

Abstract
The S3 state of the Mn4 CaO5 -cluster in photosystem II was investigated by DFT calculations and compared with EXAFS data. Considering previously proposed mechanism; a water molecule is inserted into an open coordination site of Mn upon S2 to S3 transition that becomes a substrate water, we examined if the water insertion is essential for the S3 formation, or if one cannot eliminate other possible routes that do not require a water insertion at the S3 stage. The novel S3 state structure consisting of only short 2.7–2.8 Å Mn—Mn distances was discussed.
1. Introduction
The oxygen-evolving complex (OEC) of photosystem II (PSII) is a Mn- and Ca-containing cofactor which accepts oxidizing equivalents from the photo-oxidized chlorophyll of the reaction center of PSII [1,2]. After accumulation of four oxidizing equivalents on the OEC (Si state (i = 0–4) cycle), the OEC oxidizes the water substrate to form dioxygen [3]. Reported studies to elucidate the water oxidation mechanism include X-ray diffraction (XRD) [4], site-specific mutant [5], spectroscopic method [6,7], substrate water exchange study using isotope labeling of water [8], and DFT calculations [9–15]. Most recently, the XRD study with a resolution of 1.95 Å has provided the X-ray radiation-damage free crystal structure of PSII in the dark resting S1 state and revealed the heart of the OEC, the so-called Mn4CaO5-cluster (S1-(XRD structure) of Figure 1) [16].
Figure 1.

Kok cycle and proposed structure of the Mn4 Ca-cluster, S1 -(XRD structure) [16], S2 -(theoretical model-a/b) [10], S3 -(theoretical model-a/b) [11,14,17] and S3 -(new model). Notations for each residue are similar to those in the PDB-data on the dark state [16]. Possible proton positions are not shown to emphasize the Mn—Mn distances and to respect the different proposals [9–11], except in S3 -(theoretical model-a/b) the inserted OH− is presented in red color. Throughout this Letter, a solid line is used if Mn—O bond distance is covalently bound with 2.0 Å or less, a broken line if it is non-bonding but ionically interacting with 2.0 to 2.5 Å, and no line if 2.5 Å or longer. On the hand Ca—O interaction is always ionic; a solid line is used if Ca—O bond distance is 2.8 Å or less, a broken line if it is between 2.8 and 3.0, and no line if 3.0 Å or longer. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The OEC in the S3 state is considered as one of the most critical steps, as it sets the chemical environment for the O—O bond formation to occur in the subsequent S3 to S0 transition. Interruption of the S3 state formation by various chemical and biochemical treatments therefore blocks O2 evolution activity. The two possible Mn oxidation states of the S3 state have been discussed based on the X-ray spectroscopic studies [6,7]; one is Mn4(IV4) state consisting of four Mn(IV) and another is Mn4(III,IV3)• state consisting of three Mn(IV), one Mn(III) and one non-metal radical residue oxidized in the S2 to S3 transition. The Mn4(IV4) state assignment has been supported by the EPR study [17].
The S3 state has been studied by solution EXAFS, and the metal–metal and metal–ligand distance information has been reported [7,18]. However, the presence of similar distances prevents (i.e. a few Mn—Mn) an unique solution, and Glockner et al. reported two possible EXAFS interpretations [18]; ‘Fit a’ consists of two Mn—Mn pairs with 2.72 Å distance and two Mn—Mn pairs with 2.82 Å distance, and ‘Fit b’ consists of two Mn—Mn pairs with 2.75 Å, one pair with 2.79 Å and one pair with 3.26 Å. The structure of the OEC in the S3 state has also been studied by the room temperature crystallography at X-ray free electron lasers (XFELs) [19,20]. However, the resolution is currently not sufficient to resolve the atomic distances within the Mn4CaO5 cluster. The structure has been investigated also by theoretical studies. It has been suggested that the Mn4CaO5-cluster keeps the open-cubane structure like the S1 state, and takes an additional water-derived ligand in the open-site (Mn1) to form the six-coordinate Mn atom upon oxidation in the S2 to S3 transition (S3-(theoretical model-a) proposed by Siegbahn, Cox et al., and Shoji et al., Figure 1) [11,14,17]. Consequently, the oxidation state becomes Mn4(IV4) in the S3 state with all four Mn atoms six-coordinated. A different route for the OH− inclusion in the S2 to S3 transition was suggested by other studies (see S3-(theoretical model-b) of Figure 1) [14,17]. In this case, the additional OH is located next to the dangling Mn atom (Mn4).
Several experimental results support that all Mn are Mn(IV) in the S3 state, by 5 coordinated Mn site (Mn1 or Mn4) in the S2 state becoming 6 coordination. This could happen either by having an additional water insertion to the open site, or by O5 becoming μ4-oxo. In the current study, we investigated whether μ4-oxo-like structure is possible in the S3 state that does not require an additional OH− in the Mn4CaO5-cluster. In this non-water insertion model, O5 becomes the center of the μ4-oxo that bridges Mn1, Mn3, Mn4 and Ca, and as a consequence all Mn—Mn distances will be around 2.7–2.8 Å which are characteristic of di-μ-oxo bridged Mn—Mn pair (S3-(new model) shown in Figure 1).
Two μ4-oxo structures have been reported in the literature that contain Mn and Ca; one is a planar form [21] and the other is a tetrahedral structure [22]. In order for O5 in the OEC to be μ4-oxo, however, it has to be a non-tetrahedral (seesaw) geometry as shown in Figure 2. The seesaw-like non-tetrahedral geometry can connect Mn4—O5—Mn1 linearly, although the non-tetrahedral geometry of oxo-bridged Mn has not been reported previously. Therefore, one question arises if such non-tetrahedral geometry can be realized around the O5 center of the OEC. We first examined whether O5 can form a non-tetrahedral geometry in ‘the first coordination sphere’ that includes the Mn4CaO5-cluster in the Mn4(IV4) or Mn4(III,IV3)• state with the ligands of the first coordination sphere.
Figure 2.
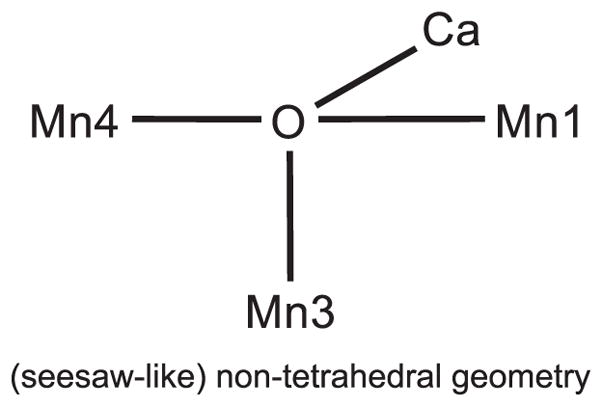
Schematic representation of the (seesaw-like) non-tetrahedral geometry which is required to form O5 as the μ4 -oxo in the OEC of the S3 state.
The structure in ‘the first coordination sphere’ was investigated with DFT calculations starting from the structure of the S2-Mn4(III,IV3) state based on our previous studies on the OEC [23]. In the first step, the ligands as well as the Mn4CaO5-cluster were not fixed so that the ligands can relax and move when the Mn4CaO5-cluster changes from S2-Mn4(III,IV4) to S3-Mn4(IV4) where the Jahn–Teller distortion of Mn(III) is no longer effective. In the second step, the ligands of the second coordination sphere are considered by fixing the terminal atoms from the crystal structure.
2. Computational details
The S2 structure (Figure S1) that we used as a starting model for optimizing the S3 structure was obtained from the 1.9 Å crystal structure [24], which is similar to the structure of the early S2 model (S2-(theoretical model-a) of Figure 1). As the first step, we carried out full optimization of ‘first coordination sphere’ that includes the Mn4Ca-cluster only with the ligands of the first coordination sphere. In the second step, we investigated the ‘second coordination sphere’ that includes the ‘first coordination sphere’, plus Asp61, Lys317, His337, Leu343, Arg354, Cl− around Glu333, and the surrounding water molecules. Notation for each residue is similar to those in the PDB-data (3ARC) on the dark state [24]. The number of atoms for the ‘second coordination sphere’ is the same as that of the early theoretical model of the S3 state [11].
The possible position of OH− was examined based on two earlier models; one is S2-(theoretical model-a/b) where W1, W3 and W4 are H2O and W2 is OH− in the S2 state (note that the protonation states are not shown in Figure 1 to emphasize the Mn—Mn distances), and the other is the S1 state where W1–W4 are all H2O as proposed by Luber et al. [9]. In the latter case, W1–W4 are present as H2O even in the S2 state, because no proton release is expected during the S1 to S2 transition. This difference in the protonation state of W2 comes from the uncertainty in the sequence of events during the S3 to S0 transition, that is, the precise sequence of O2 evolution and two-proton release have not yet been clarified. There are four possible cases as shown in Figure 3a–d. The first model shown in Figure 3a has been proposed by an earlier experimental study [25], but it is inconsistent with the recent photothermal beam deflection (PBD) study [26]. The PBD study is also inconsistent with the second model shown in Figure 3b. The third shown in Figure 3c is consistent with the PBD study and represents the release of two protons before O2 evolution. This leads one proton release from the new H2O substrate in the S0 to S1 transition, and it requires the presence of OH− in the S1 and S2 states which is consistent with the protonation state of S2-(theoretical model-a/b) (Figure 1). The fourth shown in Figure 3d represents the release of two protons via O2 evolution. Therefore, it leads to the release of two protons before the S1 state, and as a result, the S1 and S2 states become 2OH− or O2− + H2O as shown in Figure 3d.
Figure 3.

Possible details of Kok-cycle (a–d) and the possible deprotonation state of water substrates in each Si state.
In this study, we considered the two deprotonation models (Figure 3c, d), that match with the PBD study. The first one is based on the Pantazis models and assumes one of W1, W3 and W4 is OH− in addition to W2-OH−. The second model is based on the study by Luber et al. and assumes that one of W1–W4 becomes OH− in the S2 to S3 transition. The detailed discussion on the water ligand deprotonation is described in SI.
The calculations were carried out using GAUSSIAN 09 [27] and B3LYP* DFT functional which is a modification of the original B3LYP functional with a reduction of the exact exchange to 15% [28]. Grimme’s dispersion correction was used. For the structure optimization, LanL2DZ and 6-31G(d) basis sets were applied to metals (Mn, Ca) and other atoms (H, C, N, O), respectively. After that, for the energy refinement, cc-pVTZ was applied to all atoms. A dielectric constant was set as 6.0. S = 12/2 spin state was used so that Mn4(IV4) and/or Mn4(III,IV3)• states can be obtained. The Pipek–Mezey population localization was used for orbital analysis [29,30].
3. Results and discussion
The optimized structures of the ‘first coordination sphere’ in the S3 state with two of W1–W4 being OH− are shown in Figure 4. We primarily focused on two OH− cases, as this allows us to compare the energetics of current models with previously reported ones [11]. The details of the S3 models with one of W1–W4 being OH− are shown in Figure S2.
Figure 4.

Optimized structures of ‘first coordination sphere’ in the S3 state; (a) W1&W2-OH−; (b) W1&W3-OH−; (c) W1&W4-OH−; (d) W2&W3-OH−; (e) W2&W4-OH−; (f) W3&W4-OH−.
When W1 and W2 ligands are OH− (referred to as W1&W2-OH− hereafter, Figure 4a), the Mn1—Mn2, Mn1—Mn3, Mn2—Mn3 and Mn3—Mn4 pairs show distances around 2.7–2.8 Å. Mulliken’s population analysis for this indicated that all four Mn ions are Mn(IV) (Mn4(IV4), Table S1). When W1 and W3 ligands are OH− (W1&W3-OH−, Figure 4b), W3-OH− is located between Ca and Mn4 lengthening the Mn3—Mn4 pair (~3.2 Å). The oxidation state of this form is also formally Mn4(IV4). Similar Mn—Mn distances and oxidation state are obtained when W2 and W3 ligands are OH− (W2&W3-OH−, Figure 4d). When W1 and W4 ligands are OH−(W1&W4-OH−, Figure 4c), on the other hand, W4 becomes an OH-radical reducing Mn1 to Mn(III) (Mn4(III,IV3)•) due to the weak ionic interaction between W4-OH− and Ca2+. The reduced Mn1(III) stays away from Mn3(IV), showing the longer Mn1—Mn3 distance (~3.1 Å). Similar Mn—Mn distances and oxidation state are obtained when W2 and W4 ligands are OH− (W2&W4-OH−, Figure 4e). When W3 and W4 ligands are OH− (W3&W4-OH−, Figure 4f), W3 becomes an O2−, releasing one proton to W4, and as a result W4 becomes OH2. The resulting W3-O2− is located between Ca and Mn4 lengthening the Mn3—Mn4 pair (~3.1 Å).
The W1&W2-OH− structure (Figure 4a) shows the short Mn—Mn distances (2.7–2.8 Å) matching with the trend of ‘Fit a’ of Mn-EXAFS on the S3 state [18], that consists of two Mn—Mn pairs with 2.72 Å distance and two Mn—Mn pairs with 2.82 Å distance as shown in S3-(new model) of Figure 1. The W1&W2-OH− structure has O5 close to Mn1, Mn3, Mn4 and Ca (O5—Mn1: ~1.9 Å, O5—Mn3: ~1.9 Å, O5—Mn4: ~2.1 Å and O5—Ca: ~2.7 Å), showing short distances (2.7–2.8 Å) for the Mn1—Mn3 and Mn3—Mn4 pairs. Thus, the O5 position is highly asymmetric to Mn1 and Mn4, with a non-tetrahedral geometry.
The possibility of O5 being the μ4-oxo ligand is examined by analyzing localized orbitals. Two types of the localized orbitals were observed around O5; the first one has the lobe from O5 over Mn1 or over the Mn3 atom, indicating the distribution of the shared electrons (Figure 5a, b), and the second one is just distorted from O5 toward Mn4 or Ca atom indicating the unshared electrons (Figure 5c, d), where the orbitals for O5—Mn4 and O5—Ca are assigned as the non-bonding orbitals. We have also investigated the localized orbitals of a μ4-oxo bond in the Mn4CaO4 cluster reported by Zhang et al. [22], that forms a tetrahedral ligand geometry. The result shows that this μ4-oxo bond may better be explained as a covalent μ2-oxo bond plus two ionic interactions (Figure S3), similar to what we observed in the OEC.
Figure 5.

(a–d) Localized orbitals at around O5 of W1&W2-OH− structure in the S3 state; bonding with surrounding metals in (a, b) and nonbonding in (c, d), (e) schematic representations for the expected S3 -(new model) and the optimized W1&W2-OH− structure.
The above observation implies that, Mn4—O5 (distance of 2.1 Å) is non-bonded in the W1&W2-OH− structure. There are two reasons for a short Mn4—Mn3 distance of 2.8 Å. The first is the ionic interaction of O5 and Mn4, even if O5—Mn4 is non-bonding. The second reason is the ligation mode of Asp170, that bridges Mn4 and Ca with only one oxygen atom (monodentate bridge, see Figure 5e), with the second oxygen of the carboxylate being near Ca. The monodentate bridge of Asp170 takes the role resembling a μ-oxo bridge between Mn4 and Ca, bringing these two metals closer. Because Ca is bridged to O5, the monodentate bridge of Asp170 also makes the O5—Mn4 distance shorter. The formation of such a monodentate bridge was observed during the ‘first coordination sphere’ optimization. When the structure is re-optimized by restoring the initial bidentate bridge (e.g. S2-(theoretical model-a/b) of Figure 1), the monodentate-bridged structure was obtained. Thus, the monodentate bridge of Asp170 seems to be essential for this W1&W2-OH− model.
The W1&W2-OH− structure obtained from the ‘first coordination sphere’ optimization in the S3 state is energetically lower than the other structures. Thus, we further carried out the analysis by using the ‘second coordination sphere’. The total number of atoms in the ‘second coordination sphere’ model was the same as S3-(theoretical model-a) [11], where W1, W3, W4 and other water molecules are H2O, with two OH−, that comes from W2 and the water-derived ligand (OH−) inserted into the Mn4CaO5-cluster. In this ‘second coordination sphere’ analysis, we added an extra H2O in the W1&W2-OH− structure so that the total mass is consistent with the models with the inserted OH−. The location of the extra H2O was optimized based on the position of water molecules observed in the XRD structure of the S1 state [24]. When the space between Leu343 and Glu354 was filled with a H2O, the W1&W2-OH− structure shows the lowest energy.
The optimized structures for the ‘second coordination sphere’ with W1 and W2 being OH− (W1&W2-OH−) are shown in Figure 6. Two structures were examined; the first structure (Figure 6a) was obtained by fixing the α-carbon and its two nearest atoms along the backbone from the XRD structure, by following the same method taken by Siegbahn to obtain S3-(theoretical model-a) [11]. The second structure (Figure 6b) was obtained by fixing the α-carbon atoms of the selected ligands (Asp61, Lys317, His337 and Arg357) in the second coordination sphere so that the first coordination sphere of the Mn4CaO5-cluster is relaxed. The fully optimized structures are shown in Figure S4.
Figure 6.

Optimized structures of ‘second coordination sphere’ in the S3 state; (a) W1&W2-OH− fixing the α-carbon and its two nearest atoms; (b) W1&W2-OH− fixing the α-carbon atoms of Asp61, Lys317, His337 and Arg357; (c) W2&inserted water-OH− fixing the α-carbon and its two nearest atoms; (d) W2&inserted water-OH− fixing the α-carbon atoms of Asp61, Ly317, His337 and Arg357. A red broken line is used if Mn—O bond distance is between 2.0 and 2.5 Å or Ca—O bond distance is between 2.8 and 3.0 Å. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Both structures (Figure 6a, b) indicate the Mn4(IV4) oxidation state for the Mn4Ca5-cluster (Tables S2 and S3). And both structures have 2.7–2.8 Å distance for the Mn1—Mn2, Mn1—Mn3 and Mn2—Mn3 pairs, while different distances are observed for the Mn3—Mn4 pair (~2.98 Å of Figure 6a and ~2.83 Å of Figure 6b). The difference in the Mn3—Mn4 distance reflects the complicated ligand environment of Mn4. Mn4 exists in the bonding network of Asp61···(W1-OH−)···Mn4···O5, where Mn4 is attracted by O5 as well as Asp61 through the hydrogen bonding of Asp61···(W1-OH−). Mn4 is also coordinated by Asp170 which bridges Mn4 and Ca with one oxygen atom (different from that of the S1 XRD structure). It is noteworthy that the position of Mn4 and therefore the Mn3—Mn4 distance changes depending on whether the ligands of the first coordination sphere were relaxed. Thus, the second structure of W1&W2-OH− (Figure 6b) shows the trend of ‘Fit a’ of Mn-EXAFS in the S3 state (being similar to the W1&W2-OH− structure (Figure 4a) obtained by ‘first coordination sphere’ analysis) [18].
The W1&W2-OH− (Figure 6a, b) structures are also compared with that of the OH−-inserted Mn4CaO5-cluster (Figure 6c, d) with the same number of atoms and conditions. The first structure− (Figure 6a) shows +7.7 kcal/mol higher in energy than that of the OH−-inserted Mn4CaO5-cluster (Figure 6c). On the other hand, when the ligands are relaxed, the same model (Figure 6b) shows −0.1 kcal/mol energy lower than that of the OH−-inserted Mn4CaO5-cluster (Figure 6d). Namely, the W1&W2-OH− structure (Figure 6b) becomes equally stable as the OH−-inserted Mn4CaO5-cluster (Figure 6d), when the ligands in the first coordination sphere are relaxed. This implies that neither of these structures can yet be ruled out as a candidate for the actual structure in the S3 state. The W1&W2-OH− structure (Figure 6b) matches with the distances interpreted as ‘Fit-a’ in the EXAFS study, and the OH−-inserted Mn4CaO5-cluster (Figure 6c) matches with those as ‘Fit-b’ that consists of two Mn—Mn pairs with 2.75 Å, one pair with 2.79 Å and one pair with 3.26 Å [18]. The superpositions (Figures S4 and S5) of the structures shown in Figure 6 indicate that the α-carbon atoms in the first coordination sphere of the W1&W2-OH− (Figure 6b) are displaced from those of the OH−-inserted structure (Figure 6d). The OH−-inserted structure (Figure S5b) with the ligands taken from the W1&W2-OH− structure (Figure 6b) shows that the amino acids are held by the OH−-inserted Mn4CaO5-cluster (Figure 6d). The water molecules maintain their positions in the W1&W2-OH− structure. Further study is required to elucidate the structure and the bridging mode of the Mn4CaO5-cluster in the S3 state, and a QM/MM study is in progress.
A stable S3 structural model was obtained in this study, that consists of only short (2.7–2.8 Å) Mn—Mn distances. All Mn is Mn(IV), with non-bonding Mn4—O5 distance of 2.1–2.3 Å. If the OEC forms such a structure in the S3 state, we anticipate that W1 or W2 may change from OH− to O2− to increase the negative charge around Mn4 upon H+ release before e− transfer during the S3 to S4 transition [26]. Thus, W1 or W2 becomes a terminal oxo-ligand transiently and can be oxidized to a terminal oxo-radical ligand by Mn4 or tyrosyl-radical. In the first scenario (Figure 7a), W1 is expected to be the oxo radical in the S4 state before the O—O bond formation with W2. In the second scenario (Figure 7b), W2 is expected to be an oxo radical in the S4 state before the O—O bond formation with W1. These two cases (Figure 7a,b) both generate the O2 molecule from two ligands of Mn4 atom [31]. On the other hand, in the third scenario (Figure 7c) when W2 is an oxo radical, the W2 forms an O—O bond with the O5 atom. After O2 evolution and the supply of two new H2O substrates, the new O5 ligand becomes OH− in the S0 state which is in agreement with 2H+ release during the S3 to S0 transition. By H+ rearrangement, the S0 state in the lower path can convert to that in the upper path (see Figure 7, H+ rearrangement path).
Figure 7.

Possible O2 evolution paths based on the W1&W2-OH− structure in the S3 state.
4. Conclusion
The novel S3 state structure that consists only short (2.7–2.8 Å) Mn—Mn pairs (S3-(new model) of Figure 1) was investigated with DFT optimizations. The short Mn—Mn pairs are obtained when the ligands in the first coordination sphere are fully relaxed, and when W1 and W2 ligands of Mn4 are OH− (Figures 4a and 6b) and the di-μ2-oxo bridged structures are formed except for the Mn3—Mn4 pair. The Mn3—Mn4 pair is bridged by an O4 ligand, while Mn4 is non-bonding with O5. The Mn3—Mn4 pair keeps a short distance (~2.8 Å) similar to the other di-μ2-oxo bridged Mn—Mn pairs when the ligands of the first coordination sphere are relaxed. The reason for the short Mn3—Mn4 distance is the ligation mode of Asp170, that bridges Mn4 and Ca with only one oxygen atom resembling the role of a μ-oxo bridge between Mn4 and Ca. This may imply that the catalytic reaction is a process under physiological conditions where structural and electronic structure fluctuations could play an important role. The novel S3 state structure we reported here is in agreement with EXAFS data (Fit-a) [18], and its stability is compatible with the other S3 state structure (S3-(theoretical model-a)).
Supplementary Material
Acknowledgments
We thank the HOKUSAI Greatwave of RIKEN. J.Y. and V.K.Y. are supported by the Director, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences of the Department of Energy under contract DE-AC02-05CH11231, and NIH grant GM55302 (V.K.Y.).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.cplett.2016.03.010.
Footnotes
Associated content
Coordinates of ‘first coordination sphere’ in the S3 state, and ‘second coordination sphere’ in the S3 state (Table S4).
References
- 1.Tommos C, Babcock GT. Acc Chem Res. 1998;31:18. [Google Scholar]
- 2.Yano J, Yachandra VK. Chem Rev. 2014;114:4175. doi: 10.1021/cr4004874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Joliot P. Photosynth Res. 2003;76:65. doi: 10.1023/A:1024946610564. [DOI] [PubMed] [Google Scholar]
- 4.Ferreira KN, Iverson TM, Maghlaoui K, Barber J, Iwata S. Science. 2004;303:1831. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 5.Shimada Y, Suzuki H, Tsuchiya T, Tomo T, Noguchi T, Mimuro M. Biochemistry. 2009;48:6095. doi: 10.1021/bi900317a. [DOI] [PubMed] [Google Scholar]
- 6.Sauer K, Yano J, Yachandra VK. Coord Chem Rev. 2008;252:318. doi: 10.1016/j.ccr.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haumann M, Müller C, Liebisch P, Iuzzolino L, Dittmer J, Grabolle M, Neisius T, Meyer-Klaucke W, Dau H. Biochemistry. 2005;44:1894. doi: 10.1021/bi048697e. [DOI] [PubMed] [Google Scholar]
- 8.Nilsson H, Krupnik T, Kargul J, Messinger J. Biochim Biophys Acta. 2014;1837:1257. doi: 10.1016/j.bbabio.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 9.Luber S, Rivalta I, Umena Y, Kawakami K, Shen JR, Kamiya N, Brudvig G, Batista VS. Biochemistry. 2011;50:6308. doi: 10.1021/bi200681q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pantazis DA, Ames W, Cox N, Lubitz W, Neese F. Angew Chem Int Ed. 2012;51:9935. doi: 10.1002/anie.201204705. [DOI] [PubMed] [Google Scholar]
- 11.Siegbahn PEM. Phys Chem Chem Phys. 2012;14:4849. doi: 10.1039/c2cp00034b. [DOI] [PubMed] [Google Scholar]
- 12.Li X, Siegbahn PEM, Ryde U. Proc Natl Acad Sci U S A. 2015;112:3979. doi: 10.1073/pnas.1422058112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, Siegbahn PEM. Chem Eur J. 2015;21:18821. doi: 10.1002/chem.201501593. [DOI] [PubMed] [Google Scholar]
- 14.Shoji M, Isobe H, Yamaguchi K. Chem Phys Lett. 2015;636:172. [Google Scholar]
- 15.Shoji M, Isobe H, Nakajima T, Yamaguchi K. Chem Phys Lett. 2015;640:23. [Google Scholar]
- 16.Suga M, Akita F, Hirata K, Ueno G, Murakami H, Nakajima Y, Shimizu T, Yamashita K, Yamamoto M, Ago H, Shen JR. Nature. 2015;517:99. doi: 10.1038/nature13991. [DOI] [PubMed] [Google Scholar]
- 17.Cox N, Retegan M, Neese F, Pantazis DA, Boussac A, Lubitz W. Science. 2014;345:804. doi: 10.1126/science.1254910. [DOI] [PubMed] [Google Scholar]
- 18.Glöckner C, Kern J, Broser M, Zouni A, Yachandra VK, Yano J. J Biol Chem. 2013;288:22607. doi: 10.1074/jbc.M113.476622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kupitz C, et al. Nature. 2014;513:261. doi: 10.1038/nature13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kern J, et al. Nat Commun. 2014;5:4371. doi: 10.1038/ncomms5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma S, Yuan D, Chang JS, Zhou HC. Inorg Chem. 2009;48:5398. doi: 10.1021/ic900475q. [DOI] [PubMed] [Google Scholar]
- 22.Zhang C, Chen C, Dong H, Shen JR, Dau H, Zhao J. Science. 2015;348:690. doi: 10.1126/science.aaa6550. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Hatakeyama M, Ogata K, Nakamura S, Li C. J Phys Chem B. 2014;118:14215. doi: 10.1021/jp505889p. [DOI] [PubMed] [Google Scholar]
- 24.Umena Y, Kawakami K, Shen JR, Kamiya N. Nature. 2011;473:55. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 25.Schlodder E, Witt HT. J Biol Chem. 1999;274:30387. doi: 10.1074/jbc.274.43.30387. [DOI] [PubMed] [Google Scholar]
- 26.Klauss A, Haumann M, Dau H. Proc Natl Acad Sci U S A. 2012;109:16035. doi: 10.1073/pnas.1206266109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frisch MJ, et al. Gaussian 09, Revision D. 01. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 28.Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]
- 29.Pipek J, Mezey PG. J Chem Phys. 1989;90:4916. [Google Scholar]
- 30.Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA. J Comput Chem. 1993;14:1347. [Google Scholar]
- 31.Kusunoki M. Biochim Biophys Acta. 2007;1767:484. doi: 10.1016/j.bbabio.2007.03.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.