

Summary
Studies demonstrate that GRM, expressed by >60% of human melanomas, may be a therapeutic target. We performed a phase II trial of 100 mg po bid of riluzole, an inhibitor of GRM1 signaling, in patients with advanced melanoma with the primary endpoint of response rate. Thirteen patients with GRM1-positive tumors were enrolled. No objective responses were observed and accrual was stopped. Stable disease was noted in 6 (46%) patients, with 1 patient on study for 42 weeks. Riluzole was well-tolerated, with fatigue (62%) as the most common adverse event. MAPK and PI3K/AKT down regulation was noted in 33% of paired tumor biopsies. Hypothesis generating correlative studies suggested that down regulation of angiogenic markers and increased leuckocytes at the active edge of tumor correlate with clinical benefit. Pharmacokinetic analysis showed inter-patient variability consistent with prior riluzole studies. Future investigations should interrogate mechanisms of biologic activity and advance development of agents with improved bioavailability.
Keywords: riluzole, glutamate, melanoma, angiogenic factors and receptors, clinical trials, skin cancers
Background
Glutamate metabotropic receptors (GRMs) are members of the seven-transmembrane G-protein-coupled receptor (GPCR) (Pin and Duvoisin, 1995; Saugstad et al., 1996; Wang et al., 2004; Waters et al., 2004) family that bind the excitatory neurotransmitter glutamate. Divided into three groups based on sequence homology, agonist selectivity, and effector coupling, GRM signaling has been implicated in the pathogenesis of neuronal and non-neuronal diseases, particularly cancer. Indeed, high expression of GRMs 1, 3, 4, 5 and 6 has been reported in the glutamate-rich tissue environments of central nervous system tumors (Arcella et al., 2005; Aronica et al., 2003; Brocke et al., 2010; D'Onofrio et al., 2003) and in breast, prostate and melanoma tumors (Koochekpour et al., 2012; Namkoong et al., 2007a; Speyer et al., 2011). Glutamate signaling may be especially important in tumor proliferation, as the expression of glutamate receptors may influence tumor growth in, or metastases to, glutamate-rich organs and tissues such as liver and brain (Stepulak et al., 2009). GRM1 stimulation results in activation of the PLCγ-IP3-DAG cascade, which initiates extracellular signal-related kinase (ERK) 1/2 mitogen-activated protein kinase (MAPK) pathway activation, as well as PI3K-AKT-mTOR signaling. These pathways are known to be aberrantly activated in multiple cancers, particularly melanoma (Bennett, 2008; Fecher et al., 2007), and are modulated by group I GRMs in transgenic mouse melanoma models with GRM1 or GRM5 expression (Choi et al., 2011; Ohtani et al., 2008; Pollock et al., 2003b).
Our group previously reported that ectopic expression of GRM1 in melanocytes leads to melanocyte transformation in vitro and tumorigenesis in vivo. In our comprehensive examination of 25 human melanoma cell lines and 175 melanoma biopsies including primary and metastatic lesions, 80% of cell lines and 60% of tumor biopsies express GRM1 while normal melanocytes from the same patients do not express GRM1 (Namkoong et al., 2007b). Stimulation of the receptor by glutamate or agonist results in activation of the MAPK and PI3K/AKT pathways (Namkoong et al., 2007b; Pollock et al., 2003a). With these results suggesting that GRM1 is a potential target for the treatment of patients with melanoma, the clinically available agent riluzole, an inhibitor of GRM 1 signaling used for the treatment of amyotrophic lateral sclerosis, was selected for further preclinical studies (Miller, 2003; Miller et al., 2003; Mitsumoto, 1997; Noh et al., 2000).
Treatment of human melanoma cell lines and xenografts with riluzole or other GRM1 signal transduction inhibitors results in reduced cell proliferation in vitro and decreased tumor growth in vivo independent of N-RAS/B-RAF genotypes (Le et al., 2010; Namkoong et al., 2007a; Wangari-Talbot et al., 2012). Based on these findings, we conducted a 12-patient, pilot phase 0 trial of riluzole in patients with stage III and IV melanoma who were to undergo curative or palliative resection (Yip et al., 2009). Approximately one-third of patients treated with 100 mg bid of oral riluzole for 14 days showed metabolic reponses to treatment on PET-CT and evidence of suppression of pERK and pAKT signaling in the corresponding paired tumor samples. Our pilot study results led to the design of this phase II trial to determine the antitumor activity of riluzole in patients with advanced melanoma.
Results
Thirteen patients, seven male and six females, were enrolled. The majority of patients had an ECOG performance status of 1. Baseline patient characteristics including the prior treatments are summarized in Supplementary Table S1. No patients had prior treatment with inhibitors of BRAF or MEK, or checkpoint inhibitors.
Treatment delivery and discontinuation results are summarized in Supplementary Table S2. Six patients received two four-week cycles, five received between 2 and 4 cycles, and one received >4 cycles of riluzole. One patient completed only one cycle due to early disease progression. The median number of days on therapy was 54. Dose interruptions and reductions were required in two patients due to dizziness. One patient, per protocol, was permitted to receive concomitant radiation to a painful lesion present at baseline and considered not to be indicative of progression. The reasons for treatment discontinuation were disease progression in 12/13 patients, and toxicity (grade 3 dizziness) in 1/13 patients.
Adverse events noted in patients treated on this trial are summarized in Supplementary Table S3. Overall, riluzole was well tolerated. No grade 4/5 toxicities related to therapy were reported. Grade 3 toxicities possibly, probably or definitely related to treatment included dehydration in one patient (8%) and dizziness in five patients (38%). Two patients with dizziness required dose reduction for management of symptoms. One of these patients required discontinuation of protocol therapy due to persistent dizziness even with multiple dose reductions. The second patient was dose reduced to 100 mg daily and subsequently re-escalated successfully after resolution of symptoms. One patient died due to progressive disease within the first cycle; no other deaths were reported on trial or within 30 days of the last dose of riluzole.
No objective responses per RECIST criteria were observed. At the first restaging scan scheduled after six weeks of therapy, 6 of 13 patients (46%) showed stable disease as the best response. The response results along with data of correlative studies are summarized in Table 1. One patient with locally advanced disease experienced softening, flattening, and shrinkage of several locally advanced lesions in the setting of obvious growth of other lesions. A second patient developed spontaneous hemorrhage in multiple evident subcutaneous masses shortly after initiating treatment that correlated with subsequent shrinkage, but did not meet RECIST criteria for response. The duration of disease control ranged from 66 days to 300 days with a median of 132 days. One patient with a N-RAS mutation achieved prolonged disease stabilization and clinical benefit, remaining on study for 10.7 cycles (300 days). Each of the 13 patients on trial eventually developed progression, with median progression-free survival (PFS) of 63 days (95% CI = 49-77 days). The median overall survival (OS) observed was 342 days (95% CI = 148-720 days). Supplementary Figure S1 shows the Kaplan-Meier plots of PFS and OS, indicating a six-month PFS rate of 15.38% (95% CI = 2.5-38.8%) and the one-year OS rate of 46.15% (95% CI = 19.2-69.6%).
Table 1. Summary of tumor characteristics of enrolled patients.
| Patient # | Pre- and post-Tx tumor | GRM1 expression status (Pre-Tx) | Mutation B-Raf/N-Ras | RECIST at first re-evaluation | Suppression of MAPK | Suppression of PI3K/AKT | Suppression of angiogenesis | Change in CD45+ cells intra-tumoral | Change in CD45+ cells tumor/Stroma interface |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Yes | Positive | V600E/WT | SD | Yes | Yes | Yes | No change | Increase |
| 2 | Pre only | Positive | V600E/WT | SD | -- | -- | -- | -- | -- |
| 3 | Yes | Positive | V600E/WT | PD | Yes | No | No | -- | -- |
| 4 | Pre only | Positive | V600E/WT | PD | -- | -- | -- | -- | -- |
| 5 | Yes | Positive | WT/WT | SD | Yes | Yes | Yes | -- | -- |
| 6 | Yes | Positive | WT/WT | PD | No | No | No | Decrease | No change |
| 7 | Post only | Positive | WT/WT | PD | -- | -- | -- | -- | -- |
| 8 | Yes | Positive | WT/WT | PD | Yes | No | No | Decrease | Decrease |
| 9 | Yes | Positive | V600E/WT | PD | No | No | No | -- | -- |
| 10 | Post only | Positive | WT/Q61R | SD | -- | -- | -- | -- | -- |
| 11 | Yes | Positive | WT/WT | SD | Yes | Yes | Yes | No change | Increase |
| 12 | Yes | Positive | WT/WT | PD | Yes | No | Yes | -- | -- |
| 13 | Pre only | Positive | WT/WT | SD | -- | -- | -- | -- | -- |
-- Not performed due to inadequate tissue, WT = wild-type, SD = stable disease, PD = progressive disease, Tx = treatment
Imaging for tumor assessments was performed after six weeks. Response was assessed using Response Evaluation Criteria in Solid Tumors (RECIST) (Therasse et al., 2000). Tumor samples were obtained before and after treatment from 8 of the 13 enrolled patients. The procedures to determine B-RAF and N-RAS genotypes were detailed earlier (Yip et al., 2009).
Steady state plasma riluzole trough levels ranged from 8.16 - 294.4 ng/mL. Supplementary Table S4 shows the plasma riluzole trough levels on day 8 and day 10. There was a large inter-patient variation in trough plasma concentrations and the mean trough levels of riluzole were 86.99 ± 80.04 ng/mL (day 8) and 119.08 ± 108.03 ng/mL (day 10). The inter-individual variability in plasma riluzole levels in melanoma patients was similar to that of patients with ALS (Groeneveld et al., 2008; Groeneveld et al., 2001). The varied trough levels between patients is likely attributable to differences in absorption and metabolism of riluzole.
All 13 tumors (100%) at baseline were positive for GRM1 expression by IHC staining, similar to results seen in our prior phase 0 trial (Yip et al., 2009). As expected, GRM1 staining was localized to the membrane on melanoma tumor cells (Supplementary Figure S2).
Western blot analyses for the activated (phosphorylated) forms of ERK and AKT and for CD31 were performed with pre- and post-treatment tumor samples available from 8 patients (Figure 1). A decrease of 50% in protein was considered meaningful. Decreases in pERK levels were seen in 5 of 8 patients (Figure 1A and 1B). Decreased pAKT, however, was only seen in post-treatment tumor samples for patients 1, 5 and 11 (Figure 1A and 1C). Each of these three patients with decreased pERK and decreased PI3K/AKT activity achieved disease stabilization at the six-week restaging point (Table 1), with two patients showing no tumor growth and one showing a mixed pattern of growth and shrinkage of target lesions. Patients without this pattern had progressive disease. The association of MAPK and PI3K/AKT down regulation with stable disease at first restaging was statistically significant (p = 0.018; Fisher exact test).
Figure 1.
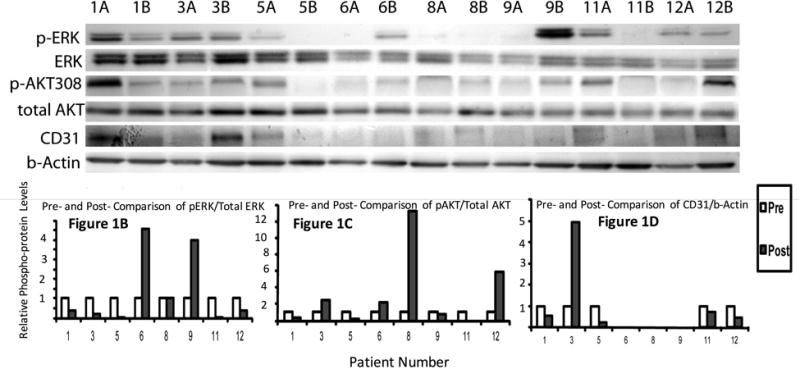
Western blots showing signaling response in paired tumor samples for ERK, pERK, pAKT308, total AKT and CD31. Tumor tissue protein lysates were prepared as described (Yip et al., 2009) and 10 μg of total protein lysate were resolved on 4-20% SDS-PAGE gels. Resolved proteins were blotted to polyvinylidene difluoride membranes by standard procedures and probed with antibodies as indicated in the figure legends. Probed and chemiluminated protein bands were exposed and captured by a Syngene G: Box Chemiluminescence imaging system with the auto-exposure mode, which is determined by the strongest band intensity on each blot. The band intensity quantities were obtained by the supporting software Gene Tools from SynGene. (A) Results from eight pairs (pre-treatment A, post-treatment B) of tumor samples from patients treated with riluzole are shown. Patients 1, 5 and 11 experienced down regulation of pERK and p-AKT and stable disease at first re-evaluation. CD31 expression was decreased in these patients as well. (B, C, and D) Quantification of activated ERK, activated AKT and CD31 from the band intensities shown in (A).
In light of our preclinical observations that GRM–over expressing xenograft tumors are larger and demonstrate higher microvessel density, and that higher levels of VEGF and IL-8 have been observed in conditioned medium from melanoma cells which over express GRM1, we hypothesized that GRM1 functions to promote angiogenesis in melanoma, with antagonism of GRM1 resulting in decreased angiogenesis (Koochekpour et al., 2012; Lee et al., 2012). To investigate this further in human samples, we performed Western blot analysis for the endothelial marker CD31 to evaluate the degree of angiogenesis in pre- and post-treatment tumor samples. CD31 levels on Western blot decreased in patients 1, 5 and 11 and also decreased in patient 12 (Figure 1A and 1D).
Tumors from patients 1, 5 and 11 whose tumors exhibited decreased pERK, pAKT and CD31 on Western blot were selected for IHC analyses. Staining was performed for pERK and CD31, as well as for cleaved caspase-3 for evidence of increased apoptosis, which was seen in the prior phase 0 trial (Yip et al., 2009). Each patient demonstrated increased activated caspase 3, decreased CD31 and decreased pERK on IHC stain in tumors post-treatment compared with pre-treatment (Figure 2A). Quantitation of the IHC results for CD31, cleaved caspase 3 and pERK (mean + SD) is shown in Figure 2B.
Figure 2.
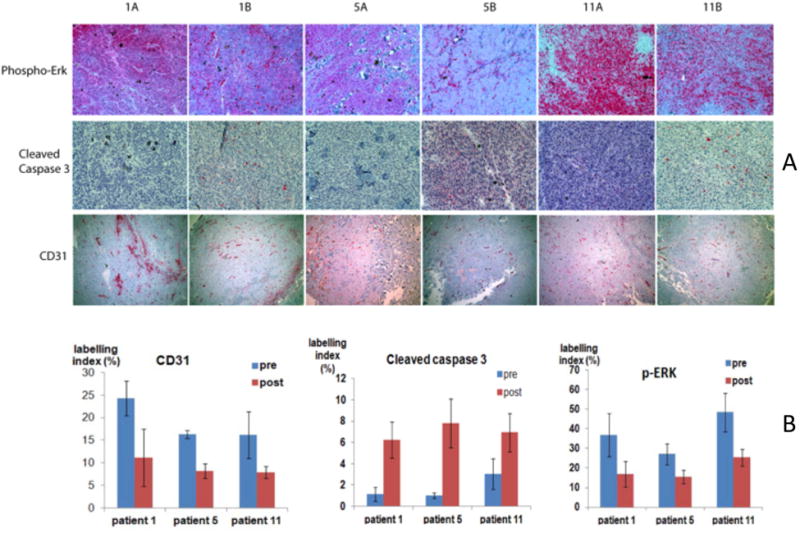
IHC staining of paired tumor samples. Slide processing and immunohistochemistry (IHC) staining was performed by the Histopathology and Imaging Shared Resource of Rutgers Cancer Institute of New Jersey. Slides were scanned by the Biomedical Imaging Center for Biomedical Imaging and Informatics. Three fields were selected from each slide for IHC staining index quantification by the online software at the site of http://153.1.200.58:8080/immunoratio. (A) Column A displays pre-treatment tumor tissue and column B displays post-treatment tumor tissue. Pre-treatment and post-treatment tumor specimens from patients 1, 5, and 11, who showed a trend of disease stabilization and decreased pERK, pAKT and CD31 on Western blot, were stained for pERK, cleaved caspase-3 and CD31. While staining intensity decreased for pERK and CD31, it increased for cleaved caspase-3 in post-treatment specimens. (B) IHC quantification of pERK, cleaved caspase-3 and CD31 (mean + SD).
To analyze the effects of riluzole on immune cells in the tumor microenvironment, 4 paired tumor samples with adequate remaining tissue (patients #1, 6, 8, and 11) were examined for leukocytes (identified using CD45) and macrophages (identified using CD68). Quantitative analysis of the data for CD45+ and CD68+ cells inside the tumor, at the tumor-stroma interface, and in the stroma in these patients is shown in Figure 3A and 3B. In 2 patients with stable disease (patient #1 and #11), CD45+ leukocytes at the tumor-stroma interface were increased in the post-treatment tumor samples (Figure 3A and 3C). In 2 patients with progressive disease (patient #6 and #8), intratumoral leukocytes were decreased in the post-treatment tumor sample (Figure 3A). Cells staining positive for CD68+ macrophages at the tumor-stroma interface were also increased in 2 samples (one coincident with increase in CD45+), decreased in 1 sample, and remained the same in 1 sample (Figure 3B).
Figure 3.
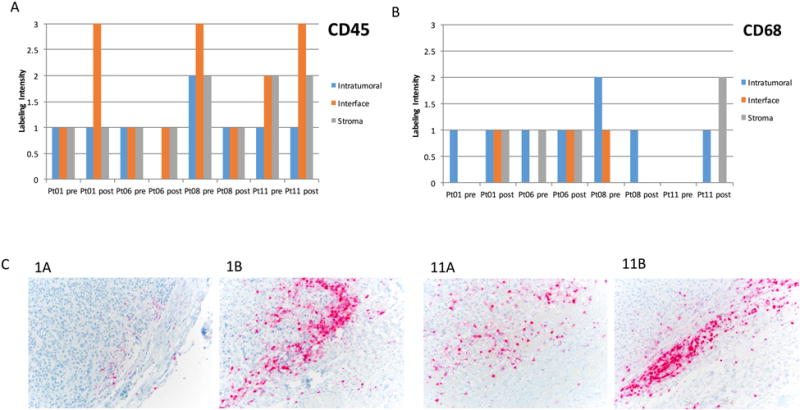
IHC quantification of (A) CD45+ cells and (B) CD68+ cells in tumor, at the tumor-stromal interface, and stroma in pre- and post-treatment samples from four patients. (C) CD45+ staining at the tumor-stromal interface increased in the post-treatment samples from patient #1 (1A, 1B) and 11 (11A, 11B). Tissue from paired biopsy pre-treatment and post-treatment samples was stained for tumor-associated leukocytes and macrophages. Intensity was graded on a 1-3 scale in a blinded manner by a clinical pathologist.
N-RAS and B-RAF mutation status determination showed that tumors from patients 1, 2, 3, 4 and 9 carried B-RAF V600E mutations and patient 10 was found to have a mutation in N-RAS (Table 1). Within this small sample size, down regulation of pERK and pAKT occurred on treatment irrespective of tumor mutation status.
Discussion
This trial was designed with the primary objective of response rate, an appropriate endpoint given our hypothesis that clinically relevant tumor shrinkage would be achieved based on our preclinical and phase 0 data (Le et al., 2010; Namkoong et al., 2007a; Yip et al., 2009). However, the best response observed was stable disease, noted in 46% of patients at first restaging. With no responses observed per RECIST criteria, the trial was terminated. While nearly half of the patients showed stable disease at first restaging, the duration of disease stabilization was, overall, relatively short and as such, is of limited clinical significance (Vidaurre et al., 2009), although prolonged disease stabilization was seen in a previosuly patient whose tumor, with N-RAS mutation status, had been previously progressing.
A dose-escalation phase I trial was not performed given the evidence of biologic activity in our phase 0 trial at 100 mg bid. This is the maximum dose utilized in clinical practice with most ALS patients treated with a schedule of 50 mg bid. We chose the 100 mg bid dose based on the assumption that a higher degree of toxicity may be acceptable in cancer patients with immediate life-threatening disease when compared to patients with more chronic illness. At this higher dose level tested in our phase II study, riluzole was, overall, well tolerated.
The potency and bioavailability of riluzole present challenges to its clinical use (Jablonski et al., 2014). Results of our pharmacokinetic analysis indicate that absorption of riluzole among subjects exhibited variability, consistent with studies of riluzole in ALS subjects (Groeneveld et al., 2001). While our sample size is too small to draw definitive conclusions, higher serum levels of riluzole did not correlate with biologic activity in tumor tissue or disease control, a phenomenon also observed in studies of riluzole in patients with ALS (Groeneveld et al., 2008). One hypothesis to explain this effect is the possibility that N-hydroxyriluzole, produced after riluzole is metabolized in the liver by cytochrome p450 and not measured in our analysis or previous analyses, exists as a pharmacologically active metabolite (Groeneveld et al., 2008). Alternatively, riluzole could be eliminated through glucuroindation or extrahepatic CYP1A1 activity (van Kan et al., 2008). The steady state concentration of riluzole in our study was highly variable, consistent with prior reports, and may be due to variable first pass effects from heterogeneous CYP1A2 expression (McDonnell et al., 2012). Efforts are underway to design more promising prodrugs that remain stable while transiting the digestive system and through first pass metabolism, becoming metabolically labile in the plasma and releasing riluzole (McDonnell et al., 2012).
The findings of this study are consistent with observations from our earlier phase 0 trial, which indicated biologic activity on PET scans and in tumor tissue in one-third of study subjects after two weeks of riluzole. Indeed, 3/8 patients with biopsiable tissue treated on the phase 2 trial showed evidence of disease stabilization on CT scans and decreased pERK and pAKT, indicating suppressed MAPK and PI3K signaling. This biochemical suppresion of active pathways is expected since it is known that activation of GRM1 leads to a dual signaling cascade that stimulates both MAPK and PI3K pathways which could be inhibited by riluzole (Chen et al., 2012; Lee et al., 2012; Marin et al., 2006).
The relationship between tumor B-RAF or N-RAS mutation status and GRM1 signaling is not clear. While it is plausible that N-RAS mutated melanoma may be particularly sensitive to GRM1 inhibition in a clinical setting, preclinical in vitro and in vivo data suggest that riluzole or other GRM1 inhibitors reduce cell proliferation and decrease tumor growth independent of N-RAS/B-RAF genotypes (Le et al., 2010; Namkoong et al., 2007; Wangari-Talbot et al., 2012). Biochemical suppression of MAPK and PI3K pathways was observed in patients whose tumors had BRAF and NRAS mutations on both the phase 0 and 2 trials. Thus our results, though limited by the very small sample size, do not demonstrate correlation between suppression of pERK/pAKT and B-RAF or N-RAS mutation status.
Correlative studies were hypothesis generating. The pattern of the correlative study outcomes suggest that the patients who had clinical benefit from riluzole treatment displayed multiple downstream markers of GRM1 signaling inhibition, including suppression of MAPK, PI3K/AKT, and markers of angiogenesis. Additionally, 2 patients (#1 and #11) who derived clinical benefit from riluzole treatment displayed an increase in CD45+ leukocytes at the tumor-stroma interface. Two patients who did not benefit from treatment did not show suppression of PI3K/AKT, angiogenesis, nor an increase in CD45+ cells at the interface. The increase in leukocytes at the interface, or “active edge,” of the tumor may be mediated by inhibition of exosomes containing MCSF and CCL-2, which normally recruit tumor-associated macrophages (TAMs) and exclude tumor infiltrating lymphocytes (TILs). In a laboratory model, GRM1 activation lead to inhibition of release of exosomes containing MCSF and CCL-2 (Li et al., 2017), which create an immunosuppressive tumor microenvironment by promoting TAMS. Inhibiting release of the exosomes with riluzole treatment may allow lymphocytes into the tumor-stroma interface and enhance the antitumor activity of immune checkpoint inhibitors in tumors void of infiltrating lymphocytes (immune desert). The apparent correlation between suppression of GRM1 pathways, increase of leukocytes at the active edge of the tumor and observed clinial benefit in patients suggests that inhibiting tumor growth and tipping the immune microenviroment towards tumor rejection is a plausible mechanism by which tumor control occurs with riluzole.
In summary, this phase II trial of riluzole demonstrated that therapy with 100 mg bid of riluzole was well tolerated in a population of patients with advanced melanoma, the majority of which exhibited M1c disease. No RECIST responses were observed and disease stabilization was overall short, but evidence for biologic activity was noted in paired tumor specimens, consistent with the initial findings in the previously published phase 0 trial. These observations suggest that while riluzole may have interesting biologic activity, further clinical study with riluzole monotherapy is not warranted. Moving forward, close attention must be paid to preclinical studies to further unravel the mechanism of action of glutamate signaling in melanoma, to examine the effects of riluzole on the immune cells in the tumor microenvironment, and to develop gluatmate antagonists with improved potency and bioavailability. A pro-drug of riluzole, trigriluzole, with improved oral bioavailability is currently undergoing clinical testing.
Supplementary Material
Significance.
The findings of our phase II trial of riluzole in patients with advanced melanoma have practical implications for future exploration of GRM1 targeting agents. With no RECIST responses and disease stabilization as the best outcome, further study of single agent riluzole was not recommended. Suppression of GRM1 related signaling pathways and increased leukocyte infiltrtaion, potential indicators of biologic activity, were noted in tumor specimens from a subset of treated patients. Moving forward, studying agents with improved bioavailability and studying the effects on the immune microenvironment, including combinatorial studies with immune modulating agents, may yield important insights.
Acknowledgments
This study was funded in part by NIH 1R21CA139473-02. The authors extend appreciation to The Cancer Institute of New Jersey Pharmacokinetics/Pharmaco dynamics Shared Resource for the PK analysis of riluzole, Biometrics Shared Resource for statistical analyses, Yu Wen, Ph.D., and Veeraswamy Manne, Ph.D. for assistance with manuscript preparation and submission.
Funded by NIH 1R21CA139473-02 and CA P30-072720
Footnotes
Disclosure statement: The authors have no potential conflicts of interest related to this work to disclose.
References
- Arcella A, Carpinelli G, Battaglia G, D'onofrio M, Santoro F, Ngomba RT, Bruno V, Casolini P, Giangaspero F, Nicoletti F. Pharmacological blockade of group II metabotropic glutamate receptors reduces the growth of glioma cells in vivo. Neuro Oncol. 2005;7:236–45. doi: 10.1215/S1152851704000961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronica E, Gorter JA, Ijlst-Keizers H, Rozemuller AJ, Yankaya B, Leenstra S, Troost D. Expression and functional role of mGluR3 and mGluR5 in human astrocytes and glioma cells: opposite regulation of glutamate transporter proteins. Eur J Neurosci. 2003;17:2106–18. doi: 10.1046/j.1460-9568.2003.02657.x. [DOI] [PubMed] [Google Scholar]
- Bennett DC. How to make a melanoma: what do we know of the primary clonal events? Pigment Cell Melanoma Res. 2008;21:27–38. doi: 10.1111/j.1755-148X.2007.00433.x. [DOI] [PubMed] [Google Scholar]
- Brocke KS, Staufner C, Luksch H, Geiger KD, Stepulak A, Marzahn J, Schackert G, Temme A, Ikonomidou C. Glutamate receptors in pediatric tumors of the central nervous system. Cancer Biol Ther. 2010;9:455–68. doi: 10.4161/cbt.9.6.10898. [DOI] [PubMed] [Google Scholar]
- Chen L, Luo A, Zhang Y, Liu F, Jiang Y, Xu Q, Chen X, Hu Q, Chen SF, Chen KJ, et al. Optimization of the single-phased white phosphor of Li2SrSiO4: Eu2+, Ce3+ for light-emitting diodes by using the combinatorial approach assisted with the Taguchi method. ACS Comb Sci. 2012;14:636–44. doi: 10.1021/co300058x. [DOI] [PubMed] [Google Scholar]
- Choi KY, Chang K, Pickel JM, Badger JD, 2nd, Roche KW. Expression of the metabotropic glutamate receptor 5 (mGluR5) induces melanoma in transgenic mice. Proc Natl Acad Sci U S A. 2011;108:15219–24. doi: 10.1073/pnas.1107304108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'onofrio M, Arcella A, Bruno V, Ngomba RT, Battaglia G, Lombari V, Ragona G, Calogero A, Nicoletti F. Pharmacological blockade of mGlu2/3 metabotropic glutamate receptors reduces cell proliferation in cultured human glioma cells. J Neurochem. 2003;84:1288–95. doi: 10.1046/j.1471-4159.2003.01633.x. [DOI] [PubMed] [Google Scholar]
- Fecher LA, Cummings SD, Keefe MJ, Alani RM. Toward a molecular classification of melanoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2007;25:1606–20. doi: 10.1200/JCO.2006.06.0442. [DOI] [PubMed] [Google Scholar]
- Groeneveld GJ, Van Kan HJ, Lie AHL, Guchelaar HJ, Van Den Berg LH. An association study of riluzole serum concentration and survival and disease progression in patients with ALS. Clin Pharmacol Ther. 2008;83:718–22. doi: 10.1038/sj.clpt.6100382. [DOI] [PubMed] [Google Scholar]
- Groeneveld GJ, Van Kan HJ, Torano JS, Veldink JH, Guchelaar HJ, Wokke JH, Van Den Berg LH. Inter- and intraindividual variability of riluzole serum concentrations in patients with ALS. J Neurol Sci. 2001;191:121–5. doi: 10.1016/s0022-510x(01)00613-x. [DOI] [PubMed] [Google Scholar]
- Jablonski MR, Markandaiah SS, Jacob D, Meng NJ, Li K, Gennaro V, Lepore AC, Trotti D, Pasinelli P. Inhibiting drug efflux transporters improves efficacy of ALS therapeutics. Annals of clinical and translational neurology. 2014;1:996–1005. doi: 10.1002/acn3.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koochekpour S, Majumdar S, Azabdaftari G, Attwood K, Scioneaux R, Subramani D, Manhardt C, Lorusso GD, Willard SS, Thompson H, et al. Serum glutamate levels correlate with Gleason score and glutamate blockade decreases proliferation, migration, and invasion and induces apoptosis in prostate cancer cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:5888–901. doi: 10.1158/1078-0432.CCR-12-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le MN, Chan JL, Rosenberg SA, Nabatian AS, Merrigan KT, Cohen-Solal KA, Goydos JS. The glutamate release inhibitor Riluzole decreases migration, invasion, and proliferation of melanoma cells. J Invest Dermatol. 2010;130:2240–9. doi: 10.1038/jid.2010.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Wall BA, Wangari-Talbot J, Chen S. Regulation of mGluR1 expression in human melanocytes and melanoma cells. Biochim Biophys Acta. 2012;1819:1123–31. doi: 10.1016/j.bbagrm.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Wall BA, Wangari-Talbot J, Shin SS, Rosenberg S, Chan JL, Namkoong J, Goydos JS, Chen S. Glutamatergic pathway targeting in melanoma: single-agent and combinatorial therapies. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:7080–92. doi: 10.1158/1078-0432.CCR-11-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wen Y, B-S J, Shah R, Isola A, Silk AW, Chen S, Goydos JS. GRM1 to induce M-CSF and CCL2 expression to lead to an upregulation of M2 macrophages in melanoma, Abstract #122; ASCO-SITC I Clinical Immuno-Oncology Symposium; February 23-25, 2017; Orlando, Florida. 2017. [Google Scholar]
- Marin YE, Namkoong J, Cohen-Solal K, Shin SS, Martino JJ, Oka M, Chen S. Stimulation of oncogenic metabotropic glutamate receptor 1 in melanoma cells activates ERK1/2 via PKCepsilon. Cell Signal. 2006;18:1279–86. doi: 10.1016/j.cellsig.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Mcdonnell ME, Vera MD, Blass BE, Pelletier JC, King RC, Fernandez-Metzler C, Smith GR, Wrobel J, Chen S, Wall BA, et al. Riluzole prodrugs for melanoma and ALS: design, synthesis, and in vitro metabolic profiling. Bioorg Med Chem. 2012;20:5642–8. doi: 10.1016/j.bmc.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. Riluzole for ALS: what is the evidence? Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:135. [PubMed] [Google Scholar]
- Miller RG, Mitchell JD, Lyon M, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND) Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:191–206. [PubMed] [Google Scholar]
- Mitsumoto H. Riluzole--what is its impact in our treatment and understanding of amyotrophic lateral sclerosis? Ann Pharmacother. 1997;31:779–81. doi: 10.1177/106002809703100619. [DOI] [PubMed] [Google Scholar]
- Namkoong J, Shin SS, Lee HJ, Marin YE, Wall BA, Goydos JS, Chen S. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007a;67:2298–305. doi: 10.1158/0008-5472.CAN-06-3665. [DOI] [PubMed] [Google Scholar]
- Namkoong J, Shin SS, Lee HJ, Marin YE, Wall BA, Goydos JS, Chen S. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer Res. 2007b;67:2298–305. doi: 10.1158/0008-5472.CAN-06-3665. [DOI] [PubMed] [Google Scholar]
- Noh KM, Hwang JY, Shin HC, Koh JY. A novel neuroprotective mechanism of riluzole: direct inhibition of protein kinase C. Neurobiol Dis. 2000;7:375–83. doi: 10.1006/nbdi.2000.0297. [DOI] [PubMed] [Google Scholar]
- Ohtani Y, Harada T, Funasaka Y, Nakao K, Takahara C, Abdel-Daim M, Sakai N, Saito N, Nishigori C, Aiba A. Metabotropic glutamate receptor subtype-1 is essential for in vivo growth of melanoma. Oncogene. 2008;27:7162–70. doi: 10.1038/onc.2008.329. [DOI] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- Pollock PM, Cohen-Solal K, Sood R, Namkoong J, Martino JJ, Koganti A, Zhu H, Robbins C, Makalowska I, Shin SS, et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat Genet. 2003a;34:108–12. doi: 10.1038/ng1148. [DOI] [PubMed] [Google Scholar]
- Pollock PM, Cohen-Solal K, Sood R, Namkoong J, Martino JJ, Koganti A, Zhu H, Robbins C, Makalowska I, Shin SS, et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat Genet. 2003b;34:108–12. doi: 10.1038/ng1148. [DOI] [PubMed] [Google Scholar]
- Saugstad JA, Segerson TP, Westbrook GL. Metabotropic glutamate receptors activate G-protein-coupled inwardly rectifying potassium channels in Xenopus oocytes. J Neurosci. 1996;16:5979–85. doi: 10.1523/JNEUROSCI.16-19-05979.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speyer CL, Smith JS, Banda M, Devries JA, Mekani T, Gorski DH. Metabotropic glutamate receptor-1 a potential therapeutic target for the treatment of breast cancer. Breast Cancer Res Treat. 2011 doi: 10.1007/s10549-011-1624-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepulak A, Luksch H, Gebhardt C, Uckermann O, Marzahn J, Sifringer M, Rzeski W, Staufner C, Brocke KS, Turski L, et al. Expression of glutamate receptor subunits in human cancers. Histochem Cell Biol. 2009;132:435–45. doi: 10.1007/s00418-009-0613-1. [DOI] [PubMed] [Google Scholar]
- Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, Van Oosterom AT, Christian MC, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- Van Kan HJ, Van Den Berg LH, Groeneveld GJ, Van Der Straaten RJ, Van Vught PW, Lie AHL, Guchelaar HJ. Pharmacokinetics of riluzole: evidence for glucuronidation as a major metabolic pathway not associated with UGT1A1 genotype. Biopharm Drug Dispos. 2008;29:139–44. doi: 10.1002/bdd.594. [DOI] [PubMed] [Google Scholar]
- Vidaurre T, Wilkerson J, Simon R, Bates SE, Fojo T. Stable disease is not preferentially observed with targeted therapies and as currently defined has limited value in drug development. Cancer J. 2009;15:366–73. doi: 10.1097/PPO.0b013e3181b9d37b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JQ, Tang Q, Parelkar NK, Liu Z, Samdani S, Choe ES, Yang L, Mao L. Glutamate signaling to Ras-MAPK in striatal neurons: mechanisms for inducible gene expression and plasticity. Mol Neurobiol. 2004;29:1–14. doi: 10.1385/MN:29:1:01. [DOI] [PubMed] [Google Scholar]
- Wangari-Talbot J, Wall BA, Goydos JS, Chen S. Functional effects of GRM1 suppression in human melanoma cells. Mol Cancer Res. 2012;10:1440–50. doi: 10.1158/1541-7786.MCR-12-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters C, Pyne S, Pyne NJ. The role of G-protein coupled receptors and associated proteins in receptor tyrosine kinase signal transduction. Seminars in Cell and Developmental Biology. 2004;15:309–323. doi: 10.1016/j.semcdb.2003.12.020. [DOI] [PubMed] [Google Scholar]
- Yip D, Le MN, Chan JL, Lee JH, Mehnert JA, Yudd A, Kempf J, Shih WJ, Chen S, Goydos JS. A phase 0 trial of riluzole in patients with resectable stage III and IV melanoma. Clin Cancer Res. 2009;15:3896–902. doi: 10.1158/1078-0432.CCR-08-3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.