

Summary
To functionally assess the oncogene xmrk co-expressed genes, and compare the functional pathways of these genes to pathways represented in the human melanoma subgroups that were characterized by high and low pigmentation function, we performed gene expression profiling of Xiphophorus melanoma tumors, characterized xmrk co-expressed genes and identified the functional pathways they are associated with. The transcriptomic features and pathways related to the xmrk expression faithfully represent the genetic differences between non-proliferative differentiated and mitogenic dedifferentiated human melanoma. This property supports Xiphophorus melanoma as an appropriate disease model of human melanoma, enabling application of melanoma etiological discovery among vertebrates.
Comparative analysis of human and animal model melanomas can uncover conserved pathways and genetic changes that are relevant for the biology of cancer cells. Spontaneous melanoma in Xiphophorus interspecies backcross hybrid progeny may be informative in identifying genes and functional pathways that are similarly related to melanoma development in all vertebrates, including humans. To assess functional pathways involved in the Xiphophorus melanoma, we performed gene expression profiling of the melanomas produced in interspecies BC1 and successive backcross generations (i.e., BC5) of the cross: X. hellerii × [X. maculatus Jp 163 A × X. hellerii]. Using RNA-Seq we identified genes that are transcriptionally co-expressed with the driver oncogene, xmrk. We determined functional pathways in the fish melanoma that are also present in human melanoma cohorts that may be related to dedifferentiation based on the expression levels of pigmentation genes. Shared pathways between human and Xiphophorus melanomas are related to inflammation, cell migration, cell proliferation, pigmentation, cancer development and metastasis. Our results suggest xmrk co-expressed genes are associated with dedifferentiation, and highlight these signaling pathways as playing important roles in melanomagenesis.
Keywords: Melanoma, Xiphophorus, Bioinformatics, Signaling transduction, Xmrk, Gene expression, Transcriptome
Introduction
While the incidence of most cancers is decreasing, skin cancer, including melanoma, continues to increase 3-7% per year, exhibiting a 6-fold increase in incidence over the past 40 years, with a 5-year survival rate of only 20% (Berger et al., 2012; Reed et al., 2012; Rigel, 2008; WE, 1982). Despite recent major advances in therapies, the prognosis for melanoma patients with advanced stage melanoma remains poor (Siegel, Naishadham, & Jemal, 2012). Approximately 50% of melanoma patients harbor a BRAF mutation (i.e., BRAF V600E), an oncogene driving the RAF/RAS/MEK signaling leading to proliferation and progression of melanoma cells (Davies et al., 2002; Gopal et al., 2010; Jakob et al., 2012; Long et al., 2011). Clinical data have shown that 50-60% of melanoma patients bearing BRAF V600E mutation respond to FDA approved BRAF inhibitor such as vemurafenib and dabrafenib (Chapman et al., 2011; Falchook et al., 2012; Flaherty et al., 2012; Flaherty et al., 2010; Hauschild et al., 2012; Lito et al., 2012; Long et al., 2012). Recently, immune checkpoint inhibitors targeting PD-1 or PD-L1 were approved by the FDA to treat advanced melanoma (e.g., Pembrolizumab). Approximately one-third of patients with advanced melanoma are responsive to PD-1 inhibitors. However, resistance to BRAF inhibitors is observed in a majority of the responsive patients. One mechanism accounting for the resistance is due to the aberrant up-regulation of EGFR and downstream PI3K/Akt signaling (Wang et al., 2015). Twenty-five percent of the immune checkpoint inhibitor responsive tumors also recur despite continued treatment. The mechanism of the resistance is still elusive although mutations in either JAK1 or JAK2 account for part of the resistance mechanism (Zaretsky et al., 2016). Therefore, mechanistic study of melanomagenesis, especially for the tumors that are not responsive to existing therapy, is needed in order to develop new effective treatments. Additionally, drug resistance observed in current melanoma therapy underscores the need for development of new therapeutic strategies that counteract therapy resistance and/or to target the disease through a different mechanism.
The Xiphophorus melanoma model, also known as “Gordon-Kosswig-Anders” melanoma model, was originally introduced in the late 1920s, as one of the first animal models leading to genetic studies of cancer (Gordon, 1927; Häussler, 1928; Kosswig, 1928). This model employs X. maculatus and X. hellerii interspecies hybrids to produce spontaneous, yet genetically controlled, melanoma. In the classical cross X. maculatus carries the spotted dorsal (Sd) macromelanophore pigmentation pattern while X. hellerii does not have such a pigmentation pattern (For reviews see: (Patton, Mathers, & Schartl, 2011; R. B. Walter & Kazianis, 2001). Neither the Sd locus, nor a functional equivalent of the X. maculatus R(Diff) locus, a hypothetical tumor suppressor mapped to linkage group 5, are present in X. hellerii. The X-chromosome and Sd linked oncogene, xmrk, is a mutant copy of the fish orthologue of the human EGFR and have been established as a melanoma driver oncogene in transgenic studies (Schartl et al., 2012; Schartl et al., 2015; Schartl et al., 2010). In contrast, the critical genetic component of the autosomal R(Diff) locus, which regulates xmrk, is still unknown. R(Diff) has been mapped to a 5.8 Mb region on linkage group 5, and is proposed to inhibit xmrk function in X. maculatus parental animals since they rarely develop melanoma tumors (Adam, Maueler, & Schartl, 1991; Kazianis et al., 1998; Kazianis et al., 1999; Lu et al., 2017). F1 interspecies hybrids between X. maculatus and X. hellerii (i.e., Sd-hellerii) exhibit enhanced dorsal fin pigmentation but do not develop melanoma, likely due to regulation by the remaining single copy of the X. maculatus R(Diff) locus. When F1 hybrids are backcrossed with X. hellerii, 25% of progeny that inherited the xmrk oncogene, but did not inherit the R(Diff) locus, develop spontaneous, lethal melanoma. Other model systems have been developed that take advantage of the xmrk driver oncogene, such as the xmrk transgenic medaka (Japanese rice fish, Oryzias latipes). In this transgenic model, xmrk is driven by pigment cell specific mitf promoter, resulting in early onset melanoma development with 100% penetrance (Schartl et al., 2012; Schartl et al., 2010).
The xmrk oncogene is capable of inducing transformation in melanocytes by maintaining sustained MAPK signaling. Similar to dedifferentiated melanocytes induced by other oncogenes (i.e., bFGF, myc, Ela, ras, or neu) dedifferentiated cells show enhanced proliferation, absence of dendrites, and a lack of melanin production (Dotto, Moellmann, Ghosh, Edwards, & Halaban, 1989; Wellbrock, Fischer, & Schartl, 1998; Wellbrock, Weisser, Geissinger, Troppmair, & Schartl, 2002; Wilson, Dooley, & Hart, 1989). The xmrk gene influences several EGFR regulated pathways that are consistent with published observations of mammalian melanomas that drive and maintain the dedifferentiated state (Ge, Fu, & Meadows, 2002; K. Smalley & Eisen, 2000; K. S. Smalley, 2003; K. S. Smalley & Eisen, 2002). These pathways include MAPK signaling, integrin signaling, PI3K signaling, STAT5 signaling, and repression of immune response (Delfgaauw et al., 2003; Geissinger, Weisser, Fischer, Schartl, & Wellbrock, 2002; Morcinek, Weisser, Geissinger, Schartl, & Wellbrock, 2002; Schartl et al., 2015; Wellbrock & Schartl, 1999, 2000; Wellbrock et al., 2002). Although some of the direct functions of xmrk driven pathways are well studied, a comprehensive understanding of genes and functional pathways that are associated with xmrk-induced dedifferentiation is lacking. As a continuation of previous studies, we used xmrk expression as a marker, and utilized contemporary RNA-Seq to perform global assessment of molecular genetic profiles in these Xiphophorus melanoma to hallmark genes that co-express or are reversely correlated with xmrk, to identify pathways that are associated with xmrk expression. We compare functional pathways associated with differentiated pigmentation related gene expression, a feature characterizing terminally differentiated pigment cells in human melanoma patients, to the functional pathways that are associated with genes that are co-expressed with xmrk in Xiphophorus melanoma. This strategy allowed identification of gene clusters representing the dedifferentiated status of Xiphophorus melanoma, and may be related to the invasive capacity of the melanoma cells. The similarity in functional pathways between the Xiphophorus melanoma and human melanoma suggest that melanomagenesis in Xiphophorus is an informative genetic representation of human melanoma etiology.
Results
Identification of an xmrk co-expression signature in Xiphophorus melanoma
The backcross of F1 hybrid (Sd-hellerii) and X. hellerii lead to spontaneous melanoma in 25% of the BC1 and BC5 progeny (Fig. 1). These tumors expand from the dorsal fin and/or caudal fin and peduncle of the interspecies hybrid progeny. To profile global gene expression, we performed RNA-Seq and assessed gene expression of these melanomas (Supplement Fig. 1). High expression of the driver oncogene, xmrk, is capable of inducing spontaneous melanoma (Schartl et al., 2010; Wittbrodt et al., 1989). The expression of the xmrk in pigment cell both suppresses differentiation and induces a transformed dedifferentiated phenotype (Wellbrock et al., 2002). However, xmrk gene expression levels vary among melanomas by 9.6 fold in both BC1 and BC5 interspecies hybrid progeny (Fig. 2a). To identify genes that correlate in expression level with xmrk, we performed co-expression analysis between each gene and xmrk. In BC1 melanoma, there are 1279 genes showing a pattern of co-expression with xmrk, and 231 genes negatively correlated with the xmrk expression pattern. In BC5 melanoma, 2631 genes co-express with xmrk and 2589 genes negatively correlate with xmrk expression (Supplement Fig. 2). Six hundred and ten genes showed co-expression and 43 genes show negative correlation patterns with xmrk expression in both BC1 and BC5 melanoma (Fig. 2b, Supplement Table 2). Nine stem cell/melanoma cancer stem cell marker, cfl, prom1 (CD133), itga6, itga8, itgb1, tbx2, cdh7, cdh20 and zeb1 are co-expressed with xmrk in the Xiphophorus melanoma (Figure 2c,d). This is in agreement with previous reports showing that xmrk is capable of dedifferentiating melanocytes in this model system (Delfgaauw et al., 2003; Wellbrock et al., 2002)
Figure 1. “Gordon-Kosswig-Anders” melanoma model.

The F1 interspecies hybrid was produced by crossing X. maculatus Jp 163 A (xmrk +/+, R(Diff) +/+) to X. hellerii (xmrk -/-, R(Diff) -/-). The F1 hybrid shows enhanced dorsal fin pigmentation but does not develop invasive melanoma due to regulation by the remaining copy of the R(Diff) locus. When an F1 hybrid is backcrossed with X. hellerii, 25% of the hybrid progeny that inherited the xmrk oncogene but did not acquire the R(Diff) locus, develop melanoma tumors.
Figure 2. Co-expression of genes with xmrk in Xiphophorus melanoma tumors.
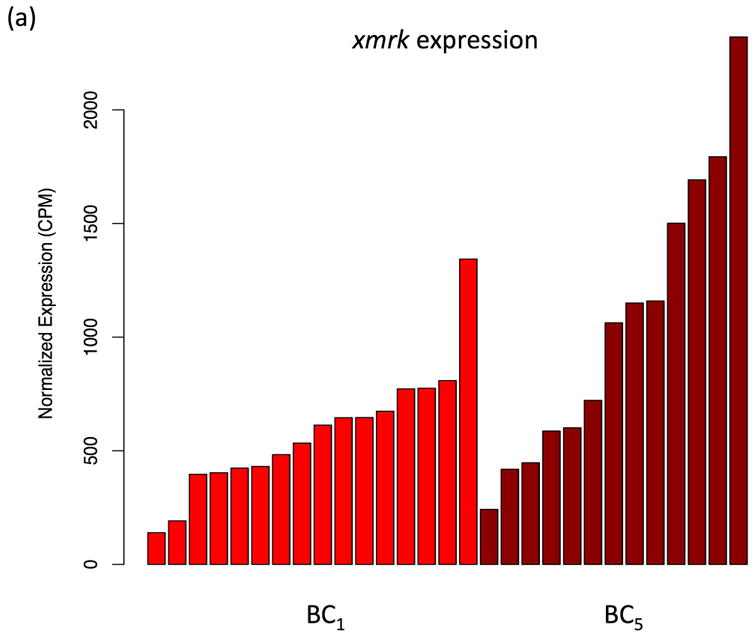

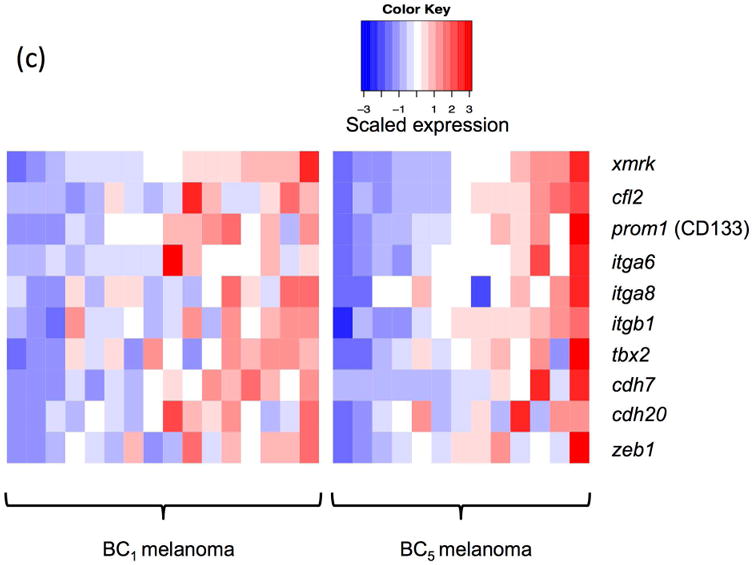
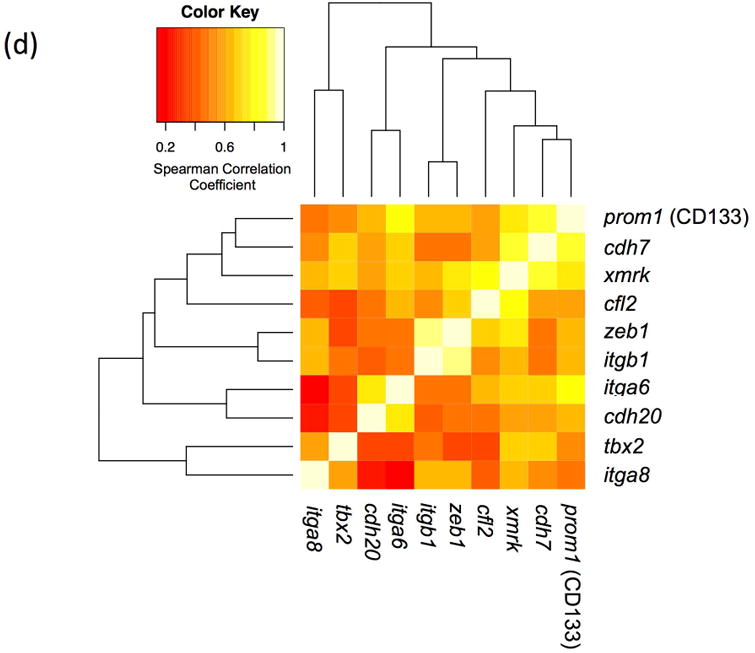
(a) The xmrk expression varies by up to 9.6 fold in BC1 and BC5. (b) 610 genes co-expressed with xmrk, of which 43 genes negatively correlate with xmrk expression in both BC1 and BC5 Xiphophorus melanoma. (c) Nine stem cell markers co-expressed with xmrk in BC1 and BC5 Xiphophorus melanoma. Heatmap represents gene expression in each melanoma tumor of one backcross individual. Melanoma samples are ordered according to xmrk expression level. (d) Correlation coefficients of stem cell markers. Spearman correlation coefficient between two stem cell makers, and between each stem cell marker and xmrk are presented in a heatmap. The dendrogram represents clustering of the coefficients.
Differential gene expression between High- and Low-MITF-axis melanoma patient cohorts
Compared to terminal differentiated melanoma cells, invasive melanoma cells are characterized by loss of pigmentation related genes, amelanotic and dedifferentiated phenotypes (Delfgaauw et al., 2003; Dotto et al., 1989; Wellbrock et al., 2002; Wilson et al., 1989). To identify genes that are co-regulated with pigmentation related genes, we next assessed the gene expression differences between melanoma patient cohorts exhibiting differentiated pigmentation pathway activities. Terminally differentiated melanoma cells show higher expression of melanin biosynthesis genes than dedifferentiated melanoma cells, and this feature was used to identify genes that are related to the melanoma proliferation and invasion. The transcription factor MITF and its direct target genes TYR, TYRP1 and DCT, exert critical control of pigmentation and melanocyte development. We identified 93 human melanoma tumors that showed lower MITF, TYR, TYRP1 and DCT expression than 50% of all 472 SKCM samples (low-MITF-axis, dedifferentiated), and 59 melanoma tumors that showed higher level expression of these genes than 50% of all SKCM tumors (high-MITF-axis, differentiated). A total of 491 genes showed differential expression between high-MITF-axis-cohort and low-MITF-axis-cohort (252 genes highly expressed, and 239 genes lowly expressed in high-MITF-axis-cohort; Fig. 3a; Supplement Table 3). Seventeen pigmentation related genes, in addition to MITF, TYR, TYRP1 and DCT, also showed up-regulation (Fig. 3b). Additionally, as expected, stem cell/melanoma cancer stem cell markers LIF, NFATC2, NGFR (CD271) showed lower expression in high-MITF-axis cohort. Metabolism genes related to dedifferentiation (i.e., S100A4, MT1X, MT1A, NNMT, NT5E, MT1G, AKR1C1, AKR1C2, GLDC) show lower expression, while metabolism genes related to differentiation (i.e., PPARGC1A and GYG2) show higher expression in high-MITF-axis cohort (Fig. 3b).
Figure 3. Differential gene expression in human melanoma.
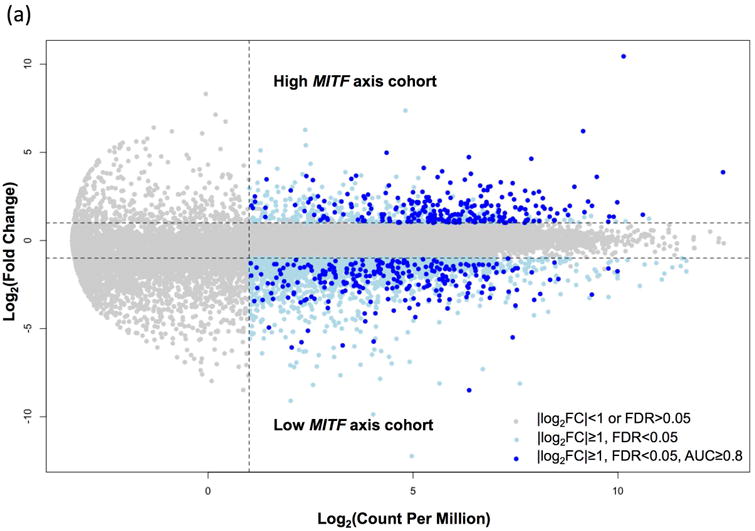
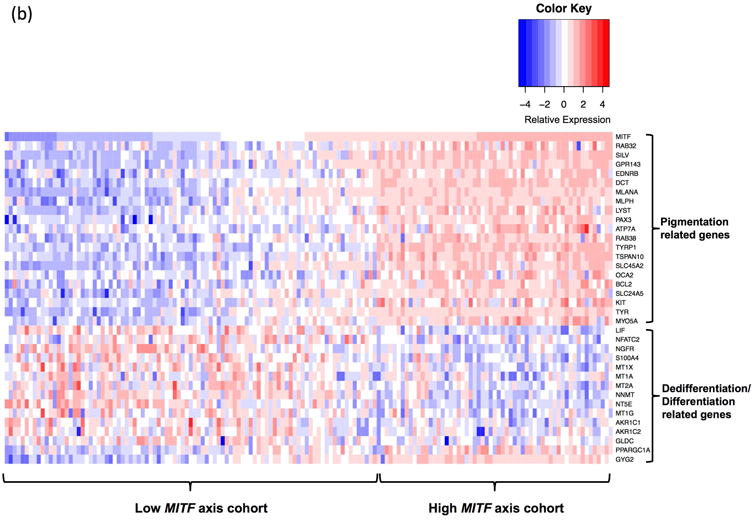
(a) To identify human melanoma patient samples with high MITF and MITF target genes, samples were categorized based on MITF, TYR, TYRP1 and DCT expression. Samples with each individual gene expressed higher than 50% of all patient samples were classified as high MITF axis cohort while samples with each MITF target gene expressed lower than 50% of all patient samples were classified as low MITF axis cohort. Differential gene expression between these two cohorts of melanoma patients showed 491 genes to be differentially expressed (|log2FC|≥1, FDR<0.05, AUC≥0.8). (b) In addition to MITF, TYR, TYRP1 and DCT, 17 other pigmentation related genes were also higher expressed in patients that over-expressed MITF and its target genes. Thirteen dedifferentiation related genes showed lower expression, and 2 differentiation related genes showed higher expression in high MITF axis cohort.
Comparison of functional pathways between Xiphophorus melanoma and human melanoma
As both the xmrk co-expressed genes, and the low-MITF-axis cohort show dedifferentiation markers, we next attempted to identify functional pathways that are associated with the observed gene expression signature that may represent dedifferentiation. The xmrk co-expressed genes, and genes that show negative correlation to the xmrk expression pattern, were analyzed using Gene Set Enrichment Analysis implemented within the Ingenuity Pathway Analysis software to identify over-represented signaling pathways. Pathway enrichment analysis of the genes co-expressed and negatively correlated with the xmrk expression in the Xiphophorus melanoma are represented by 91 functional pathways (-log10(enrichment p-value) > 2; Supplement Fig. 4a, b; Supplement Table 4). Similarly, pathway enrichment analysis was also performed on differentially expressed genes between the two melanoma patient cohorts. These genes were clustered into 29 functional pathways (-log10(enrichment p-value) > 2; Supplement Fig. 4a, c; Supplement Table 4). Twelve signaling pathways (Axonal Guidance Signaling, Colorectal Cancer Metastasis Signaling, Epithelial Adherence Junction Signaling, Eumalanin Biosynthesis, IL8 Signaling, ILK Signaling, Melanocyte Development and Pigmentation Signaling, Molecular Mechanism of Cancer, Ovarian Cancer Signaling, Pancreatic Adenocarcinoma Signaling, Semaphorin Signaling in Neurons, and Superpathway of Inositol Phosphate Compounds) are shared between the Xiphophorus melanoma and human melanoma (Fig. 4a, b). Three of the 12 shared pathways (Colorectal Cancer Metastasis Signaling, IL-8 Signaling and Pancreatic Adenocarcinoma Signaling) are repressed in the patient cohorts that highly express pigmentation related genes and activated in the patient cohorts that lowly express pigmentation related genes. The same 3 pathways are activated in the Xiphophorus melanoma with highly expressed xmrk and its co-expressed genes (Supplement Table 4).
Figure 4. Comparison of Signaling pathways enriched in Xiphophorus and human melanoma.
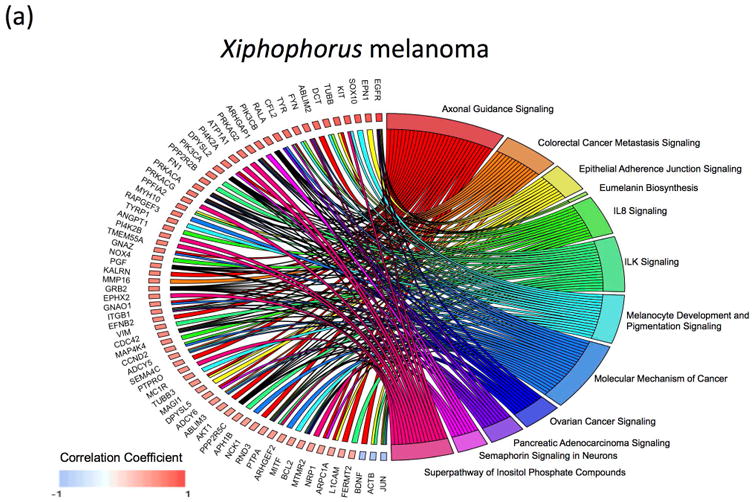
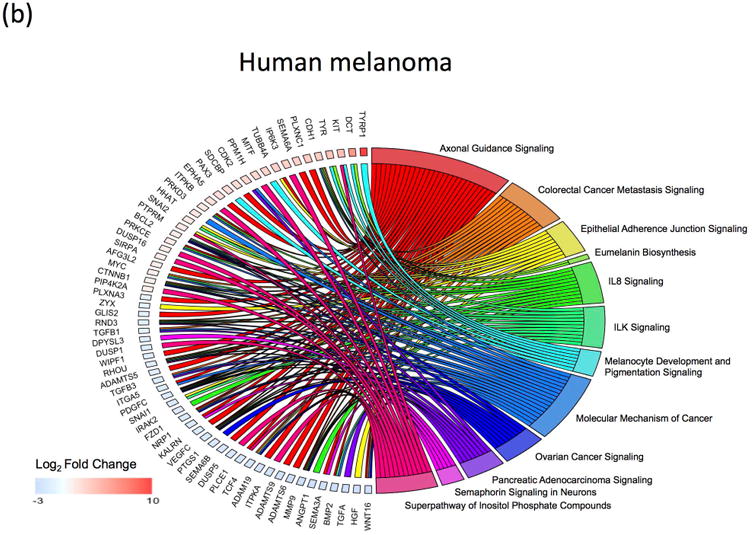
(a) Genes enriched in the 12-shared functional pathways in Xiphophorus melanoma. (b) Genes enriched in the 12-shared functional pathways in human melanoma.
Discussion
The xmrk oncogene is a mutant copy of fish egfr gene encoding a Receptor Tyrosine Kinase that forms constitutively homodimers and thereby becomes activated in a ligand-independent manner. Although some downstream signaling pathways that are directly regulated by xmrk in Xiphophorus melanoma have been well studied (i.e., MAPK signaling, STAT5 signaling, PI3K/Akt signaling; Meierjohann, Schartl, & Volff, 2004; Morcinek et al., 2002; Wellbrock, Fischer, & Schartl, 1999; Wellbrock & Schartl, 2000; Wellbrock et al., 2002), other genes that are co-expressed with xmrk and the pathways they associate with, have not been well defined. In this study, to test whether xmrk expression is associated with transcriptomic features of malignant melanoma and to further investigate the ability of Xiphophorus melanoma to model human melanoma, we identified genes and associated functional pathways involved in xmrk driven melanomagenesis in Xiphophorus and compared these to functional pathways associated with human melanoma. We analyzed Xiphophorus melanomas from both BC1 and BC5 hybrid fish, and observed more xmrk-correlating genes in BC5 (5220 genes in BC5) than in BC1 tumors (1510 genes in BC1). This is not due to different sequencing platforms or other technical differences. First, co-expression analyses were performed within tumors from BC1 and BC5 animals, respectively. Second, the gene expression values are normalized to library size (total read counts per sample). Thus, the different numbers of xmrk correlating genes is a result of differences in the genetic background between BC1 and BC5 hybrid fish. BC1 hybrids have 75% of the genome inherited from the recurrent parent (i.e., X. hellerii), while BC5 hybrids have over 98% of the genome derived from the recurrent parent. This difference in genome constitution leads to larger expression variation among BC1 individuals, and likely accounts for the lower number of xmrk correlating genes in BC1 than BC5 due to a greater degree of interspecies allele interactions from the 25% of the X. maculatus genome (non-recurrent parent) present in the BC1 genetic background. We included both BC1 and BC5 samples to capture the most conserved gene set associated with xmrk expression, regardless of the complexity of the genetic backgrounds.
Phenotype plasticity is an essential feature of melanoma. This feature is derived from neural crest progenitor cells that respond to morphogenetic cues from tissue microenvironments and give rise to respective lineages, including melanocytes (Simoes-Costa & Bronner, 2015; Takahashi, Sipp, & Enomoto, 2013). Studies have revealed that melanoma is organized and driven by a subpopulation of cancer cells that have the properties of dedifferentiated stem cells, such as disruption of dendricity, enhanced cell proliferation, and loss of pigmentation (Bracalente et al., 2016; Frank, Schatton, & Frank, 2010; Kalluri & Weinberg, 2009; Lee & Vasioukhin, 2008; Royer & Lu, 2011; Saez-Ayala et al., 2013; Schiaffino, 2010; Serafino et al., 2004). The xmrk oncogene can induce transformation of differentiated melanocytes (Delfgaauw et al., 2003; Wellbrock et al., 1998; Wellbrock et al., 2002). It has been shown to repress the MITF differentiation signal, implying MITF functional suppression partially accounts for the mechanism by which xmrk drives dedifferentiation (Delfgaauw et al., 2003). We found that xmrk expression varied in the Xiphophorus melanoma (Fig. 2a), even in those tumors that had been derived from successive backcrossing (i.e. BC5). This variation suggests that different levels of xmrk signaling activity may be associated with variable target gene regulation among melanomas in individual fish. The melanoma tumor mass is comprised of a large proportion of differentiated non-proliferating melanoma cells, and a small portion of dedifferentiated proliferating melanoma cells (i.e., malignant melanoma with high fraction of well differentiated, non-malignant pigment cells; Goodall et al., 2008). Considering xmrk is capable of transforming pigment cells to a dedifferentiated status, we hypothesized the level of xmrk expression may be an indicator of the relative size of the dedifferentiated melanoma cell population within the tumor mass. We used relatively high xmrk expression levels to hallmark a dedifferentiation state of Xiphophorus melanoma cells. Melanoma stem cell makers and metastasis related genes prom1 (CD133), itga6, itgab, itgb1, tbx2, zeb1, cdh7, cdh20 and cfl (Fig. 2c; (Argaw-Denboba et al., 2017; Bosserhoff, Ellmann, & Kuphal, 2011; Bracalente et al., 2016; Madjd et al., 2016; Moore et al., 2004; Zhao et al., 2015; Zimmerer et al., 2016) were found to be co-expressed with xmrk. These observations show xmrk is associated with a cluster of genes that are capable of maintaining the cells in an undifferentiated state. Functional pathways that are known to associate with xmrk oncogenic effect are identified among the xmrk co-expressed genes, including EGF signaling, ERK/MAPK signaling, integrin signaling, PI3K/Akt signaling and PI3K related pathways, PTEN signaling, Melanocyte Development and Pigmentation Signaling (Supplement Fig. 4b; Supplement Table 4; Meierjohann et al., 2004; Schartl et al., 2015; Wellbrock et al., 1999; Wellbrock & Schartl, 2000; Wellbrock et al., 2002).
Since expression of MITF driven pigmentation related genes and melanogenesis hallmark the differentiated status of pigment cells, we used MITF and its target genes related to pigment synthesis TYR, TYRP, and DCT to represent different transcriptomic features of disease subtypes in human melanoma (i.e., dedifferentiation and differentiation; Carreira et al., 2005; Carreira et al., 2006; Cheli et al., 2011; Cheli et al., 2012; Cheli, Ohanna, Ballotti, & Bertolotto, 2010; Garraway et al., 2005; Hoek & Goding, 2010; Loercher, Tank, Delston, & Harbour, 2005; Pinner et al., 2009). As expected, we identified two melanoma patient sample cohorts: a cohort that lowly expressed melanin synthesis genes (low-MITF-axis), and a cohort that highly expressed melanin synthesis genes (high-MITF-axis). Along with the lower expression of pigmentation related genes, the low-MITF-axis cohort shows higher expression of stem cell, neural-crest progenitor cell and melanoma cells dedifferentiation markers LIF, NGFR (CD271), and NFATC2 (Fig. 3b; Bernhardt et al., 2017; Boiko et al., 2010; Landsberg et al., 2012; Martello & Smith, 2014; Perotti et al., 2016; Riesenberg et al., 2015). Their higher expression suggested that melanomas in low-MITF-axis cohort have a higher percentage of dedifferentiated melanoma cells. Low-MITF-axis cohort also highly expresses S100A4, a metastasis-promoting microenvironment factor (Berge et al., 2011; Schmidt-Hansen et al., 2004), as well as several dedifferentiation related metabolism genes MT1X, MT1A, MT2A, NNMT, NT5E, MT1G, AKR1C1, AKR1C2, GLDC. Additionally, Low-MITF-axis cohort lowly expresses differentiation related metabolism genes PPARGC1 and GYG2 (Bettum et al., 2015). These observations suggest the low-MITF-axis cohort represents dedifferentiated melanoma tumors that are characterized by stem cell-like transcriptional features, while the high-MIFT-axis cohort is associated with differentiated non-invasive melanoma (Fig. 3b). Genes co-regulated with MITF and its target genes in human melanoma are mainly related to expected pigmentation, inflammation, cell migration and proliferation, cancer development and metastasis, and stem cell (Supplement Fig. 4c; Supplement Table 4). The presence of these signaling pathways is consistent with the dedifferentiation status of low-MITF-axis melanoma cohort, suggesting the low expression of MITF and its target genes are indicative of the dedifferentiation expression signature and the phenotype of a subtype of melanoma cells.
To test whether dedifferentiation of Xiphophorus melanoma and human melanoma involved similar signaling pathways, we next compared the functional pathways associated with genes that were co-expressed with xmrk in Xiphophorus melanoma to pathways associated with genes that were differentially expressed between the high- and low-MITF-cohort. We found 12 functional pathways that are shared between Xiphophorus melanoma and human melanoma (Fig. 4a, b). These pathways involved in inflammation (IL-8 signaling), cell migration (Axonal guidance signaling, Epithelial adherence junction signaling, ILK signaling, Semaphorin signaling, Superpathway of Inositol phosphate compounds), pigmentation (Eumelanin biosynthesis, Melanocyte development and pigmentation signaling), proliferation, cancer development and metastasis (Colorectal cancer metastasis signaling, Ovarian cancer signaling, Pancreatic adenocarcinoma signaling, Molecular mechanism of cancer). To summarize the genetic signature comparisons between Xiphophorus and human melanoma correspond to very similar groups of functional pathways and suggest that all vertebrate melanomas may share disease specific genetic signatures reflecting common developmental mechanisms (Fig. 5). Additionally, IL8 signaling and two signaling pathways related to cancer metastasis (Colorectal cancer metastasis signaling, Pancreatic adenocarcinoma signaling) are activated by xmrk co-expressed genes. The same pathways are also activated in melanoma cohorts that lowly expressed pigmentation related genes (Supplement Table 4). This consistency in functional changes indicates the high expressing xmrk Xiphophorus melanoma share transcriptomic features, and molecular functions of highly proliferative, dedifferentiated human melanoma. These results further substantiate the Xiphophorus melanoma model as representing melanoma cancer cell plasticity at the genetic level, and its potential utility as a model to delineate the genetic etiology of select states in melanoma progression.
Figure 5. Model for the relation melanoma cell dedifferentiation with xmrk expression.
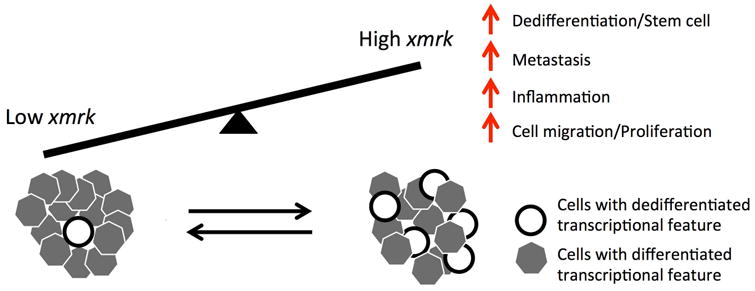
In conclusion, the transcriptomic features and tumorigenic pathways related to the xmrk expression faithfully represent the genetic differences between non-proliferative differentiated and mitogenic dedifferentiated human melanoma. This property supports Xiphophorus melanoma as an appropriate disease model of human melanoma, enabling application of melanoma etiological discovery among vertebrates. Additionally, delineating the mechanism of xmrk-driven melanomagenesis and identifying compounds that are able to repressing the xmrk-initiated transcriptional changes that may be applicable to human melanoma treatment.
Materials and Methods
Animal model
A total of 16 first generation backcross (BC1) animals used in this study were supplied by the Xiphophorus Genetic Stock Center (Fig. 1. For contact information see: http://www.xiphophorus.txstate.edu/). Specifically, a X. maculatus Jp 163 A female was artificially inseminated with sperm from a male X. hellerii (Sarabia) to produce F1 interspecies hybrids. F1 interspecies hybrid males were then backcrossed to X. hellerii females to generate the BC1 hybrid progeny. About 25% of the BC1 progeny developed melanoma tumors. At dissection, fish were anesthetized in an ice bath and upon loss of gill movement sacrificed by cranial resection. Organs were either dissected directly into TRI-Reagent (Sigma Inc. St. Louis) placed in a dry ice-ethanol bath if the RNA was isolated at the time of dissection, or dissected into RNAlater (Ambion Inc.) and kept at -80°C for later use. All BC1 fish were maintained and samples taken in accordance with protocol approved by IACUC (IACUC2015107711).
A total of 13 fifth generation melanoma tumor-bearing backcross hybrids (BC5) were produced in an independent series of successive crosses, utilizing F1 hybrids originating from a reciprocal cross in the Biocenter fish facilities (University of Würzburg, Würzburg, Germany). These BC5 progeny were produced from X. maculatus Jp 163 A males mated to X. hellerii (Rio Lancetilla) females. The F1 hybrid females, which developed benign pigment cell precursor lesions, were then successively backcrossed to X. hellerii males to produce BC5. All BC5 fish used in this study were from laboratory stocks maintained in the governmentally certified animal facilities of the Biocenter. All BC5 fish were maintained and samples taken as described above in accordance with the applicable EU and national German legislation governing animal experimentation. Fish were sacrificed by over-anesthetization with MS222. All animal experimentation was done under authorization (AZ 568/300-1870/13) of the Veterinary Office of the District Government of Lower Franconia, Germany, in accordance with the German Animal Protection Law (TierSchG).
RNA isolation and RNA sequencing
RNA from a total of 16 melanoma tumors and skin dissected from BC1 interspecies hybrid progeny, as well as 13 BC5 melanoma tumors was isolated as previously detailed (Lu et al., 2015; D. J. Walter et al., 2014) using TRI-Reagent (Sigma Inc., St. Louis, MO, USA). Briefly, samples were homogenized in TRI-reagent followed by addition of 200 μl/ml chloroform and the samples vigorously shaken and subjected to centrifugation at 12,000 g for 15 min at 4°C. Total RNA was further purified using RNeasy mini RNA isolation kit (Qiagen, Valencia, CA, USA). Residual DNA was eliminated by incubating RNA samples with DNase for DNA digestion at 25deg;C for 15 min. Total RNA concentration was determined using a Qubit 2.0 fluorometer (Life Technologies, Grand Island, NY, USA). RNA quality was verified on an Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA) to confirm that RIN scores were above 8.0 prior to sequencing.
RNA sequencing of BC1 fish was performed upon libraries constructed using the Illumina TruSeq library preparation system (Illumina, Inc., San Diego, CA, USA). RNA libraries were sequenced as 125 bp paired-end fragments using Illumina Hi-Seq 2000 system (Illumina, Inc., San Diego, CA, USA). RNA libraries of BC5 fish were sequenced as 100 bp pair-end fragments using the Illumina Hi-Seq 4000 system (Illumina, Inc., San Diego, CA, USA) by the Beijing Genomics Institute (BGI, Hong Kong, China). Sequencing adaptors were removed from raw sequencing reads. The processed reads were subsequently trimmed and filtered based on quality scores by using a custom filtration algorithm that removed low-scoring sections of each read and preserved the longest remaining fragment (Garcia et al., 2012). For RNA-Seq statistics, see Supplement Table 1.
Gene expression profiling and co-expression analysis in Xiphophorus melanoma model
To fully represent gene expression profiles of the Xiphophorus melanomas, a concatenated reference transcriptome was constructed by combining the Ensembl X. maculatus transcriptome (ftp.ensembl.org/pub/release-80/fasta/xiphophorus_maculatus/cdna/Xiphophorus_maculatus.Xipmac4.4.2.cdna.all.fa.gz), X. hellerii transcriptome and xmrk sequence (GenBank: X16891.2; Schartl et al., 2013; Shen et al., 2016; Wittbrodt et al., 1989). The trimmed and filtered short sequencing reads were aligned to the custom transcriptome using Bowtie2 (Langmead & Salzberg, 2012). Custom Perl scripts were developed to count short sequencing reads with either a perfect alignment to one transcript or a perfect secondary alignment to include all short reads mapped to both X. maculatus and X. hellerii alleles of a given gene (Lu et al., 2015; Shen et al., 2013). Sequencing read counts of each gene were normalized to the corresponding library size. BC1 and BC5 melanomas were ranked on their xmrk expression, respectively. A gene expression correlation coefficient was calculated for each coding gene using Spearman Ranking Correlation Analysis. A gene with correlation coefficient ≥ 0.5 or ≤ -0.5 was classified as xmrk co-expressed gene or xmrk negative correlated gene. Only genes that showed co-expression or negative correlation with xmrk in both BC1 and BC5 melanoma are further analyzed. The workflow of sample collection and data processing is given in Supplement Fig. 1.
Differential gene expression analysis in human melanoma
A total of 473 gene expression profiles from human skin cutaneous melanoma (SKCM) were retrieved from The Cancer Genome Atlas (TCGA, tcga-data.nci.nih.gov) SKCM dataset through TCGA data portal. A custom perl script was used to combine the dataset and append a patient-specific sample name to corresponding expression profiles. To separate tumor samples with high pigmentation pathway gene expression and low pigmentation pathway gene expression, tumor samples were ranked on expression levels of MITF, TYR, TYRP1 and DCT. Tumor samples with these 4 genes expressed in lower than 50% of all samples were classified as low pigmentation pathway activity samples (low-MITF-axis-cohort). Tumor samples with these 4 genes expressed in higher than 50% of all samples were classified as high pigmentation pathway activity samples (high-MITF-axis-cohort). The Low-MITF-axis-cohort consists of 93 tumor samples, and the High-MITF-axis-cohort of 59 tumor samples. Differential gene expression analyses were performed between high and low pigmentation pathway activity samples using edgeR (Log2FC ≥ 1 or Log2FC ≤ -1, FDR ≤ 0.05; (Robinson, McCarthy, & Smyth, 2010). To identify the most diagnostic differentially expressed genes in human SKCM dataset, a receiver operating characteristic (ROC) curve was plotted for each differentially expressed gene using R/Bioconductor package pROC. Only differentially expressed genes with ROC Area Under Curve (AUC) ≥ 0.8 were kept for further analysis.
Gene set enrichment analysis
Gene Ontology (GO) analysis was conducted using R package “GOstats” (Falcon & Gentleman, 2007). All genes with designated GO term in the GO database (GO.db) were used as background genes, and an enrichment p-value of 0.001 was used to determine statistically significant enrichment. Pathway analysis of xmrk co-regulated genes in the Xiphophorus melanoma, and differentially expressed genes in human melanoma were conducted by implementing Ingenuity Pathway Analysis (IPA, Qiagen, Redwood City, California). Pathway enrichment was determined by a p-value < 0.01 (or -log10 p-value>2). Signaling pathways that share genes are connected to form functional network using R package “igraph”. Node size represents number of genes belonging to certain pathways. Width of edges represents number of shared genes of connected pathways. Functional networks were formed using a force-directed layout algorithm.
Quantitative real time PCR
Xiphophorus melanoma derived gene expression was compared to paired normal skin for identification of differential gene expression (Log2FC ≥ 1 or Log2FC ≤ -1, FDR ≤ 0.05, Log2CPM ≥ 1). A total of 2044 genes differentially expressed (1057 genes down-regulated, 987 genes up-regulated) between BC1 tumors and paired normal skin tissue. Ten genes were chosen to be validated using QRT-PCR. QRT-PCR was performed by SYBR Green-based detection with an Applied Biosystems 7500Fast system (Applied Bioscience, Carlsbad, CA, USA). Each reaction was subjected to 40 cycles each at 95 °C for 20 s, 95 °C for 15 s, and 60 °C for 30 s. The 18S gene was selected for normalization of all samples. The mean CT values from triplicate runs were used to calculate relative expression levels between tumors and paired skin samples.
Data Availability
All sequencing files are submitted to Gene Expression Omnibus (GEO).
Supplementary Material
Supplement Figure 1. Flow chart of data analysis For Xiphophorus samples, RNA samples from both normal skin and tumor samples were collected, purified and sequenced. Raw sequencing results were quality checked and filtered using standard pipelines that remove sequencing adaptors, clip off low sequencing base calls at the end of sequencing reads, and filter out sequencing read with low overall quality scores. Sequencing reads were mapped to reference transcriptome sequences and expression values were subsequently quantified. For Xiphophorus melanoma, gene expression correlation coefficient was calculated between xmrk and all coding genes. Genes positively and negatively correlated with xmrk were identified. Human SKCM dataset, in the format of expression profiles, was downloaded from TCGA. Patient samples with high MITF, DCT, TYR and TYRP1 were assigned as high-MITF-axis-cohort, and patient samples with low MITF, DCT, TYR and TYRP1 were assigned as low-MITF-axis-cohort. Differentially expressed genes (DEGs) between high- and low- MITF-axis-cohort were analyzed using R/Bioconductor package edgeR. Functional and signaling pathway enrichment analyses were performed using IPA.
Supplement Figure 2. Histogram of co-expression correlation coefficient Co-expression analyses was performed between each coding gene and xmrk. Genes with expression correlation coefficient ≥ 0.5 or ≤ -0.5 are positively or negatively correlated to xmrk expression.
Supplement Figure 3. Validation of RNA-Seq identified differentially expressed genes Xiphophorus melanoma derived gene expression was compared to paired normal skin for identification of differential gene expression (Log2FC ≥ 1 or Log2FC ≤ -1, FDR ≤ 0.05, Log2CPM ≥ 1). Ten genes were chosen to be validated using qPCR. All of the 10 genes showed consistent up-regulation or down-regulation between qPCR and RNA-Seq.
Supplement Figure 4. Signaling pathways enriched in Xiphophorus and human melanoma, (a) 91 functional pathways are over-represented by xmrk co-expressed and negatively correlated genes. 29 functional pathways are over-represented by low-MITF-axis human cohorts. 12 functional pathways are shared by Xiphophorus and human melanoma. (b) Ninety-one signaling pathways were identified to represent genes that co-express or are negatively correlated with xmrk (red) in Xiphophorus melanoma tumors. Pathways including functional pathways known to associate with xmrk oncogenic effect are identified among the xmrk co-expressed genes, including activation of EGF signaling, ERK/MAPK signaling, integrin signaling, PI3K/Akt signaling, and repression of PTEN signaling, Melanocyte Development and Pigmentation Signaling, and several PI3K related pathways (ILK Signaling, eNOS Signaling, Renin-Angiotensin Signaling, Docosahexaenoic Acid Signaling, CTLA4 Signaling in Cytotoxic T Lymphocytes, Virus Entry via Endocytic Pathways and Leukocyte Extravasation Signaling, inflammation (gray; IL-8 signaling), cell migration (light blue; Axonal guidance signaling, Epithelial adherence junction signaling, ILK signaling, Semaphorin signaling, Superpathway of Inositol phosphate compounds), pigmentation (blue; Eumelanin biosynthesis, Melanocyte development and pigmentation signaling), cancer development and metastasis (pink; Colorectal cancer metastasis signaling, Ovarian cancer signaling, Pancreatic adenocarcinoma signaling, Molecular mechanism of cancer). Pathways of a function group are used to set boundaries of color cloud. (c) Twenty-nine signaling pathways representing genes co-regulated with pigmentation related genes in human cohorts. These pathways are related to inflammation (gray; IL-8 signaling), cell migration and proliferation (light blue; Axonal guidance signaling, Epithelial adherence junction signaling, ILK signaling, Semaphorin signaling, Superpathway of Inositol phosphate compounds, Regulation of the Epithelial-Mesenchymal Transition Pathway), pigmentation (blue; Eumelanin biosynthesis, Melanocyte development and pigmentation signaling), cancer development and metastasis (pink; Colorectal cancer metastasis signaling, Ovarian cancer signaling, Pancreatic adenocarcinoma signaling, Basal Cell Carcinoma Signaling, Bladder Cancer Signaling, Thyroid Cancer Signaling, Molecular mechanism of cancer, Glioblastoma Multiforme Signaling, Protein Kinase A Signaling, TGF-β Signaling, Wnt/β-catenin Signaling), stem cell (green; Human Embryonic Stem Cell Pluripotency, Mouse Embryonic Stem Cell Pluripotency). Twelve signaling pathways (Axonal Guidance Signaling, Colorectal Cancer Metastasis Signaling, Epithelial Adherence Junction Signaling, Eumelanin Biosynthesis, IL8 Signaling, ILK Signaling, Melanocyte Development and Pigmentation Signaling, Molecular Mechanism of Cancer, Ovarian Cancer Signaling, Pancreatic Adenocarcinoma Signaling, Semaphorin Signaling in Neurons, and Superpathway of Inositol Phosphate Compounds) are present in both Xiphophorus and human melanoma. See Supplement Table 4 for pathway names.
Supplement Table 1. RNA-Seq results statistics
Supplement Table 2. xmrk co-expressed and negatively correlated genes in Xiphophorus melanoma
Supplement Table 3. Differentially expressed genes between high-MITF-axis-cohort and low-MITF-axis-cohort melanoma tumors
Supplement Table 4. Signaling pathways enriched by the differentially expressed genes in the Xiphophorus BC1 and BC5 melanoma tumors, and differentially expressed genes between high-MITF-axis-cohort and low-MITF-axis-cohort human melanoma tumors
Significance.
Our observation that the transcriptomic feature of Xiphophorus melanoma represents dedifferentiated, proliferative human melanoma suggests that Xiphophorus melanoma model is an appropriate model system of human melanoma, enabling application of melanoma etiological discovery among vertebrates. Additionally, delineating the mechanism of xmrk-driven melanomagenesis and identifying compounds that are able to repressing the xmrk-initiated transcriptional changes may be applicable to human melanoma treatment.
Acknowledgments
The authors express their gratitude to the employees of the Xiphophorus Genetic Stock Center, Texas State University, and the fish facilities of the Biocenter, University of Würzburg for maintaining the pedigreed fish lines, performing interspecies crosses, and caring for the hybrid animals used in this study. This work was supported by the National Institutes of Health, Division of Comparative Medicine, R24-OD-011120, R24-OD-011199 and R24-OD-018555, subaward number 215420C.
Footnotes
Conflict of Interest Statement: The authors declare that there is no conflict of interest regarding the publication of this article.
References
- Adam D, Maueler W, Schartl M. Transcriptional activation of the melanoma inducing Xmrk oncogene in Xiphophorus. Oncogene. 1991;6(1):73–80. [PubMed] [Google Scholar]
- Argaw-Denboba A, Balestrieri E, Serafino A, Cipriani C, Bucci I, Sorrentino R, et al. Matteucci C. HERV-K activation is strictly required to sustain CD133+ melanoma cells with stemness features. J Exp Clin Cancer Res. 2017;36(1):20. doi: 10.1186/s13046-016-0485-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge G, Pettersen S, Grotterod I, Bettum IJ, Boye K, Maelandsmo GM. Osteopontin--an important downstream effector of S100A4-mediated invasion and metastasis. Int J Cancer. 2011;129(4):780–790. doi: 10.1002/ijc.25735. [DOI] [PubMed] [Google Scholar]
- Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, et al. Garraway LA. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485(7399):502–506. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt M, Novak D, Assenov Y, Orouji E, Knappe N, Weina K, et al. Utikal J. Melanoma-Derived iPCCs Show Differential Tumorigenicity and Therapy Response. Stem Cell Reports. 2017;8(5):1379–1391. doi: 10.1016/j.stemcr.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettum IJ, Gorad SS, Barkovskaya A, Pettersen S, Moestue SA, Vasiliauskaite K, et al. Prasmickaite L. Metabolic reprogramming supports the invasive phenotype in malignant melanoma. Cancer Lett. 2015;366(1):71–83. doi: 10.1016/j.canlet.2015.06.006. [DOI] [PubMed] [Google Scholar]
- Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, et al. Weissman IL. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466(7302):133–137. doi: 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosserhoff AK, Ellmann L, Kuphal S. Melanoblasts in culture as an in vitro system to determine molecular changes in melanoma. Exp Dermatol. 2011;20(5):435–440. doi: 10.1111/j.1600-0625.2011.01271.x. [DOI] [PubMed] [Google Scholar]
- Bracalente C, Salguero N, Notcovich C, Muller CB, da Motta LL, Klamt F, et al. Duran H. Reprogramming human A375 amelanotic melanoma cells by catalase overexpression: Reversion or promotion of malignancy by inducing melanogenesis or metastasis. Oncotarget. 2016;7(27):41142–41153. doi: 10.18632/oncotarget.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira S, Goodall J, Aksan I, La Rocca SA, Galibert MD, Denat L, et al. Goding CR. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature. 2005;433(7027):764–769. doi: 10.1038/nature03269. [DOI] [PubMed] [Google Scholar]
- Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KS, et al. Goding CR. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006;20(24):3426–3439. doi: 10.1101/gad.406406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Group BS. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheli Y, Giuliano S, Botton T, Rocchi S, Hofman V, Hofman P, et al. Ballotti R. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene. 2011;30(20):2307–2318. doi: 10.1038/onc.2010.598. [DOI] [PubMed] [Google Scholar]
- Cheli Y, Giuliano S, Fenouille N, Allegra M, Hofman V, Hofman P, et al. Ballotti R. Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene. 2012;31(19):2461–2470. doi: 10.1038/onc.2011.425. [DOI] [PubMed] [Google Scholar]
- Cheli Y, Ohanna M, Ballotti R, Bertolotto C. Fifteen-year quest for microphthalmia-associated transcription factor target genes. Pigment Cell Melanoma Res. 2010;23(1):27–40. doi: 10.1111/j.1755-148X.2009.00653.x. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Delfgaauw J, Duschl J, Wellbrock C, Froschauer C, Schartl M, Altschmied J. MITF-M plays an essential role in transcriptional activation and signal transduction in Xiphophorus melanoma. Gene. 2003;320:117–126. doi: 10.1016/s0378-1119(03)00817-5. [DOI] [PubMed] [Google Scholar]
- Dotto GP, Moellmann G, Ghosh S, Edwards M, Halaban R. Transformation of murine melanocytes by basic fibroblast growth factor cDNA and oncogenes and selective suppression of the transformed phenotype in a reconstituted cutaneous environment. J Cell Biol. 1989;109(6 Pt 1):3115–3128. doi: 10.1083/jcb.109.6.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, et al. Kefford RF. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012;379(9829):1893–1901. doi: 10.1016/S0140-6736(12)60398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23(2):257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Weber J. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, et al. Chapman PB. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363(9):809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest. 2010;120(1):41–50. doi: 10.1172/JCI41004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia TI, Shen Y, Catchen J, Amores A, Schartl M, Postlethwait J, Walter RB. Effects of short read quality and quantity on a de novo vertebrate transcriptome assembly. Comp Biochem Physiol C Toxicol Pharmacol. 2012;155(1):95–101. doi: 10.1016/j.cbpc.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, et al. Sellers WR. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436(7047):117–122. doi: 10.1038/nature03664. [DOI] [PubMed] [Google Scholar]
- Ge X, Fu YM, Meadows GG. U0126, a mitogen-activated protein kinase kinase inhibitor, inhibits the invasion of human A375 melanoma cells. Cancer Lett. 2002;179(2):133–140. doi: 10.1016/s0304-3835(02)00004-6. [DOI] [PubMed] [Google Scholar]
- Geissinger E, Weisser C, Fischer P, Schartl M, Wellbrock C. Autocrine stimulation by osteopontin contributes to antiapoptotic signalling of melanocytes in dermal collagen. Cancer Res. 2002;62(16):4820–4828. [PubMed] [Google Scholar]
- Goodall J, Carreira S, Denat L, Kobi D, Davidson I, Nuciforo P, et al. Goding CR. Brn-2 represses microphthalmia-associated transcription factor expression and marks a distinct subpopulation of microphthalmia-associated transcription factor-negative melanoma cells. Cancer Res. 2008;68(19):7788–7794. doi: 10.1158/0008-5472.CAN-08-1053. [DOI] [PubMed] [Google Scholar]
- Gopal YN, Deng W, Woodman SE, Komurov K, Ram P, Smith PD, Davies MA. Basal and treatment-induced activation of AKT mediates resistance to cell death by AZD6244 (ARRY-142886) in Braf-mutant human cutaneous melanoma cells. Cancer Res. 2010;70(21):8736–8747. doi: 10.1158/0008-5472.CAN-10-0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon M. The Genetics of a Viviparous Top-Minnow Platypoecilus; the Inheritance of Two Kinds of Melanophores. Genetics. 1927;12(3):253–283. doi: 10.1093/genetics/12.3.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Chapman PB. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–365. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- Häussler G. Über Melanombildungen bei Bastarden von Xiphophorus Helleri und Platypoecilus Maculatus var. Rubra. Journal of Molecular Medicine. 1928;7 [Google Scholar]
- Hoek KS, Goding CR. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010;23(6):746–759. doi: 10.1111/j.1755-148X.2010.00757.x. [DOI] [PubMed] [Google Scholar]
- Jakob JA, Bassett RL, Jr, Ng CS, Curry JL, Joseph RW, Alvarado GC, et al. Davies MA. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118(16):4014–4023. doi: 10.1002/cncr.26724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazianis S, Gutbrod H, Nairn RS, McEntire BB, Della Coletta L, Walter RB, et al. Morizot DC. Localization of a CDKN2 gene in linkage group V of Xiphophorus fishes defines it as a candidate for the DIFF tumor suppressor. Genes Chromosomes Cancer. 1998;22(3):210–220. [PubMed] [Google Scholar]
- Kazianis S, Morizot DC, Coletta LD, Johnston DA, Woolcock B, Vielkind JR, Nairn RS. Comparative structure and characterization of a CDKN2 gene in a Xiphophorus fish melanoma model. Oncogene. 1999;18(36):5088–5099. doi: 10.1038/sj.onc.1202884. [DOI] [PubMed] [Google Scholar]
- Kosswig C. Über Bastarde der Teleostier Platypoecilus und Xiphophorus. Z Indukt Abstamm Vererbungsl. 1928;44 [Google Scholar]
- Landsberg J, Kohlmeyer J, Renn M, Bald T, Rogava M, Cron M, et al. Tuting T. Melanomas resist T-cell therapy through inflammation-induced reversible dedifferentiation. Nature. 2012;490(7420):412–416. doi: 10.1038/nature11538. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Vasioukhin V. Cell polarity and cancer--cell and tissue polarity as a non-canonical tumor suppressor. J Cell Sci. 2008;121(Pt 8):1141–1150. doi: 10.1242/jcs.016634. [DOI] [PubMed] [Google Scholar]
- Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, et al. Rosen N. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22(5):668–682. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loercher AE, Tank EM, Delston RB, Harbour JW. MITF links differentiation with cell cycle arrest in melanocytes by transcriptional activation of INK4A. J Cell Biol. 2005;168(1):35–40. doi: 10.1083/jcb.200410115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long GV, Menzies AM, Nagrial AM, Haydu LE, Hamilton AL, Mann GJ, et al. Kefford RF. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29(10):1239–1246. doi: 10.1200/JCO.2010.32.4327. [DOI] [PubMed] [Google Scholar]
- Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, et al. Schadendorf D. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012;13(11):1087–1095. doi: 10.1016/S1470-2045(12)70431-X. [DOI] [PubMed] [Google Scholar]
- Lu Y, Boswell M, Boswell W, Kneitz S, Hausmann M, Klotz B, et al. Walter R. Molecular genetic analysis of the melanoma regulatory locus in Xiphophorus interspecies hybrids. Mol Carcinog. 2017;56(8):1935–1944. doi: 10.1002/mc.22651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Bowswell M, Bowswell W, Yang K, Schartl M, Walter RB. Molecular genetic response of Xiphophorus maculatus-X. couchianus interspecies hybrid skin to UVB exposure. Comp Biochem Physiol C Toxicol Pharmacol. 2015;178:86–92. doi: 10.1016/j.cbpc.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madjd Z, Erfani E, Gheytanchi E, Moradi-Lakeh M, Shariftabrizi A, Asadi-Lari M. Expression of CD133 cancer stem cell marker in melanoma: a systematic review and meta-analysis. Int J Biol Markers. 2016;31(2):e118–125. doi: 10.5301/jbm.5000209. [DOI] [PubMed] [Google Scholar]
- Martello G, Smith A. The nature of embryonic stem cells. Annu Rev Cell Dev Biol. 2014;30:647–675. doi: 10.1146/annurev-cellbio-100913-013116. [DOI] [PubMed] [Google Scholar]
- Meierjohann S, Schartl M, Volff JN. Genetic, biochemical and evolutionary facets of Xmrk-induced melanoma formation in the fish Xiphophorus. Comp Biochem Physiol C Toxicol Pharmacol. 2004;138(3):281–289. doi: 10.1016/j.cca.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Moore R, Champeval D, Denat L, Tan SS, Faure F, Julien-Grille S, Larue L. Involvement of cadherins 7 and 20 in mouse embryogenesis and melanocyte transformation. Oncogene. 2004;23(40):6726–6735. doi: 10.1038/sj.onc.1207675. [DOI] [PubMed] [Google Scholar]
- Morcinek JC, Weisser C, Geissinger E, Schartl M, Wellbrock C. Activation of STAT5 triggers proliferation and contributes to anti-apoptotic signalling mediated by the oncogenic Xmrk kinase. Oncogene. 2002;21(11):1668–1678. doi: 10.1038/sj.onc.1205148. [DOI] [PubMed] [Google Scholar]
- Patton EE, Mathers ME, Schartl M. Generating and analyzing fish models of melanoma. Methods Cell Biol. 2011;105:339–366. doi: 10.1016/B978-0-12-381320-6.00014-X. [DOI] [PubMed] [Google Scholar]
- Perotti V, Baldassari P, Molla A, Vegetti C, Bersani I, Maurichi A, et al. Mortarini R. NFATc2 is an intrinsic regulator of melanoma dedifferentiation. Oncogene. 2016;35(22):2862–2872. doi: 10.1038/onc.2015.355. [DOI] [PubMed] [Google Scholar]
- Pinner S, Jordan P, Sharrock K, Bazley L, Collinson L, Marais R, et al. Sahai E. Intravital imaging reveals transient changes in pigment production and Brn2 expression during metastatic melanoma dissemination. Cancer Res. 2009;69(20):7969–7977. doi: 10.1158/0008-5472.CAN-09-0781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed KB, Brewer JD, Lohse CM, Bringe KE, Pruitt CN, Gibson LE. Increasing incidence of melanoma among young adults: an epidemiological study in Olmsted County, Minnesota. Mayo Clin Proc. 2012;87(4):328–334. doi: 10.1016/j.mayocp.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesenberg S, Groetchen A, Siddaway R, Bald T, Reinhardt J, Smorra D, et al. Holzel M. MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat Commun. 2015;6:8755. doi: 10.1038/ncomms9755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigel DS. Cutaneous ultraviolet exposure and its relationship to the development of skin cancer. J Am Acad Dermatol. 2008;58(5 Suppl 2):S129–132. doi: 10.1016/j.jaad.2007.04.034. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer C, Lu X. Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ. 2011;18(9):1470–1477. doi: 10.1038/cdd.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez-Ayala M, Montenegro MF, Sanchez-Del-Campo L, Fernandez-Perez MP, Chazarra S, Freter R, et al. Rodriguez-Lopez JN. Directed phenotype switching as an effective antimelanoma strategy. Cancer Cell. 2013;24(1):105–119. doi: 10.1016/j.ccr.2013.05.009. [DOI] [PubMed] [Google Scholar]
- Schartl M, Kneitz S, Wilde B, Wagner T, Henkel CV, Spaink HP, Meierjohann S. Conserved expression signatures between medaka and human pigment cell tumors. PLoS One. 2012;7(5):e37880. doi: 10.1371/journal.pone.0037880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schartl M, Shen Y, Maurus K, Walter R, Tomlinson C, Wilson RK, et al. Warren WC. Whole Body Melanoma Transcriptome Response in Medaka. PLoS One. 2015;10(12):e0143057. doi: 10.1371/journal.pone.0143057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schartl M, Walter RB, Shen Y, Garcia T, Catchen J, Amores A, et al. Warren WC. The genome of the platyfish, Xiphophorus maculatus, provides insights into evolutionary adaptation and several complex traits. Nat Genet. 2013;45(5):567–572. doi: 10.1038/ng.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schartl M, Wilde B, Laisney JA, Taniguchi Y, Takeda S, Meierjohann S. A mutated EGFR is sufficient to induce malignant melanoma with genetic background-dependent histopathologies. J Invest Dermatol. 2010;130(1):249–258. doi: 10.1038/jid.2009.213. [DOI] [PubMed] [Google Scholar]
- Schiaffino MV. Signaling pathways in melanosome biogenesis and pathology. Int J Biochem Cell Biol. 2010;42(7):1094–1104. doi: 10.1016/j.biocel.2010.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Hansen B, Klingelhofer J, Grum-Schwensen B, Christensen A, Andresen S, Kruse C, et al. Grigorian M. Functional significance of metastasis-inducing S100A4(Mts1) in tumor-stroma interplay. J Biol Chem. 2004;279(23):24498–24504. doi: 10.1074/jbc.M400441200. [DOI] [PubMed] [Google Scholar]
- Serafino A, Sinibaldi-Vallebona P, Lazzarino G, Tavazzi B, Rasi G, Pierimarchi P, et al. Garaci E. Differentiation of human melanoma cells induced by cyanidin-3-O-beta-glucopyranoside. FASEB J. 2004;18(15):1940–1942. doi: 10.1096/fj.04-1925fje. [DOI] [PubMed] [Google Scholar]
- Shen Y, Chalopin D, Garcia T, Boswell M, Boswell W, Shiryev SA, et al. Walter RB. X. couchianus and X. hellerii genome models provide genomic variation insight among Xiphophorus species. BMC Genomics. 2016;17:37. doi: 10.1186/s12864-015-2361-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Garcia T, Pabuwal V, Boswell M, Pasquali A, Beldorth I, et al. Walter RB. Alternative strategies for development of a reference transcriptome for quantification of allele specific expression in organisms having sparse genomic resources. Comp Biochem Physiol Part D Genomics Proteomics. 2013;8(1):11–16. doi: 10.1016/j.cbd.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- Simoes-Costa M, Bronner ME. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142(2):242–257. doi: 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalley K, Eisen T. The involvement of p38 mitogen-activated protein kinase in the alpha-melanocyte stimulating hormone (alpha-MSH)-induced melanogenic and anti-proliferative effects in B16 murine melanoma cells. FEBS Lett. 2000;476(3):198–202. doi: 10.1016/s0014-5793(00)01726-9. [DOI] [PubMed] [Google Scholar]
- Smalley KS. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? Int J Cancer. 2003;104(5):527–532. doi: 10.1002/ijc.10978. [DOI] [PubMed] [Google Scholar]
- Smalley KS, Eisen TG. Differentiation of human melanoma cells through p38 MAP kinase is associated with decreased retinoblastoma protein phosphorylation and cell cycle arrest. Melanoma Res. 2002;12(3):187–192. doi: 10.1097/00008390-200206000-00001. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Sipp D, Enomoto H. Tissue interactions in neural crest cell development and disease. Science. 2013;341(6148):860–863. doi: 10.1126/science.1230717. [DOI] [PubMed] [Google Scholar]
- Walter DJ, Boswell M, Volk de Garcia SM, Walter SM, Breitenfeldt EW, Boswell W, Walter RB. Characterization and differential expression of CPD and 6-4 DNA photolyases in Xiphophorus species and interspecies hybrids. Comp Biochem Physiol C Toxicol Pharmacol. 2014;163:77–85. doi: 10.1016/j.cbpc.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter RB, Kazianis S. Xiphophorus interspecies hybrids as genetic models of induced neoplasia. ILAR J. 2001;42(4):299–321. doi: 10.1093/ilar.42.4.299. [DOI] [PubMed] [Google Scholar]
- Wang J, Huang SK, Marzese DM, Hsu SC, Kawas NP, Chong KK, et al. Hoon DS. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J Invest Dermatol. 2015;135(2):532–541. doi: 10.1038/jid.2014.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WE H. Genetics: Animal Tumors. In: Becker FF, editor. Cancer: a comprehensive treatise. 2. New York: Plenum Press; 1982. pp. 47–70. [Google Scholar]
- Wellbrock C, Fischer P, Schartl M. Receptor tyrosine kinase Xmrk mediates proliferation in Xiphophorus melanoma cells. Int J Cancer. 1998;76(3):437–442. doi: 10.1002/(sici)1097-0215(19980504)76:3<437::aid-ijc24>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Fischer P, Schartl M. PI3-kinase is involved in mitogenic signaling by the oncogenic receptor tyrosine kinase Xiphophorus melanoma receptor kinase in fish melanoma. Exp Cell Res. 1999;251(2):340–349. doi: 10.1006/excr.1999.4580. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Schartl M. Multiple binding sites in the growth factor receptor Xmrk mediate binding to p59fyn, GRB2 and Shc. Eur J Biochem. 1999;260(1):275–283. doi: 10.1046/j.1432-1327.1999.00180.x. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Schartl M. Activation of phosphatidylinositol 3-kinase by a complex of p59fyn and the receptor tyrosine kinase Xmrk is involved in malignant transformation of pigment cells. Eur J Biochem. 2000;267(12):3513–3522. doi: 10.1046/j.1432-1327.2000.01378.x. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Weisser C, Geissinger E, Troppmair J, Schartl M. Activation of p59(Fyn) leads to melanocyte dedifferentiation by influencing MKP-1-regulated mitogen-activated protein kinase signaling. J Biol Chem. 2002;277(8):6443–6454. doi: 10.1074/jbc.M110684200. [DOI] [PubMed] [Google Scholar]
- Wilson RE, Dooley TP, Hart IR. Induction of tumorigenicity and lack of in vitro growth requirement for 12-O-tetradecanoylphorbol-13-acetate by transfection of murine melanocytes with v-Ha-ras. Cancer Res. 1989;49(3):711–716. [PubMed] [Google Scholar]
- Wittbrodt J, Adam D, Malitschek B, Maueler W, Raulf F, Telling A, et al. Schartl M. Novel putative receptor tyrosine kinase encoded by the melanoma-inducing Tu locus in Xiphophorus. Nature. 1989;341(6241):415–421. doi: 10.1038/341415a0. [DOI] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Ribas A. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375(9):819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, He X, Wang Y, Shi F, Wu D, Pan M, et al. Dou J. Decrease of ZEB1 expression inhibits the B16F10 cancer stem-like properties. Biosci Trends. 2015;9(5):325–334. doi: 10.5582/bst.2015.01106. [DOI] [PubMed] [Google Scholar]
- Zimmerer RM, Matthiesen P, Kreher F, Kampmann A, Spalthoff S, Jehn P, et al. Tavassol F. Putative CD133+ melanoma cancer stem cells induce initial angiogenesis in vivo. Microvasc Res. 2016;104:46–54. doi: 10.1016/j.mvr.2015.12.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement Figure 1. Flow chart of data analysis For Xiphophorus samples, RNA samples from both normal skin and tumor samples were collected, purified and sequenced. Raw sequencing results were quality checked and filtered using standard pipelines that remove sequencing adaptors, clip off low sequencing base calls at the end of sequencing reads, and filter out sequencing read with low overall quality scores. Sequencing reads were mapped to reference transcriptome sequences and expression values were subsequently quantified. For Xiphophorus melanoma, gene expression correlation coefficient was calculated between xmrk and all coding genes. Genes positively and negatively correlated with xmrk were identified. Human SKCM dataset, in the format of expression profiles, was downloaded from TCGA. Patient samples with high MITF, DCT, TYR and TYRP1 were assigned as high-MITF-axis-cohort, and patient samples with low MITF, DCT, TYR and TYRP1 were assigned as low-MITF-axis-cohort. Differentially expressed genes (DEGs) between high- and low- MITF-axis-cohort were analyzed using R/Bioconductor package edgeR. Functional and signaling pathway enrichment analyses were performed using IPA.
Supplement Figure 2. Histogram of co-expression correlation coefficient Co-expression analyses was performed between each coding gene and xmrk. Genes with expression correlation coefficient ≥ 0.5 or ≤ -0.5 are positively or negatively correlated to xmrk expression.
Supplement Figure 3. Validation of RNA-Seq identified differentially expressed genes Xiphophorus melanoma derived gene expression was compared to paired normal skin for identification of differential gene expression (Log2FC ≥ 1 or Log2FC ≤ -1, FDR ≤ 0.05, Log2CPM ≥ 1). Ten genes were chosen to be validated using qPCR. All of the 10 genes showed consistent up-regulation or down-regulation between qPCR and RNA-Seq.
Supplement Figure 4. Signaling pathways enriched in Xiphophorus and human melanoma, (a) 91 functional pathways are over-represented by xmrk co-expressed and negatively correlated genes. 29 functional pathways are over-represented by low-MITF-axis human cohorts. 12 functional pathways are shared by Xiphophorus and human melanoma. (b) Ninety-one signaling pathways were identified to represent genes that co-express or are negatively correlated with xmrk (red) in Xiphophorus melanoma tumors. Pathways including functional pathways known to associate with xmrk oncogenic effect are identified among the xmrk co-expressed genes, including activation of EGF signaling, ERK/MAPK signaling, integrin signaling, PI3K/Akt signaling, and repression of PTEN signaling, Melanocyte Development and Pigmentation Signaling, and several PI3K related pathways (ILK Signaling, eNOS Signaling, Renin-Angiotensin Signaling, Docosahexaenoic Acid Signaling, CTLA4 Signaling in Cytotoxic T Lymphocytes, Virus Entry via Endocytic Pathways and Leukocyte Extravasation Signaling, inflammation (gray; IL-8 signaling), cell migration (light blue; Axonal guidance signaling, Epithelial adherence junction signaling, ILK signaling, Semaphorin signaling, Superpathway of Inositol phosphate compounds), pigmentation (blue; Eumelanin biosynthesis, Melanocyte development and pigmentation signaling), cancer development and metastasis (pink; Colorectal cancer metastasis signaling, Ovarian cancer signaling, Pancreatic adenocarcinoma signaling, Molecular mechanism of cancer). Pathways of a function group are used to set boundaries of color cloud. (c) Twenty-nine signaling pathways representing genes co-regulated with pigmentation related genes in human cohorts. These pathways are related to inflammation (gray; IL-8 signaling), cell migration and proliferation (light blue; Axonal guidance signaling, Epithelial adherence junction signaling, ILK signaling, Semaphorin signaling, Superpathway of Inositol phosphate compounds, Regulation of the Epithelial-Mesenchymal Transition Pathway), pigmentation (blue; Eumelanin biosynthesis, Melanocyte development and pigmentation signaling), cancer development and metastasis (pink; Colorectal cancer metastasis signaling, Ovarian cancer signaling, Pancreatic adenocarcinoma signaling, Basal Cell Carcinoma Signaling, Bladder Cancer Signaling, Thyroid Cancer Signaling, Molecular mechanism of cancer, Glioblastoma Multiforme Signaling, Protein Kinase A Signaling, TGF-β Signaling, Wnt/β-catenin Signaling), stem cell (green; Human Embryonic Stem Cell Pluripotency, Mouse Embryonic Stem Cell Pluripotency). Twelve signaling pathways (Axonal Guidance Signaling, Colorectal Cancer Metastasis Signaling, Epithelial Adherence Junction Signaling, Eumelanin Biosynthesis, IL8 Signaling, ILK Signaling, Melanocyte Development and Pigmentation Signaling, Molecular Mechanism of Cancer, Ovarian Cancer Signaling, Pancreatic Adenocarcinoma Signaling, Semaphorin Signaling in Neurons, and Superpathway of Inositol Phosphate Compounds) are present in both Xiphophorus and human melanoma. See Supplement Table 4 for pathway names.
Supplement Table 1. RNA-Seq results statistics
Supplement Table 2. xmrk co-expressed and negatively correlated genes in Xiphophorus melanoma
Supplement Table 3. Differentially expressed genes between high-MITF-axis-cohort and low-MITF-axis-cohort melanoma tumors
Supplement Table 4. Signaling pathways enriched by the differentially expressed genes in the Xiphophorus BC1 and BC5 melanoma tumors, and differentially expressed genes between high-MITF-axis-cohort and low-MITF-axis-cohort human melanoma tumors
Data Availability Statement
All sequencing files are submitted to Gene Expression Omnibus (GEO).