF GISTs had a Notch pathway alteration (NOTCH2; P < .001).
Fig 2.

Driver mutations are representative of the genomic landscape of GI stromal tumors. Of the 62 patients with GI stromal tumors with available next generation sequencing, 65 known driver mutations were identified: 41 in KIT, seven in PDGFRA, six in NF1, seven in succinate dehydrogenase (SDH) subunits (A, B, C, or D), two in BRAF, and two in KRAS.
NF1 Genomic Alterations Are Associated With DJF GIST in a Validation Cohort
To ensure that the findings were not limited to a single institutional experience, we validated these observations with another cohort of 115 patients with GISTs with available NGS data from the MSKCC. Seven (6.1%) had identifiable NF1 genomic alterations. These tumors were unifocal in five (71%). Two of the unifocal NF1 mutant GISTs were located within the DJF (Fig 1B), of which neither patient had clinical NF-1. Demographic and clinicopathologic characteristics of these two patients are listed in Table 3, with the characteristics of the remaining five patients without DJF and/or multifocal GISTs listed in the Data Supplement. In addition to an NF1 mutation, one patient had a concomitant mutation in EP300, which encodes a histone acetyltransferase/transcriptional coactivator that has been previously reported to interact with MAML1 and MAML2 to potentiate Notch signaling.24 This supports our earlier observation that unifocal NF1 mutant DJF GISTs also can have impaired Notch signaling. In summary, 40% of unifocal NF1 mutant GISTs in the MSKCC cohort were localized at the DJF, which corroborates the high rate (83.3%) of similar tumors in the UCSD cohort. Taken together, these data demonstrate that NF1 mutant GISTs frequently occur at the DJF in adults and that these tumors also harbor concurrent activating KIT and/or inactivating Notch pathway mutations (Fig 3).
Table 3.
Clinicopathologic Data of Two Duodenal-Jejunal Flexure GI Stromal Tumors in the Second Cohort

Fig 3.

Summary of duodenal-jejunal flexure genomic alterations. Tumor size, mitotic index (MI), and genomic alterations for patients with duodenal-jejunal flexure GI stromal tumors (GISTs) from both cohorts are presented and organized by known driver genes and Notch pathway alterations. Somatic and germline mutations are annotated by color for mutation type. SNP, single nucleotide polymorphism.
DISCUSSION
GISTs associated with NF1 genomic alterations usually are believed to be multifocal and have been presumed to arise only in the setting of clinical NF-1.25 Before the widespread use of NGS, clinical NF-1 was believed to occur in only 1.5% of GISTs (6% of duodenal GISTS; 4% of jejunal-ileal GISTs).19 A recent Italian study reported NF1 mutations in 13 (59%) of 22 quadruple-negative (ie, KIT/PDGFRA/BRAF mutation–negative/SDH-intact) GISTs,11 but the frequency of NF1 mutations in the entire Italian GIST population was not reported. Moreover, we recently analyzed NGS data from an international cohort of 186 patients with GISTs that included some patients from our institution (17 of whom have NGS data within the current study).7 In that study, the frequency of NF1 mutations was 9.7% (18 of 186). We now show in two single-institution cohorts of patients with GISTs that 6.1% (MSKCC, seven of 115) to 9.7% (UCSD, six of 62) have NF1 alterations. If we exclude the 17 patients who overlap with our previous study (two of whom have NF1 mutations [UCSD patients 3 and 5 in the current study]), we show that 8.9% (four of 45) of patients with GISTs have NF1 alterations. Taken together, NF1 mutation frequencies range from 6% to 10% in GIST and are higher than previously appreciated but similar to our recent analysis.7 For the first time in our knowledge, we show that broad genomic profiling of DJF or ligament of Treitz GISTs in adults reveals frequent NF1 alterations (somatic and/or germline) that occur even in the absence of clinical NF-1. This discovery suggests a unique mechanism of oncogenesis whereby both acquired and germline NF1 gene alterations can lead to GIST development at this specific anatomic location. Moreover, it represents a previously unappreciated presentation of clinical NF-1 in adults who may have a low-penetrance germline mutation.
The data support investigations of NF1 testing (somatic and/or germline) in all patients with DJF GISTs, the rationale for which is threefold: characterization of GISTs with NF1 genomic alterations may have implications for imatinib resistance in the absence of KIT mutations and may lead to potential alterations in personalized therapy; diagnosis of an otherwise clinically silent, heritable condition (ie, patients with NF-1 who present with isolated DJF GIST) provides earlier awareness of the risk of additional tumors (eg, dermal neurofibromas, nervous system tumors [including MPNSTs, pancreatic neuroendocrine tumors, pheochromocytomas, and other sarcomas])10 and allows for earlier screening of these NF-1–associated tumors, although whether different malignancies and GISTs have unique presentation patterns remains to be determined; and diagnosis of NF-1 allows for genetic counseling and testing of potentially affected family members. Of note, individuals can have segmental mosaicism of NF-1, which occurs when an NF1 somatic mutation occurs early in embryonic development such that only the tissues derived from the NF1-mutated cell carry the mutation while the remaining tissues are wild type.26 As with UCSD patient 2, a clinical genetics work-up may be indicated for patients with NF1 mutations on tumor profiling that could indicate a germline rather than a somatic origin.
Tumor development in the setting of NF1 genomic alterations is associated with additional second-hit mutations.23 We now have shown that DJF tumors with NF1 genomic alteration also can harbor concurrent KIT mutations. In general, most KIT mutations portend imatinib sensitivity, whereas NF1 mutations do not.3,27 Consistent with this aforementioned drug sensitivity, tumors with a high risk of recurrence are recommended for adjuvant imatinib on the basis of the American College of Surgeons Oncology Group Z900128 and Scandinavian Sarcoma Group XVIII/AIO29 trials where cumulative results demonstrated improved relapse-free survival (RFS) and overall survival with 36 months of adjuvant imatinib in patients with high-risk disease. In our cohort, three patients with DJF GISTs had a high risk of recurrence by validated risk assessment models.30,31 UCSD patient 4 lacked a KIT mutation and was predicted to not respond to imatinib therapy; instead, this patient could experience toxicity. On the basis of the tumor mutation, this patient was not offered adjuvant imatinib. UCSD patient 1 had a KIT exon 9 mutation and was treated with adjuvant imatinib 800 mg daily for 5 years. This approach was supported by EORTC-62005 and Southwest Oncology Group S0033/Cancer and Leukemia Group B 150105 phase III trials that collectively demonstrated improved response rates and RFS in patients with advanced GISTs and KIT exon 9 mutations treated with high-dose imatinib (800 mg daily) compared with standard-dose imatinib (400 mg daily).32 The patient also had a germline NF1 variant and ultimately developed a local recurrence after 5 years of adjuvant imatinib therapy. UCSD patient 3 had a known KIT exon 11 mutation and was treated with dose-escalated imatinib followed by the multikinase inhibitor sunitinib for metastatic disease. NGS tumor profiling revealed the presence of both a somatic NF1 mutation and an ARID1A loss. A brief trial of the mammalian target of rapamycin inhibitor everolimus in combination with imatinib was attempted to block parallel signaling pathways, but the patient rapidly succumbed to the disease. Much like patients with metastatic colorectal cancer who are tested for pan-RAS mutations before treatment targeting upstream cell surface receptors (ie, epidermal growth factor receptor),33,34 we propose that patients with DJF GISTs be tested for NF1 (which would activate RAS) and BRAF V600E genomic alterations (which two of the patients with DJF GISTs also harbored) before being offered imatinib therapy, which targets upstream KIT.
We have recently shown that NOTCH1 can be mutated in both wild-type and non–wild-type GISTs.7 We now add evidence that implicates the Notch signaling pathway in GISTs. The Notch receptors (NOTCH1-4) bind to canonical ligands on adjacent cells (eg, Delta-like and Jagged). In turn, Notch undergoes sequential proteolytic cleavage, which releases the Notch intracellular domain (NICD). The NICD then translocates to the nucleus and leads to activation of a transcriptional complex (including mastermind-like proteins 1 and 2 [MAML1 and MAML2] and E1A binding protein p300 [EP300]) that regulates expression of target genes, such as HES1.35 Herein, we found inactivating mutations in both NOTCH2 and MAML2 that co-occur with NF1 mutations. We identified another tumor with a mutation in CDC73, which encodes the RNA polymerase II–associated factor complex protein parafibromin and was recently reported to potentiate Notch signaling by binding to the NICD.36 We also found an EP300 mutation in the validation cohort that could abrogate signaling of the Notch transcriptional complex. Of note, Notch signaling was reported to have a tumor suppressor function in GIST by downregulating KIT mRNA expression.37 This prior study also demonstrated that patients with GISTs with low HES1 expression had shorter RFS times than those with high HES1 expression. Whether loss-of-function mutations in Notch pathway genes that lead to decreased HES1 expression contribute to GIST tumorigenesis remains to be determined. Moreover, data in neural stem-cell differentiation suggest that the Notch pathway intersects with NF1 signaling. NF1 regulates MEK/Smad3/Jagged1/Hes1–dependent glial and neuronal differentiation.38 Notch has also been reported to mediate transformation of benign plexiform neurofibromas into MPNSTs in the setting of NF-1,39 which suggests an alternate oncogenic role of Notch in NF-1. More studies are required to elucidate the biologic significance of Notch pathway mutations in GIST, including NF-1–associated GIST.
Finally, the mechanism by which NF1 mutations lead to GISTs that specifically arise at the DJF remains unclear. Expression of the NF1 gene product is ubiquitous throughout all tissue types,40 but an analysis of open reading frames adjacent to the NF1 locus on chromosome 17q reveals that the SLC6A4 gene that encodes a high-affinity, sodium-dependent serotonin (5-hydroxytryptamine-3) transporter may have some tissue specificity for the small intestine, particularly the dudodenum.41 In addition, interstitial cells of Cajal, which are the precursor cells to GIST, express 5-hydroxytryptamine-3 receptors, and their activation enhances gut pacemaker activity.42 One could postulate that this region of chromatin on 17q is open and transcriptionally active at the DJF. Therefore, NF1 may be more susceptible to mutations at this site. However, the exact mechanism behind the specific association of NF1 genomic alterations with DJF GIST will require additional investigation.
In conclusion, this work provides new insights into the pathobiology of NF1 mutant GISTs. We continue to stratify patients with GISTs into distinct groups on the basis of their tumor genomics and now provide evidence that anatomic location at the DJF may have genetic and clinical implications. The data support NF1 genetic testing in all patients with DJF GISTs. Taken together, this study demonstrates how implementation of genomically driven precision oncology care can guide personalized treatments and counseling for patients with GISTs as well as for their family members.
Footnotes
Supported by the Tower Cancer Research Foundation (to A.M.B.); National Institutes of Health (NIH) R01CA102613 (to R.P.D.); GIST Cancer Research Fund and Young Gastrointestinal Research Fund (to M.v.M.); NIH R21CA177519, R21CA192072, P30CA023100, and U54HL108460 (to O.H.); and NIH K08CA168999, R21CA192072, and P30CA023100 (to J.K.S.).
AUTHOR CONTRIBUTIONS
Conception and design: Adam M. Burgoyne, Martina De Siena, Maha Alkhuziem, Jason K. Sicklick
Financial support: Jason K. Sicklick
Administrative support: Jason K. Sicklick
Provision of study materials or patients: Paul T. Fanta, Margaret von Mehren, Jason K. Sicklick
Collection and assembly of data: Adam M. Burgoyne, Benjamin Medina, Paul T. Fanta, Martin G. Belinsky, Margaret von Mehren, John A. Thorson, Timothy Bowler, Francesco D’Angelo, Olivier Harismendy, Ronald P. DeMatteo, Jason K. Sicklick
Data analysis and interpretation: Adam M. Burgoyne, Maha Alkhuziem, Chih-Min Tang, Paul T. Fanta, Margaret von Mehren, John A. Thorson, Lisa Madlensky, Dwayne G. Stupack, Olivier Harismendy, Ronald P. DeMatteo, Jason K. Sicklick
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or po.ascopubs.org/site/ifc.
Adam M. Burgoyne
No relationship to disclose
Martina De Siena
No relationship to disclose
Maha Alkhuziem
No relationship to disclose
Chih-Min Tang
No relationship to disclose
Benjamin Medina
No relationship to disclose
Paul T. Fanta
No relationship to disclose
Martin G. Belinsky
No relationship to disclose
Margaret von Mehren
Consulting or Advisory Role: CytRx, Blueprint Medicines, Janssen Pharmaceuticals, Deciphera Pharmaceuticals
Research Funding: ArQule
Travel, Accommodations, Expenses: Janssen Pharmaceuticals, Blueprint Medicines, Novartis, Arog Pharmaceuticals
Other Relationship: National Comprehensive Cancer Network
John A. Thorson
No relationship to disclose
Lisa Madlensky
Employment: Janssen Pharmaceuticals (I)
Patents, Royalties, Other Intellectual Property: Husband has patents through his employment with Janssen Pharmaceuticals; these are methods patents and not related to any therapeutics (I)
Timothy Bowler
Employment: REGENXBIO (I)
Stock and Other Ownership Interests: REGENXBIO (I)
Francesco D’Angelo
No relationship to disclose
Dwayne G. Stupack
Travel, Accommodations, Expenses: Grifols
Olivier Harismendy
No relationship to disclose
Ronald P. DeMatteo
No relationship to disclose
Jason K. Sicklick
Consulting or Advisory Role: bioTheranostics
Research Funding: Novartis, Foundation Medicine, Blueprint Medicines
REFERENCES
- 1.Ma GL, Murphy JD, Martinez ME, et al. : Epidemiology of gastrointestinal stromal tumors in the era of histology codes: Results of a population-based study. Cancer Epidemiol Biomarkers Prev 24:298-302, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ordog T, Zörnig M, Hayashi Y: Targeting disease persistence in gastrointestinal stromal tumors. Stem Cells Transl Med 4:702-707, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Corless CL, Barnett CM, Heinrich MC: Gastrointestinal stromal tumours: Origin and molecular oncology. Nat Rev Cancer 11:865-878, 2011 [DOI] [PubMed] [Google Scholar]
- 4.Rubin BP, Heinrich MC, Corless CL: Gastrointestinal stromal tumour. Lancet 369:1731-1741, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Liegl-Atzwanger B, Fletcher JA, Fletcher CD: Gastrointestinal stromal tumors. Virchows Arch 456:111-127, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Agaram NP, Wong GC, Guo T, et al. : Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 47:853-859, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi E, Chmielecki J, Tang CM, et al. : FGFR1 and NTRK3 actionable alterations in “wild-type” gastrointestinal stromal tumors. J Transl Med 14:339, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. doi: 10.1002/path.4677. Brenca M, Rossi S, Polano M, et al: Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J Pathol 238:543-549, 2016. [DOI] [PubMed] [Google Scholar]
- 9.Hechtman JF, Zehir A, Mitchell T, et al. : Novel oncogene and tumor suppressor mutations in KIT and PDGFRA wild type gastrointestinal stromal tumors revealed by next generation sequencing. Genes Chromosomes Cancer 54:177-184, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burgoyne AM, Somaiah N, Sicklick JK: Gastrointestinal stromal tumors in the setting of multiple tumor syndromes. Curr Opin Oncol 26:408-414, 2014 [DOI] [PubMed] [Google Scholar]
- 11. doi: 10.1158/1078-0432.CCR-16-0152. Gasparotto D, Rossi S, Polano M, et al: Quadruple-negative GIST is a sentinel for unrecognized neurofibromatosis type 1 syndrome. Clin Cancer Res 23:273-282, 2017. [DOI] [PubMed] [Google Scholar]
- 12.Anderson JL, Gutmann DH: Neurofibromatosis type 1. Handb Clin Neurol 132:75-86, 2015 [DOI] [PubMed] [Google Scholar]
- 13.Uusitalo E, Rantanen M, Kallionpää RA, et al. : Distinctive cancer associations in patients with neurofibromatosis type 1. J Clin Oncol 34:1978-1986, 2016 [DOI] [PubMed] [Google Scholar]
- 14.Mitin N, Rossman KL, Der CJ: Signaling interplay in Ras superfamily function. Curr Biol 15:R563-R574, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Rad E, Tee AR: Neurofibromatosis type 1: Fundamental insights into cell signalling and cancer. Semin Cell Dev Biol 52:39-46, 2016 [DOI] [PubMed] [Google Scholar]
- 16.Lavoie H, Therrien M: Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 16:281-298, 2015 [DOI] [PubMed] [Google Scholar]
- 17.Belinsky MG, Rink L, Cai KQ, et al. : Somatic loss of function mutations in neurofibromin 1 and MYC associated factor X genes identified by exome-wide sequencing in a wild-type GIST case. BMC Cancer 15:887, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schottenfeld D, Beebe-Dimmer JL, Vigneau FD: The epidemiology and pathogenesis of neoplasia in the small intestine. Ann Epidemiol 19:58-69, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miettinen M, Fetsch JF, Sobin LH, et al. : Gastrointestinal stromal tumors in patients with neurofibromatosis 1: A clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol 30:90-96, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Frampton GM, Fichtenholtz A, Otto GA, et al. : Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 31:1023-1031, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng DT, Mitchell TN, Zehir A, et al. : Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 17:251-264, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quintáns B, Pardo J, Campos B, et al. : Neurofibromatosis without neurofibromas: Confirmation of a genotype-phenotype correlation and implications for genetic testing. Case Rep Neurol 3:86-90, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maertens O, Prenen H, Debiec-Rychter M, et al. : Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet 15:1015-1023, 2006 [DOI] [PubMed] [Google Scholar]
- 24.McElhinny AS, Li JL, Wu L: Mastermind-like transcriptional co-activators: Emerging roles in regulating cross talk among multiple signaling pathways. Oncogene 27:5138-5147, 2008 [DOI] [PubMed] [Google Scholar]
- 25.Agaimy A, Vassos N, Croner RS: Gastrointestinal manifestations of neurofibromatosis type 1 (Recklinghausen’s disease): Clinicopathological spectrum with pathogenetic considerations. Int J Clin Exp Pathol 5:852-862, 2012 [PMC free article] [PubMed] [Google Scholar]
- 26.Messiaen L, Vogt J, Bengesser K, et al. : Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1). Hum Mutat 32:213-219, 2011 [DOI] [PubMed] [Google Scholar]
- 27.Sicklick JK, Lopez NE: Optimizing surgical and imatinib therapy for the treatment of gastrointestinal stromal tumors. J Gastrointest Surg 17:1997-2006, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.DeMatteo RP, Ballman KV, Antonescu CR, et al. : Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: A randomised, double-blind, placebo-controlled trial. Lancet 373:1097-1104, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joensuu H, Eriksson M, Sundby Hall K, et al. : One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: A randomized trial. JAMA 307:1265-1272, 2012 [DOI] [PubMed] [Google Scholar]
- 30.Miettinen M, Lasota J: Gastrointestinal stromal tumors: Pathology and prognosis at different sites. Semin Diagn Pathol 23:70-83, 2006 [DOI] [PubMed] [Google Scholar]
- 31.Joensuu H: Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol 39:1411-1419, 2008 [DOI] [PubMed] [Google Scholar]
- 32.Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST) : Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: A meta-analysis of 1,640 patients. J Clin Oncol 28:1247-1253, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pietrantonio F, Cremolini C, Petrelli F, et al. : First-line anti-EGFR monoclonal antibodies in panRAS wild-type metastatic colorectal cancer: A systematic review and meta-analysis. Crit Rev Oncol Hematol 96:156-166, 2015 [DOI] [PubMed] [Google Scholar]
- 34.Sorich MJ, Wiese MD, Rowland A, et al. : Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: A meta-analysis of randomized, controlled trials. Ann Oncol 26:13-21, 2015 [DOI] [PubMed] [Google Scholar]
- 35.Yuan X, Wu H, Xu H, et al. : Notch signaling: An emerging therapeutic target for cancer treatment. Cancer Lett 369:20-27, 2015 [DOI] [PubMed] [Google Scholar]
- 36.Kikuchi I, Takahashi-Kanemitsu A, Sakiyama N, et al. : Dephosphorylated parafibromin is a transcriptional coactivator of the Wnt/Hedgehog/Notch pathways. Nat Commun 7:12887, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dumont AG, Yang Y, Reynoso D, et al. : Anti-tumor effects of the Notch pathway in gastrointestinal stromal tumors. Carcinogenesis 33:1674-1683, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen YH, Gianino SM, Gutmann DH: Neurofibromatosis-1 regulation of neural stem cell proliferation and multilineage differentiation operates through distinct RAS effector pathways. Genes Dev 29:1677-1682, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y, Rao PK, Wen R, et al. : Notch and Schwann cell transformation. Oncogene 23:1146-1152, 2004 [DOI] [PubMed] [Google Scholar]
- 40.Gutmann DH, Wood DL, Collins FS: Identification of the neurofibromatosis type 1 gene product. Proc Natl Acad Sci U S A 88:9658-9662, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gill RK, Pant N, Saksena S, et al. : Function, expression, and characterization of the serotonin transporter in the native human intestine. Am J Physiol Gastrointest Liver Physiol 294:G254-G262, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu HN, Ohya S, Nishizawa Y, et al. : Serotonin augments gut pacemaker activity via 5-HT3 receptors. PLoS One 6:e24928, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]