

Abstract
The intestinal epithelium provides a critical barrier that separates the gut microbiota from host tissues. Non-steroidal anti-inflammatory drugs (NSAIDs) are efficacious analgesics and antipyretics, and are among the most frequently used drugs worldwide. In addition to gastric damage, NSAIDs are toxic to the intestinal epithelium, causing erosions, perforations and longitudinal ulcers in the gut. Here we use a unique in vitro human primary small intestinal cell monolayer system to pinpoint the intestinal consequences of NSAID treatment. We found that physiologically relevant doses of the NSAID diclofenac (DCF) are cytotoxic because they uncouple mitochondrial oxidative phosphorylation and generate reactive oxygen species. We also find that DCF induces intestinal barrier permeability, facilitating the translocation of compounds from the luminal to the basolateral side of the intestinal epithelium. The results we outline here establish the utility of this novel platform, representative of the human small intestinal epithelium to understand NSAID toxicity, which can be applied to study multiple aspects of gut barrier function including defense against infectious pathogens and host-microbiota interactions.
Keywords: Small intestine, leaky gut, bacterial translocation, NSAIDs, mitochondria, superoxide
Graphical Abstract
Primary human small intestinal monolayers for modelling NSAID-induced damage
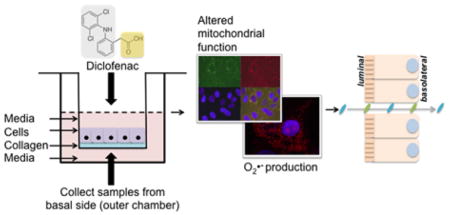
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most commonly used medications in the world, consumed daily by over 30 million adults worldwide. In the United States alone, nearly a quarter of adults take a dose of prescription or over the counter NSAIDs each day1. NSAIDs effectively manage inflammation and pain caused by a range of etiologies, from arthritis and cancer to sports-related injuries. NSAIDs are inhibitors of the cyclooxygenase (COX) enzymes (COX-1 and 2), which synthesize prostaglandins (PGs) that are critical mediators of inflammation, potent vasodilators, and inhibitors of platelet aggregation. The widespread use of NSAIDs comes with a downside, however – NSAIDs damage both the gastric and intestinal mucosa and are associated with a range of gastrointestinal (GI) complications. The elderly and chronic NSAID users, such as those afflicted with rheumatoid or osteoarthritis, are particularly affected by NSAID-associated side effects. NSAID usage is expected to continue rising to mitigate aging-related degenerative and inflammatory conditions; concurrently, the preponderance of NSAID-associated side effects is also expected to increase2. NSAIDs can cause relatively benign, nuisance-type symptoms such as heartburn, nausea, dyspepsia and abdominal pain2. However, in as many as 80% of NSAID users, acute hemorrhages and mucosal erosions are detected in the gastroduodenal mucosa upon endoscopic examination3; a subset of these can lead to serious complications such as bleeding and perforation, erosions, ulcers, strictures, perforation and bowel obstruction3. Chronic NSAID usage can cause colitis, and exacerbate preexisting conditions such as inflammatory bowel disease (IBD), irritable bowel syndrome (IBS), and diverticular disease4. Long-term users suffer from diminished absorptive capacity and increased intestinal permeability.5
The bulk of clinical studies have focused on the pathophysiology of upper GI tract damage inflicted by NSAIDs, likely due to self-reporting, and the relative ease with which damage may be clinically confirmed in this accessible location. It is clear that NSAIDs can also inflict damage elsewhere in the GI tract, particularly in the small intestine. In 1979, it was reported that erythrocyte extrusion and focal erosion of jejunal villi occurs within five minutes of treatment with aspirin in healthy volunteers3, 6. Enterohepatic circulation, microvascular events and disruption of cellular oxidative processes have all been implicated in the pathophysiology of NSAIDs. It has also been shown that NSAID-induced lesions are diminished by antibiotic treatment7–8, and germ-free animals are resistant to indomethacin-induced intestinal damage9. These results implicate a role for the intestinal microbiota and/or microbial metabolites in mediating NSAID GI toxicity6, 10. Indeed, we have previously reported that inhibiting gut microbial β-glucuronidase enzymes reduces reactivation of NSAID-glucuronide metabolites by microbiome-encoded β-glucuronidases, thus reducing small intestinal ulceration resulting from acute treatment with diclofenac, indomethacin and ketoprofen11–12. Together, these observations indicate that NSAID-induced damage results from a range of factors.
Despite the overwhelming evidence that NSAIDs can damage the GI tract, prostaglandins, the prostaglandin (PG) products of COX enzymes are important for repairing the intestinal epithelium. The prostaglandin PGE2 is crucial for regenerating and promoting survival of small intestinal crypts following intestinal injury resulting from radiation or the dextran sodium sulfate-induced colitis model13–14. Additionally, without PGE2, Rag2−/− knockout mice lacking immunomodulatory TRegs develop severe colitis15. Thus, NSAID-mediated inhibition of prostaglandin secretion can have multifactorial effects on intestinal homeostasis.
The small intestine is the primary site of digestion, nutrient absorption and secretion of digestive enzymes; it is also critical for the metabolism and absorption of pharmacopeia, particularly those that are orally administered. Diverse differentiated cells including enterocytes, Paneth, goblet, enterochromaffin and tuft cells, as well as undifferentiated transit amplifying cells and stem cells reside along a crypt and villus axis; stem cells are located at the base of crypts and proliferating cells differentiate along the crypt-villus axis. Gradients of growth factors, morphogens and microbial metabolites govern the patterning and polarity of intestinal cells, fluctuations of which can impact the health and disease states of the small intestine. The polarized, absorptive enterocytes maintain a tight barrier against the intestinal lumen, protecting the periphery from invading pathogens and toxic substances. Loss of barrier integrity can lead to activated immune signaling and intestinal inflammation, which can further affect the intestinal barrier, and facilitate the progression of various systemic pathologies; this syndrome is colloquially referred to as “leaky gut”16. Compromised intestinal integrity can facilitate the translocation of immunostimulatory microbial products that can cause chronic systemic inflammation in individuals infected with the Human Immunodeficiency Virus (HIV), resulting in the progression to AIDS. Moreover, disruptions in intestinal integrity can lead to dysbiosis, an alteration in the composition of intestinal commensal microbes observed in AIDS17, respiratory diseases18, irritable bowel disease19, cancer20 etc. Moreover, dysbiosis affects the immune system in myriad ways21 that can further amplify the sequelae of a number of diseases.
NSAID toxicity has been previously modeled in vitro using transformed cell lines22. However, the normal intestine clearly differs from cancer cell lines. In fact, drug permeability and transport are miscorrelated between the widely used Caco2 colon carcinoma cell lines and the human intestine23. The in vitro growth of primary, small-intestinal epithelial cells as enteroids with a luminal compartment surrounded by a cell monolayer has transformed the field of intestinal biology24–26. These multicellular structures possess all cell lineages found in vivo including stem cells. Despite their advantages, these enclosed structures pose significant limitations. The enteroid luminal surface is not accessible to added drugs or toxins, making assay of the cellular response to luminal chemical exposure (as occurs in vivo) challenging. Enteroids are formed within a thick hydrogel so that measuring molecular transport across the cell monolayer is difficult. Human non-transformed27 and primary28 monolayer intestinal epithelial cells have been cultured on stiff surfaces such as a Transwell membrane. However, these systems demonstrate a limited subset of cells present in the intestinal epithelium due to the nonphysiologic substrate. Recently, we demonstrated a self-renewing monolayer derived from primary human small intestine comprised of both differentiated and undifferentiated cells on a collagen-based scaffolding29. Cells within the monolayer replicated key features of the in vivo small intestine including appropriate basal to luminal cell polarity and differentiation into goblet cells and enterocytes. More importantly, the system possessed an open and accessible monolayer surface permitting luminal chemical exposure followed by facile assay of cell response. The permeable hydrogel supporting the monolayer coupled to exchangeable luminal and basal fluid reservoirs enabled measurement of molecular transport across the monolayer. This primary, epithelial monolayer is an excellent model system for understanding the impact of drugs such as NSAIDs on the human small intestine.
RESULTS AND DISCUSSION
We recently developed a self-renewing, in vitro human small intestinal monolayer cultured on a collagen scaffold which provided the necessary biophysical and biochemical cues to support both stem/proliferative and differentiated cells29. The porous scaffold was formed within a modified Transwell apparatus so that growth factors and other reagents could be applied to either the luminal or basal cell surfaces (FIG 1A). The accessible luminal and basal reservoirs above and below the scaffold permit sampling of these fluids. The cells within the monolayer display the appropriate markers, with intense actin staining the apex of the epithelial cells, corresponding to the absorptive microvilli that are adjacent to the intestinal lumen (FIG 1B). The basolateral side of the epithelial cells was marked by Integrin β4 staining, an adhesion protein by which epithelial cells attach to and interact with the underlying extracellular matrix (FIG 1B). The intestinal epithelium forms a protective barrier against damaging agents and pathogenic microbes by forming tight intercellular junctions that are reinforced by E-cadherin proteins. The typical flagstone E-cadherin staining pattern (FIG 1C), and the transepithelial electrical resistance (TEER) of >79 Ω cm2 confirms that our HSI monolayers exclude the movement of molecules from the luminal to the basolateral compartments. Finally, this human small intestine model cultured in the presence of Wnt3A, Noggin and R-spondin1 is composed primarily of proliferative cells, as evidenced by the high degree of incorporation of 5-ethynyl-2-deoxyuridine (EdU) over the course of 24 hours (FIG 1D). Thus, our system contains fully polarized monolayers largely comprised of proliferating cells, with the necessary integrity for drug toxicity and permeability studies. While the present study lacks immune cells, this platform is amenable to the incorporation of various lineages of immune cells, which will facilitate the investigation of cytokine function and inflammation, further improving the utility of this platform.
Figure 1. Overview of the two-dimensional human small intestine (HSI) monolayer utilized.

(A) Schematic of the Transwell apparatus containing a collagen scaffold, upon which human small intestinal monolayers are cultured. (B) Longitudinal cross section of monolayers stained with F-actin and Integrin β4 to label the luminal and basolateral regions, respectively. (C) En face view of monolayers stained with F-actin and E-cadherin reveals characteristic flagstone staining pattern. Scale bar, 25 μm. (D) HSI monolayers consist primarily of proliferating, EdU+ cells. Colors used: EdU, red; DNA, blue.
We sought to employ this small intestine culture monolayer to study the sequelae of NSAID treatment using diclofenac, a widely prescribed analgesic for both human and veterinary use. Diclofenac (2-(2,6-dichloroanilino) phenylacetic acid (DCF) has an approximately 10-fold higher preference for the COX-2 isoenzyme versus COX-1, and is primarily ingested orally, although it may also be administered via topical, intravenous, intramuscular and intrarectal routes. While DCF is better tolerated than non-selective NSAIDs such as indomethacin, it is still associated with a variety of toxic small intestinal side effects30–31. A typical therapeutic dose of DCF (50 mg orally) translates into local concentrations between 300–1600 μM of DCF within the intestinal lumen (assuming an intestinal fluid volume of between 0.1–0.5 L)32. Therefore, we employed a range of DCF between 500 μM and 1000 μM to recapitulate an intraluminal physiologically relevant concentration. Initial studies included a lower concentration of 250 μM DCF, which was found to not be cytotoxic (data not shown).
Diclofenac, like many other non-selective NSAIDs, is amphipathic, containing lipophilic and charged moieties, and is capable of donating a proton. These properties allow it to interact with a variety of biological membranes. It has been proposed that DCF and other NSAIDs inflict mitochondrial damage by uncoupling oxidative phosphorylation, increasing reactive oxygen species (ROS), diminishing ATP production, and thus reducing cell viability33–34. Thus, we used Mitotracker Red CMH2-XRos (abbreviated as CMH2-XRos) to assess the impact of DCF on mitochondrial potential. CMH2-XRos is non-fluorescent until it is oxidized in respiring cells, whereupon it accumulates in the mitochondrial matrix. CMH2-XRos fluorescence intensity is directly correlated with cell health, with increased fluorescence observed in actively respiring cells35. We found that DCF sharply reduces the fluorescence intensity of CMH2-XRos in a dose-dependent manner (FIG 2A, FIG 2B). These results suggest that the amphipathic properties of DCF can decouple oxidative phosphorylation, leading to decreased mitochondrial function. Moreover, DCF exerted a dose-dependent cytotoxic effect in our human small intestinal cell monolayer culture system (HSI) within a 24-hour treatment period, as determined using endpoint CellTox Green cytotoxicity reagent (FIG 2C).
Figure 2. Diclofenac exerts cytotoxicity by reducing mitochondrial membranepotential and inducing O2•−.

(A) DCF-treated cells have reduced Mitotracker CMH2-XRos staining compared to vehicle-treated control. Images were acquired at 20x and are representative images from three independent experiments. (B) Quantification of the mean fluorescence intensity of (A) demonstrates that DCF significantly reduces CMH2-XRos staining. (C) Compared to untreated control, 24h treatment with DCF exerts significant dose-dependent cytotoxicity, as measured by CellTox Green endpoint assay. ** p<0.01 by one-way ANOVA with Dunnett’s test for multiple comparisons to the control. (D) MitoSOX staining reveals high levels of O2•− generated in response to DCF, within 24h. Cells are counterstained in both (A) and (D) with Hoechst to label nuclei, and Mitotracker Green to label mitochondria. Scale bar, 25 μm.
The mitochondria of most mammalian cells produce ROS such as O2•−, H2O2, and −•OH, which can be important for intracellular redox signaling, but more often result in oxidative stress, damage to cellular macromolecules, depletion of antioxidants, and a range of pathologies. The presence of mitochondrial superoxide dismutase underscores the importance of the superoxide (O2•−) anion among other ROS species. In mitochondria with an appropriate mitochondrial transmembrane potential and proton motive force, electron transport chain shuttles electrons along a series of complexes (I–IV), coupling electron transport with proton transport from the mitochondrial matrix into the intermembrane space. O2•− is produced by one-electron reduction of the terminal electron acceptor O2. As described above, DCF-mediated uncoupling of the electron transport chain causes an accumulation of electrons in the matrix, leading to an increase in O2•−, and subsequent mitochondrial and cellular injury36. We found that 24h DCF treatment induces high levels of O2•− in a dose-dependent manner, as evident from the fluorescent sensor MitoSOX Red (FIG 2D).
To determine whether cell proliferation was influenced by DCF, we incubated HSI in media containing EdU, either 500 or 1000 μM DCF, or vehicle control. At the end of 24h, cells were stained, mounted on slides and images were acquired and quantified using Definiens software37–38(FIG 3A). We quantified cell density as number of nuclei per mm2, the absolute number of intact nuclei, and the EdU fluorescent intensity of Hoechst positive cells, indicative of proliferative (stem/progenitor) cells. Integrating the weighted EdU+ fluorescent intensity with the corresponding number of cells led to the generation of the histological score, a parameter that reflects the total proliferative subset of a given sample. We found that within 24h, DCF reduced the total number of cells in a dose dependent manner (FIG 3A). Both cell cell density (FIG 3B) and absolute cell number (FIG 3C) were diminished, albeit not significantly, upon DCF treatment. Additionally, DCF diminished replicative potential, as seen by the reduced number of EdU+, Hoechst+ double positive cells (FIG 3D). Overall, DCF acutely reduced the histological score of HSI within 24h (FIG 3E). Similar results were seen after a 48h treatment (SUPP FIG 1), with a larger magnitude of effect.
Figure 3. DCF reduces the proliferation of stem/progenitor cells in HSI monolayers.

(A) DCF-treated HSI monolayers have a smaller proportion of EdU+ nuclei compared to controls. Scale bar = 200 μm. Following 24h treatment, while (B) cellular density (number of cells per square millimeter) is unchanged, DCF decreases (C) the total number of nuclei in HSI monolayers. (D) DCF reduces EdU fluorescence intensity of HSI, and (E) reduces the histological score, which is described in Methods. ** p<0.01, ***p< 0.001 by one-way ANOVA with Dunnett’s test for multiple comparisons to the control.
Tight junctions and low permeability are hallmarks of our HSI monolayer system. The diffusion of Lucifer Yellow (LY), an aqueous, fluorescent dye from the luminal to the basolateral compartments is a surrogate marker for assaying barrier permeability39. The optimal permeability for a DCF toxicity assay was investigated by measuring the relationship of TEER and LY leakage in a series of monolayers. Monolayers with TEER < 42 Ω cm2 showed an LY apparent permeability (Papp) of 7.36 ± 5.1 cm s−1 (n=4), while monolayers with TEER > 79 Ω cm2 showed a Papp of LY 2.35 ± 1.05 cm s−1 (n=3). Although the monolayers show moderate TEER values, LY permeability measurements indicate that the monolayers were not leaky and were below the permeability cutoff of 5 x10−7 cm s−1 used by others for drug transport/absorption assays40. Further, the previously reported TEER of rat ileum of 35 ± 4.9 Ω cm2 and rat colon of 100 ± 26 Ω cm2 is similar to that measured in our primary cell culture system41–42. Caco-2, a colonic carcinoma cell line, is well known to have higher TEER values (400–600 Ω cm2) relative to primary cells of the small intestine41–42. After measuring TEER, we incubated confluent HSI monolayers with increasing concentrations of DCF with 500 μM LY placed in the luminal compartment (outlined in FIG 4A). The TEER difference over time is presented as percent change (ΔTEER%) from initial TEER, thus each monolayer was normalized to its initial TEER, which is set to 100% (see Methods for details).. There was a significant decrease in ΔTEER% when monolayers were treated with 1000 or 2000 μM DCF for 24 h compared to control (p<0.02) (FIG 4B), whereas 200 or 500 μM DCF did not appreciably alter the TEER compared to control (p>0.6). LY apparent permeability (Papp) in monolayers that were untreated or treated with 200 μM DCF for 24 h, was 2.8 ± 1.16 (x 10−7) cm s−1 and 2.28 ± 2.56 (x 10−7) cm s−1 respectively. That is, Papp of these two conditions is lower than 5 x 10−7 cm s−1, the common upper bound of Papp considered to be a non-compromised monolayer40, confirming that the barrier is intact in these conditions. However, for monolayers treated with DCF 1000 μM or 2000 μM for 24 h, the average LY Papp is significantly higher than 5 x 10−7 cm s−1 (Papp, 1000 μM = 10. 84 ± 6.38 (x 10−7) cm s−1, Papp, 2000 μM =18.66 ± 4.63 (x 10−7) cm s−1). The ΔTEER % reduction was accompanied by increased Papp of LY for monolayers incubated with DCF at >1000 μM for 24 h (FIG 4C) confirming the disruption of tight junctions in the monolayers. The barrier integrity of the monolayers was also evaluated at 3h by measuring LY permeability; the average LY Papp of 2000 μM DCF-treated samples (Papp 2000 μM = 4. 70 ± 2.78 (x 10−7) cm s−1) was quite close to the permeability for a compromised monolayer. Thus at 2000 μM, DCF could disrupt cell-cell interactions in as little as 3h.
Figure 4. DCF breaches barrier integrity and causes permeability in HSI monolayers.

(A) Sequence of the experimental workflow detailing points at which TEER was measured. (B) 1000 and 2000 μM of DCF significantly reduces ΔTEER (%) within 24h. Mean ΔTEER (%) of DCF-treated samples were compared to mean of vehicle-treated cells using one-way ANOVA with Dunnett test for multiple comparisons. *p<0.05. (C) DCF increases the Papp of LY through HSI within 24h, with significant differences observed for 1000 and 2000 μM DCF. Means of treated cells were compared to vehicle-cells using one-way ANOVA with Dunnet test for multiple comparisons. *p<0.05, ***p<0.001. (D) 24h treatment with LPS along did not significantly reduces ΔTEER (%) (p=0.266) but DCF significantly reduces ΔTEER%, and this reduction is also observed when LPS is also added to cells, indicating an overall loss of barrier integrity. Means of experimental groups were compared to each other using two-way ANOVA with Sidak’s correction for multiple comparisons. **p<0.01, ***p<0.001. (E) Immunofluorescence of F-actin and E-cadherin demonstrates a loss of flagstone pattern staining with DCF treatment, which is also observed with LPS co-treatment. Scale bar, 25 μm.
Lipopolysaccharide (LPS) is an integral outer-membrane component of Gram-negative bacteria. LPS elicits the secretion of pro-inflammatory cytokines and signaling molecules such as nitric oxide and eicosanoids, which collectively mount a robust inflammatory response against suspected bacterial invaders. In humans, endotoxin levels range between 50–150 endotoxin units (EU) per gram of feces, resulting from the normal turnover of microbiota the intestinal lumen43. We repeated the translocation studies described above using LPS instead of Lucifer Yellow (FIG 4D). Consistent with previous reports44, LPS alone did not diminish TEER, indicating that in the absence of a secondary damaging agent the intestinal epithelium maintains an intact barrier against LPS translocation in our model. However, DCF addition significantly reduced TEER in both LPS-treated and LPS-free HSI monolayers (p = 0.001). Further, staining LPS-treated monolayers with F-actin and E-cadherin shows maintenance of the flagstone pattern characteristic of non-treated monolayers, confirming that LPS alone does not disrupt barrier integrity of the intestinal epithelium (FIG 4E). DCF disrupts this regular staining both with and without LPS treatment, suggestive of increased barrier permeability (FIG 4E).
In summary, using a novel primary human small intestine monolayer system, we have demonstrated that the amphipathic NSAID diclofenac disrupts mitochondrial potential, and exerts oxidative stress by generation of the ROS species O2•−. The cumulative effect of NSAIDs results in cytotoxicity, and reduction in the proliferative potential of intestinal epithelial cells. Moreover, DCF causes intestinal barrier permeability, as evidenced from diffusion of LY from the luminal to the basolateral compartment. DCF also disrupts the tight junctions that are necessary for excluding LPS and other pathogenic molecules from systemic circulation. Thus, our HSI monolayer system provides a useful platform with which to study intestinal permeability and cytotoxicity resulting from a variety of xenobiotics, and to examine the role the intestinal microbiota play in these processes.
METHODS
Detailed lists of materials used and methods are available as Supporting Information.
Supplementary Material
Acknowledgments
This work was supported by NIH grants CA098468 (MRR), USDA 055336 (SJB), CA207416 (MRR, SJB), DK109559 (NLA, SJB, SM), T32DK007737 (APB). The UNC Translational Pathology Laboratory is supported, in part, by grants from the NCI (CA016086), NIEHS (P30ES010126-15A1), NCBT (2015-IDG-1007), and the University Cancer Research Fund (UCRF). The authors thank Dr. Eric Brustad for the use of his plate reader, the UNC Microscopy Services Lab and Dr. George Glekas (Olympus) for their technical expertise and advice, and members of the Allbritton & Redinbo laboratories for helpful discussions.
Footnotes
CONFLICTS OF INTEREST
SJB, STM, NLA have a financial interest in Altis Biosystems, LLC, which is commercializing human intestinal cell platforms for drug and microbiome screening. MRR is a founder of Symberix, Inc, which is developing microbiome-targeting therapeutics. The other authors have no conflicts to disclaim.
BIBLIOGRAPHY
- 1.Kaufman DW, Kelly JP, Rosenberg L, Anderson TE, Mitchell AA. Recent patterns of medication use in the ambulatory adult population of the United States: the Slone survey. JAMA. 2002;287(3):337–44. doi: 10.1001/jama.287.3.337. [DOI] [PubMed] [Google Scholar]
- 2.Wolfe MM, Lichtenstein DR, Singh G. Gastrointestinal toxicity of nonsteroidal antiinflammatory drugs. N Engl J Med. 1999;340(24):1888–99. doi: 10.1056/NEJM199906173402407. [DOI] [PubMed] [Google Scholar]
- 3.Ehsanullah RS, Page MC, Tildesley G, Wood JR. Prevention of gastroduodenal damage induced by non-steroidal anti-inflammatory drugs: controlled trial of ranitidine. BMJ. 1988;297(6655):1017–21. doi: 10.1136/bmj.297.6655.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davies NM. Toxicity of nonsteroidal anti-inflammatory drugs in the large intestine. Dis Colon Rectum. 1995;38(12):1311–21. doi: 10.1007/BF02049158. [DOI] [PubMed] [Google Scholar]
- 5.Sigthorsson G, Tibble J, Hayllar J, Menzies I, Macpherson A, Moots R, Scott D, Gumpel MJ, Bjarnason I. Intestinal permeability and inflammation in patients on NSAIDs. Gut. 1998;43(4):506–11. doi: 10.1136/gut.43.4.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whittle BJ. Mechanisms underlying intestinal injury induced by anti-inflammatory COX inhibitors. Eur J Pharmacol. 2004;500(1–3):427–39. doi: 10.1016/j.ejphar.2004.07.042. [DOI] [PubMed] [Google Scholar]
- 7.Whittle BJ. Cyclooxygenase and nitric oxide systems in the gut as therapeutic targets for safer anti-inflammatory drugs. Curr Opin Pharmacol. 2004;4(6):538–45. doi: 10.1016/j.coph.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 8.Syer SD, Blackler RW, Martin R, de Palma G, Rossi L, Verdu E, Bercik P, Surette MG, Aucouturier A, Langella P, Wallace JL. NSAID enteropathy and bacteria: a complicated relationship. J Gastroenterol. 2015;50(4):387–93. doi: 10.1007/s00535-014-1032-1. [DOI] [PubMed] [Google Scholar]
- 9.Robert A, Asano T. Resistance of germfree rats to indomethacin-induced intestinal lesions. Prostaglandins. 1977;14(2):333–41. doi: 10.1016/0090-6980(77)90178-2. [DOI] [PubMed] [Google Scholar]
- 10.Boelsterli UA, Redinbo MR, Saitta KS. Multiple NSAID-induced hits injure the small intestine: underlying mechanisms and novel strategies. Toxicol Sci. 2013;131(2):654–67. doi: 10.1093/toxsci/kfs310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LoGuidice A, Wallace BD, Bendel L, Redinbo MR, Boelsterli UA. Pharmacologic targeting of bacterial beta-glucuronidase alleviates nonsteroidal anti-inflammatory drug-induced enteropathy in mice. J Pharmacol Exp Ther. 2012;341(2):447–54. doi: 10.1124/jpet.111.191122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saitta KS, Zhang C, Lee KK, Fujimoto K, Redinbo MR, Boelsterli UA. Bacterial beta-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: mode of action and pharmacokinetics. Xenobiotica. 2014;44(1):28–35. doi: 10.3109/00498254.2013.811314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohn SM, Schloemann S, Tessner T, Seibert K, Stenson WF. Crypt stem cell survival in the mouse intestinal epithelium is regulated by prostaglandins synthesized through cyclooxygenase-1. J Clin Invest. 1997;99(6):1367–79. doi: 10.1172/JCI119296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tessner TG, Cohn SM, Schloemann S, Stenson WF. Prostaglandins prevent decreased epithelial cell proliferation associated with dextran sodium sulfate injury in mice. Gastroenterology. 1998;115(4):874–82. doi: 10.1016/s0016-5085(98)70259-8. [DOI] [PubMed] [Google Scholar]
- 15.Chinen T, Komai K, Muto G, Morita R, Inoue N, Yoshida H, Sekiya T, Yoshida R, Nakamura K, Takayanagi R, Yoshimura A. Prostaglandin E2 and SOCS1 have a role in intestinal immune tolerance. Nat Commun. 2011;2:190. doi: 10.1038/ncomms1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Odenwald MA, Turner JR. Intestinal permeability defects: is it time to treat? Clin Gastroenterol Hepatol. 2013;11(9):1075–83. doi: 10.1016/j.cgh.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vujkovic-Cvijin I, Dunham RM, Iwai S, Maher MC, Albright RG, Broadhurst MJ, Hernandez RD, Lederman MM, Huang Y, Somsouk M, Deeks SG, Hunt PW, Lynch SV, McCune JM. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci Transl Med. 2013;5(193):193ra91. doi: 10.1126/scitranslmed.3006438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budden KF, Gellatly SL, Wood DL, Cooper MA, Morrison M, Hugenholtz P, Hansbro PM. Emerging pathogenic links between microbiota and the gut-lung axis. Nat Rev Microbiol. 2017;15(1):55–63. doi: 10.1038/nrmicro.2016.142. [DOI] [PubMed] [Google Scholar]
- 19.Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol. 2017;14(10):573–584. doi: 10.1038/nrgastro.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhatt AP, Redinbo MR, Bultman SJ. The role of the microbiome in cancer development and therapy. CA Cancer J Clin. 2017;67(4):326–344. doi: 10.3322/caac.21398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levy M, Kolodziejczyk AA, Thaiss CA, Elinav E. Dysbiosis and the immune system. Nat Rev Immunol. 2017;17(4):219–232. doi: 10.1038/nri.2017.7. [DOI] [PubMed] [Google Scholar]
- 22.Omatsu T, Naito Y, Handa O, Hayashi N, Mizushima K, Qin Y, Hirata I, Adachi S, Okayama T, Kishimoto E, Takagi T, Kokura S, Ichikawa H, Yoshikawa T. Involvement of reactive oxygen species in indomethacin-induced apoptosis of small intestinal epithelial cells. J Gastroenterol. 2009;44(Suppl 19):30–4. doi: 10.1007/s00535-008-2293-3. [DOI] [PubMed] [Google Scholar]
- 23.Larregieu CA, Benet LZ. Drug discovery and regulatory considerations for improving in silico and in vitro predictions that use Caco-2 as a surrogate for human intestinal permeability measurements. AAPS J. 2013;15(2):483–97. doi: 10.1208/s12248-013-9456-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zachos NC, Kovbasnjuk O, Foulke-Abel J, In J, Blutt SE, de Jonge HR, Estes MK, Donowitz M. Human Enteroids/Colonoids and Intestinal Organoids Functionally Recapitulate Normal Intestinal Physiology and Pathophysiology. J Biol Chem. 2016;291(8):3759–66. doi: 10.1074/jbc.R114.635995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.In JG, Foulke-Abel J, Estes MK, Zachos NC, Kovbasnjuk O, Donowitz M. Human mini-guts: new insights into intestinal physiology and host-pathogen interactions. Nat Rev Gastroenterol Hepatol. 2016;13(11):633–642. doi: 10.1038/nrgastro.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merker SR, Weitz J, Stange DE. Gastrointestinal organoids: How they gut it out. Dev Biol. 2016;420(2):239–250. doi: 10.1016/j.ydbio.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 27.Trapecar M, Goropevsek A, Gorenjak M, Gradisnik L, Slak Rupnik M. A co-culture model of the developing small intestine offers new insight in the early immunomodulation of enterocytes and macrophages by Lactobacillus spp. through STAT1 and NF-kB p65 translocation. PLoS One. 2014;9(1):e86297. doi: 10.1371/journal.pone.0086297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noel G, Baetz NW, Staab JF, Donowitz M, Kovbasnjuk O, Pasetti MF, Zachos NC. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci Rep. 2017;7:45270. doi: 10.1038/srep45270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Gunasekara DB, Reed MI, DiSalvo M, Bultman SJ, Sims CE, Magness ST, Allbritton NL. A microengineered collagen scaffold for generating a polarized crypt-villus architecture of human small intestinal epithelium. Biomaterials. 2017;128:44–55. doi: 10.1016/j.biomaterials.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scholer DW, Boettcher I, Ku EC, Schweizer A. Pharmacology of diclofenac sodium (Voltaren) Semin Arthritis Rheum. 1985;15(2 Suppl 1):61–4. doi: 10.1016/s0049-0172(85)80012-3. [DOI] [PubMed] [Google Scholar]
- 31.Seager JM, Cullen DJ, Pearson G, Holmes S, Doherty M, Wilson JV, Garrud P, Garner S, Maynard A, Logan RF, Hawkey CJ. Ibuprofen versus other non-steroidal anti-inflammatory drugs: use in general practice and patient perception. Aliment Pharmacol Ther. 2000;14(2):187–91. doi: 10.1046/j.1365-2036.2000.00699.x. [DOI] [PubMed] [Google Scholar]
- 32.Niu X, de Graaf IA, Langelaar-Makkinje M, Horvatovich P, Groothuis GM. Diclofenac toxicity in human intestine ex vivo is not related to the formation of intestinal metabolites. Arch Toxicol. 2015;89(1):107–19. doi: 10.1007/s00204-014-1242-6. [DOI] [PubMed] [Google Scholar]
- 33.Whitehouse MW, Haslam JM. Ability of some antirheumatic drugs to uncouple oxidative phosphorylation. Nature. 1962;196:1323–4. doi: 10.1038/1961323a0. [DOI] [PubMed] [Google Scholar]
- 34.Somasundaram S, Sigthorsson G, Simpson RJ, Watts J, Jacob M, Tavares IA, Rafi S, Roseth A, Foster R, Price AB, Wrigglesworth JM, Bjarnason I. Uncoupling of intestinal mitochondrial oxidative phosphorylation and inhibition of cyclooxygenase are required for the development of NSAID-enteropathy in the rat. Aliment Pharmacol Ther. 2000;14(5):639–50. doi: 10.1046/j.1365-2036.2000.00723.x. [DOI] [PubMed] [Google Scholar]
- 35.Cottet-Rousselle C, Ronot X, Leverve X, Mayol JF. Cytometric assessment of mitochondria using fluorescent probes. Cytometry A. 2011;79(6):405–25. doi: 10.1002/cyto.a.21061. [DOI] [PubMed] [Google Scholar]
- 36.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mulrane L, Rexhepaj E, Penney S, Callanan JJ, Gallagher WM. Automated image analysis in histopathology: a valuable tool in medical diagnostics. Expert Review of Molecular Diagnostics. 2008;8(6):707–725. doi: 10.1586/14737159.8.6.707. [DOI] [PubMed] [Google Scholar]
- 38.Torphy RJ, Naim R, Midkiff B, Volmar K, Collisson E, Yeh JJ. Abstract B80: Stroma in pancreatic cancer: Automated quantification and its prognostic significance. Cancer Research. 2016;76(24 Supplement):B80–B80. doi: 10.1158/1538-7445.panca16-b80. [DOI] [Google Scholar]
- 39.Yamaura Y, Chapron BD, Wang Z, Himmelfarb J, Thummel KE. Functional Comparison of Human Colonic Carcinoma Cell Lines and Primary Small Intestinal Epithelial Cells for Investigations of Intestinal Drug Permeability and First-Pass Metabolism. Drug Metab Dispos. 2016;44(3):329–35. doi: 10.1124/dmd.115.068429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hubatsch I, Ragnarsson EG, Artursson P. Determination of drug permeability and prediction of drug absorption in Caco-2 monolayers. Nat Protoc. 2007;2(9):2111–9. doi: 10.1038/nprot.2007.303. [DOI] [PubMed] [Google Scholar]
- 41.Le Ferrec E, Chesne C, Artusson P, Brayden D, Fabre G, Gires P, Guillou F, Rousset M, Rubas W, Scarino ML. In vitro models of the intestinal barrier. The report and recommendations of ECVAM Workshop 46. European Centre for the Validation of Alternative methods. Altern Lab Anim. 2001;29(6):649–68. doi: 10.1177/026119290102900604. [DOI] [PubMed] [Google Scholar]
- 42.Artursson P, Ungell AL, Lofroth JE. Selective paracellular permeability in two models of intestinal absorption: cultured monolayers of human intestinal epithelial cells and rat intestinal segments. Pharm Res. 1993;10(8):1123–9. doi: 10.1023/a:1018903931777. [DOI] [PubMed] [Google Scholar]
- 43.Park SH, Kim KA, Ahn YT, Jeong JJ, Huh CS, Kim DH. Comparative analysis of gut microbiota in elderly people of urbanized towns and longevity villages. BMC Microbiol. 2015;15:49. doi: 10.1186/s12866-015-0386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moon C, VanDussen KL, Miyoshi H, Stappenbeck TS. Development of a primary mouse intestinal epithelial cell monolayer culture system to evaluate factors that modulate IgA transcytosis. Mucosal Immunol. 2014;7(4):818–28. doi: 10.1038/mi.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.