

Abstract
Background
Atopy and viral respiratory tract infections synergistically promote asthma exacerbations. IgE cross-linking inhibits critical virus-induced IFN-α responses of plasmacytoid dendritic cells (pDCs), which can be deficient in patients with allergic asthma.
Objective
We sought to determine whether reducing IgE levels in vivo with omalizumab treatment increases pDC antiviral IFN-α responses in inner-city children with asthma.
Methods
PBMCs and pDCs isolated from children with exacerbation-prone asthma before and during omalizumab treatment were stimulated ex vivo with rhinovirus and influenza in the presence or absence of IgE cross-linking. IFN-α levels were measured in supernatants, and mRNA expression of IFN-α pathway genes was determined by using quantitative RT-PCR (qRT-PCR) in cell pellets. FcεRIα protein levels and mRNA expression were measured in unstimulated cells by using flow cytometry and qRT-PCR, respectively. Changes in these outcomes and associations with clinical outcomes were analyzed, and statistical modeling was used to identify risk factors for asthma exacerbations.
Results
Omalizumab treatment increased rhinovirus- and influenza-induced PBMC and rhinovirus-induced pDC IFN-α responses in the presence of IgE cross-linking and reduced pDC surface FcεRIα expression. Omalizumab-induced reductions in pDC FcεRIα levels were significantly associated with a lower asthma exacerbation rate during the outcome period and correlated with increases in PBMC IFN-α responses. PBMC FcεRIα mRNA expression measured on study entry significantly improved an existing model of exacerbation prediction. Conclusions: These findings indicate that omalizumab treatment augments pDC IFN-α responses and attenuates pDC FcεRIα protein expression and provide evidence that these effects are related. These results support a potential mechanism underlying clinical observations that allergic sensitization is associated with increased susceptibility to virus-induced asthma exacerbations.
Keywords: Plasmacytoid dendritic cells, rhinovirus, IFN-α, asthma, IgE, omalizumab, FcεRα
Allergic sensitization is a risk factor in the development of acute asthma exacerbations with viral respiratory infections. Heymann et al1 demonstrated that allergic sensitization and increased serum IgE levels to house dust mite enhance the likelihood that a rhinovirus respiratory tract infection will cause an asthma exacerbation. Furthermore, allergen exposure in allergic patients increases the risk for respiratory virus–induced wheezing and asthma exacerbations.2,3
A critical link between IgE levels and suppression of Toll-like receptor (TLR) 9–induced plasmacytoid dendritic cell (pDC) IFN-α responses was first described by Schroeder et al,4 suggesting that pDC antiviral responses might be suppressed similarly in the setting of atopy. We observed that the magnitude of pDC IFN-α responses to ex vivo viral challenge is inversely related to serum IgE levels.5 In addition, we showed that stimulation of the IgE/FcεRI pathway in pDCs through IgE cross-linking abrogates viral and TLR7-induced pDC IFN-α production,5,6 a finding that could explain why pDCs isolated from patients with allergic asthma have impaired IFN-α responses to viruses. Furthermore, surface expression of FcεRI on pDCs significantly correlates with serum IgE concentrations7 and is associated with diminished virus-induced IFN-α responses in these cells.5 Reduction of IgE levels with omalizumab can significantly reduce pDC FcεRI expression,8,9 a finding that could translate to improved antiviral responses in these cells. Collectively, these data suggest a significant interaction between IgE level, FcεRI expression, and asthma exacerbations and also that reducing IgE and FcεRI expression in vivo could restore IFN-α responses and possibly contribute to the prevention of virus-provoked asthma exacerbations.
The Preventative Omalizumab or Step-up Therapy for Severe Fall Exacerbations (PROSE) study (clincaltrials.gov no. NCT01430403) was designed to evaluate the effect of reducing IgE levels with omalizumab in vivo on asthma exacerbations, as well as on ex vivo pDC antiviral IFN-α responses. Children treated with omalizumab had a significant restoration of ex vivo IFN-α responses in PBMCs, and the group with a greater restoration in IFN-α responses had a lower asthma exacerbation rate.10 In this report we tested the hypothesis that omalizumab treatment would (1) boost antiviral IFN-α responses of both PBMCs and purified pDCs in the presence and absence of IgE cross-linking and (2) attenuate pDC FcεRIα expression. We also investigated whether these omalizumab-induced cellular changes were associated with clinical outcomes. Finally, we used multivariate modeling to determine whether the cellular phenotypes and IFN-α responses in our study would improve the predictive value of a previously developed model for asthma exacerbations in this population of high-risk children.
METHODS
Mechanistic study design
In participants from 2 of the 8 sites of the PROSE clinical trial (UT Southwestern Medical Center, Dallas, Texas, and National Jewish Health, Denver, Colorado), blood for ex vivo assays was drawn before randomization and 12 to 16 weeks after initiation of treatment. These assays were designed to measure the effect of IgE cross-linking on virus (rhinovirus and influenza)– induced and TLR7 agonist (gardiquimod)–induced IFN-α in cultures of PBMCs (all participants) and pDCs (in a subset of participants) and to determine the effect of omalizumab versus placebo treatment on these IFN-α responses.
Isolation of PBMCs, pDCs, culture conditions, and reagents
PBMCs were isolated by means of density centrifugation with Ficoll-Paque (GE Healthcare, Fairfield, Conn). In a subset of participants, pDCs were purified from PBMCs by means of negative selection with antibody-coated magnetic particles (EasySep Human Plasmacytoid DC Enrichment Kit, Catalog #19062; STEMCELL Technologies, Vancouver, British Columbia, Canada), according to the manufacturer’s Manuel EasySep Protocol and as previously used in our studies.11 Purity of the isolated pDCs (defined as Lin−HLA-DR+CD11c−CD123+ events) was greater than 80%. pDCs were distinguished from basophils by means of HLA-DR expression (because pDCs express high levels of HLA-DR, whereas basophils lack HLA-DR expression). Basophil contamination in purified pDC samples was minimal (median of <0.1% in pDC samples obtained during both the prerandomization and postrandomization phase of the study). Aliquots of PMBCs and pDCs were preserved in Cyto-Chex solution (Streck, Omaha, Neb) for subsequent antibody staining and flow cytometric analysis. Cells were cultured at 0.5 × 106/0.2 mL (PBMCs) or 0.1 × 105/0.2 mL (pDCs) in 96-well round-bottom plates in complete RPMI 1640 medium supplemented with 10% heat-inactivated FBS, 1% penicillin-streptomycin, 1% sodium pyruvate, 1% HEPES buffer solution, 1% nonessential amino acids, 1% glutamate, 100 μmol/L β-mercaptoethanol, and 10 ng/mL IL-3. Cells were cultured for 18 hours in the presence or absence of an IgE cross-linking antibody (rabbit anti-human IgE, 1 μg/mL; Bethyl Laboratories, Montgomery, Tex). This antibody differs from omalizumab in its ability to bind and cross-link IgE on cell-surface FcεRIα receptors, whereas omalizumab only binds to free IgE. After 18 hours, PBMCs were stimulated with RV-A16 (106 plaque-forming units/mL; a gift from Wai-Ming Lee and Yury Bochkov, University of Wisconsin-Madison, Madison, Wis), influenza virus (A/PR/8/34 [H1N1], 0.1 plaque-forming units/cell; Charles River Laboratories, Malvern, Pa), or gardiquimod (a TLR7 agonist, 1 μg/mL; InvivoGen, San Diego, Calif) for 24 hours. Cells and supernatants were then harvested by means of centrifugation and stored at −80°C for RNA extraction/qRT-PCR and IFN-α ELISA, respectively.
Flow cytometry
PBMCs were stained5 with the following fluorochrome-conjugated anti-human antibodies: HLA-DR allophycocyanin (APC)–Cy7, lineage–fluorescein isothiocyanate (FITC) mixture, CD14–Pacific Blue, CD123-PeCy5 (BD Biosciences), and FcεRIα-PE (eBioscience, San Diego, Calif); acquired on a BD LSR II flow cytometer (BD Biosciences); and analyzed by using FlowJo software (TreeStar, Ashland, Ore). pDCs were identified as lineage-negative, HLA-DR+, and CD123+ cells. The mean fluorescence intensity of FcεRIα was determined by using flow cytometry and subsequently converted to molecules of equivalent soluble fluorochrome by using the Ultra Rainbow Calibration Kit (BD Biosciences).
IFN-α quantification
IFN-α levels were measured by using the Human IFN-α (panspecific to detect 12 IFN-α subtypes) ELISA Kit (MabTech, Cincinnati, Ohio) and analyzed on an ELISA reader DTX 880 Multimode detector (Beckman Coulter, Fullerton, Calif).
RNA isolation and qRT-PCR for detection of FcεRIα and mRNA of interferon signaling and response genes
Total pDC RNAwas extracted from 2 × 104 pDCs by using RNA-Bee (Tel-Test, Friendswood, Tex) and chloroform, as previously described, and stored at −80°C.5 Total PBMC RNAwas extracted from 1.5 to 2 × 106 PBMCs with the Qiagen RNeasy Mini Kit (Qiagen, Valencia, Calif). cDNA synthesis was carried with a High Capacity cDNA Reverse Transcriptase Kit (Applied Bio-systems, Foster City, Calif). Expression of TLR7 and FCER1A mRNA in unstimulated pDCs and PBMCs and FCER1G mRNA in unstimulated PBMCs was quantified by performing quantitative RT-PCR with the CFX384 Real Time system (Bio-Rad Laboratories, Hercules, Calif). TLR7, IFNB1, IFNA1, IFIT1, interferon regulatory factor 7 (IRF7), and DDX58 expression was also measured in cultured PBMCs. Sequences for TLR7 and FcεRIα probes/primers (TaqMan) shown in Table E1 in this article’s Online Repository at www.jacionline.org were used, as previously reported.5 The reporter dye and quencher were 6-fluorescein amidite and tetramethylrhodamine, respectively. Applied Biosystems TaqMan Assays (Thermo Fisher Scientific, Waltham, Mass) shown in Table E2 in this article’s Online Repository at www.jacionline.org were used for IRF7, IFNB1, IFNA1, DDX58, and IFIT1. mRNA expression for all genes in cultured PBMCs was normalized to hypoxanthine-guanine phosphoribosyltransferase mRNA (Thermo Fisher Scientific). mRNA expression for all genes in unstimulated PBMCs and pDCs was normalized to hypoxanthine-guanine phosphoribosyltransferase mRNA or PPIA mRNA (Thermo Fischer Scientific). Results were based on 2−ΔΔct values.
Statistical analysis
Cell values were skewed and therefore log-transformed for statistical tests and models. To compare differences between prerandomization and postrandomization within treatment groups (placebo and omalizumab), paired t tests were used, and ratio of geometric means were reported. Change between prerandomization and postrandomization was calculated by using a log difference. Change was dichotomized at the median value to compare exacerbation rates by the level of prerandomization versus postrandomization change. Logistic regression was performed (Firth method was used in the case of zero counts) by using dichotomized change as the predictor and any exacerbation as the outcome; associated odds ratios were reported. Pearson correlations were used to compare continuous change between variables.
Because of the larger sample size required for multivariate modeling of exacerbations, participants from the third arm of the PROSE clinical trial, an arm that received an inhaled corticosteroid boost, were pooled together with placebo-treated participants. Notably, the primary outcome in the inhaled corticosteroid arm did not differ from placebo in the PROSE trial.10 The multivariate modeling process consisted of 3 steps. First, prerandomization mechanistic variables were compared between exacerbators and nonexacerbators by using t tests within treatment group (non-omalizumab and omalizumab). Because of the exploratory nature of the analysis, we did not account for multiple comparisons. Next, an elastic net procedure was performed to select the most relevant baseline mechanistic outcomes after adjusting for known baseline predictors of exacerbations.12 Finally, logistic models were fit by using known baseline predictors of exacerbations (age, total IgE, atopy, blood eosinophils, exacerbations in the previous 90 days, FEV1/forced vital capacity ratio, and treatment step)13 stratified by treatment group. Similar models were fit by using the known predictors of exacerbations and the PBMC mechanistic outcomes selected by using the elastic net procedure.14 All cellular phenotype and IFN-α response variables with less than 40% missing data were included as candidate predictors. Before multivariate modeling, any missing data among the candidate predictors (which amounted to 5% of the values) were imputed by using an imputation based on a random forest approach. Nested models were compared by using likelihood ratio tests and area under the receiver operating characteristic curve. SAS 9.3 (SAS Institute, Cary, NC) and R version 3.3.2 software were used for analyses.
RESULTS
IFN-α responses were evaluated in participants receiving omalizumab and placebo treatment (n = 92) at 2 clinical sites. Participants had similar demographic and laboratory characteristics as those in the multisite clinical trial, including low income, predominant Hispanic ethnicity, increased peripheral blood eosinophil counts, and total serum IgE levels (see Table E3 in this article’s Online Repository at www.jacionline.org). No significant differences in baseline characteristics were found between subjects from the 2 sites (data not shown).
Effect of omalizumab treatment on ex vivo antiviral IFN-α responses in PBMCs and purified pDCs
Ex vivo antiviral IFN-α responses were first measured in PBMC cultures (n = 92) obtained at 2 time points in the study: before randomization and 12 to 16 weeks after treatment was initiated. As previously reported, PBMC generation of IFN-α to rhinovirus in the presence of IgE cross-linking was significantly increased in the postomalizumab treatment group compared with the posttreatment placebo group.10 We now report that omalizumab treatment significantly increased IFN-α responses after IgE cross-linking and stimulation with either rhinovirus (ratio of geometric means, 2.06; 95% CI, 1.20-3.55; P = .01) or influenza virus (ratio of geometric means, 1.57; 95% CI, 1.09-2.25; P = .02; Fig 1 and Table I). In contrast, omalizumab treatment did not significantly affect IFN-α responses to rhinovirus, influenza virus, or gardiquimod in the absence of ex vivo IgE cross-linking.
FIG 1.
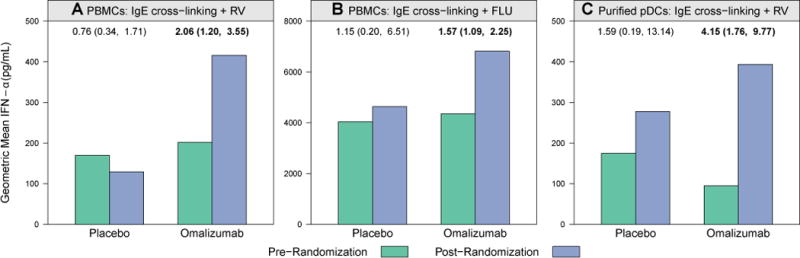
Enhanced PMBC and pDC IFN-α responses after omalizumab. PBMC and pDC IFN-α responses to rhinovirus (RV) and influenza virus (FLU) significantly increased during the intervention phase of the study in the omalizumab group. Bars represent geometric mean IFN-α concentrations (in picograms per milliliter) measured in supernatants. Ratio of geometric means comparing postintervention versus preintervention values within each group are annotated above the bars, along with associated 95% CIs. Boldface indicates a P value of less than .05, as determined by using the paired t test. A, A 2.06-fold increase in PBMC IFN-α response to rhinovirus plus IgE cross-linking (P = .01). B, A 1.57-fold increase in PBMC IFN-α response to influenza virus plus IgE cross-linking (P = .02). C, A 4.15-fold increase in pDC IFN-α response to rhinovirus plus IgE cross-linking (P = .003) was observed between prerandomization (green bars) and postrandomization (blue bars) values in the omalizumab group.
TABLE I.
| Mechanistic parameter |
Omalizumab
|
Placebo
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell type |
Ex vivo stimulus |
No. | Pre- randomization, mean (SD) |
Post- randomization, mean (SD) |
Ratio of geometric means (95% CI) |
P value |
No. | Pre- randomization, mean (SD) |
Post- randomization, mean (SD) |
Ratio of geometric means (95% CI) |
P value |
| PBMC | Rhinovirus | 64 | 1365.8 (6.1) | 1035.8 (7.0) | 0.76 (0.47–1.22) | .25 | 23 | 1443.1 (6.3) | 764.2 (7.9) | 0.53 (0.21–1.32) | .16 |
|
| |||||||||||
| Anti-IgE plus rhinovirus | 56 | 201.7 (6.9) | 415.7 (8.2) | 2.06 (1.20–3.55) | .01 | 23 | 169.7 (5.5) | 129.0 (6.4) | 0.76 (0.34–1.71) | .49 | |
|
| |||||||||||
| Influenza virus | 64 | 7304.7 (2.2) | 7301.8 (2.4) | 1.00 (0.80–1.24) | >.99 | 23 | 6864.2 (3.7) | 6371.6 (2.6) | 0.93 (0.50–1.74) | .81 | |
|
| |||||||||||
| Anti-IgE plus influenza virus | 38 | 4352.1 (2.6) | 6814.9 (2.3) | 1.57 (1.09–2.25) | .02 | 10 | 4043.6 (5.6) | 4637.1 (3.9) | 1.15 (0.20–6.51) | .86 | |
|
| |||||||||||
| Gardiquimod | 64 | 856.4 (3.8) | 1167.3 (4.1) | 1.36 (1.00–1.87) | .05 | 23 | 1314.3 (4.7) | 985.4 (3.4) | 0.75 (0.49–1.16) | .18 | |
|
| |||||||||||
| pDC | Rhinovirus | 25 | 125.2 (5.3) | 237.5 (7.1) | 1.90 (0.83–4.34) | .12 | 12 | 216.3 (7.6) | 260.4 (8.0) | 1.2 (0.26–5.64) | .80 |
|
| |||||||||||
| Anti-IgE plus rhinovirus | 14 | 94.8 (4.4) | 393.6 (6.4) | 4.15 (1.76–9.77) | .003 | 7 | 174.8 (7.2) | 277.7 (6.7) | 1.59 (0.19–13.14) | .61 | |
|
| |||||||||||
| Influenza | 25 | 236.6 (3.7) | 317.5 (3.5) | 1.34 (0.87–2.08) | .18 | 11 | 171.6 (4.7) | 340.2 (3.4) | 1.98 (0.84–4.70) | .11 | |
|
| |||||||||||
| pDC | Gardiquimod | 21 | 936.6 (6.5) | 1900.2 (4.7) | 2.03 (0.85–4.83) | .10 | 11 | 1280.3 (6.6) | 1955.4 (2.7) | 1.53 (0.33–7.14) | .55 |
Boldface indicates P < .05.
IFN-α concentrations (in picograms per milliliter) measured in supernatants after depicted ex vivo stimulations.
Means are geometric means, and SDs are geometric SDs.
pDCs were purified from a subset of participants (n = 43) to determine whether omalizumab affected pDC antiviral IFN-α responses. Similar to effects noted with PMBCs, omalizumab significantly enhanced rhinovirus-induced IFN-α responses of purified pDCs in the presence of IgE cross-linking treatment (Fig 1 and Table I; ratio of geometric means, 4.15; 95% CI, 1.76-9.77; P = .003). In contrast, omalizumab treatment had no significant effects on pDC-secreted IFN-α responses in the absence of IgE cross-linking. Because of the low frequency of pDCs in blood samples, insufficient numbers of pDCs were available to evaluate influenza virus– or gardiquimod-stimulated pDC IFN-α responses in the presence of IgE cross-linking.
We reported previously that the risk for asthma exacerbation after omalizumab treatment was related to restoration of rhinovirus-induced IFN-α responses of PBMCs.10 Here we investigated whether omalizumab’s effects on IFN-α responses, as induced by other ex vivo stimulation conditions in PBMCs or pDCs, had a similar relationship to asthma exacerbations (see Table E4 in this article’s Online Repository at www.jacionline. org). We found no additional significant relationships between IFN-α restoration and proportions of exacerbations.
Effect of omalizumab on TLR7 mRNA expression in unstimulated PBMCs and pDCs
TLR7, a sensor of single-stranded viral RNA, plays an important role in virus-induced pDC IFN-α secretion,15,16 and IgE cross-linking has been shown to impair virus-induced TLR7 expression.15 We hypothesized that omalizumab can induce TLR7 expression and that this effect could contribute to observed increases in IFN-α responses. Unexpectedly, treatment with omalizumab resulted in decreased expression of TLR7 mRNA in unstimulated PMBCs (P = .002) and pDCs (P = .008), as shown in Table II. In addition, there were nonsignificant trends for lower exacerbation rates in the group with the greatest omalizumab-associated change/decrease in PBMC and pDC TLR7 expression (see Table E5 in this article’s Online Repository at www.jacionline.org). Omalizumab did not affect baseline PBMC expression of IRF7, a master regulator of pDC IFN-α secretion (data not shown).17
TABLE II.
| Mechanistic parameter
|
Omalizumab
|
Omalizumab
|
Placebo
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell type | Method measured |
Readout* | No. | Prerandomization, mean (SD) |
Postrandomization, mean (SD) |
Ratio of geometric means (95% CI) |
P value | No. | Prerandomization, mean (SD) |
Postrandomization, mean (SD) |
Ratio of geometric means (95% CI) |
P value |
| PBMC | RT-PCR | TLR7 mRNA | 64 | 1.2 (1.6) | 1.0 (1.8) | 0.81 (0.71–0.92) | .002 | 23 | 1.3 (1.6) | 1.2 (2.0) | 0.92 (0.72–1.18) | .50 |
|
| ||||||||||||
| Purified pDC | RT-PCR | TLR7 mRNA | 18 | 0.3 (1.7) | 0.2 (1.5) | 0.79 (0.66–0.93) | .008 | 7 | 0.2 (1.4) | 0.2 (1.8) | 0.89 (0.62–1.29) | .47 |
Boldface indicates P < .05.
TLR7 mRNA values represent expression measured before ex vivo stimulation and normalized to HPRT expression (2−Δct).
Means are geometric means, and SDs are geometric SDs.
Effect of omalizumab on transcription of genes related to IFN-α responses
We next tested the hypothesis that omalizumab was associated with upregulation of other genes involved in type I interferon signaling, including TLR7; IRF717; IFNB1, the first member of the type I interferon (IFN-α/β) family to be secreted; IFNA1, one of at least 12 distinct IFN-α subtypes18; IFIT1, an interferon-stimulated gene18; and DDX58 (retinoic acid–inducible gene I [RIG-I]), a cytoplasmic sensor of pathogen-associated molecular patterns within viral RNA. Cells obtained before and during omalizumab treatment were stimulated with rhinovirus and IgE cross-linking. Omalizumab significantly increased transcription of IFNB1 (ratio of geometric means, 1.95; 95% CI, 1.04-3.66; P = .04) and DDX58 (RIG-I; ratio of geometric means, 1.43; 95% CI, 1.03-1.99; P = .04) mRNA (Table III). Transcription of TLR7, IRF7, IFNA1, and IFIT1 was not significantly affected.
TABLE III.
Effect of omalizumab on rhinovirus-induced PBMC expression of IFN-α signaling and responsiveness genes*
| Omalizumab
|
Placebo
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| mRNA expression† |
No. | Prerandomization, mean (SD) |
Postrandomization, mean (SD) |
Ratio of geometric means (95% CI) |
P value |
No. | Prerandomization, mean (SD) |
Postrandomization, mean (SD) |
Ratio of geometric means (95% CI) |
P value |
| TLR7 | 56 | 2.5 (2.2) | 3.0 (2.3) | 1.21 (0.94–1.55) | .13 | 22 | 2.6 (2.1) | 2.4 (2.0) | 0.94 (0.66–1.34) | .73 |
|
| ||||||||||
| IRF7 | 56 | 7.1 (3.4) | 7.6 (2.7) | 1.07 (0.75–1.54) | .70 | 22 | 8.0 (2.8) | 5.9 (2.8) | 0.74 (0.45–1.21) | .21 |
|
| ||||||||||
| IFNB1 | 55 | 23.8 (11.6) | 46.4 (8.8) | 1.95 (1.04–3.66) | .04 | 20 | 27.2 (7.2) | 20.7 (8.2) | 0.76 (0.28–2.05) | .57 |
|
| ||||||||||
| IFNA1‡ | 32 | 17.9 (11.5) | 32.0 (12.5) | 1.79 (0.65–4.94) | .25 | 13 | 65.4 (6.5) | 24.2 (5.6) | 0.37 (0.12–1.15) | .08 |
|
| ||||||||||
| DDX58 (RIG-I) | 56 | 6.2 (3.6) | 8.9 (2.8) | 1.43 (1.03–1.99) | .04 | 22 | 9.1 (2.7) | 6.4 (3.3) | 0.7 (0.41–1.22) | .20 |
|
| ||||||||||
| IFIT1 | 56 | 44.8 (13.2) | 75.5 (8.5) | 1.69 (0.85–3.34) | .13 | 22 | 65.9 (8.0) | 24.5 (10.9) | 0.37 (0.14–1.01) | .05 |
Boldface indicates P < .05.
Means are geometric means, and SDs are geometric SDs.
Cells were stimulated ex vivo with anti-IgE plus rhinovirus and mRNA expression of demonstrated genes normalized to HPRT expression (2−Δct).
Measuring transcriptional levels of total IFN-α was not feasible given the existence of at least 12 distinct IFN-α subtypes.
Effects of omalizumab treatment on surface FcεRIα expression on pDCs
As previously demonstrated,8 omalizumab treatment significantly reduced FcεRIα surface expression on pDCs (Fig 2, A, and Table IV). We next tested whether omalizumab affected FcεRIα mRNA levels in either PBMCs or pDCs and found that this was not the case (Table IV). Similarly, omalizumab treatment did not affect mRNA levels of the FcεRI receptor γ chain (FcεRIγ), a transmembrane adaptor of the FcεRI complex (data not shown). FCER1G mRNA expression was measured only in PBMCs because of the limited availability of pDC RNA.
FIG 2.
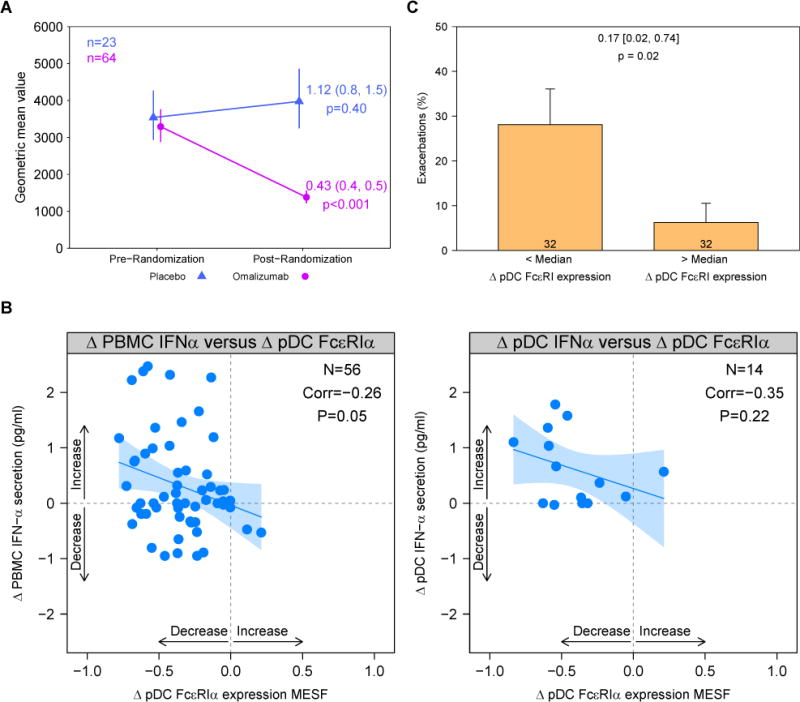
Loss of pDC FcεRIα surface expression is associated with restoration of IFN-α responses and decreased exacerbations. A, pDC surface FcεRIα expression is expressed as mean equivalent soluble fluorochrome values in the omalizumab group (represented in pink) and placebo group (represented in blue). Points represent geometric mean values at corresponding time points; vertical segments cover the 95% CI around the geometric mean. B, Correlation between change in IFN-α level and change in pDC FcεRIα surface expression in the omalizumab group. IFN-α levels were measured in PBMC (left) or pDC (right) supernatants after stimulation with anti-IgE plus rhinovirus; pDC FcεRIα expression was measured in unstimulated cells. Change is calculated as a log difference. Blue line and shading represent a linear model fit and its 95% CI. C, Proportion of exacerbations in the omalizumab group is associated with FcεRIα change. Bars represent the proportion of exacerbations during the study for each group (less than and greater than the median level of change in FcεRIα), and error bars represent SEs. Numbers annotated on the bars represent the number of subjects. Odds ratios, associated 95% CIs, and P values are annotated above the bars.
TABLE IV.
| Mechanistic parameter
|
Omalizumab
|
Omalizumab
|
Placebo
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell type | Method measured |
Readout* | No. | Prerandomization, mean (SD) |
Postrandomization, mean (SD) |
Ratio of geometric means (95% CI) |
P value |
No. | Prerandomization, mean (SD) |
Postrandomization, mean (SD) |
Ratio of geometric means (95% CI) |
P value |
| pDCs‡ | Flow cytometry | Surface FcεRIα expression | 64 | 3231.2 (1.7) | 1381.7 (1.6) | 0.43 (0.37–0.49) | <.001 | 23 | 3540.0 (1.6) | 3974.9 (1.6) | 1.12 (0.85–1.48) | .40 |
|
| ||||||||||||
| PBMCs | RT-PCR | FCER1A mRNA | 64 | 1.9 (1.8) | 1.9 (1.7) | 1.02 (0.91–1.13) | .78 | 23 | 1.5 (1.8) | 1.3 (2.1) | 0.85 (0.67–1.08) | .17 |
|
| ||||||||||||
| Purified pDCs | RT-PCR | FCER1A mRNA | 18 | 0.4 (1.7) | 0.4 (1.9) | 1.07 (0.80–1.42) | .62 | 7 | 0.3 (2.1) | 0.2 (1.7) | 0.69 (0.53–0.9) | .02 |
Boldface indicates P < .05.
FCER1A mRNA values represent expression measured before ex vivo stimulation and normalized to HPRT expression (2−Δct).
Means are geometric means, and SDs are geometric SDs.
pDCs and basophils gated in total PBMCs for flow cytometric analysis (unstimulated cells).
We next tested whether omalizumab-induced reductions in pDC FcεRIα surface expression were related to increased rhinovirus-induced IFN-α secretion in the presence of IgE cross-linking and found a modest inverse correlation (Fig 2, B, left panel; Pearson correlation = −0.26; P = .05). There was a similar trend in parallel studies with purified pDCs (Fig 2, B, right panel). Finally, among omalizumab-treated participants, a significant association between changes in pDC FcεRIα surface expression and exacerbation rate was observed (odds ratio, 0.17; 95% CI, 0.02-0.74; P = .02; Fig 2, C, and see Table E5).
Sample sizes for all of the mechanistic variables included in Tables I–IV and Figs 1 and 2, A-C, are displayed in Table E6 in this article’s Online Repository at www.jacionline.org by treatment group and asthma exacerbation status. Fig E1 in this article’s Online Repository at www.jacionline.org provides a visual representation of the overlap between participants with available mechanistic data and exacerbation status.
Prediction of asthma exacerbations
To investigate the association between mechanistic variables and exacerbations within treatment groups (omalizumab and non-omalizumab), we first explored univariate associations (see Table E7 in this article’s Online Repository at www.jacionline.org). Mechanistic variables assessed during the prerandomization period included PBMC IFN-α responses; unstimulated PBMC expression of FCER1A, TLR7, and IRF7 mRNA; and pDC surface FcεRIα expression. Only variables with less than 40% missing data were included. Two variables were identified as risk factors for exacerbations in the non-omalizumab group, including PBMC IRF7 mRNA after stimulation with rhinovirus and IgE cross-linking (ratio of geometric means exacerbators/nonexacerbators, 0.44; P =.04) and FCER1A mRNA expression in unstimulated PBMCs (ratio of geometric means, 0.61; P = .03).
We next investigated whether addition of these baseline mechanistic measurements could improve an existing predictive model for asthma exacerbations. Using a multivariate model that includes clinical and laboratory parameters,12,13 we obtained a significant area under the curve (0.89 and 0.87 and P = .01 and .008 for the “non-omalizumab” group [combined placebo and inhaled corticosteroid boost] and the omalizumab group, respectively; model 1, Table V).
TABLE V.
Multivariate modeling* to predict asthma exacerbations
| Non-omalizumab†
|
Omalizumab
|
|||||
|---|---|---|---|---|---|---|
| Model | Variables‡ | Area under the ROC curve | P value | Variables‡ | Area under the ROC curve | P value |
| 1: Known clinical predictors | Age, total IgE, atopy, blood eosinophils, exacerbations in previous 90 d, FEV1/FVC ratio, treatment step | 0.89 | .01 | Age, total IgE, atopy, blood eosinophils, exacerbations in previous 90 d, FEV1/FVC ratio, treatment step | 0.87 | .008 |
|
| ||||||
| 2: Known clinical predictors plus new selected mechanistic variables | Known clinical predictors plus PBMC FCER1A mRNA expression | 0.97 | .002 | NA | NA | NA |
FVC, Forced vital capacity; NA, no variables were selected by the feature selection algorithm; ROC, receiver operating characteristic.
The set of mechanistic variables considered by using the feature selection procedure included data obtained from unstimulated PMBCs (FcεRIα protein and mRNA expression and TLR7 and IRF7 mRNA expression) and from ex vivo rhinovirus-stimulated PBMC assays (IFN-α response). The new mechanistic variables selected by using the feature selection procedure were then included in model 2.
Includes participants receiving placebo (n = 23) and inhaled corticosteroid boost (n = 36) treatments.
Known clinical predictors include age, total IgE level, atopy, blood eosinophil count, exacerbations in previous 90 days, FEV1/forced vital capacity ratio, and treatment step.
Using the variable selection procedure, we next added mechanistic variables measured at prerandomization (baseline) to this model. In the non-omalizumab group baseline PBMC FCER1A mRNA expression was found to be a significant predictor of exacerbations after accounting for the set of clinical and laboratory parameters in model 1. In particular, addition of baseline PBMC FCER1A mRNA expression increased the area under the curve by 0.08 to 0.97 (P = .002) and reduced the proportions of false-positive and false-negative predictions by 50% (see Table E8 in this article’s Online Repository at www.jacionline.org).19 No mechanistic parameters were found to be significant additions to known clinical predictors of exacerbations in the omalizumab group.
DISCUSSION
Overexpression of FcεRIα and cross-linking of this receptor can disrupt virus-induced IFN-α responses,5,6 and this represents a potential mechanism for more severe viral respiratory illnesses in patients with allergic asthma. We demonstrated previously that omalizumab treatment of urban children with moderate asthma restores ex vivo PBMC IFN-α responses and that these improved responses were related to a lower risk for asthma exacerbation.10 In the present study we extend these findings by showing that omalizumab treatment in vivo restores IFN-α responses to both rhinovirus and influenza. Furthermore, we demonstrated omalizumab’s effects on pDCs, including reduced expression of FcεRIα on the cell surface and increased virus-induced IFN-α responses in the presence of IgE cross-linking. These effects on pDCs, which are the major source of virus-induced IFN-α among immune cells,20 were inversely correlated. Type I interferons suppress development and stability of TH2 lymphocytes; increasing IFN-α levels could thus diminish TH2-mediated allergic inflammation associated with asthma pathogenesis.21 In support of this, inhaled IFN-β has shown promise as a potential treatment for virus-induced asthma exacerbations.22 These findings provide additional insights into IgE-mediated mechanisms that suppress antiviral responses.
Our findings suggest that omalizumab improves IFN-α responses through reduction of FcεRIα, the α-chain of the high-affinity IgE receptor, on pDC surfaces. Elegant studies of pDC signaling performed by Cao et al23,24 suggest that the mechanism for this effect might involve FcεRIγ, an immunoreceptor tyrosine–based activation motif–containing transmembrane adaptor of the FcεRI complex that can transduce inhibitory signals through pDC surface receptors ILT7 (immunoglobulin-like transcript 7) and BDCA-2 (blood dendritic cell antigen-2). FcεRIγ signaling through these receptors inhibits TLR7/9-induced pDC interferon responses through a B-cell receptor–like pathway involving Lyn/BLK, Syk, and BLNK.24 This immunoreceptor tyrosine–based activation motif–mediated inhibition of pDC interferon is effectively reversed by Syk inhibition in primary human pDCs23 and by knockdown of Syk and FcεRIγ in a pDC cell line25 and is likely also involved in IgE-mediated inhibition of pDC IFN-α responses through FcεRIα-induced FcεRIγ signaling. Therefore reduction of surface FcεRIα could diminish subsequent FcεRIγ signaling in the presence of IgE cross-linking conditions and lead to increased capacity of pDCs to secrete IFN-α in an allergic environment, although we were unable to assess this in our study. The increased rhinovirus-induced PBMC IFNB1 and DDX58 (RIG-I) mRNA levels observed in the posttreatment omalizumab group suggest that pathways downstream of FcεRIγ and upstream of IFN-α signaling might be affected. Although we observed no differences in IRF7 transcription in the posttreatment omalizumab group, it is possible that critical signaling events, such as IRF7 phosphorylation, which is required for IRF7 activation, nuclear translocation, and induction of type I interferon expression, could be enhanced in the setting of reduced IgE and FcεRIα. Other post-translational modifications known to regulate IRF7, including ubiquitination,26 could contribute similarly to the observed effects but were not assessed in our study. Because RIG-I (DDX58) signaling also promotes type I interferon production,27 increased rhinovirus-induced RIG-I mRNA levels after omalizumab could also contribute to enhanced IFN-α responses. Proof of this concept would require demonstration of diminished FcεRIγ signaling in pDCs from omalizumab-treated participants, which was not feasible given the limited numbers of pDCs available for these analyses. Investigating the effect of omalizumab on pDC FcεRIγ signaling could be tested in future omalizumab treatment studies in adults, from whom higher blood volumes could be obtained.
The finding that omalizumab reduces pDC surface FcεRIα protein levels without regulating FcεRIα transcription suggests that this effect is likely related to reduction of free serum IgE levels. Consistent with this idea, free IgE levels have been shown to correlate with pDC surface FcεRIα expression both before and after omalizumab treatment.9 The duration of reduced pDC FcεRIα expression after discontinuation of omalizumab was not measured in our study. Basophil surface FcεRIα protein expression has been shown to increase after omalizumab withdrawal in atopic subjects, paralleling the increase in free serum IgE levels.19 Given that serum IgE similarly regulates surface FcεRIα protein expression on pDCs, it is reasonable to hypothesize that pDC FcεRIα levels would also increase after omalizumab withdrawal as free serum IgE levels increase to preomalizumab levels. Whether such a return to preomalizumab pDC FcεRIα protein expression levels would result in loss of the observed improved IFN-α responses also remains undetermined and represents an area for future investigation.
In addition to FcεRIα- and FcεRIγ-mediated effects, other mechanisms of allergic inflammation–induced inhibition of antiviral responses have been reported. Aeroallergen-induced IL-33 can suppress pDC IRF7 expression and impair IFN-α production and promote a respiratory virus–induced asthma phenotype in a murine model of asthma.28 TLR7 hyporesponsiveness and reduced function have been reported in both murine and human studies of allergic disease28–30 and likely contribute to associated defective IFN-α antiviral responses. Reduced rhinovirus-induced PBMC expression of other critical signaling molecules, including IRF1, IRF7, nuclear factor κB family members, and signal transducer and activator of transcription 1, have also been reported in patients with allergic asthma30 and suggest roles for both interferon-dependent and independent pathways in the synergistic contributions of allergens and viruses to exacerbations of atopic asthma. In addition, the TH2 cytokines IL-4 and IL-13 inhibit TLR3 expression and IRF3 activation, resulting in impaired rhinovirus-induced interferon responses in human bronchial epithelial cells.31
The clinical relevance of omalizumab’s effect on pDC surface FcεRIα expression is underscored by association of a lower exacerbation rate with omalizumab-induced change in FcεRIα surface protein expression. Based on this, one might expect that higher baseline FcεRIα expression would have predicted exacerbations in this population of high-risk children. Surprisingly, this was not the case; pDC surface FcεRIα expression did not improve the ability to predict exacerbations when added to an existing prediction model, which can be attributed to its high correlation with total IgE levels. In contrast, baseline FCER1A mRNA expression in unstimulated PBMCs significantly enhanced the accuracy of an existing predictive model of asthma exacerbations12 in our study population, although this factor was not influenced by omalizumab treatment. The finding that mRNA levels for FcεRIα fit and improve this asthma exacerbation prediction model supports the concept that the high affinity IgE receptor plays an important role in exacerbations.
We have demonstrated that omalizumab therapy results in improved virus-induced pDC IFN-α responses and have linked omalizumab-induced changes in surface pDC FcεRIα expression to improved clinical outcomes (decreased asthma exacerbation rate). Our finding that FCER1A mRNA expression is not affected by omalizumab yet can be used to improve prediction of asthma exacerbations is intriguing and underscores the complexity of FcεRIα regulation in immune cells.32 Future studies investigating the connection between virus-induced pDC IFN-α responses and FcεRIα regulation are needed to further define the mechanisms underlying the connection between atopy, antiviral response, and allergic disease. Our findings support the concept that targeting the IgE pathway in pDCs represents a promising strategy to increase interferon responses, improve antiviral activity in the airways, and reduce exacerbations of allergic asthma.
Supplementary Material
Key messages.
Treatment with omalizumab restores ex vivo IFN-α responses to rhinovirus and influenza virus in the presence of IgE cross-linking and decreases pDC surface FcεRIα expression.
The relationship between increases in rhinovirus-induced IFN-α levels and reduced pDC surface FcεRIα expression in the omalizumab group suggests one potential mechanism underlying the link between atopy and viruses in promoting asthma exacerbations.
The enhancement of exacerbation prediction by addition of baseline PBMC FCER1A mRNA measurements to an existing model of known clinical risk factors highlights the critical role of the IgE high-affinity receptor FcεRIα in promoting asthma exacerbations in children.
Acknowledgments
Supported in whole or in part with Federal funds from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Department of Health and Human Services, under contract and grant numbers NIH NIAID 5R01AI098077, HHSN272200900052C, HHSN272201000052I, 1UM1AI114271-01, and UM2AI117870. Additional support was provided by the National Center for Research Resources and National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, under grants NCATS/NIH UL1TR000150, NCRR/NCAT/NIH UL1TR000077-04, UL1TR000451, UL1TR001105, UL1TR000040, UM1AI109565, UL1TR000075, 1UL1RR025780, UL1TR000154, and UL1TR001082. The following were donated: omalizumab and matching placebo by Novartis and fluticasone and matching placebo by GlaxoSmithKline under a clinical trial agreement with the University of Wisconsin-Madison; EpiPens by Mylan; and Ayr nasal rinse by B.F. Ascher & Company.
M. A. Gill’s institution received a grant from the National Institutes of Health (NIH)/National Institute of Allergy and Infectious Diseases (NIAID), consulting fees and support for travel from the American Academy of Allergy, Asthma & Immunology (AAAAI) for this work and received lecture fees from the American Academy of Pediatrics (AAP) for other works. A. H. Liu received a grant from the NIH/NIAID for this work; served on the Data Safety Monitoring Board for GlaxoSmithKline; and received speakers’ fees from Merck Sharp & Dohme for other works. A. Calatroni received a grant from the NIH/NIAID for this work. R. Z. Krouse’s, B. Shao’s, and A. Schiltz’s institutions received grants from the NIH/NIAID for this work. J. E. Gern’s institution received a grant from the NIH/NIAID for this work and personally received consultancy fees from Janssen, Regeneron, and PReP Biosciences and travel expenses from Boehringer Ingelheim. W. W. Busse received a grant from the NIH/NIAID for this work; board membership from Boston Scientific and ICON; and consultancy fees from Novartis, Glaxo SmithKline, Genentech, Roche, Boehringer Ingelheim, Sanofi Genzyme, AstraZeneca, Teva, 3M, PrEPBiopharm, Circassia, Regeneron, Peptinnovate, Knopp Bio, and Elsevier.
Abbreviations
- IRF
Interferon regulatory factor
- pDC
Plasmacytoid dendritic cell
- PROSE
Preventative Omalizumab or Step-Up Therapy for Fall Exacerbations
- RIG-I
Retinoic acid–inducible gene I
- TLR
Toll-like receptor
Footnotes
Disclosure of potential conflict of interest: A. Togias declares no relevant conflicts of interest.
References
- 1.Heymann PW, Carper HT, Murphy DD, Platts-Mills TA, Patrie J, McLaughlin AP, et al. Viral infections in relation to age, atopy, and season of admission among children hospitalized for wheezing. J Allergy Clin Immunol. 2004;114:239–47. doi: 10.1016/j.jaci.2004.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green RM, Custovic A, Sanderson G, Hunter J, Johnston SL, Woodcook A. Synergism between allergens and viruses and risk of hospital admission with asthma: case-control study. BMJ. 2002;324:763. doi: 10.1136/bmj.324.7340.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray CS, Poletti G, Kebadze T, Morris J, Woodcook A, Johnston SL, et al. Study of modifiable risk factors for asthma exacerbations: virus infection and allergen exposure increase the risk of asthma hospital admissions in children. Thorax. 2006;61:376–82. doi: 10.1136/thx.2005.042523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schroeder JT, Bieneman AP, Xiao H, Chichester KL, Vasagar K, Saini S, et al. TLR9- and FcepsilonRI-mediated responses oppose one another in plasmacytoid dendritic cells by down-regulating receptor expression. J Immunol. 2005;175:5724–31. doi: 10.4049/jimmunol.175.9.5724. [DOI] [PubMed] [Google Scholar]
- 5.Gill MA, Bajwa G, George TA, Dong CC, Dougherty II, Jiang N, et al. Counter-regulation between the FcepsilonRI pathway and antiviral responses in human plasmacytoid dendritic cells. J Immunol. 2010;184:5999–6006. doi: 10.4049/jimmunol.0901194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Durrani SR, Montville DJ, Pratt AS, Sahu S, DeVries MK, Rajamanickam V, et al. Innate immune responses to rhinovirus are reduced by the high-affinity IgE receptor in allergic asthmatic children. J Allergy Clin Immunol. 2012;130:489–95. doi: 10.1016/j.jaci.2012.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foster B, Metcalfe DD, Prussin C. Human dendritic cell 1 and dendritic cell 2 subsets express FcepsilonRI: correlation with serum IgE and allergic asthma. J Allergy Clin Immunol. 2003;112:1132–8. doi: 10.1016/j.jaci.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Prussin C, Griffith DT, Boesel KM, Lin H, Foster B, Casale TB. Omalizumab treatment downregulates dendritic cell FcepsilonRI expression. J Allergy Clin Immunol. 2003;112:1147–54. doi: 10.1016/j.jaci.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 9.Schroeder JT, Bieneman AP, Chichester KL, Hamilton RG, Xiao H, Saini SS, et al. Decreases in human dendritic cell-dependent T(H)2-like responses after acute in vivo IgE neutralization. J Allergy Clin Immunol. 2010;125:896–901.e6. doi: 10.1016/j.jaci.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 10.Teach SJ, Gill MA, Togias A, Sorkness CA, Arbes SJ, Jr, Calatroni A, et al. Preseasonal treatment with either omalizumab or an inhaled corticosteroid boost to prevent fall asthma exacerbations. J Allergy Clin Immunol. 2015;136:1476–85. doi: 10.1016/j.jaci.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bajwa G, DeBerardinis RJ, Shao B, Hall B, Farrar JD, Gill MA. Cutting edge: critical role of glycolysis in human plasmacytoid dendritic cell antiviral responses. J Immunol. 2016;196:2004–9. doi: 10.4049/jimmunol.1501557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teach SJ, Gergen PJ, Szefler SJ, Mitchell HE, Calatroni A, Wildfire J, et al. Seasonal risk factors for asthma exacerbations among inner-city children. J Allergy Clin Immunol. 2015;135:1465–73.e5. doi: 10.1016/j.jaci.2014.12.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoch HE, Calatroni A, West JB, Liu AH, Gergen PJ, Gruchalla RS, et al. Predicting fall seasonal asthma exacerbations in inner-city children in the absence and presence of omalizumab therapy. J Allergy Clin Immunol. 2017 Epub ahead of print. [Google Scholar]
- 14.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1–22. [PMC free article] [PubMed] [Google Scholar]
- 15.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–31. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 16.Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 17.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–7. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 18.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 19.Saini SS, MacGlashan DW, Jr, Sterbinskym SA, Togias A, Adelman DC, Lichtenstein LM, et al. Down-regulation of human basophil IgE and FC epsilon RI alpha surface densities and mediator release by anti-IgE-infusions is reversible in vitro and in vivo. J Immunol. 1999;162:5624–30. [PubMed] [Google Scholar]
- 20.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 21.Huber JP, Ramos HJ, Gill MA, Farrar JD. Cutting edge: type I IFN reverses human Th2 commitment and stability by suppressing GATA3. J Immunol. 2010;185:813–7. doi: 10.4049/jimmunol.1000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Djukanovic R, Harrison T, Johnston SL, Gabbay F, Wark P, Thomson NC, et al. The effect of inhaled IFN-beta on worsening of asthma symptoms caused by viral infections. A randomized trial. Am J Respir Crit Care Med. 2014;190:145–54. doi: 10.1164/rccm.201312-2235OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao W, Rosen DB, Ito T, Bover L, Bao M, Watanabe G, et al. Plasmacytoid dendritic cell-specific receptor ILT7-Fc epsilonRI gamma inhibits Toll-like receptor-induced interferon production. J Exp Med. 2006;203:1399–405. doi: 10.1084/jem.20052454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao W, Zhang L, Rosen DB, Bover L, Watanabe G, Bao M, et al. BDCA2/Fc epsilon RI gamma complex signals through a novel BCR-like pathway in human plasmacytoid dendritic cells. PLoS Biol. 2007;5:e248. doi: 10.1371/journal.pbio.0050248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bao M, Hanabuchi S, Facchinetti V, Du Q, Bover L, Plumas J, et al. CD2AP/SHIP1 complex positively regulates plasmacytoid dendritic cell receptor signaling by inhibiting the E3 ubiquitin ligase Cbl. J Immunol. 2012;189:786–92. doi: 10.4049/jimmunol.1200887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ning S, Pagano JS, Barber GN. IRF7: activation, regulation, modification and function. Genes Immun. 2011;12:399–414. doi: 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–92. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lynch JP, Werder RB, Simpson J, Loh Z, Zhang V, Haque A, et al. Aeroallergen-induced IL-33 predisposes to respiratory virus-induced asthma by dampening antiviral immunity. J Allergy Clin Immunol. 2016;138:1326–37. doi: 10.1016/j.jaci.2016.02.039. [DOI] [PubMed] [Google Scholar]
- 29.Roponen M, Yerkovich ST, Hollams E, Sly PD, Holt PG, Upham JW. Toll-like receptor 7 function is reduced in adolescents with asthma. Eur Respir J. 2010;35:64–71. doi: 10.1183/09031936.00172008. [DOI] [PubMed] [Google Scholar]
- 30.Pritchard AL, White OJ, Burel JG, Carroll ML, Phipps S, Upham JW. Asthma is associated with multiple alterations in anti-viral innate signalling pathways. PLoS One. 2014;9:e106501. doi: 10.1371/journal.pone.0106501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Contoli M, Ito K, Padovani A, Poletti D, Marku B, Edwards MR, et al. Th2 cytokines impair innate immune responses to rhinovirus in respiratory epithelial cells. Allergy. 2015;70:910–20. doi: 10.1111/all.12627. [DOI] [PubMed] [Google Scholar]
- 32.MacGlashan D., Jr IgE and FcepsilonRI regulation. Clin Rev Allergy Immunol. 2005;29:49–60. doi: 10.1385/criai:29:1:049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.