

Abstract
Building additional functionality into both the membrane and the internal compartments of biocompatible liposomes by self-assembly can provide ways of enhancing colloidal stability and spatial and temporal control of contents release. An interdigitation-fusion process is used to encapsulate near infrared light absorbing copper sulfide nanoparticles in the interior compartments of dipalmitoylphosphatidylcholine and dipalmitoylphosphatidylglycerol liposomes. Once formed, the liposome membrane is modified to include lysolipids and polyethylene glycol lipids by partitioning from lysolipid and PEG-lipid micelles in solution. This results in sterically stable, thermosensitive liposomes with a permeability transition near physiological temperature that can be triggered by NIR light irradiation. Rapid changes in local concentration can be induced with spatial and temporal control using NIR laser light.
Graphical abstract
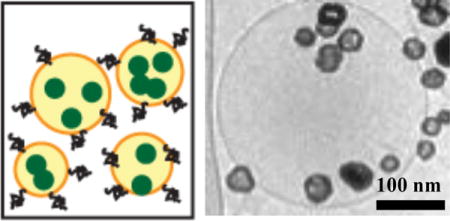
Schematic and TEM image of thermosensitive liposomes with NIR-absorbing nanoparticles
1. Introduction
A challenging problem for liposome applications is the conflict between contents retention and contents release in various environments. An optimal carrier would retain its contents without loss until the time and place that release is desired at some user-defined optimal rate. However, for a single bilayer liposome, it has proven difficult to switch from contents retention to varied levels of contents release, or switch from fast release back to contents retention on command1–9. In drug delivery applications, for example, liposomes begin distributing into various organs and the tumor and are also eliminated from the blood following intravenous injection12. Liposomes preferentially enter tumors through their leaky vasculature, an effect known as “enhanced permeation”, but also return to the blood through the same leaky vasculature, although not necessarily at the same rate due to the poor lymphatic drainage of tumor, i.e. the “enhanced retention” effect12. The combination is known as enhanced permeation and retention (EPR). However, as liposome levels in the blood decrease, liposomes and drug from the tissues and tumor start returning to the blood and the liposome tumor concentration decreases. This results in a transient maximum liposome tumor concentration that can be significantly greater than surrounding tissue due to the EPR effect. However, if enough drug is not released during the time the liposomes reside in the tumor, the benefit from the EPR effect is decreased.
Less than optimal release caused Doxil efficacy not to improve as expected from the increased liposomal doxorubicin accumulation at the tumor site13. Cisplatin incorporated into PEGylated liposomes extended systemic half-life to 40 – 55 hrs,14 in comparison to the 15 – 20 minute clearance of free cisplatin. However, the formulation failed in human trials because cisplatin was not released at a therapeutic rate from the liposomes, even though 5- to 10-fold greater liposome encapsulated cisplatin accumulated in the tumors. However, if the timing and rate of liposome contents release can be controlled, it should be possible to release the liposome contents at the time and place where they would do the most good. This requires a mechanism to induce contents release with spatial and temporal control.
“Thermosensitive liposomes” (TSL) and other stimuli-sensitive liposomes were developed to help resolve this problem3–5, 15. TSL have a membrane composition that induces a step-change in permeability in response to modest changes in temperature above 37 °C4, 16–22. Needham, Dewhirst and coworkers18–21 pioneered lysolipid-TSL formulations (LTSL), which in addition to dipalmitoylphosphatidylcholine (DPPC), contain the lysolipid monopalmitoylphosphatidylcholine (MPPC) and lipid-conjugated PEG2000 (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] or DSPE-PEG2000). Experimental and computer simulations suggest that the permeability transition is due to the formation of lysolipid-stabilized transient pores that occur near the phase transition temperature of DPPC21, 23–27. Figure 1 shows that for DPPC liposomes with 4 mol% DSPE-PEG2000, there is minimal change in the rate of CF release at the main phase transition temperature of 41°C; the high-temperature, fluid Lα phase bilayer is only marginally more permeable than the lower-temperature, ordered Lβ′ bilayer (Fig. 1A)23, 28. Adding 10 mol% MPPC dramatically increases CF release rates for temperatures ≥ 39°C; release was undetectable at 37°C for more than 3 hours. Membrane permeability continues to increase with temperature beyond 39°C to a maximum permeability between 40 and 41°C. At 40°C, all of the encapsulated CF was released within 2.5 minutes. LTSL have shown improved efficacy with a number of cancer cell lines in vitro and in vivo, and clinical trials are ongoing for treatment of liver and breast cancers with combined hyperthermia and LTSL21, 23. Release from LTSL is typically done via bulk heating with water baths, radiofrequency (RF) or microwave (MW) radiation, etc.19–22, 29. However, external control of the temperature profile and extent of heating are difficult, especially in highly perfused tissues.
Figure 1.
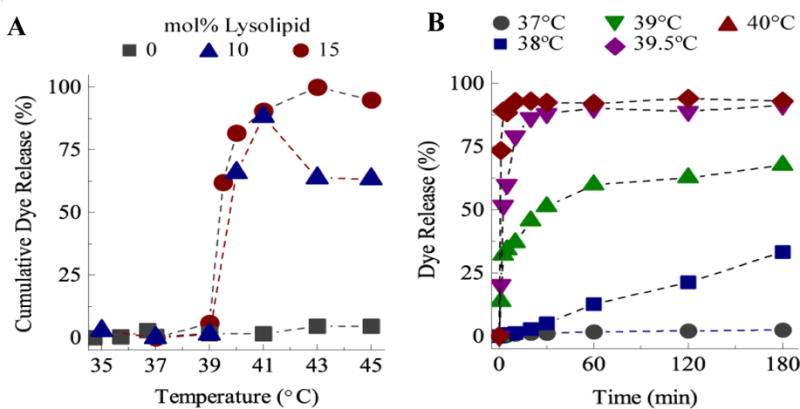
A) Cumulative carboxyfluorescein (CF) dye release in 2.5 minutes as a function of MPPC lysolipid mole fraction in DPPC liposomes. Without MPPC, the permeability does not change with temperature. With MPPC, the permeability undergoes a step-change at 39 – 40 °C. B) CF dye release as a function of time at a given bulk sample temperature for 15:85 mol% MPPC: DPPC liposomes. At 37 °C, there is negligible release, with complete release occurring in < 5 minutes at 40 °C.
Here, we demonstrate a sequential, self-assembly process to create a composite nanoparticle/inter-digitation fusion liposome (IFL) carrier that uses continuous wave near infrared (NIR) laser light to initiate and control contents release. Copper sulfide is a p-type semiconductor that strongly absorbs NIR light30–32; unlike plasmon-resonant gold nanoshells or nanorods, the absorption does not depend on the nanoparticle shape or size33–38. This structure-independent absorption enables the synthesis of smaller diameter nanoparticles, which can be readily encapsulated within a liposome interior. 5–10 nm copper sulfide (CuS) nanoparticles can be encapsulated into DPPC liposomes using the ethanol induced interdigitated phase transition of saturated phospholipids8, 39. However, the interdigitated phase transition is inhibited if lysolipids or PEG-lipids are added to the DPPC bilayers, so a second self-assembly step is necessary. The CuS-DPPC liposomes are made thermosensitive by contacting the liposomes first with a micellar solution of MPPC followed by contact with a micellar solution of PEG-lipid10. By controlling the ratio of MPPC and PEG-lipid to DPPC, the liposome membrane composition can be tailored to provide a permeability transition at ~ 40 °C (Fig. 1) and a sterically stabilized polymer layer to prevent flocculation or opsonization.
We show that irradiation with low intensity NIR light causes a sufficient temperature rise in the CuS nanoparticles and the liposome membrane to induce the permeability transition and rapidly release the liposome contents19, 20, 23, 40, 41. The great advantage of using NIR light to induce release is that tissue, blood, etc. are relatively transparent to 650–950 nm wavelength light, allowing NIR transmission in soft tissues at depths up to several cm22, 42. Laser heating induces a near instantaneous response, allowing the liposome contents to be released in seconds. The liposome temperature reverts to ambient quickly when NIR irradiation stops, allowing the liposomes to re-seal which stops drug release10. Only lysolipid-containing, thermosensitive CuS-DPPC liposomes irradiated by the laser release their contents, which provides a targeting mechanism for spatial and temporal control of drug release. This inside-outside self-assembly process can be used to encapsulate almost any nanoparticle within a liposome membrane, the composition of which can be modified to include lysolipids for thermosensitivity and PEG-lipids for steric stability.
Experimental
Materials
DPPC, methoxy-terminated DSPE-PEG2000 and DSPE-PEG750, carboxyfluorescein-labeled DPPE-PEG2000 and MPPC were purchased from Avanti Polar Lipids (Alabaster, Al). The lipophilic carbocyanine lipid (1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine perchlorate; DiD) was purchased from Invitrogen and used to label DPPC liposomes as needed. The NBD-labeled lysolipid, 1-{12-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]dodecanoyl}-2-hydroxy-sn-glycero-3-phosphocholine, was purchased from Avanti and used to label the lysolipid fraction. Sodium citrate, copper chloride, sodium sulfide, carboxyfluorescein 5,6 (CF), buffers, solvents and other chemicals were purchased from Sigma-Aldrich Chemical Inc. (St Louis, MO) and used as received. The water used in the experiments was of Milli-Q grade with a resistance higher than 18.2 M-ohms-cm.
Copper Sulfide Nanoparticle Synthesis
CuS nanoparticles were synthesized by stirring 2 mM CuCl2 with 1.4 mM sodium citrate in water at room temperature. The pale blue CuCl2 solution immediately turns a dark brown upon addition of an equivalent volume of 2 mM Na2S. The solution was stirred for 5 minutes at room temperature followed by 15 minutes at 85–90°C. Reaction completion is indicated by the solution turning dark green. After cooling the citrate-stabilized CuS nanoparticles to room temperature, thiol-terminated 750 Da molecular weight polyethylene glycol (SH-PEG MW750) was added at a concentration of 1.6 mM and stirred overnight at room temperature to coat the nanoparticles with PEG to stabilize the CuS against flocculation and sedimentation. PEGylated CuS nanoparticles were stored at 4°C until use. CuS absorbance was measured using a Jasco V-530 UV-vis spectrometer and the size distribution determined by conventional transmission electron microscopy imaging after spreading the CuS nanoparticles on formvar-covered TEM grids and drying.
Interdigitation-Fusion Liposomes
DPPC was dissolved in chloroform in glass vials and the solvent removed by evaporation. If needed, 0.1 – 1 mol% diD dye could be added to the DPPC in chloroform. The lipid was hydrated overnight at 55 °C in PBS at 25mg/ml DPPC. 50 – 100 nm diameter unilamellar liposomes were prepared by performing at least five freeze-thaw cycles, followed by extrusion in an Avanti Mini-Extruder (Alabaster, Al) using 100 nm pore diameter filters. The DPPC (or modified DPPC) liposomes were transformed into interdigitated bilayer sheets by dropwise addition of ethanol (3 molar net ethanol concentration) to the liposome suspension at room temperature6, 43. The interdigitated sheets were centrifuged at low speed to pellet the sheets, and then washed with buffer. Carboxyfluorescein 5,6 (CF) was dissolved in sodium hydroxide and the volume was adjusted with 65 mM PBS at pH 7.4 buffer to achieve a final 50mM CF solution with physiological osmolarity. PEGylated CuS nanoparticles were mixed with the CF solution and added to the interdigitated sheets and the solution was held at 55 °C for 20 minutes to induce encapsulation of the CuS nanoparticles and CF and form closed liposomes. If needed, 30 mM MPPC in buffer was added to incorporate lysolipid into the liposomes during the heating process; the desired ratio of MPPC to DPPC in the final liposomes was set by the mole ratio of MPPC:DPPC in solution. Liposomes were separated from unencapsulated CuS nanoparticles and unincorporated MPPC by repeated slow speed centrifugation followed by exchange of the supernatant with fresh PBS. To sterically stabilize the liposomes, DSPE-PEG2000 was added to the solution at 5 mol% of the total liposome lipid concentration at 37°C and the mixture allowed to equilibrate for 48 hours. Excess DSPE-PEG2000 was removed by centrifugation and repeated washing with buffer. The average size of the liposomes was determined using cryo-TEM imaging as described below or using single-particle tracking with a Nanosight NTA 2.3 particle-tracking device.
NIR Irradiation and Dye Release
CF release from the liposomes was measured in semi-micro optical glass cuvettes (Starna) within a custom, temperature-controlled fluorescence spectrometer that was coupled to a continuous wave laser diode for irradiation. 200 μl of sample was placed within a cuvette of 4 mm width and 10 mm path length. The temperature was controlled using a qpod 2e® Peltier sample compartment (Quantum Northwest). Irradiation was performed with a continuous wave laser diode at 797 nm (Coherent F6 Series) with the beam diameter adjusted to 5 mm to ensure that the entire sample volume was irradiated. The incident laser power was varied using an ITC4000 controller (ThorLabs) and calibrated using a PM30 Optical Power Meter (ThorLabs). Sample heating was measured with a thermocouple in solution linked to an Omegaette H306 digital thermometer (Omega). At discrete intervals, fluorescence was measured by exciting the sample with a 475 nm LED source (Ocean Optics, LS-475 Mikropack) and measuring the emission spectra with a Maya2000 Pro Spectrometer (Ocean Optics). Fluorescence emission spectra were integrated over the range of 510 to 530 nm, and dye release was calculated as , where I(t) was the intensity at a given time, Io was the intensity prior to heating or irradiation, and ILysis was the intensity accompanying complete release following liposomal lysis with Triton X-100.
Zeta Potential Measurements
A Malvern ZetaSizer Nano ZS (Westborough MA) instrument was used to determine zeta potentials. About 750 μl of sample liquid was deposited into the sample cuvette. A laser beam within the instrument is split to provide a reference and incident beam. The incident beam passes through the center of the sample cell and the scattered light at an angle of about 13° is detected. An electric field of optimal intensity determined by the instrument software is applied to the cell and the particle movement causes the intensity of light to fluctuate with a frequency proportional to the particle speed. This information is passed to a digital signal processor and then to a computer to produce a frequency spectrum from which the electrophoretic mobility and zeta potential are calculated.
TEM Characterization
Aqueous suspensions were spread as a thin film (0.5 – 10 μm) onto formvar-coated electron microscopy grids (SPI Supplies, West Chester, PA) within a Vitrobot Mark IV (FEI, Hillsboro, OR) to ensure a reproducible sample thickness, minimal sample evaporation prior to cooling (and potential concentration or reorganization of the sample), and an optimal cooling rate. Following equilibration, the samples were rapidly plunged into liquid ethane cooled in a bath of liquid nitrogen. After vitrification, samples remain submerged under liquid nitrogen until transfer via a GATAN (Pleasanton, CA) cryo-transfer unit to a FEI Technai Sphera G2 transmission electron microscope to maintain sample temperature below −170° C. “Low-Dose” imaging conditions were used to prevent sample disruption due to melting, chemical reactions and other forms of radiation damage44.
Lysolipid/PEG Partitioning
Unilamellar liposomes were synthesized at 25 mg/ml using the thin film hydration technique and then extruded to 100 nm in diameter as described above. A red carbocyanine membrane dye (DiD) was included to track the liposome population. A 30 mM micellar solution of lysolipid was prepared by hydrating a dried film of lysolipid with PBS. The micellar solution contained 10 mol% NBD-labeled lysolipid (green). To track PEG partitioning, 2.5 mol% of fluorescently labeled DSPE-PEG2000 was added to DSPE-PEG2000. An aliquot of the appropriate labeled micellar solution was incubated overnight (18–20 hours) with the labeled liposomes at either 37° C or 55° C. Uptake of the lysolipid by the gel or liquid crystalline phase membrane was assessed by isolating the liposome fraction using a gravity size exclusion column and quantifying and comparing the red liposome and green lysolipid fluorescence signals of the elution fractions. The much larger liposomes (50 – 100 nm) elute more rapidly from the column than the smaller micellar (5 nm) or monomeric lysolipid or DSPE-PEG2000.
Results and Discussion
Thiol-PEG stabilized CuS nanoparticles absorb strongly in the near-infrared region, with a broad absorption peak from 800 – 1000 nm (Fig. 2). The synthesized CuS nanoparticles are small, with mean diameter less than 10 nm as measured from TEM images. A concentration of 1015 nanoparticles/ml was calculated based on the average size determined from TEM, assuming complete reaction.
Fig. 2.
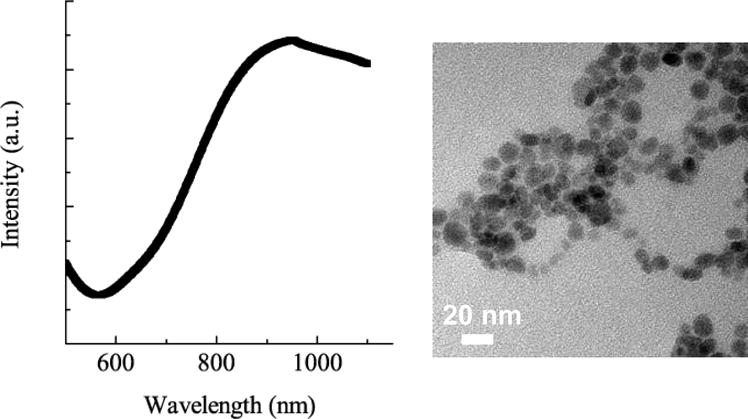
CuS nanoparticles have a broad absorption peak from 850 – 100 nm in the NIR. TEM images show relatively monodisperse particles with a mean size of 9 ± 2 nm and a concentration of ~ 1015 particles/ml.
Efficient encapsulation of the CuS nanoparticles into liposomes was done by taking advantage of the interdigitated phase of DPPC and similar saturated phospholipids6, 8, 9, 39 (Fig. 3). Cooling Lα phase dipalmitoylphosphatidylcholine (DPPC) liposomes from above the gel-liquid crystal temperature, Tc, of 41 °C to room temperature causes the acyl chains of the lipids to crystallize and tilt to accommodate the area mismatch between the phosphocholine headgroups and the acyl chains, leading to the L β′ or gel phase (Fig. 3)45. Adding 3M ethanol to the L β′ phase swells the headgroup region, further increasing the area mismatch between the headgroups and acyl chains, resulting in the interdigitated LβI phase9, 39, 46–49. Wide angle X-ray diffraction of the interdigitated sheets shows a single reflection at q ≈ 15.3 nm−1 indicating an untilted hexagonal lattice with d ≈ 0.41 nm, consistent with the interdigitated LβI phase9, 45, 46. Interdigitation of the lipid bilayer results in both a decrease in membrane thickness and an increase in membrane rigidity9, 50. Small (100 nm) unilamellar vesicles rupture in the LβI phase as the bending energy exceeds the free energy cost of exposing membrane edges to the aqueous environment9, 47. The resulting open bilayer sheets then fuse into larger sheets to lower edge energy, resulting in interdigitated lipid sheets of 1 – 10 microns in extent. The transition to interdigitated sheets is accompanied by a significant increase in viscosity and the liposome suspension changes from a translucent fluid to an opaque, milky-white gel.
Fig. 3.
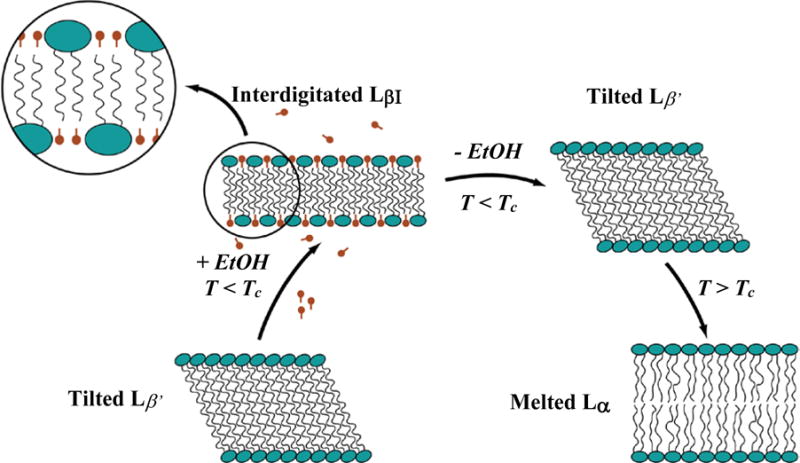
Phase progression used to encapsulate CuS nanoparticles inside liposomes. At temperatures below the gel-liquid crystal transition Tc, the bilayer is in the L β′ phase with the molecules tilted to accommodate the area mismatch between heads and tails. Ethanol swells the headgroups leading to the interdigitated LβI phase. The bending modulus increases, leading to the formation of stacks of open bilayer sheets. Removing the ethanol causes the reversion to the L β′ phase, but the sheets remain open. Raising the temperature above Tc melts and softens the bilayers, and they close to form liposomes, passively encapsulating nanoparticles in the solution.
The open stacks of bilayers remain after the replacement of the ethanol-aqueous buffer mixture with pure buffer as long as T < Tc. However, X-ray diffraction shows that the bilayer structure changes; the single reflection of the interdigitated phase separates into a sharp reflection at q ≈ 14.8 nm−1 with a broad shoulder at q ≈ 15.25 nm−1 consistent with the tilted L β′ phase9. When the planar lipid sheets are heated above 41 °C into the liquid crystalline, or Lα phase, the hydrocarbon chains melt and the reduced membrane bending energy makes closed liposomes the minimal energy state. In the process of forming closed liposomes, CuS nanoparticles suspended with the bilayer sheets (or any nanometer scale particles in the suspension) are encapsulated6, 8, 9, 39. On cooling to room temperature, the bilayers re-enter the L β′ phase, but the liposomes remain closed and retain their contents. This metastable phase progression can accommodate small fractions (~ 3–5 mol%) of fluorescently labeled lipids or cholesterol, or larger fractions of saturated dipalmitoylphosphatidylglycerol in the final liposomes if needed8, 9, 39
However, this interdigitation – fusion process cannot accommodate lysolipids and PEG-lipids at the mole fractions necessary to promote rapid permeability changes (Fig. 1) and steric stabilization. Interdigitation has been observed for DPPC bilayers with low levels (< 5 mol%) of 750 Da molecular weight PEG-lipids, but lysolipid and DSPE-PEG2000 at the necessary combined molar fraction of 12–15 mol% prevent the interdigitation transition9.
The necessary lysolipid and PEG-lipid fractions can be added to the CuS liposomes by the spontaneous partitioning of micellar lysolipid and PEG-lipid into the bilayer. MPPC and DSPE-PEG2000 are relatively soluble in aqueous solution and form micelles that can rapidly exchange and partition into the liposome bilayer51, 52. We explored the rate and extent of equilibrium partitioning of MPPC and DSPE-PEG2000 micelles into interdigitation-fusion liposomes. Partitioning into the membrane below the permeability transition temperature is preferred to minimize leakage of encapsulated small molecules at the phase transition temperature (Fig. 1) and may be advantageous for incorporating temperature sensitive biological ligands attached to the PEG-lipids. Fluorescently labeled lysolipid and PEG-lipids were used to evaluate the uptake of lysolipid and PEG-lipid from a micellar solution into pre-formed fluorescently labeled liposomes. Release of encapsulated CF was used to determine the impact on permeability and determine the limits of liposome stability.
Figure 4 shows a schematic of lysolipid (or DSPE-PEG2000) insertion into a liposome bilayer. In the external solution, the lysolipid exists both in micelles and in its monomeric form at its critical micelle concentration (CMC, 4 μM for MPPC)52, 53. Lysolipid monomers rapidly diffuse throughout the solution and partition into the liposome bilayer. Adsorption of MPPC monomers into the outer bilayer leaflet occurs at a rate of 0.2 sec−1 as measured using micropipette techniques.52, 53 Lysolipid micelles can also fuse with the membrane, but this process happens more slowly due to the larger size of the micelles relative to the monomers; the micelles primarily act as a depot to keep the monomer concentration at the CMC54. As the lysolipid partitions into the outer bilayer leaflet, the unequal distribution between the outer and inner leaflet leads to an increase in the surface area of the outer monolayer relative to the inner monolayer. The area per molecule of the inner monolayer necessarily must increase to match the outer monolayer, creating tension across the membrane, and defects that promote the exchange (flip-flop) of lysolipid across the membrane53.
Figure 4.
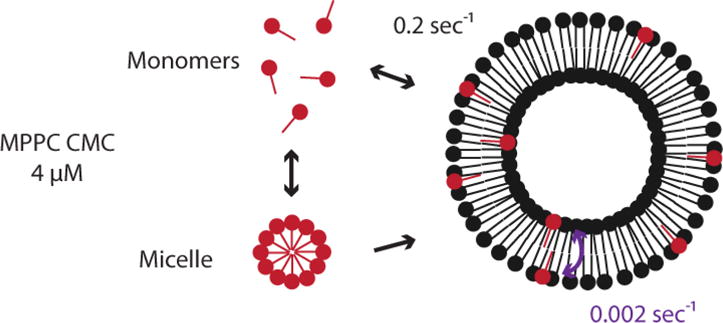
Mechanism of lysolipid transfer into pre-formed liposomes from micellar lysolipid. Lysolipid micelles provide a reservoir to maintain monomeric lysolipid at the CMC. Lysolipid monomers rapidly partition into the outer leaflet of the liposome bilayer. Lysolipid is exchanged from the outer to the inner bilayer leaflet through lipid flip-flop or transient defect structures.
Unilamellar DPPC liposomes of 100 nm diameter at 25 mg/ml with 0.1 mol% of the red carbocyanine membrane dye, diD, were mixed with various amounts of a 30 mM micellar solution of MPPC lysolipid in PBS. The MPPC was labeled with 10 mol% of green NBD-MPPC analog; our results are consistent with the NBD-lysolipid partitioning in a similar fashion as the unlabeled lysolipid (as determined by CF release as a function of temperature, Fig. 5). The mixtures were incubated either one hour or overnight (18–20 hours) at 37°C (gel phase, below Tc) or 55°C (fluid phase, above Tc). The partitioning of the lysolipid between gel or fluid phase liposomes and micelles was determined by separating the liposomes and micelles using a gravity size exclusion column followed by quantifying the relative DiD and NBD fluorescence signals of the elution fractions corresponding to the liposomes. The liposomes, being an order of magnitude larger than the micelles, eluted first.
Fig. 5.
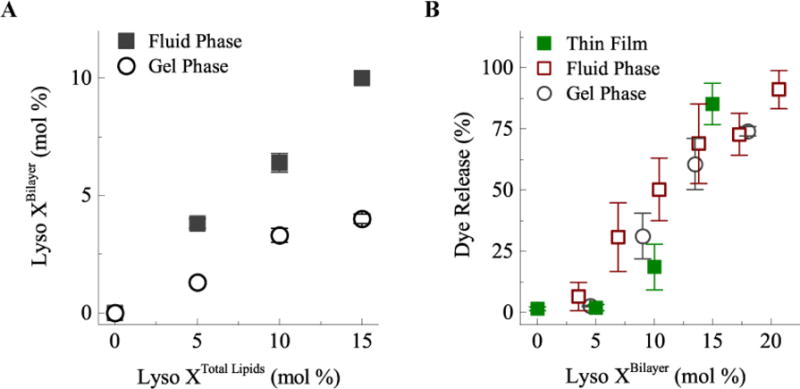
A) Lysolipid partitioning into DPPC liposomes as a function of total lysolipid concentration. Lysolipid (MPPC with 10 mol% NBD-lysolipid) is added to the DPPC liposome; Lyso XTotal Lipids is the lysolipid fraction of the total lipid concentration (lyso+DPPC) in solution. Lyso XBilayer is the mole fraction of MPPC in the bilayer. Partitioning is higher in the fluid phase relative to the gel phase of DPPC. (B) Membrane permeability determined by carboxyfluorescein dye release from the lysolipid-containing liposomes at 40°C after 2.5 minutes. Membrane permeability only depends on the lysolipid mole fraction in the bilayer and not on how the lysolipid – liposomes were prepared. Thin film refers to mixing the lysolipid and DPPC before extruding the liposomes as in Fig. 1.
MPPC partitions into both the low temperature gel or high temperature fluid phase DPPC bilayers, although the partitioning was twice as great for the high temperature, fluid Lα phase (Fig. 5A). Membrane uptake (Lyso XBilayer) was proportional to the concentration of MPPC in solution (Lyso XTotal Lipids) relative to DPPC. Equilibrium partitioning was reached in one hour, as samples incubated overnight had negligible additional uptake. To evaluate the effect of the lysolipid on the permeability, the bulk sample temperature was rapidly increased to 40°C and held for 2.5 minutes. No difference in fractional dye release and hence, membrane permeability, was observed between liposomes made with the lysolipid present in the initial lipid mixture (Thin Film in Fig. 5B) versus liposomes that had lysolipid added from micellar solution following formation either at low or high temperature. Adding lysolipid into interdigitation-fusion liposomes allows for the same enhanced membrane permeability at the transition temperature, making the interdigitation-fusion liposomes thermosensitive. We determined that DPPC interdigitation-fusion liposomes with an MPPC fraction of 8–10 mol% in combination with 4 mol% DSPE-PEG2000 provides an optimal combination of fast contents release on heating and long-term stability prior to heating. Liposomes destabilized, that is, were not capable of retaining internalized CF, at MPPC concentrations exceeding 20 mol%. Fig. 6C shows that DPPC forms lysolipid-stabilized bilayer discs coexisting with ruptured liposomes at higher MPPC mole fractions10.
Fig. 6.
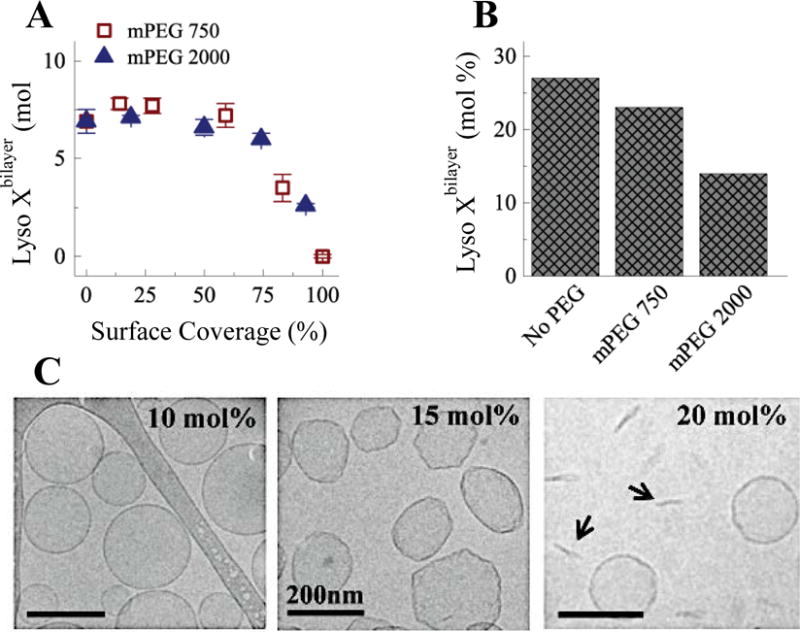
(A) Micellar lysolipid was added to PEGylated liposomes at 10 mol% of the total lipid concentration. 100% surface coverage for methoxy-terminated DSPE-PEG2000 (mPEG 2000) was 5 mol% and 17 mol% for methoxy-terminated DSPE-PEG750 (mPEG 750). Lyoslipid partitioning into the liposome membranes decreased at 50% surface coverage to zero at 100% coverage. (B) The maximum concentration of lysolipid needed to destabilize liposomes decreased with PEGylation. 50% surface coverage of mPEG 750) has minor effects, but 50% surface coverage of mPEG 2000 reduces the allowable lysolipid mole fraction by half. (C) Cryo-TEM images of DPPC with 4 mol% DSPE-PEG2000 with increasing lysolipid mole fraction. Lysolipid plus DSPE-PEG2000 leads to faceted liposomes that break up into discs (arrows).10, 11
Liposomes require a layer of polyethylene glycol to stabilize the liposomes against aggregation and fusion55, 56. When tethered to a surface at low grafting density, the hydrophilic PEG polymer chains extend into the aqueous solution in a random coil conformation in the “mushroom” regime. As the grafting density increases, the PEG chains repel each other laterally, causing the PEG polymer chains to elongate and extend further into the solution, forming an extended polymer “brush”55, 56 configuration. The extent of PEG surface coverage depends both on the molecular weight and the grafting density of the PEG. The radius of gyration of a PEG chain in water scales as:57
| (1) |
in which ls is the length of the polymer monomer (0.35 nm for the ethylene oxide repeat units in PEG) and N is the number of monomers in each PEG chain. DSPE-PEG2000 has 45 ethylene-oxide monomers, so RF ~ 3.5 nm, while DSPE-PEG750 has 17 monomers giving an RF ~ 2 nm. The PEG-lipid headgroup area is proportional to the length of the PEG chain according to:57
| (2) |
giving a headgroup area of 9 nm2 for DSPE-PEG2000 and 3 nm2 for DSPE-PEG750. The percentage of PEG-lipids leading to complete surface coverage of random coils can be estimated from the ratio of the area of the DPPC headgroup (~ 0.5 nm2) to the area of the PEG-lipid headgroup (Eqn. 2). This estimate gives the transition coverage from the mushroom to brush regime at ~ 5 mol% DSPE-PEG2000 and ~17 mol% DSPE-PEG750. Clinically used Doxil (liposomal doxorubicin), contains ~ 4 mol% of DSPE-PEG2000,58 so steric stabilization and prevention of opsonization and clearance in the circulation correlates with a near-complete mushroom concentration in which the liposome surface is covered by PEG57.
Fig. 6A shows the shows that DSPE-PEG on the bilayer also inhibits lysolipid transfer into the bilayer. 100 nm unilamellar DPPC liposomes with various mole fractions of either DSPE-PEG2000 or DSPE-PEG750 were mixed with dye-labeled MPPC micellar solutions at 10 mol% of the total DPPC concentration. A 100% surface coverage for DSPE-PEG2000 was 5 mol% and 17 mol% for DSPE-PEG750. Fig. 6A shows that PEG surface coverage exceeding 50% reduces lysolipid transfer into the bilayer; 100% surface coverage prevents any lysolipid incorporation into the liposomes. No difference was observed between DSPE-PEG2000 liposomes and DSPE-PEG750 liposomes at equal surface coverage despite the difference in the radius of gyration for these two molecular weights. This suggests that lysolipid accesses the bilayer through gaps between PEG molecules on the surface, as the thickness of the PEG coating is determined by the molecular weight while the surface coverage determines the extent of free space between the polymer chains.
PEG-lipids, similar to lysolipids, have relatively large headgroup to tail group areas, form micelles in aqueous solution, and also have the potential to destabilize the liposome membrane at concentrations above the overlap concentration10, 11. Fig. 6B shows that the total mole fraction of lysolipid plus PEG-lipids within the bilayer determines liposome stability. Liposome stability was assessed by measuring CF retention; a complete lack of CF retention was taken to be an indication of liposome destabilization. In the absence of PEG-lipid, liposomes remained stable up to 25 mol% MPPC. 50% surface coverage of DSPE-PEG750 slightly reduced the total amount of MPPC that could be incorporated prior to destabilization. However, 50% surface coverage of DSPE-PEG2000 reduced the amount of MPPC to < 15 mol% for stable liposomes.
In addition to a PEG-lipid coating, it may be useful to add a net negative charge to the liposomes to minimize aggregation or adsorption to biological surfaces, which are predominantly negatively charged. The DSPE-PEG2000 used here is terminated with an anionic methoxy group; liposomes with 5 mol% of methoxy-terminated DSPE-PEG2000 have a zeta-potential of −8.9 mV. In Fig. 7, DPPC liposomes with various mole fractions of dipalmitoylphosphatidylglycerol (DPPG) were compared to confirm that the steric effect of DSPE-PEG2000, rather than the negative surface charge, impact lysolipid uptake into the membrane. The inset of Fig. 7A shows the negatively charged phosphatidylglycerol headgroup. The zeta potential of DPPC liposomes is ~ 0 mV in the absence of DPPG but decreases to −35 mV with 60 mol% DPPG. However, within experimental error, the surface charge of the liposomes did not change the partitioning of MPPC into the bilayer.
Fig. 7.
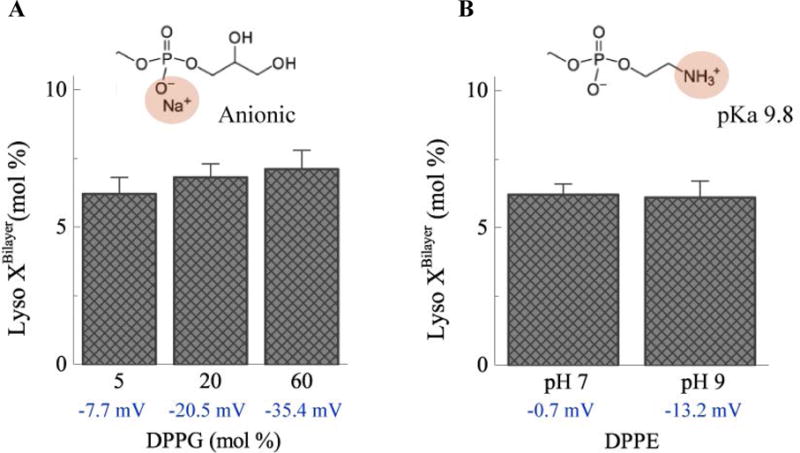
A) The zeta potential of DPPC liposomes becomes more negative as the fraction of negatively-charged DPPG (inset) increases. However, partitioning of MPPC lysolipid into the membrane is independent of the zeta potential. B) Deprotonation of the terminal amine group (inset) of DPPE at pH 9 leads to a decrease in zeta potential for 5:95 DPPE:DPPC liposomes. However, the surface charge did not impact MPPC lysolipid partitioning into the membrane.
The headgroup charge of dipalmitoylphosphatidylethanolamine (DPPE) varies with pH over the range of pH 6 to pH 10. The DPPE headgroup contains a terminal amine group with a pKa of 9.8. The amine group is protonated and the headgroup is net neutral at pH 7; increasing the pH leads to deprotonation of the headgroup and a negative surface charge. MPPC added to DPPC:DPPE liposomes at either pH 7 or pH 9 did not alter lysolipid partitioning over the range of zeta potentials and pH that might be encountered for typical liposome formulations.
Although DSPE-PEG2000 has two fully saturated stearoyl chains, its large hydrophilic headgroup leads to its self-assembly into micelles (CMC = 5.8 μM51) in aqueous solutions, similar to the lysolipids55, 59. A 15 mM micellar solution of DSPE-PEG2000 with 2.5 mol% of fluorescently labeled DSPE-PEG2000 was added to a suspension of 40 mM, 200 nm diameter DPPC liposomes. Following separation of the liposomes in a gravity size exclusion column, the relative liposome and DSPE-PEG2000 fluorescence were compared. Fig. 8 shows that the DSPE-PEG2000 transfers into both fluid and gel phase lipid bilayers, similar to the lysolipids. Transfer into the fluid phase membrane was more rapid than into the gel phase bilayer. In contrast to lysolipid uptake, which reached equilibrium within an hour, complete uptake of PEG-lipid occurred more slowly. Within the fluid phase, about 50% of the DSPE-PEG2000 transferred into the membrane within the first hour, however, DSPE-PEG2000 continued to partition into the membrane for 48 hours. In the fluid phase, liposome bilayer concentrations exceeding 5 mol% DSPE-PEG2000 led to liposome destabilization, as measured by release of CF from the liposomes. In the gel phase, the liposomes remained stable at 5 mol% DSPE-PEG2000. Fig. 8B shows that a final concentration of 4–5 mol% DSPE-PEG2000 can be achieved within ~ 1 hour at 55°C in the fluid phase or after 48 hours at 37°C with an initial DSPE-PEG2000 concentration of 10 mol% of the total lipids.
Fig. 8.
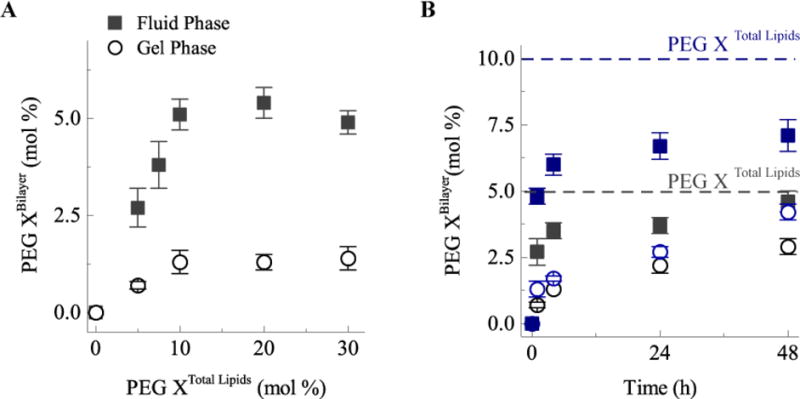
A) Transfer of DSPE-PEG2000 into the liposomes at either 37°C (gel phase, open circles) or 55°C (fluid phase, filled squares) during 1 hour of incubation with solutions containing various DSPE-PEG2000 mol% relative to the total lipids. The maximum amount of DSPE-PEG2000 incorporation after 1 hr is about 5 mol%, which corresponds to the fully covered “mushroom” state. B) Slow evolution of DSPE-PEG2000 transfer for 5 mol% DSPE-PEG2000 (black symbols) or 10 mol% (blue symbols) in solution. After 48 hours in the fluid phase (filled squares), most of the DSPE-PEG2000 has incorporated into the liposomes. About 50% of DSPE-PEG2000 incorporates into the liposomes in the gel phase (open symbols).
Optimized Construction
From our preliminary work, optimized thermosensitive, sterically stabilized interdigitation-fusion liposomes should contain 8–10 mol% MPPC and 4 mol% DSPE-PEG2000, with the remainder being DPPC. Fig. 9 shows a schematic of the optimized self-assembly process we used to make lysolipid-containing thermosensitive liposomes with encapsulated CuS nanoparticles stabilized by DSPE-PEG2000. Interdigitated DPPC sheets were prepared by adding 3M ethanol to extruded 50 nm DPPC liposomes in PBS buffer at room temperature, which converted the translucent blue liposome suspension into an opaque, milky-white gel6, 43. The interdigitated sheets were centrifuged at low speed to concentrate the sheets and washed repeatedly with fresh buffer to remove the ethanol. After washing, the pellet of DPPC sheets were mixed with an aqueous suspension of freshly prepared, thiol-PEG stabilized CuS nanoparticles (1015 CuS NP/ml) and carboxyfluorescein dye (as needed) for a final concentration of 50 mg/ml DPPC, 0.41× 1015 CuS NP/ml, and 20 mM CF. Sufficient 30 mM micellar MPPC was added to reach a solution ratio of about 6:1 DPPC:MPPC (see Fig. 5A), and the mixture was heated for 1 hour at 55°C to incorporate 8–10 mol% lysolipid within the liposome bilayer. After heating, the lysolipid-containing, CuS encapsulated liposomes were concentrated at low centrifugal force (1200×g) and the supernatant exchanged for buffer. Approximately 10-fold higher centrifugation speeds are required to sediment the PEGylated CuS NP. By repeating the centrifugation and washing, the final sample contained only the lysolipid-DPPC liposomes with encapsulated CuS NP and CF.
Fig. 9.
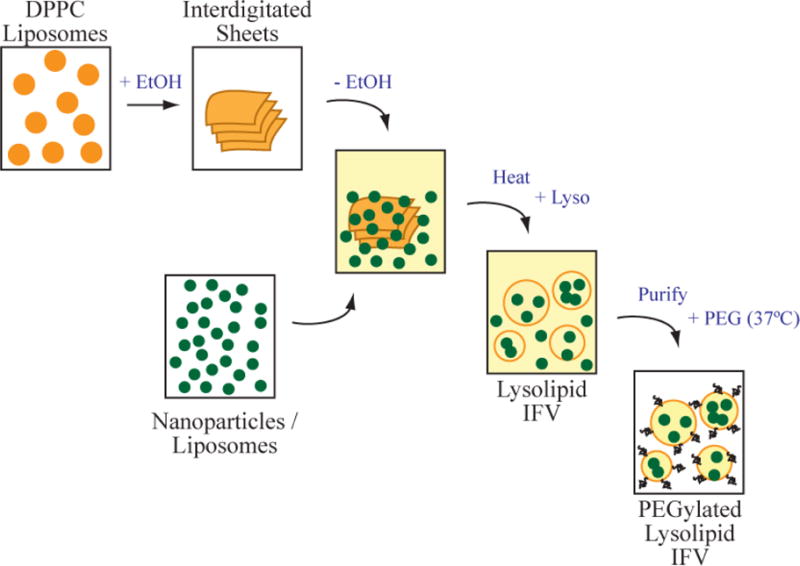
Schematic diagram of the optimized process to form thermosensitive liposomes encapsulating CuS nanoparticles (or other liposomes). Interdigitated bilayer sheets are made by ethanol treatment of small DPPC liposomes, followed by repeated washing to remove the ethanol. The sheets are mixed with thiol-PEG stabilized copper sulfide nanoparticles and carboxyfluorescein dye in saline buffer and heated above the transition temperature of the lipid. The bilayers melt and revert to closed liposome form, trapping the nanoparticles and dye. Centrifugation and repeated washing with buffer removes the unencapsulated nanoparticles and dye. A micellar solution of DSPE-PEG2000 is added and partitions into the liposome bilayers. Excess PEG-lipid is removed by centrifugation and washing.
Following purification, CuS NP liposomes were re-suspended in 75 mM PBS at a final concentration of 9 mg/ml DPPC. Assuming a liposome internal volume fraction of 60%, the concentration of CuS NP in the final sample is [Initial concentration × 0.07 dilution × 60% encapsulation] or ~ 80 μM CuS. To sterically stabilize the liposomes, DSPE-PEG2000 was added at 5 mol% of the total liposome lipid concentration at 37°C and the mixture allowed to equilibrate for 48 hours. Excess DSPE-PEG2000 was removed by centrifugation and repeated washing with buffer. Introduction of DSPE-PEG2000 as the final stage of the process simplifies purification during the synthesis as the PEGylated liposomes sediment much more slowly than non-PEGylated liposomes.
NIR-Triggered Dye Release
The optimized interdigitation-fusion liposomes (IDL) contained CuS nanoparticles within the liposome interiors with MPPC and DSPE-PEG2000 incorporated into the bilayer membrane by self-assembly. The IDL were irradiated by continuous, 800 nm NIR light within a cuvette controlled at 37°C. The absorption and photothermal conversion of the NIR light by the CuS nanoparticles in the IDL leads to an increase in the sample temperature as measured with a thermocouple (Fig. 10). After 5 minutes of irradiation at 7 W/cm2, the liposomes with encapsulated CuS reached 40°C (ΔT=3°C), while the buffer only reached 38°C (ΔT=1°C). This is consistent with the strong absorption of the CuS and the relatively weak absorption by water at 800 nm. Lower power densities would be required for a NIR light source in the range of 900 – 950 nm, over which CuS has more than twice the specific absorption (Fig. 2).
Fig. 10.
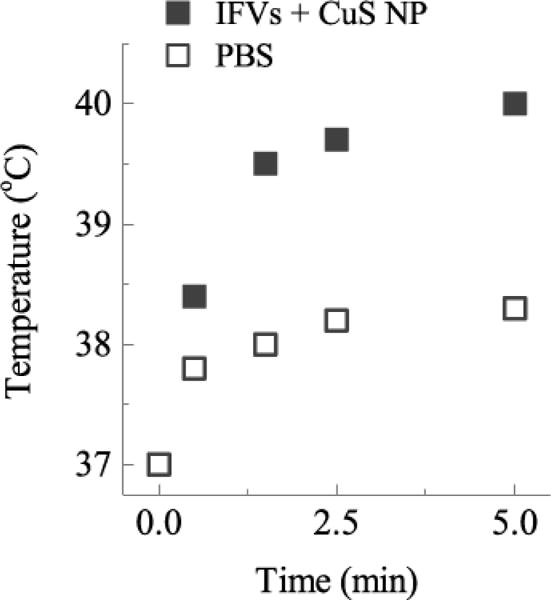
Sample temperature at various times on irradiation of liposomes encapsulating CuS NP (IFV + CuS NP) or buffer (PBS) with 800 nm NIR light at 7 W/cm2. Sample temperature after 5 min is 40°C for samples containing CuS NP, which is 2°C higher than buffer irradiated under the same conditions.
Both encapsulated CuS nanoparticles and MPPC in the liposome bilayer are required for rapid contents release (Fig. 11). Heating under NIR light irradiation initiates near-complete dye release from the lysolipid-containing IDL at laser power densities ≥ 5 W/cm2 within 5 minutes. In contrast, negligible dye is released from DPPC or DPPC plus MPPC IDL at any laser power used without encapsulated CuS. Without the specific absorption of NIR light by the CuS NP, the sample temperature remains below the ~ 40°C membrane transition temperature, leading to minimal dye release. With CuS laser power up to a 3°C increase (40°C final sample temperature) at the highest laser power tested of 7 W/cm2. This power intensity is well below the 12 W/cm2 threshold observed in previous studies that caused skin irritation in animals, which is the maximum power that could be safely be used in vivo30, 31, 60. Without MPPC in the bilayer, even though the temperature increases the same for liposomes containing CuS, no dye release is observed. Comparable release may be achieved at lower laser power if the drug delivery carrier were irradiated with longer wavelength light, which would improve the photothermal conversion efficiency of the CuS NP. At 800 nm, the absorption of CuS is approximately 40% of the absorption at 900 nm (Fig. 2).
Fig. 11.
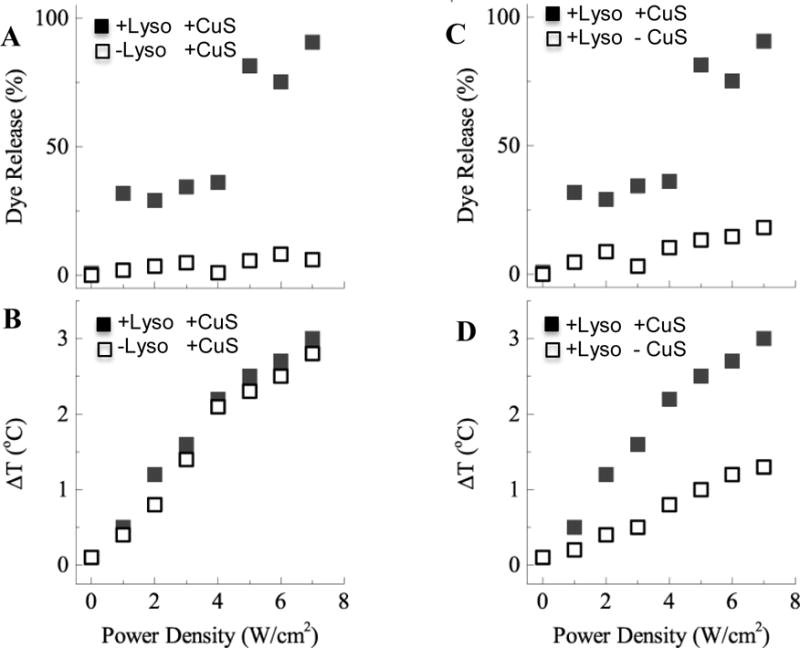
Measured carboxyfluorescein (CF) dye release and sample temperature increase after 5 minutes of NIR light exposure. A) Increasing dye release with NIR light power density was observed from liposomes with encapsulated CuS containing lysolipid in the bilayer (+Lyso+CuS), but not from liposomes encapsulating CuS without lysolipid (−Lyso+CuS). (B) However, the temperature increase of the sample after 5 minutes was the same for both samples. (C) Lysolipid containing liposomes with CuS (+Lyso+CuS) release dye but liposomes with lysolipid but without CuS do not release dye (+Lyso-CuS),. (D) Without encapsulated CuS NP, lysolipid-containing liposomes did not heat sufficiently to reach the permeability transition temperature. Both lysolipid and CuS are necessary for rapid contents release.
Conclusions
We present an inside-outside self-assembly process that only requires sequential mixing and simple washing and centrifugation steps to create thermosensitive, sterically stable liposome carriers with rapid contents release triggered by physiologically friendly near infra-red (NIR) light. Ethanol-induced interdigitation of DPPC (or mixed DPPC and DPPG) bilayers is used to encapsulate copper sulfide nanoparticles. The metastable phase progression used first takes advantage of the greatly increased membrane stiffness in the interdigitated phase of DPPC with added ethanol. Heating the interdigitated DPPC bilayers with CuS nanoparticles in suspension induces a phase change that softens the interdigitated bilayers, causing them to revert to closed bilayer liposomes, and in the process, capture CuS nanoparticles in the liposome interior. This co-localizes the CuS and the liposomes, so that the local heating induced by the NIR light can raise the liposome membrane temperature. The DPPC membrane is modified to include 8 – 10 mol% MPPC lysolipid and 3–5 mol% DSPE-PEG2000 by incubating these micelle-forming lipids with the liposomes to create a permeability transition in the membrane at ~ 40°C, as well as sterically stabilize the liposomes against flocculation or opsonization in biological environments. Irradiating the CuS-lysolipid- DSPE-PEG2000 -DPPC liposomes with NIR laser light power at levels well below that known to damage skin causes complete contents release from the liposomes within a few minutes. Without irradiation, contents are held for days. Previous work has shown that rapid drug release plus slight hyperthermia provides synergistic cell killing10 that could soon be translated into new photo-triggered and targeted nanocarriers for drug release in the body based on NIR light-addressable liposomes. The new liposomes can be used to provide a rapid, localized concentration change with the spatial and temporal control provided by physiologically friendly NIR light.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant R01 EB012637. The authors thank I. Shieh for help in setting up the laser system.
References
- 1.Zasadzinski JA, Wong B, Forbes N, Braun GB, Wu G. Current Opinion in Colloid and Interface Science. 2011;16:203–214. doi: 10.1016/j.cocis.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Charrois GJR, Allen TM. Biochimica et Biophyisca Acta. 2004;1663:167–177. doi: 10.1016/j.bbamem.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Torchilin VP. Eru J Pharmaceutics and Biopharmaceutics. 2009;71:431–444. doi: 10.1016/j.ejpb.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ganta S, Devalapally H, Shahiwala A, Amiji M. J Controlled Release. 2008;126:187–204. doi: 10.1016/j.jconrel.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 5.Zhu L, Torchilin VP. Integr Biol. 2013;5:96–107. doi: 10.1039/c2ib20135f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boyer C, Zasadzinski JA. ACS Nano. 2007;1:176–182. doi: 10.1021/nn7002025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walker SA, Kennedy MT, Zasadzinski JA. Nature. 1997;387:61–64. doi: 10.1038/387061a0. [DOI] [PubMed] [Google Scholar]
- 8.Kisak ET, Coldren B, Evans CA, Boyer C, Zasadzinski JA. Current Medicinal Chemistry. 2004;11:199–219. doi: 10.2174/0929867043456197. [DOI] [PubMed] [Google Scholar]
- 9.Wong B, Boyer C, Steinbeck C, Peters D, van Zanten R, Chmelka B, Zasadzinski JA. Adv Materials. 2011;23:2320–2325. doi: 10.1002/adma.201003578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Forbes N, Pallaoro A, Reich NO, Zasadzinski JA. Particles and Particle Systems Characterization. 2014;31:1158–1167. doi: 10.1002/ppsc.201400035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnsson M, Edwards K. Biophys J. 2003;85:3839–3847. doi: 10.1016/S0006-3495(03)74798-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen TM, Cullis PR. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 13.O’Brien MER, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DC, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C, C. Group Ann Oncol. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 14.Zamboni WC, Gervais AC, Egorin MJ, Schellens JHM, Zuhowski EG, Pluim D, Joseph E, Hamburger DR, Working P, Colbern G, Tonda ME, Potter DM, Eiseman JL. Cancer Chemother Pharmacol. 2004;53:329–336. doi: 10.1007/s00280-003-0719-4. [DOI] [PubMed] [Google Scholar]
- 15.Allen TM, Cullis PR. Adv Drug Delivery Reviews. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 16.Yatvin MB, Weinstein JN, Dennis WH, Blumenthal R. Science. 1978;202:1290–1293. doi: 10.1126/science.364652. [DOI] [PubMed] [Google Scholar]
- 17.Weinstein JN, Magin RL, Yatvin MB, Zaharko DS. Science. 1979;204:188–191. doi: 10.1126/science.432641. [DOI] [PubMed] [Google Scholar]
- 18.Anyarambhatia GR, Needham D. J Liposome Research. 1999;9:491–506. [Google Scholar]
- 19.Needham D, Anyarambhatla G, Kong G, Dewhirst MW. Cancer Research. 2000;60:1197–1201. [PubMed] [Google Scholar]
- 20.Needham D, Dewhirst MW. Advanced Drug Delivery Reviews. 2001;53:285–305. doi: 10.1016/s0169-409x(01)00233-2. [DOI] [PubMed] [Google Scholar]
- 21.Dewhirst MW, Landon CD, Hofmann CL, Stauffer PR. Surg Oncol Clin N Am. 2013;22:545–561. doi: 10.1016/j.soc.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agrawal A, Mackey MA, El-Sayed MA, Bellamkondo RV. ACS Nano. 2011;5:4919–4926. doi: 10.1021/nn201010q. [DOI] [PubMed] [Google Scholar]
- 23.Landon CD, Park JY, Needham D, Dewhirst MW. Open Nanomedicine Journal. 2011;3:38–64. doi: 10.2174/1875933501103010038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang Y, Wang H, Kindt JT. Biophys J. 2010;98:2895–2903. doi: 10.1016/j.bpj.2010.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jung HT, Coldren B, Zasadzinski JA, Iampietro D, Kaler EW. Proceedings of the National Academy of Sciences. 2001;98:1353–1357. doi: 10.1073/pnas.041420998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung HT, Lee SY, Coldren B, Zasadzinski JA, Kaler EW. Proceedings of the National Academy of Sciences. 2002;99:15318–15322. doi: 10.1073/pnas.242374499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fromherz P. Chem Phys Letters. 1983;94:259–266. [Google Scholar]
- 28.Li L, ten Hagen TLM, Hossann M, van Rhoon GC, Eggermont AM, Haemmerich D, Koning GA. J Controlled Release. 2013;168:142–150. doi: 10.1016/j.jconrel.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 29.Stauffer PR, G SN. Int J Hyperthermia. 2004;20:671–677. doi: 10.1080/02656730400007220. [DOI] [PubMed] [Google Scholar]
- 30.Zhou M, Zhang R, Huang M, Lu W, Song S, Melancon MP, Tian M, Liang D, Li C. J Am Chem Soc. 2010;132:15351–15358. doi: 10.1021/ja106855m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramadan S, Guo L, Li Y, Yan B, Lu W. Small. 2012;8:3143–3150. doi: 10.1002/smll.201200783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Huang Q, Huang M, Li C, Chen W. Nanomedicine. 2010;5:1161–1171. doi: 10.2217/nnm.10.85. [DOI] [PubMed] [Google Scholar]
- 33.Prevo BG, Esakoff SA, Mikhailovsky A, Zasadzinski JA. Small. 2008;4:1183–1195. doi: 10.1002/smll.200701290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Proc Natl Acad Sci U S A. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Cancer Letters (Amsterdam, Netherlands) 2004;209:171–176. doi: 10.1016/j.canlet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 36.Link S, Burda C, Nikoobakht B, El-Sayed MA. Chem Phys Letters. 1999;315:12–18. [Google Scholar]
- 37.Huang X, El-Sayed IH, Qian W, El-Sayed MA. Journal of the American Chemical Society. 2006;128:2115–2120. doi: 10.1021/ja057254a. [DOI] [PubMed] [Google Scholar]
- 38.Jain PK, Qian W, El-Sayed MA. J Am Chem Soc. 2006;128:2426–2433. doi: 10.1021/ja056769z. [DOI] [PubMed] [Google Scholar]
- 39.Kisak ET, Coldren B, Zasadzinski JA. Langmuir. 2002;18:284–288. [Google Scholar]
- 40.Manzour AA, Lindner LH, Landon CD, Park J-Y, Simnick AJ, Dreher MR, Das S, Hanna G, Park W, Chilkoti A, Koning GA, ten Hagen TLM, Needham D, Dewhirst MW. Cancer Research. 2012;72:5566–5575. doi: 10.1158/0008-5472.CAN-12-1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mills JK, Needham D. Methods in Enzymology. 2004;387:82–113. doi: 10.1016/S0076-6879(04)87006-X. [DOI] [PubMed] [Google Scholar]
- 42.Weissleder R. Nature Biotechnology. 2001;19:316–317. doi: 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- 43.Ahl PL, Perkins WR. Methods in Enzymology. 2003;367:80–98. doi: 10.1016/S0076-6879(03)67007-2. [DOI] [PubMed] [Google Scholar]
- 44.Coldren B, van Zanten R, Mackel MJ, Zasadzinski JA, Jung HT. Langmuir. 2003;19:5632–5639. [Google Scholar]
- 45.Tardiue A, Luzzati V, Reman FC. Journal of Molecular Biology. 1973;75:711–733. doi: 10.1016/0022-2836(73)90303-3. [DOI] [PubMed] [Google Scholar]
- 46.Adachi T, Takahashi H, Ohki K, Hatta I. Biophys J. 1995;68:1850–1855. doi: 10.1016/S0006-3495(95)80361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boni LT, Minchey SR, Perkins WR, Ahl PL, Slater JL, Tate MW, Gruner SM, Janoff AS. Biochimica et Biophyisca Acta. 1993;1146:247–257. doi: 10.1016/0005-2736(93)90363-5. [DOI] [PubMed] [Google Scholar]
- 48.McIntosh TJ, McDaniel RV, Simon SA. Biochimica et Biophyisica Acta. 1983;731:109–114. [Google Scholar]
- 49.Simon SA, McIntosh TJ. Biochimica et Biophyisca Acta. 1984;773:169–172. doi: 10.1016/0005-2736(84)90562-5. [DOI] [PubMed] [Google Scholar]
- 50.Ahl PL, Chen L, Perkins WR, Minchey SR, Boni LT, Taraschi TF, Janoff AS. Biochim Biophys Acta. 1994;1995:237–244. doi: 10.1016/0005-2736(94)90262-3. [DOI] [PubMed] [Google Scholar]
- 51.Uster PS, Allen TM, Daniel BE, Mendez CJ, Newman MS, Zhu GZ. FEBS Letters. 1996;386:243–246. doi: 10.1016/0014-5793(96)00452-8. [DOI] [PubMed] [Google Scholar]
- 52.Needham D, Stoicheva N, Zhelev DV. Biophys J. 1997;73:2615–2629. doi: 10.1016/S0006-3495(97)78291-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Needham D, Zhelev DV. Ann Biomed Eng. 1995;23:287–298. doi: 10.1007/BF02584429. [DOI] [PubMed] [Google Scholar]
- 54.Israelachvili JN. Intermolecular and surface forces. 2nd. Academic Press; London: 1992. [Google Scholar]
- 55.Zalipsky S, Hansen CB, Lopes de Menezes DE, Allen TM. J Controlled Release. 1996;39:153–161. [Google Scholar]
- 56.Klibanov AL, Maruyama K, Torchilin VP, Huang L. FEBS Letters. 1990;268:235–237. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]
- 57.Kenworthy AK, Hristova K, Needham D, McIntosh TJ. Biophys J. 1995;68:1921–1936. doi: 10.1016/S0006-3495(95)80369-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allen TM. Current Opinion in Colloid and Interface Science. 1996;1:645–651. [Google Scholar]
- 59.Zalipsky S, Brandeis E, Newman MS, Woodle MC. FEBS Letters. 1994;353:71–74. doi: 10.1016/0014-5793(94)01013-7. [DOI] [PubMed] [Google Scholar]
- 60.Timko BP, Dvir T, Kohane DS. Adv Materials. 2010;22:4925–4943. doi: 10.1002/adma.201002072. [DOI] [PubMed] [Google Scholar]