

Abstract
Stress-induced structural remodeling in the adult hippocampus, involving debranching and shortening of dendrites and suppression of neurogenesis, provides a cellular basis for understanding the impairment of neural plasticity in the human hippocampus in depressive illness. Accordingly, reversal of structural remodeling may be a desirable goal for antidepressant therapy. The present study investigated the effect of tianeptine, a modified tricyclic antidepressant, in the chronic psychosocial stress model of adult male tree shrews (Tupaia belangeri), a model with high validity for research on the pathophysiology of major depression. Animals were subjected to a 7-day period of psychosocial stress to elicit stress-induced endocrine and central nervous alterations before the onset of daily oral administration of tianeptine (50 mg/kg). The psychosocial stress continued throughout the treatment period of 28 days. Brain metabolite concentrations were determined in vivo by proton magnetic resonance spectroscopy, cell proliferation in the dentate gyrus was quantified by using BrdUrd immunohistochemistry, and hippocampal volume was measured post mortem. Chronic psychosocial stress significantly decreased in vivo concentrations of N-acetyl-aspartate (−13%), creatine and phosphocreatine (−15%), and choline-containing compounds (−13%). The proliferation rate of the granule precursor cells in the dentate gyrus was reduced (−33%). These stress effects were prevented by the simultaneous administration of tianeptine yielding normal values. In stressed animals treated with tianeptine, hippocampal volume increased above the small decrease produced by stress alone. These findings provide a cellular and neurochemical basis for evaluating antidepressant treatments with regard to possible reversal of structural changes in brain that have been reported in depressive disorders.
Keywords: neurogenesis‖proton magnetic resonance spectroscopy‖depression‖hippocampus‖tree shrew
Depressive disorders are among the most common and life-threatening illnesses and represent a significant public health problem (1). Despite extensive preclinical and clinical investigations, the exact neurobiological processes leading to depression and the mechanisms responsible for the therapeutic effects of antidepressant drugs are not completely understood (2).
The hippocampus is one of the brain structures that has been extensively studied with regard to the actions of stress, depression and antidepressant actions (3, 4). Recent imaging studies in humans revealed that the hippocampus undergoes selective volume reduction in stress-related neuropsychiatric disorders such as recurrent depressive illness (5–7). Within the hippocampal formation, the dentate gyrus is one of the few brain structures where production of new neurons occurs even in the adult mammalian brain (8–10). Several experiential, neuroendocrine, and genetic factors that regulate neurogenesis in the adult dentate gyrus have been identified (11). One factor that potently suppresses adult granule cell proliferation is stress (12, 13). Besides reduced neurogenesis, the hippocampal formation has also been shown to undergo another morphological change in response to stress, namely the retraction of apical dendrites of CA3 pyramidal neurons (14). Based on these findings in animals, the clinically observed hippocampal volume loss can be partially explained by dendritic retraction and reduced cell proliferation (3, 4).
In rats, treatment with the antidepressant tianeptine prevented stress-induced retraction of apical dendrites of CA3 pyramidal neurons (15, 16). Moreover, chronic antidepressant treatment, including electroshock, increased granule cell proliferation in rats' hippocampal dentate gyrus (17). These and other findings led to the hypothesis that antidepressants could oppose the stress-induced loss of neural plasticity, by blocking or reversing the retraction of hippocampal neurons and by increasing cell survival and function (18–20).
So far, the effects of antidepressants on neurogenesis have been investigated in normal animals (17, 20, 21); however, antidepressants are commonly given to sufferers of affective disorders. To establish whether any plasticity changes are related to the neurobiology of depression, valid animal models should be investigated. Based on the clinical evidence that links stressful life events with depressive episodes (22), several animal models have been developed (23). These models have been extremely useful in elaborating and detecting the effects of antidepressant drugs, but one has to consider that animal models for psychiatric disorders should fulfill three major criteria (24). The first is “face validity” and assesses how well the symptoms observed in the animals resemble those in human patients. The second criterion, “predictive validity,” addresses the question how well animals in the model respond favorably to the same drugs as humans do under the same treatment conditions. The third criterion, “construct validity,” assesses to what extent the model is consistent with the theoretical rationale. One of the models with high face, predictive, and construct validity for research on the pathophysiology of major depression is the chronic psychosocial stress paradigm in male tree shrews (25, 26).
Given the clinical significance of stimulating neurogenesis in adulthood, especially in therapy of neuropsychiatric diseases involving the hippocampus, we investigated whether antidepressant administration would oppose stress-induced adverse effects in the hippocampal formation. In the present study we used the tricyclic antidepressant tianeptine, which is known to prevent and even to reverse stress-induced changes in cerebral morphology (16, 27, 28). Tianeptine is devoid of the classic actions of antidepressants on central monoamine activities but has a well documented therapeutic activity compared with other antidepressants (28). In contrast to most antidepressants, tianeptine is thought to enhance serotonin uptake as suggested by studies performed ex vivo (28). Despite a growing number of studies focusing on tianeptine's properties (28), the exact mechanisms underlying its therapeutic effects have to be clarified.
To mimic a realistic situation of antidepressant intervention, we started treating the animals after stress-induced alterations had been established. Tianeptine was administered orally, while the psychosocial stress continued during the whole treatment period. At the end of the experiment, we measured brain metabolite concentrations in vivo with proton magnetic resonance spectroscopy, and determined hippocampal volume and cell proliferation in the dentate gyrus post mortem.
Materials and Methods
Animals and Antidepressant Treatment.
Experimentally naive adult male tree shrews (Tupaia belangeri, average age 12 ± 4 months; n = 24) were obtained from the breeding colony at the German Primate Center (Göttingen, Germany). The day-active tree shrews are regarded as an intermediate between insectivores and primates (29). Animals were housed individually in air-conditioned facilities on a 12-h/12-h light/dark cycle and with free access to food and water (for details see ref. 30). All animal experiments were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Government of Lower Saxony, Germany. Tree shrews received tianeptine (Stablon; Servier, Courbevoie, France) dissolved in tap water via light-protected bottles. In a pilot study, we determined that a solution of 20 mg tianeptine per 100 ml water resulted in serum concentrations ranging from 50 to 220 ng tianeptine/ml, and the same is reported for patients under tianeptine treatment (C. Muñoz, personal communication). Based on a daily mean water intake of 50 ml (31) and a mean body weight of 200 g, the mean intake of tianeptine was 50 mg/kg per day.
Experimental Procedure.
Animals were divided into four groups: namely, the Control, the Control + Tianeptine, the Stress, and the Stress + Tianeptine groups (n = 6 in each). During the first 7 days, the activity of the hypothalamus-pituitary-adrenocortical axis was determined by measuring cortisol in the daily morning urine. The second phase of the experiment was a 7-day-long period, during which the animals of the Stress and the Stress + Tianeptine groups were submitted to daily psychosocial conflict. The induction of psychosocial conflict was carried out according to our standard procedure (25). Briefly, one naive male was introduced into the cage of a socially experienced male. This co-housing resulted in active competition for control over the territory, and after establishment of a clear dominant/subordinate relationship, the two animals were separated by a wire mesh barrier. The barrier was removed every day for ≈1 h, allowing physical contact between the two males only during this time. By this procedure, the subordinate animal was protected from repeated attacks, but it was constantly exposed to olfactory, visual, and acoustic cues from the dominant. Under these conditions, subordinate animals displayed characteristic subordination behavior. They reduced their level and sphere of activity in their cages, showed tail ruffling, and elicited alarm cries. The third experimental phase consisted of the tianeptine treatment, lasting 28 days. During this time, the subordinate animals remained in the psychosocial conflict situation and were treated daily with the antidepressant (50 mg/kg body weight per day; Stress + Tianeptine group) or received tap water (Stress group). Animals of the Control + Tianeptine group remained singly housed in their cages and received tianeptine (50 mg/kg body weight per day) via the drinking water for 28 days. Animals of the Control group remained singly housed in their cages and received normal tap water. Throughout the whole experiment, from all animals morning urine samples were collected daily.
Localized Proton Magnetic Resonance Spectroscopy (MRS).
At the end of the experiment, all animals underwent proton MRS measurements to evaluate in vivo brain metabolite concentrations of N-acetyl-aspartate (NAA), creatine and phosphocreatine (Cr), choline-containing compounds (Cho), and myo-inositol (Ins). For detailed description of the measurements, see ref. 31. In brief, fully relaxed short echo-time proton MR spectra [stimulated echo acquisition mode (STEAM), TR/TE/TM = 6000/20/10 ms, 64 averages] were acquired from a 0.245-ml (7 × 5 × 7 mm3) volume-of-interest (VOI) at 2.35 T by using a MRBR 4.7/400 mm magnet (Magnex Scientific, Abingdon, England) driven by DBX electronics (Bruker, Karlsruhe, Germany). Radiofrequency excitation and signal reception were accomplished by a 14-cm Helmholtz coil and a 2-cm surface coil, respectively. Anesthetized animals (70:30 N2O:O2, 0.5–1.0% halothane; see ref. 31) were measured in a prone position with their head firmly fixed between a plastic-made holder and the surface coil. The position of the VOI was carefully selected from multislice sagittal and coronal T1-weighted gradient-echo images [fast low angle shot (FLASH), TR/TE = 150/5 ms, 20° flip angle, 50 mm field-of-view, 256 × 256 data matrix, 1-mm sections] and centrally placed in the forebrain, including parasagittal neocortex, subjacent white matter, and portions of subcortical forebrain structures (caudate-putamen, hippocampus, thalamus, ventricles; see also ref. 31). Metabolite quantification involved fully automated and user-independent spectral evaluation by lcmodel (32) and calibration with respect to the brain water concentration (33).
Histological Procedures.
Bromodeoxyuridine injection and immunocytochemistry.
On the last experimental day, animals received a single i.p. injection of 5-bromo-2′-deoxyuridine (BrdUrd; 100 mg/kg; Sigma) to label dividing cells and were perfused 24 h later. This survival time allows for the completion of at least one cell cycle by cells in S phase at the time of BrdUrd injection (34). In deep anesthesia, the animals were perfused transcardially with 4% paraformaldehyde. A freezing microtome was used to collect serial coronal 50-μm sections through the septo-temporal extent of the hippocampus. Every fifth section was slide-mounted and coded before processing for immunocytochemistry to ensure objectivity. According to the standard protocol (12), BrdUrd labeling requires the following pretreatment steps: DNA denaturation (0.01 M citric acid (pH 6.0), 95°C, 20 min), membrane permeabilization (0.1% trypsin, 10 min), and acidification (2 M HCl, 30 min). Primary antibody concentration was mouse anti-BrdUrd (DAKO, 1:100), and immunocytochemistry was completed by using the avidin-biotin/diaminobenzidine visualization method (Vector Laboratories) followed by counterstaining with hematoxylin.
BrdUrd quantification.
A modified unbiased stereology protocol was used that has been reported to successfully quantify BrdUrd labeling (17, 35). Every fifth section (an average of 11) through the rostral/caudal extent of the left hippocampus was examined (between A 1.0 and A 3.0; ref. 36). All BrdUrd-labeled cells in the granule cell layer together with the subgranular zone, defined as a two-cell-body-wide zone along the border of the granule cell layer, were counted regardless of size or shape. To enable counting of cell clusters, cells were examined under ×400 and ×1,000 magnification, omitting cells in the outermost focal plane. The total number of BrdUrd-labeled cells was estimated by multiplying the number of cells counted in every 5th section by 5.
Volume measurement.
To determine the volume of the entire hippocampus (hippocampus proper, dentate gyrus) and the granule cell layer, adjacent sections were analyzed by using the Neurolucida system (Microbrightfield, Colchester, VT). Volumes were estimated on the basis of the Cavalieri principle; starting at a random position, every 5th section was used (an average of 11). Briefly, the specific structures were outlined, and the computed areas were then summed and multiplied with the thickness of the sections and with the intersection distance. Volume was reported as mm3.
Analysis of Urinary Cortisol.
Urinary free cortisol was measured by a scintillation proximity RIA (as described recently in ref. 25). To correct for physiological alteration in urine dilutions, the resulting concentrations were related to creatinine concentrations, which were determined with a Beckman Creatinine Analyzer 2.
Data Analysis.
Results are presented as the mean ± SEM. Treatment effects were assessed with a two-way ANOVA (stress × treatment), followed by Tukey's honestly significantly different (HSD) test as post hoc analysis for further examination of group differences.
Results
Stress-Induced Changes in Brain Metabolites Are Normalized by Tianeptine Treatment.
Proton MRS was applied to measure major brain metabolites such as the neuroaxonal marker N-acetyl-aspartate, which is found only in neuroaxonal tissue of the adult brain and putatively indicates neuronal viability and function (37), creatine and phosphocreatine as important energy metabolites, and choline-containing compounds representing formation and degradation products of cell membranes such as (glycero-)phosphocholine, and myo-inositol as a marker for glial cells (38). Mean proton MR spectra are shown in Fig. 1, and the quantitative data are summarized in Table 1. Relative to the Control group, chronically stressed animals revealed significantly decreased concentrations of N-acetyl-aspartate (−13%; q = 4.08; P < 0.05), creatine and phosphocreatine (−15%; q = 4.07; P < 0.05), and choline-containing compounds (−13%; q = 4.05; P < 0.05). Only the concentration of the glial marker myo-inositol remained unchanged. Administration of tianeptine to stressed animals (Stress + Tianeptine) resulted in normal brain metabolite concentrations. Antidepressant treatment alone (Control + Tianeptine) had no effect on the brain metabolites. These findings suggest that tianeptine administration can preserve brain metabolism from certain effects of psychosocial stress.
Figure 1.

Proton magnetic resonance spectroscopy showing the effect of long term psychosocial stress and concomitant tianeptine application on functional brain neurochemistry (mean spectra for three experimental groups; n = 6 animals per group): Control (A), Stress (B), Stress + Tianeptine (C). The spectra (STEAM, TR/TE/TM = 6000/20/10 ms, 0.245 ml volume of interest, 64 accumulations per animal) include resonances from N-acetyl-aspartate (NAA), creatine and phosphocreatine (Cr), choline-containing compounds (Cho), and myo-inositol (Ins). Individual spectra were scaled in proportion to the brain water concentration before averaging across animals. Arrows indicate statistically significant differences in comparison with animals from the Control group. Note that the decreases of Cho, Cr, and NAA in the Stress group are prevented in the Stress + Tianeptine group.
Table 1.
Cerebral metabolite concentrations (mM/VOI) of N-acetyl-aspartate (NAA), creatine and phosphocreatine (Cr), choline-containing compounds (Cho), and myo-inositol (Ins)
| Control | Control + tianeptine | Stress | Stress + tianeptine | |
|---|---|---|---|---|
| NAA | 10.0 ± 0.3 | 9.4 ± 0.2 | 8.7 ± 0.4* | 9.1 ± 0.3 |
| Cr | 7.3 ± 0.2 | 7.2 ± 0.3 | 6.2 ± 0.3* | 7.1 ± 0.3 |
| Cho | 2.4 ± 0.1 | 2.3 ± 0.1 | 2.1 ± 0.1* | 2.3 ± 0.1 |
| Ins | 5.8 ± 0.2 | 5.6 ± 0.3 | 5.9 ± 0.5 | 6.4 ± 0.2 |
Data are given as mean ± SEM (*, P < 0.05, vs. Control).
Chronic Tianeptine Treatment Prevents Stress-Induced Suppression of Adult Hippocampal Cell Proliferation.
BrdUrd injection was given on the last day of the experiment, 1 day before sacrifice, to measure proliferation after 5 weeks of treatment. Proliferating cells were found predominantly in the subgranular zone along the subpyramidal and infrapyramidal blades of the dentate gyrus, and only occasionally BrdUrd-positive cells were observed in the hilus. BrdUrd-labeled cells generally occurred singly or in small clusters of three to five cells. Psychosocial stress for 35 days resulted in a significant 33% decrease in the number of BrdUrd-positive cells relative to unstressed controls (Fig. 2). Two-way ANOVA (stress × treatment) revealed significant main effects of stress [F(1,20) = 7.79; P < 0.05] and of treatment [F(1,20) = 8.48; P < 0.01]. Tukey's post hoc comparisons showed significant difference comparing the Control and the Stress group (q = 4.57; P < 0.05). Tianeptine treatment of stressed tree shrews resulted in a significant increase in the number of BrdUrd-labeled cells in the dentate gyrus of the Stress + Tianeptine group relative to the Stress group (q = 4.55; P < 0.05). This result indicates that—despite elevated cortisol levels (see below)—chronic treatment with the antidepressant tianeptine overcomes the stress-induced reduction of cell proliferation in the adult dentate gyrus. Comparison of cell proliferation between animals from the Control group and unstressed animals treated with tianeptine revealed no differences (q = 0.69; P = 0.96). These findings indicate that chronic tianeptine treatment affects cell proliferation only in stress-based conditions.
Figure 2.
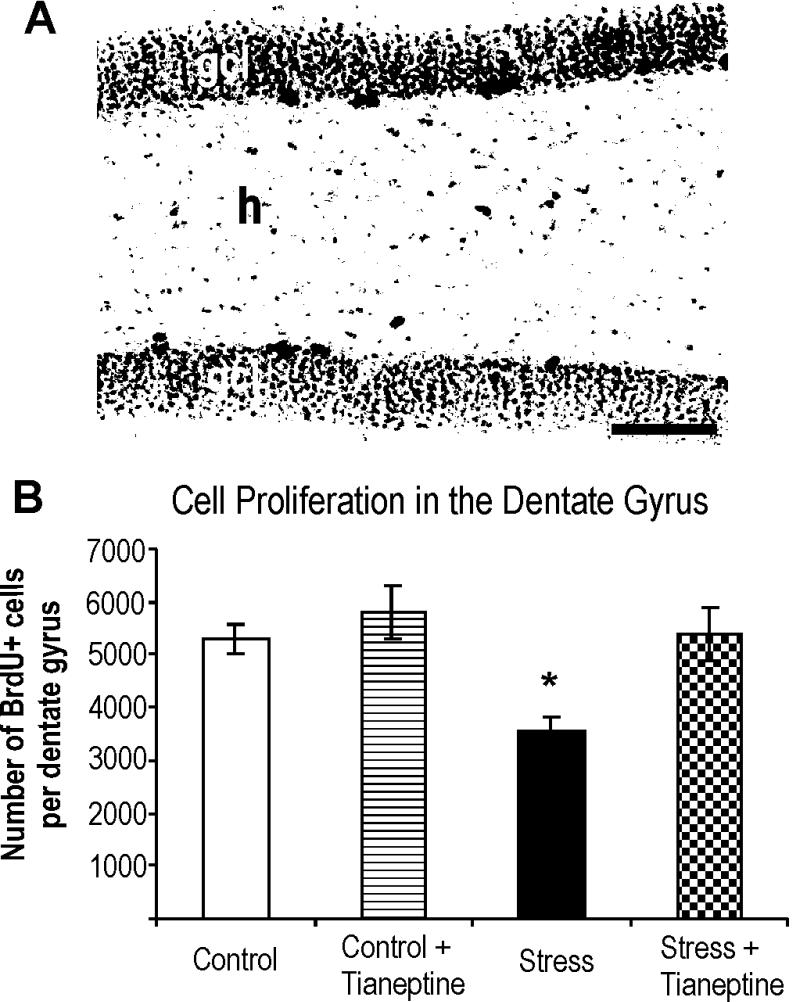
Cell proliferation in the hippocampal dentate gyrus of the tree shrew was quantified with the use of BrdUrd immunohistochemistry. (A) A representative photomicrographs of a Control animal. The majority of proliferating cells was found dominantly in the subgranular zone defined as a two-cell-body-wide zone between the granule cell layer (gcl) and hilus (h). Positively labeled cells generally occurred singly or in small clusters comprised of three to five cells. Scale bar = 100 μm. (B) Chronic psychosocial stress significantly suppressed cell proliferation in the hippocampal dentate gyrus (Stress), whereas chronic tianeptine treatment reversed the stress-induced effect (Stress + Tianeptine). Antidepressant treatment alone had an insignificant effect on hippocampal cell proliferation (Control + Tianeptine). Results are given as mean ± SEM number of BrdUrd-positive cells in the hippocampal dentate gyrus. *, P < 0.05 vs. Controls.
Stress-Induced Change in Hippocampal Volume Is Prevented by Tianeptine Treatment.
To determine whether chronic psychosocial stress and antidepressant treatment influence hippocampal volume, volumetry of the entire left hippocampus (Cornu Ammonis, gyrus dentatus) was performed post mortem. Chronic exposure to psychosocial stress resulted in a trend for an insignificant decrease (−7%) of the hippocampal volume in the subordinate tree shrews (Stress) compared with nonstressed Controls (Fig. 3). Despite elevated cortisol levels in the chronically stressed animals treated with tianeptine (see below), the hippocampal volume was significantly larger (q = 4.22; P < 0.05) compared with stressed animals not treated with the antidepressant. This increase produced by tianeptine suggests that hippocampal volume loss in depressed humans could possibly be prevented by antidepressants. A comparison of the Stress + Tianeptine group with the Control group revealed no statistical difference (q = 1.97; P = 0.51).
Figure 3.

Postmortem volumetry of the hippocampus. Chronic psychosocial stress resulted in a decrease (7%) of the hippocampal volume compared with unstressed controls (P = 0.40). This decrease was prevented by tianeptine treatment (Stress + Tianeptine vs. Stress; *, P < 0.05).
Furthermore, the volume of the granule cell layer was estimated to evaluate whether the decreased proliferation rate in the dentate gyrus contributes to the hippocampal volume reduction. No changes in the volume of the granule cell layer (in mm3) were observed in any of the groups: Control, 1.9 ± 0.1; Control + Tianeptine, 2.0 ± 0.1; Stress, 1.9 ± 0.1; Stress + Tianeptine, 1.9 ± 0.1 (two-way ANOVA: stress F(1,20) = 0.45, P = 0.50; treatment F(1,20) = 0.13, P = 0.72). This finding indicates that suppressed cell proliferation in the dentate gyrus does not contribute to the observed hippocampal volume loss.
Stress-Induced Activation of the Hypothalamic-Pituitary-Adrenal (HPA) Axis.
Psychosocial stress induced a sustained activation of the HPA axis, as indicated by the nonadapting elevation of urinary cortisol excretion in the two stress groups (Table 2). No statistical differences between the Stress and Stress + Tianeptine group were found, except on week 4 yielding significantly higher cortisol excretion (P < 0.02) in stressed animals treated with tianeptine. Despite the daily treatment of subordinate animals with tianeptine (Stress + Tianeptine), cortisol did not return to basal levels (Table 2). This finding is in line with one study in rats showing that stress-elicited elevations in plasma corticosterone were not reduced by tianeptine treatment (39). Other studies, however, demonstrated that tianeptine clearly reduces HPA response to stress (40). No effects on urinary cortisol excretion were observed in animals of the two control groups (Control; Control + Tianeptine).
Table 2.
Effects of chronic psychosocial stress and concomitant tianeptine treatment on urinary free cortisol
| Experimental week | Control | Control + tianeptine | Stress | Stress + tianeptine |
|---|---|---|---|---|
| 1 | 88.5 ± 9.7 | 95.8 ± 11.0 | 91.1 ± 16.0 | 72.9 ± 11.2 |
| 2 | 89.8 ± 14.9 | 103.0 ± 12.1 | 146.3 ± 31.7** | 114.2 ± 16.4** |
| 3 | 83.6 ± 13.2 | 99.5 ± 8.1 | 132.8 ± 20.7* | 169.7 ± 19.1** |
| 4 | 93.7 ± 12.6 | 100.5 ± 11.1 | 143.7 ± 20.5* | 203.2 ± 22.7** |
| 5 | 81.1 ± 11.9 | 96.1 ± 12.9 | 139.9 ± 18.8* | 158.1 ± 18.7** |
| 6 | 83.6 ± 11.9 | 104.3 ± 16.5 | 135.4 ± 18.4* | 172.4 ± 19.3** |
Control: animals remained undisturbed from week 1 to 6. Control + tianeptine: animals were untreated between week 1 and 2; from week 3 onwards, they received tianeptine in the drinking water (50 mg/kg per day). Stress: animals were undisturbed during week 1 and were daily stressed from week 2 to 6. Stress + tianeptine: animals were undisturbed during week 1 and were daily stressed from week 2 to 6; from week 3 onwards they received tianeptine in the drinking water (50 mg/kg per day).
Data are expressed as pg cortisol/μmol creatinine and are given as mean ± SEM of six data blocks representing experimental weeks 1–6; n = 6 animals per group:
, P = 0.054;
, P < 0.05;
, P < 0.01, vs. week 1.
Discussion
The results of this study show that chronic psychosocial stress decreases the in vivo concentrations of cerebral metabolites, the proliferation rate of the granule precursor cells in the dentate gyrus. Both of these alterations can be prevented by treating the stressed animals with the antidepressant tianeptine, which also increased hippocampal volume above the nonsignificant decrease found in stressed animals. Collectively, these findings extend and reinforce novel theories claiming that restoring plasticity of neural circuits at cellular and molecular levels can lead to antidepressant effects (2, 18–20, 41).
Normalization of Cerebral Metabolites.
Taking advantage of the chronic psychosocial stress paradigm in male tree shrews, an animal model that has a high validity to investigate the pathophysiology of depressive disorders (25, 26), we mimicked a realistic situation of antidepressant intervention with daily oral tianeptine application that started after the stress-induced neurobiological and physiological alterations had been established. The action of the drug was followed across a clinically relevant time period of 4 weeks while the psychosocial stress continued during the whole treatment period. As revealed by in vivo measurements of brain metabolites with quantitative proton MRS, stress-induced metabolic changes were prevented by tianeptine. These findings rely on the ability to determine in vivo metabolite concentrations rather than ratios thereof. In fact, concomitant reductions of NAA, Cho, and Cr would have escaped detection if the study had been based on mere alterations of metabolite ratios. This approach might explain why the use of metabolite ratios in clinical proton MRS studies of depressed subjects led to conflicting results in frontal cortex and basal ganglia (42–45).
Alterations of the Cho level have been discussed to result from changes of cytosolic choline compounds because of disturbances in the formation and degradation of cell membranes. Because Cho is highly concentrated in glia cells (46), one may speculate about a respective reduction of glia cell number, size, and density as found in postmortem brains of depressed patients (47). However, this interpretation is not supported by the observation of a normal Ins concentration, which is considered to be a glial marker not present in neurons (38). Further experiments are necessary to determine a possible involvement of glial cells in the pathophysiology of depression.
A reduction of the cerebral NAA concentration is commonly understood as a reduction of neuroaxonal cellular density and/or dysfunction. Interestingly, a recent clinical study reported an increase of the brain NAA concentration during lithium treatment (48). Our preclinical data provide support for the contention that mood disorders and stressful life events are associated with reductions of neuronal viability and/or that function and antidepressant treatment reestablishes neuronal resilience (2, 41).
Stress-Induced Reduction in Cell Proliferation Is Prevented by Tianeptine Treatment.
Decreased granule cell proliferation has been reported in subordinate tree shrews in response to acute psychosocial stress (12). In the current experiments, animals were stressed for 7 days before tianeptine treatment started, and stress was continued during the whole treatment period. Thus, it is very likely that the cell proliferation rate was initially suppressed because of stress and reversed across the antidepressant application for 28 days. This finding indicates that the time for up-regulation of BrdUrd labeling is within the “therapeutical lag” of antidepressant action. In line with this idea are recent studies in unchallenged rats, showing that increased BrdUrd labeling is observed after chronic but not short-term treatment with the different classes of antidepressant drugs, such as the monoamino oxidase inhibitor tranylcypromine, the serotonin-selective reuptake inhibitor fluoxentine, and the norepinephrine-selective reuptake inhibitor reboxetine (17).
Regulation of neurogenesis occurs on several levels, such as cell proliferation, differentiation, migration, and survival. Previously, we demonstrated that, 3 weeks after BrdUrd injection, about 80% of BrdUrd-labeled cells expressed the neuronal marker neuron-specific enolase, and were incorporated into the granule cell layer (12). Animals of the present study were perfused 24 h after the BrdUrd injection; thus, histological analysis could evaluate only the rate of cell proliferation. Survival and future cellular phenotype of these newly generated cells were not investigated. We can only speculate that the majority of these cells would have differentiated into mature neurons that are integrated into the hippocampal circuitry.
Potential Mechanisms Regulating Cell Proliferation.
Stress and depression are often associated with a hyperactivity of the HPA axis (49), resulting in elevated levels of adrenal steroid hormones (cortisol in man and tree shrews; corticosterone in rats), and adrenal steroids have been shown to inhibit the granule cell proliferation (50). The absence of either type 1 or type 2 adrenal steroid receptors on granule cell precursors suggests that the effects on cell proliferation are transmitted throughout other factors, such as N-methyl-d-aspartate (NMDA) receptors (51). Indeed, blockade of NMDA receptors increases the number of newly generated cells (9, 12, 52). However, the cellular sites and mechanisms by which NMDA receptor antagonists act on cell proliferation remain to be elucidated because proliferating cells in the adult dentate gyrus do not exhibit immunoreactivity for the NMDA receptor subunit NR1 (53).
The neurotransmitter serotonin is thought to stimulate granule cell production whereas depletion of serotonin reduces neurogenesis (54, 55). Therapeutic interventions, such as treatment with the selective serotonin reuptake inhibitor fluoxetine, augmented neurogenesis (17, 21). In line with the growing body of evidence suggesting that mood stabilizers and antidepressants exert neurotrophic effects is a recent report that lithium treatment positively affected neurogenesis in mice (56).
Although hormones and neurotransmitter signals trigger proliferation, the direct mitogenic stimulus to the progenitor cells appears to be mediated via growth factors. Selective induction of neurogenesis can be achieved by using insulin-like growth factor (IGF)-1 (57, 58). Yet another candidate is the brain-derived neurotrophic factor (BDNF), which has been shown to be up-regulated in the hippocampus after antidepressant treatment (59). These results indicate that regulation of neurogenesis is a complex process. Obviously, signal transduction cascades connecting surface and/or intracellular receptors with the intracellular machinery are necessary to regulate the coordinated transcription of genes finally contributing to the proliferation, differentiation, migration, and integration of granule cells into the dentate gyrus neural network.
Prevention of Hippocampal Volume Loss.
The present studies provide a basis for evaluating antidepressant treatments both at the cellular level and by in vivo imaging with regard to possible reversal of the structural changes in brain that have been reported in depressive illness (5, 6, 60). At the cellular level, we have reported previously that 4 weeks of social conflict induced in subordinate tree shrews a retraction of dendritic trees in hippocampal CA3 pyramidal neurons that could be prevented by daily treatment with phenytoin, an antiepileptic drug that has shown mood stabilizing efficacy (14, 61). This finding emphasizes the fact that excitatory amino acids play a major role in driving the dendritic remodeling (3). However, tianeptine treatment does prevent and even reverses dendritic remodeling in rats caused by stress and by glucocorticoid treatment and thus its action needs to be explored vis-à-vis excitatory amino acid action in brain. In the present study, tianeptine treatment also prevented the suppression of neurogenesis caused by psychosocial stress, a process in which excitatory amino acids are known to mediate suppression (9).
By using in vivo imaging with magnetic resonance spectroscopy, the present study showed that psychosocial stress caused a reduction in the neuroaxonal marker NAA and that this change could be prevented by tianeptine treatment, providing a neurochemical endpoint for antidepressant treatments in vivo. Previously, in the tree shrew model, we could demonstrate a mild stress-induced hippocampal volume loss in vivo by magnetic resonance imaging (62), and we found that this loss is due neither to reduction in neuron number nor to increased apoptotic cells in the Ammon's horn (63, 64).
At present, we are unaware of any previous evidence that antidepressant treatment can prevent/reverse stress-induced hippocampal volume loss, although one type of hippocampal volume loss is reversible as demonstrated in patients with spontaneous Cushing's syndrome (65). Besides the possibility that hippocampal atrophy may result from neuronal loss, it is also plausible to assume that the reversibility is due to alterations in glia cells, as well as in the dendritic, axonal, and synaptic components of the hippocampal neural network (66). This idea is in accord with previous results, suggesting that tianeptine, in addition to preventing reduction in number of apical dendritic branch points and length of apical dendrites, also reverses already established dendritic retractions (15, 16), whereas other antidepressants such as fluoxetine, fluvoxamine, or desimipramine failed to block shortening of dendrites (16).
Conclusions
The present data show that stress-induced alterations in brain metabolism, hippocampal volume, and cell proliferation are counteracted by tianeptine treatment that started 7 days after the onset of psychosocial stress. Thus, these findings provide experimental evidence for recent theories that impairments of brain structural plasticity are important features of depressive illness and suggest that pharmacological treatments should be sought to reverse structural changes in brain.
Acknowledgments
This work was in part supported by I.R.I. Servier, Courbevoie, France and by a grant of the Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie (0311467B).
Abbreviations
- MRS
magnetic resonance spectroscopy
- NAA
N-acetyl-aspartate
- Cr
creatine and phosphocreatine
- Cho
choline-containing compounds
- Ins
myo-inositol
- HPA
hypothalamic-pituitary-adrenal
- NMDA
N-methyl-d-aspartate
Footnotes
See commentary on page 12320.
References
- 1.Murray C J, Lopez A D. Lancet. 1997;349:1498–1504. doi: 10.1016/S0140-6736(96)07492-2. [DOI] [PubMed] [Google Scholar]
- 2.Manji H K, Drevets W C, Charney D S. Nat Med. 2001;7:541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- 3.McEwen B S. Annu Rev Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- 4.McEwen B S. Biol Psychiatry. 2000;48:721–731. doi: 10.1016/s0006-3223(00)00964-1. [DOI] [PubMed] [Google Scholar]
- 5.Sheline Y, Wang P, Gado M, Csernansky J, Vannier M. Proc Natl Acad Sci USA. 1996;93:3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bremner J D, Narayan M, Anderson E R, Staib L H, Miller H L, Charney D S. Am J Psychiatry. 2000;157:115–117. doi: 10.1176/ajp.157.1.115. [DOI] [PubMed] [Google Scholar]
- 7.Sapolsky R M. Arch Gen Psychiatry. 2000;57:925–935. doi: 10.1001/archpsyc.57.10.925. [DOI] [PubMed] [Google Scholar]
- 8.Altman J, Das G D. J Comp Neurol. 1965;124:319–335. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]
- 9.Cameron H A, McEwen B S, Gould E. J Neurosci. 1995;15:4687–4692. doi: 10.1523/JNEUROSCI.15-06-04687.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eriksson P S, Perfilieva E, Bjork-Eriksson T, Alborn A M, Nordborg C, Peterson D A, Gage F H. Nat Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- 11.Fuchs E, Gould E. Eur J Neurosci. 2000;12:2211–2214. doi: 10.1046/j.1460-9568.2000.00130.x. [DOI] [PubMed] [Google Scholar]
- 12.Gould E, McEwen B S, Tanapat P, Galea L A, Fuchs E. J Neurosci. 1997;17:2492–2498. doi: 10.1523/JNEUROSCI.17-07-02492.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gould E, Tanapat P, McEwen B S, Flügge G, Fuchs E. Proc Natl Acad Sci USA. 1998;95:3168–3171. doi: 10.1073/pnas.95.6.3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magariños A M, McEwen B S, Flügge G, Fuchs E. J Neurosci. 1996;15:3534–3540. doi: 10.1523/JNEUROSCI.16-10-03534.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watanabe Y, Gould E, Daniels D C, Cameron H, McEwen B S. Eur J Pharmacol. 1992;222:157–162. doi: 10.1016/0014-2999(92)90830-w. [DOI] [PubMed] [Google Scholar]
- 16.Magariños A M, Deslandes A, McEwen B S. Eur J Pharmacol. 1999;371:113–122. doi: 10.1016/s0014-2999(99)00163-6. [DOI] [PubMed] [Google Scholar]
- 17.Malberg J E, Eisch A J, Nestler E J, Duman R S. J Neurosci. 2000;20:9104–1910. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duman R S, Heninger G R, Nestler E J. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 19.Duman R S, Malberg J, Thome J. Biol Psychiatry. 1999;46:1181–1191. doi: 10.1016/s0006-3223(99)00177-8. [DOI] [PubMed] [Google Scholar]
- 20.Jacobs B L, Praag H, Gage F H. Mol Psychiatry. 2000;5:262–269. doi: 10.1038/sj.mp.4000712. [DOI] [PubMed] [Google Scholar]
- 21.Manev H, Uz T, Smalheiser N R, Manev R. Eur J Pharmacol. 2001;411:67–70. doi: 10.1016/s0014-2999(00)00904-3. [DOI] [PubMed] [Google Scholar]
- 22.Kendler K S, Karkowski L M, Prescott C A. Am J Psychitary. 1999;156:837–841. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- 23.Yadid G, Nakash R, Deri I, Tamar G, Kinor N, Gispan I, Zangen A. Prog Neurobiol. 2000;62:353–378. doi: 10.1016/s0301-0082(00)00018-6. [DOI] [PubMed] [Google Scholar]
- 24.Willner P. Psychopharmacology. 1984;83:1–16. doi: 10.1007/BF00427414. [DOI] [PubMed] [Google Scholar]
- 25.Fuchs E, Kramer M, Hermes B, Netter P, Hiemke C. Pharmacol Biochem Behav. 1996;54:219–228. doi: 10.1016/0091-3057(95)02166-3. [DOI] [PubMed] [Google Scholar]
- 26.van Kampen M, Schmitt U, Hiemke C, Fuchs E. Pharmacol Biochem Behav. 2000;65:539–546. doi: 10.1016/s0091-3057(99)00190-2. [DOI] [PubMed] [Google Scholar]
- 27.McEwen B S, Conrad C D, Kuroda Y, Frankfurt M, Magarinos A M, McKittrick C. Eur Neuropsychopharmacol. 1997;7:S323–S328. doi: 10.1016/s0924-977x(97)00064-3. [DOI] [PubMed] [Google Scholar]
- 28.Wagstaff A J, Ormrod D, Spencer C M. CNS Drugs. 2001;15:231–259. doi: 10.2165/00023210-200115030-00006. [DOI] [PubMed] [Google Scholar]
- 29.Martin R D. Primate Origins and Evolution. London: Chapman & Hall; 1990. [Google Scholar]
- 30.Fuchs E. UFAW Handbook on the Care and Management of Laboratory Animals. Oxford: Blackwell; 1999. pp. 235–245. [Google Scholar]
- 31.Michaelis, T., de Biurrun, G., Watanabe, T., Frahm, J., Ohl, F. & Fuchs, E. (2001) J. Psychiatr. Res., in press. [DOI] [PubMed]
- 32.Provencher S W. Magn Reson Med. 1993;30:672–679. doi: 10.1002/mrm.1910300604. [DOI] [PubMed] [Google Scholar]
- 33.Michaelis T, Wick M, Fujimori H, Matsumura A, Frahm J. NMR Biomed. 1999;12:309–314. doi: 10.1002/(sici)1099-1492(199908)12:5<309::aid-nbm572>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 34.Takahashi T, Nowakowski R S, Caviness V S. J Neurocytol. 1992;21:185–197. doi: 10.1007/BF01194977. [DOI] [PubMed] [Google Scholar]
- 35.Eisch A J, Barrot M, Schad C A, Self D W, Nestler E J. Proc Natl Acad Sci USA. 2000;97:7579–7584. doi: 10.1073/pnas.120552597. . (First Published June 6, 2000; 10.1073/pnas.120552597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tigges J, Shanta T R. A Stereotaxic Atlas of the Tree Shrew (Tupaia glis) Baltimore: Williams & Wilkins; 1969. [Google Scholar]
- 37.Birken D L, Oldendorf W H. Neurosci Biobehav Rev. 1989;13:23–31. doi: 10.1016/s0149-7634(89)80048-x. [DOI] [PubMed] [Google Scholar]
- 38.Brand A, Richter-Landsberg C, Leibfritz D. Dev Neurosci. 1993;15:289–298. doi: 10.1159/000111347. [DOI] [PubMed] [Google Scholar]
- 39.Broqua P, Baudrie V, Laude D, Chaouloff F. Biol Psychiatry. 1992;31:391–400. doi: 10.1016/0006-3223(92)90232-o. [DOI] [PubMed] [Google Scholar]
- 40.Delbende C, Bunel D T, Tarozzo G, Grino M, Olivier C, Mocaër E, Vaudry H. Eur J Pharmacol. 1994;251:245–251. doi: 10.1016/0014-2999(94)90406-5. [DOI] [PubMed] [Google Scholar]
- 41.Manji H K, Moore G J, Chen G. Biol Psychiatry. 2000;48:740–754. doi: 10.1016/s0006-3223(00)00979-3. [DOI] [PubMed] [Google Scholar]
- 42.Renshaw P F, Lafer B, Babb S M, Fava M, Stoll A L, Christensen J D, Moore C M, Yurgelun-Todd D A, Bonello C M, Pillay S S, et al. Biol Psychiatry. 1997;41:837–843. doi: 10.1016/S0006-3223(96)00256-9. [DOI] [PubMed] [Google Scholar]
- 43.Frey R, Metzler D, Fischer P, Heiden A, Scharfetter J, Moser E, Kasper S. J Psychiatr Res. 1998;32:411–420. doi: 10.1016/s0022-3956(98)00033-8. [DOI] [PubMed] [Google Scholar]
- 44.Winsberg M E, Sachs N, Tate D L, Adalsteinsson E, Spielman D, Ketter T A. Biol Psychiatry. 2000;47:475–481. doi: 10.1016/s0006-3223(99)00183-3. [DOI] [PubMed] [Google Scholar]
- 45.Steingard R J, Yurgelun-Todd D A, Hennen J, Moore J C, Moore C M, Vakili K, Young A D, Katic A, Beardslee W R, Renshaw P F. Biol Psychiatry. 2000;48:1053–1061. doi: 10.1016/s0006-3223(00)00942-2. [DOI] [PubMed] [Google Scholar]
- 46.Urenjak J, Williams S R, Gadian D G, Noble M. J Neurosci. 1993;13:981–989. doi: 10.1523/JNEUROSCI.13-03-00981.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajkowska G. Biol Psychiatry. 2000;48:766–777. doi: 10.1016/s0006-3223(00)00950-1. [DOI] [PubMed] [Google Scholar]
- 48.Moore G J, Bebchuk J M, Hasanat K, Chen G, Seraji-Bozorgzad N, Wilds I B, Faulk M W, Koch S, Glitz D A, Jolkovsky L, et al. Biol Psychiatry. 2000;48:1–8. doi: 10.1016/s0006-3223(00)00252-3. [DOI] [PubMed] [Google Scholar]
- 49.Holsboer F. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 50.Gould E, Cameron H A, Daniels D C, Woolley C S, McEwen B S. J Neurosci. 1992;12:3642–3650. doi: 10.1523/JNEUROSCI.12-09-03642.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cameron H A, Woolley C S, Gould E. Brain Res. 1993;611:342–346. doi: 10.1016/0006-8993(93)90524-q. [DOI] [PubMed] [Google Scholar]
- 52.Nacher J, Rosell D R, Alonso-Llosa G, McEwen B S. Eur J Neurosci. 2001;13:512–520. doi: 10.1046/j.0953-816x.2000.01424.x. [DOI] [PubMed] [Google Scholar]
- 53.Cameron H A, Gould E. Receptor Dynamics in Neural Development. Boca Raton, FL: CRC; 1996. pp. 141–157. [Google Scholar]
- 54.Brezun J M, Daszuta A. Eur J Neurosci. 1999a;12:391–396. doi: 10.1046/j.1460-9568.2000.00932.x. [DOI] [PubMed] [Google Scholar]
- 55.Brezun J M, Daszuta A. Neuroscience. 1999b;89:999–1002. doi: 10.1016/s0306-4522(98)00693-9. [DOI] [PubMed] [Google Scholar]
- 56.Chen G, Rajkowska G, Du F, Seraji-Bozorgzad N, Manji H K. J Neurochem. 2000;75:1729–1734. doi: 10.1046/j.1471-4159.2000.0751729.x. [DOI] [PubMed] [Google Scholar]
- 57.Aberg M A, Aberg N D, Hedbacker H, Oscarsson J, Eriksson P S. J Neurosci. 2000;20:2896–2903. doi: 10.1523/JNEUROSCI.20-08-02896.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trejo J L, Carro E, Torres-Alemán I. J Neurosci. 2001;21:1628–1634. doi: 10.1523/JNEUROSCI.21-05-01628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nibuya M, Morinobu S, Duman R S. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lupien S J, De Leon M, De Santi S, Convit A, Tarshish C, Nair N P V, Thakur M, McEwen B S, Hauger R L, Meaney M J. Nat. Neurosci. 1998. 169–73. [DOI] [PubMed] [Google Scholar]
- 61.Gareri P, Falconi U, De Fazio P, De Sarro G. Prog Neurobiol. 2000;61:353–396. doi: 10.1016/s0301-0082(99)00050-7. [DOI] [PubMed] [Google Scholar]
- 62.Ohl F, Michaelis T, Vollmann-Honsdorf G K, Kirschbaum C, Fuchs E. Psychoneuroendocrinology. 2000;25:357–363. doi: 10.1016/s0306-4530(99)00062-1. [DOI] [PubMed] [Google Scholar]
- 63.Vollmann-Honsdorf G K, Flügge G, Fuchs E. Neurosci Lett. 1997;233:121–124. doi: 10.1016/s0304-3940(97)00647-2. [DOI] [PubMed] [Google Scholar]
- 64.Lucassen P, Vollmann-Honsdorf G K, Gleisberg M, Czeh B, de Kloet E R, Fuchs E. Eur J Neurosci. 2001;14:161–166. doi: 10.1046/j.0953-816x.2001.01629.x. [DOI] [PubMed] [Google Scholar]
- 65.Starkman M, Giordani B, Gebraski S S, Berent S, Schork M A, Schteingart D E. Biol Psychiatry. 1999;46:1595–1602. doi: 10.1016/s0006-3223(99)00203-6. [DOI] [PubMed] [Google Scholar]
- 66.Sousa N, Lukoyanov N V, Madeira M D, Almeida O F X, Paula-Barbosa M M. Neuroscience. 2000;97:253–266. doi: 10.1016/s0306-4522(00)00050-6. [DOI] [PubMed] [Google Scholar]