

Abstract
Premise of the study
Although asexual taxa are generally seen as evolutionary dead ends, asexuality appears to provide a short-term benefit in some taxa, including a wider geographic distribution compared to sexual relatives. However, this may be an illusion created by multiple, morphologically cryptic, asexual lineages, each occupying a relatively small area. In this study we investigate the role of multiple lineages in the biogeography of Myriopteris gracilis Fée (Pteridaceae), a North American apomictic triploid fern species with a particularly large range.
Methods
Rangewide asexuality was assessed by counting spores/sporangium in 606 M. gracilis specimens from across the species range, and lineage structure was assessed with both plastid DNA sequence and Genotyping By Sequencing (GBS) SNP datasets.
Key Results
Spore counting of >600 specimens identified no sexual populations, establishing that M. gracilis is exclusively asexual. The plastid data estimated the crown age of M. gracilis at ca. 2.5 mya and identified two lineages, each largely confined to the eastern or western portions of the range. These groups were further subdivided by the GBS data, revealing at least seven asexual lineages of varying geographic distributions, each occupying a relatively small portion of the total range of M. gracilis.
Conclusions
Although maintained exclusively through asexual reproduction, the broad distribution of M. gracilis is a compilation of numerous, independently formed asexual lineages. Since no single asexual lineage occupies the full extent of the species distribution, recurrent lineage formation should be considered when evaluating the short-term benefit of asexuality in this taxon and others.
Keywords: asexual reproduction, Döpp-Manton sporogenesis, geographic parthenogenesis, genotyping by sequencing, Pipeline for Untangling Reticulate Complexes, Pteridaceae, trnG-trnR intergenic spacer
The dominance of sex among eukaryotes is surprising given its numerous demographic and transmission disadvantages relative to asexual reproduction (Williams, 1975; Maynard-Smith, 1978; Bell, 1982). This so-called “paradox of sex” remains one of the most compelling topics in evolutionary biology, and multiple hypotheses have been proposed in order to explain the rarity of asexuality among multicellular organisms (reviewed in Otto, 2009). Most explanations focus on the role of sex in providing a diversity of genotypes and preventing the buildup of deleterious genetic load. Conversely, the failure of asexual lineages is attributed to their inherent lack of adaptive flexibility as well as an inability to rid themselves of deleterious mutations (reviewed in Schwander and Crespi, 2009; Hojsgaard et al., 2015). Despite the characterization of asexuality as an evolutionary dead end (Wagner, 1970; Schwander and Crespi, 2009; Janko, 2013), there is evidence to suggest that the transition to asexuality may confer some degree of short-term success. The most frequently cited examples involve the phenomenon known as geographical parthenogenesis, in which asexual species exhibit geographic ranges substantially different from those of their sexual progenitors (Hörandl, 2006; Tilquin and Kokko 2016). Asexual species often occupy higher latitudes and elevations compared to related sexual diploids (Glesner and Tilman, 1978; Kearney, 2005; Lundmark and Saura, 2006), and many exhibit broader geographic distributions than their sexual progenitors (Bierzychudek, 1985; Ross et al., 2013). Although these unique and/or more extensive asexual ranges suggest the possibility of a short-term benefit, the recurrent formation of asexual lineages can create the illusion of geographic success. Asexual plant species (excluding those that bypass sex via vegetative reproduction) are typically also polyploid (Silvertown, 2008; Van Dijk, 2009), and the recurrent formation of polyploid lineages is the rule rather than the exception (Soltis et al., 2003). To the extent that the transition to asexuality and polyploidization are linked (Whitton et al., 2008), this recurrent formation of asexual lineages could result in a broadly distributed asexual species comprising a mosaic of independent lineages. Each one of these could potentially then be less geographically successful than the total species range suggests.
One of the more dramatic North American examples of a geographically successful asexual plant is the slender lip fern, Myriopteris gracilis Fée (Pteridaceae). This species, until recently known as Cheilanthes feei T. Moore (see Grusz and Windham, 2013), grows on calcareous rocks throughout much of the United States (Windham and Rabe, 1993). Outside of its core range in the southwestern U.S., M. gracilis extends as far north as southern Canada (Brunton, 1979) and as far east as North Carolina (Rothfels et al., 2012a). This broad distribution is thought to exclusively comprise obligate asexuals, as sexual populations have never been documented (Windham and Rabe, 1993). The few published chromosome counts indicate that the species is an apomictic triploid (Windham and Yatskievych, 2003), and it has been considered an autotriploid due to the lack of a morphologically plausible combination of parental species. This autopolyploid hypothesis is further supported by nuclear gapCp "short" sequencing (data not shown). This locus (Schuettpelz et. al., 2008) was evaluated in two M. gracilis samples and 87 samples representing 50 Myriopteris species or putative hybrids using the amplicon sequencing and Pipeline for Untangling Reticulate Complexes (PURC) approach outlined in Rothfels et al. (2017). A maximum-likelihood phylogeny identified a single, strongly supported clade of exclusively M. gracilis alleles (100% ML bootstrap support). The slender lip fern therefore represents a singular opportunity to investigate the apparent success of a widely distributed non-hybrid asexual polyploid. Here we ask two basic questions: 1) Is M. gracilis exclusively asexual? and 2) Does M. gracilis comprise a single successful asexual lineage or a mosaic of less widely distributed genotypes?
MATERIALS AND METHODS
Field collections and spore counting
Establishing that M. gracilis is exclusively asexual is critical, since the recurrent formation of asexual lineages by cryptic sexuals undermines the idea of a geographically successful asexual species. As an autopolyploid, M. gracilis presumably co-existed with its sexual diploid progenitor for a time, and sexual populations may still be present. Fortunately, details of the life cycle in leptosporangiate ferns greatly simplify the diagnosis of reproductive mode. In these ferns, non-vegetative asexual reproduction (= apomixis) involves the alternation of generations without any change in chromosome number (Haufler et al., 2016). A modified version of meiosis produces unreduced spores that germinate and form a similarly unreduced gametophyte. A sporophyte then develops directly from this gametophyte without the formation of archegonia (Walker, 1979; reviewed in Grusz, 2016). The production of viable, unreduced spores in M. gracilis involves a common apomictic pathway known as Döpp-Manton sporogenesis (Döpp, 1939; Manton, 1950). Due to an incomplete mitotic division immediately prior to meiosis, the Döpp-Manton pathway consistently produces 32 spores in a given sporangium, half the number resulting from the typical sexual pathway. The reproductive mode of any fertile specimen can therefore be quickly ascertained by determining the number of spores per sporangium. Hundreds of fertile M. gracilis specimens are available, as this commonly collected species is well represented in both regional and national herbaria.
For this study, a total of 120 specimens (Appendix S1 - see Supplemental Data with the online version of this article) were field-collected from 20 states, including disjunct eastern populations in Kentucky and Virginia. At each site, material sufficient for voucher specimens was obtained and leaf tissue from a single individual was preserved in silica gel for subsequent molecular analyses. Reproductive mode was determined for each sample by counting the number of spores per sporangium. Counts were obtained from mature, unopened sporangia which were removed from the underside of a leaf and placed in a small droplet of glycerol. Each sporangium was then carefully opened with a dissecting needle, its contents teased apart, and individual spores counted on a dissecting microscope under low magnification. Typically a single sporangium was examined per specimen, although multiple sporangia were consulted in cases where completely intact sporangia were difficult to find. Additional spore counts were obtained in the same manner from 488 herbarium specimens archived in 17 herbaria (Appendix S1). All herbarium specimens providing spore counts were manually georeferenced using Google Earth (Google, Mountain View, California, USA) and Earth Point (Clark 2015), and tissue samples were obtained from select specimens for inclusion in our plastid sequence dataset.
Assessing genetic variability
Determining whether M. gracilis comprises one or multiple lineages requires data from individuals scattered across its large range. Assuming that the formation of multiple lineages would capture different portions of the genetic variation initially present in the sexual progenitor, asexual lineages should be genetically distinct from one another, while individuals from the same lineage should be relatively similar. Genetic variability of M. gracilis was initially assessed by sequencing a highly variable plastid genome region. While this single locus dataset could provide only a coarse estimation of lineage diversity, these data could be obtained rapidly from both freshly collected and herbarium tissues. A genomic Genotyping by Sequencing (GBS) dataset was then generated in order to obtain a finer-scale estimate of lineage diversity. This method identifies single nucleotide polymorphisms (SNPs) at thousands of locations across the genome by massive parallel sequencing of a reduced representation library (Elshire et al., 2011). While GBS has predominantly been applied to sexual species, the genomic fingerprint it provides is potentially well suited for distinguishing clonal lineages (Reitzel et al., 2013).
DNA extraction and plastid sequencing
Genomic DNA was extracted from silica-dried samples using the DNeasy Plant Minikit (Qiagen, Valencia, California, USA), and from herbarium-acquired tissues following the protocol detailed in Beck et al. (2012). A section of the plastid genome spanning a portion of the trnG intron, one of two trnG exons, and nearly half of the trnG-trnR intergenic spacer (herein referred to as trnGR) was PCR-amplified under standard conditions using previously published primers (Beck et al., 2010). Sanger sequencing was performed at the University of Chicago Comprehensive Cancer Center DNA Sequencing and Genotyping Facility.
Plastid phylogeny and divergence time estimation
Sequence data were assembled into contigs and edited with CLC Workbench (CLC Bio Aarhus, Denmark). All trnGR sequences have been deposited in the European Nucleotide Archive (study# PRJEB 21093; Appendix S1), and all alignments have been deposited on the Dryad Digital Repository (doi:10.5061/dryad.cs635). Following alignment, a haplotype network for the 130 M. gracilis trnGR sequences was generated with TCS version 1.18 (Clement et al., 2000). The phylogenetic position of M. gracilis and divergence times among intraspecific lineages were then estimated by analyzing M. gracilis trnGR sequence variation in the context of a genus-wide phylogeny reconstructed by Grusz et al. (2014) using two different strategies.
In the first, M. gracilis node ages were estimated directly in the context of the full Grusz et al. (2014) dataset. Representatives of the three unique slender lip fern trnGR sequences observed in this study (see results) were added to the complete Grusz et al. dataset, along with a single additional sequence from each of the closely related species Myriopteris lanosa (Michx.) Grusz & Windham and Myriopteris parryi (D. C. Eaton) Grusz & Windham (Appendix S1). Following manual alignment and the exclusion of regions of uncertain homology, the best-fitting models of sequence evolution were estimated using the Akaike Information Criterion in PartitionFinder v.1.1 (Lanfear et al., 2012) for each of four partitions- the (1) first, (2) second, and (3) third positions of the trnG exon and (4) the remaining non-coding portion of the trnGR region. Phylogeny and divergence times were then co-estimated in BEAST v1.8 (Drummond et al., 2012). Three non-truncated, normally distributed priors were utilized: 1) (37.1 ± 3.71 mya) on the root height; 2) (28.2 ± 2.82 mya) on the crown age of the pellaeid outgroup clade; and 3) (27.2 ± 2.7 mya) on the crown age of Myriopteris. These mean node ages were obtained from Schuettpelz and Pryer (2009), and standard deviations of ten percent of the mean age were implemented (Rothfels et al., 2012b; Sigel et al., 2014). Three independent runs were performed in BEAST, each assuming the node age priors discussed above and the following parameters: partitions with unlinked substitution models; an estimated linked lognormal relaxed uncorrelated clock model; a linked birth-death speciation tree prior; and an untruncated, gamma distributed (shape = 0.001, scale = 1000) clock mean (ucld.mean) (Drummond and Bouckaert 2015). Each run comprised 1 × 107 generations, sampling every 1000 generations. Inter-run convergence was assessed with TRACER v1.6 (Rambaut et al., 2014). After discarding the first 1 × 106 generations of each run as burnin the remaining trees were compiled and annotated using LogCombiner v1.8 and TreeAnnotator v.1.8 (Drummond et al., 2012). Mean and 95% highest probability density (HPD) on node height were then evaluated in FigTree v1.4 (Rambaut, 2012).
The second strategy involved estimating trnGR mutation rate in the context of a trimmed Myriopteris phylogeny, and subsequently using this rate to estimate node ages within and among M. gracilis and closely related species. The Grusz et al. (2014) dataset was trimmed to include a single representative of each species. The best-fitting model of sequence evolution was identified as described above, and three independent runs were performed in BEAST with run parameters and convergence assessment as described above. After discarding the first 1 × 106 generations of each run as burnin, the mean rate of the combined 27,000 trees was used as a prior in a coalescent-based analysis among M. gracilis and immediate relatives. This dataset comprised one representative of each of the three unique M. gracilis trnGR sequences, the two unique Myriopteris lanosa (Michx.) Grusz & Windham sequences obtained by Grusz et al. (2014), and the Myriopteris longipila (Baker) Grusz & Windham sequence from that study. Best-fitting models of sequence evolution were identified as before, and three BEAST runs were performed implementing the following parameters: partitions with unlinked substitution models; an estimated linked lognormal relaxed uncorrelated clock model (lognormal ucld.mean prior 0.00127 ± 0.145); and a linked constant size coalescent tree prior. Run length parameters, convergence assessment, burnin removal, and tree annotation were implemented as described above.
Reference genome sequencing and assembly
High molecular weight genomic DNA from M. gracilis specimen Wickell 18 was used to generate a standard Illumina TruSeq DNA sequencing library. The library was subjected to paired-end 100-bp sequencing on a single lane of an Illumina HiSeq2500 instrument at the University of Kansas Genome Sequencing Core, and raw read data (FASTQ) have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (www.ncbi.nlm.nih.gov/sra) under BioProject ID PRJNA388661. Raw reads were preprocessed by removing adaptors using Scythe v. 0.991 (Buffalo, 2015), and trimming low-quality sequences using Sickle v. 1.200 (Joshi and Fass, 2011). Trimming involved removing sequences with average quality values below 20 and reads less than 80 bp in length. Trial assemblies revealed a number of scaffolds with relatively high coverage, some of which exhibited high BLAST (Altschul et al., 1990) matches to fungal endophyte sequences. Fungal endophytes have been previously documented in M. gracilis (Palmieri, 2001), and in order to remove potential contaminants, trimmed reads were aligned to the genomes of the ascomycete fungi Alternaria brassicicola (Schwein.) Wiltshire (GenBank Accession Number: ACIW00000000.1) and Alternaria arborescens E. G. Simmons (AIIC00000000.1) using Bowtie2 v. 2.1.0 (Langmead and Salzburg, 2012) with default settings. The 349,763,030 high-quality reads that did not align to either endophyte genome were assembled using ABySS v. 1.5.2 (Simpson et al., 2009). Multiple assemblies were generated using a range of k-mer values, and the optimal k-mer size (k = 71) was empirically determined based on contig N50 values and the size of the largest contig generated.
Genotyping by sequencing (GBS)
Genomic DNA was quantified using a Quant-iT™ dsDNA HS Assay on the Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, California, USA). All extracts were then visually evaluated by running 100 ng of DNA against a λ DNA-HindIII digest size ladder on a 1% agarose gel. A set of 95 geographically representative samples exhibiting sufficient high quantity/quality genomic DNA were chosen for GBS. Library preparation and sequencing were conducted at the Genomic Diversity Facility (GDF) at Cornell University’s Biotechnology Resource Center. Test libraries were constructed from a single genomic DNA sample using two restriction enzymes (EcoT221, PstI). Libraries were visualized with a Bio-Rad Experion (Bio-Rad, Hercules, California, USA) in order to determine which restriction enzyme yielded the library with the smallest number of fragments. EcoT221 was chosen given its slightly smaller fragment pool, one that should result in increased sequencing depth of each fragment. Each sample was pooled with a barcode and common adapter, digested with EcoT22I, and subsequently ligated together. The barcode adapter contained a 4–8 bp barcode sequence that was subsequently used to identify each sample, allowing subsequent reactions to be carried out in multiplex. All samples were then pooled and purified using a Qiagen QIAquick PCR Purification Kit (Qiagen, Valencia, California, USA). Ligated fragments were then combined with a pair of primers for PCR amplification. In addition to priming amplification, these oligos facilitated the addition of a short (~15 nucleotide) sequence at the 5′ end to bind amplification products to the lanes of the Illumina (Illumina, San Diego, California, USA) flowcell. Following amplification, the resulting reduced representation library was subjected to 100-bp single end sequencing on two lanes of an Illumina HiSeq 2500.
GBS filtering and analysis
Initial data processing employed the TASSEL-GBS "Discovery" pipeline (Glaubitz et al., 2014), with default pipeline settings except for those noted in Appendix S2. Prior to alignment, TASSEL 4.0 (Bradbury et al., 2007) was used to assemble 64 bp consensus sequences or “tags” by aligning identical reads. Unique tags were identified by searching the raw data for a combination of the restriction site along with DNA barcodes from each sample found in the “master key file.” Unique tags were then merged to create a master tag file. The preliminary M. gracilis genome assembly noted above was indexed using the Burrows-Wheeler alignment tool (BWA; Li and Durbin, 2009), and BWA was subsequently used to align all tags found in the master tag file to the indexed reference genome and export this alignment in SAM format. Following alignment, the SAM file was converted into a “tags on physical map” (TOPM) file, and TASSEL was used to output an HDF5 or “tags by taxa” file, used to assign SNPs to individuals according to their DNA barcode. Data from both the TOPM file and the HDF5 file were then used to call SNPs in TASSEL. Tags that mapped to the same location on the reference genome (TOPM) were considered a “locus,” and differences between samples at a given locus represented SNPs. The resulting HapMap SNP file is a matrix providing full codominant information at each locus in each sample. Original read data (FASTQ) have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive under BioProject ID PRJNA389100, and the final filtered HapMap file is archived on the Dryad Digital Repository (doi:10.5061/dryad.cs635).
The HapMap matrix was aggressively filtered for missingness both by sample and site in TASSEL. Samples for which >80% of sites were missing were first excluded, and sites not present in all remaining samples (>0% missingness) were then removed. This highly filtered HapMap matrix was analyzed with both multivariate clustering and phylogenetic approaches. Principal coordinates analysis with modal clustering (PCO-MC) (Reeves and Richards, 2009) was used to identify significant clusters and rank them according to how tightly individuals are associated within each cluster across the entire range of axes defined in a standard principal coordinate analysis (PCoA). In order to visualize PCO-MC results, a Principal Components Analysis (PCA) was conducted in TASSEL, and a plot of individual scores on the three most informative components was plotted using the “matplotlib” package (Hunter, 2007) in Python 2.7. A maximum-likelihood phylogeny was inferred using RAxML version 8.2.8 (Stamatakis, 2014) implemented through the CIPRES Science Gateway (Miller et al., 2010). The General Time Reversal model of nucleotide substitution with the gamma model of rate heterogeneity (GTRGAMMA) was implemented in both a standard search and 1000 bootstrap replicates. A maximum-parsimony phylogeny was inferred using PAUP version 4.0a150 (Swofford, 2002). Given the relatively high level of heterozygosity observed, a cost-matrix incorporating 10 states (4 standard nucleotides plus 6 bi-nucleotide ambiguity states- see Potts et al., 2014) was implemented in both a heuristic search and 1000 bootstrap replicates.
Morphological analyses
Our molecular data clearly establish the genomic distinctiveness of groups of M. gracilis individuals (see below), and we attempted to discover morphological character-states that would allow for the discrimination of these lineages. Numerous characters were evaluated in pilot studies, including leaf division, leaf length/width, petiole/leaf length ratio, pinna pubescence, rachis pubescence, inter-pinna-pair rachis length, and pinna length/width. Rachis pubescence and pinna width were identified as the strongest candidate characters and were scored in each of the 73 individuals included in the final GBS analysis. Rachis pubescence was scored as: 1) completely glabrous; 2) exhibiting widely scattered hairs; 3) generally hairy; and 4) densely hairy so as to largely obscure the rachis itself. Pinna width was measured as the distance between the tips of the two ultimate (proximate) pinnules of the proximal-most pinnae. The relationship between these two characters was visualized on a bivariate scatterplot in RStudio (RStudio team, 2015).
RESULTS
Geographic distribution of asexual lineages
Spore counts were obtained from 606 M. gracilis specimens from 24 U.S. states, 1 Canadian province, and 2 states of Mexico (Appendix S1; Fig. 1). Of these, 602 individuals exhibited ca. 32 spores per sporangium. It is uncommon to observe all 32 spores, as a few are typically crushed or otherwise lost during disruption/spreading of the sporangium contents. In these 602 individuals 25–32 spores were seen, far below the 50+ spores one would observe in a sporangium of a sexual individual. Four individuals exhibited sporangia with >32 spores, but in all cases the spores were strikingly shriveled and misshapen. These misshapen spores are of sporadic occurrence among asexual triploid ferns, and indeed approx. 2% of M. gracilis individuals we examined exhibited both sporangia with >32 misshapen spores and those with ca. 32 well-formed spores. In contrast, the 64 spores of a sporangium in a sexual individual are obviously different- quite uniform in both size and shape. We therefore interpreted these occasional misshapen spores as also the result of unsuccessful completion of Döpp-Manton sporogenesis, and spore phenotypes therefore established that all 606 counted specimens were asexual.
Fig. 1.
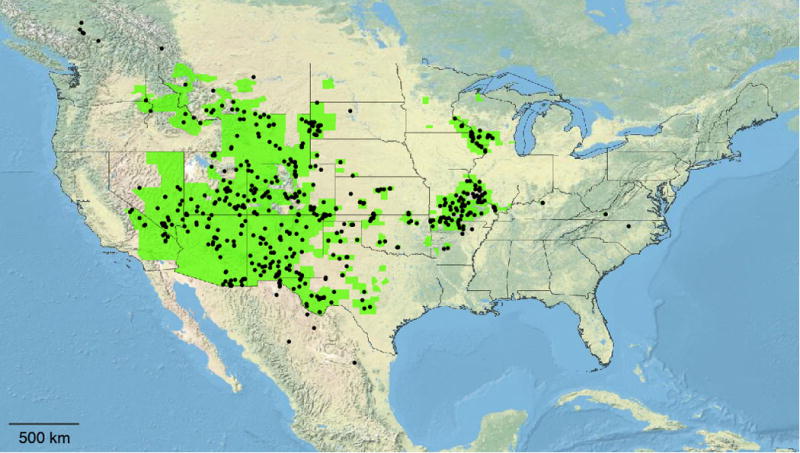
Location of 606 spore-counted individuals of Myriopteris gracilis in the context of the U.S. county-level distribution (adapted from Kartesz, 2015). Individuals exhibited 32 spores per sporangium (rarely >32 irregular spores), indicating rangewide asexuality.
Plastid phylogeny and divergence times
The plastid trnGR region was successfully amplified and sequenced from 130 M. gracilis individuals, including 17 herbarium specimens (Appendix S1). The haplotype network resulting from the 1,052 site M. gracilis-only alignment indicated that 128/130 individuals exhibited one of two common haplotypes that differed at six sites (Fig. 2 inset). Two individuals exhibited a rare haplotype that differed from one of the common haplotypes by a single substitution. The geographic array of these three haplotypes is highly structured, as the two common haplotypes occupy the eastern or western portions of the M. gracilis range with overlap in AR, KS, and MO (Fig. 2). Clear exceptions to this distribution involved individuals exhibiting the “eastern” haplotype in Eddy Co., NM (Worthington 35037) and Weld Co., CO (Beck 1445), and an individual exhibiting the “western” haplotype in Iowa Co., WI (Beck 1312). The two individuals with the rare “northern” haplotype were both found in MN (Beck 1323, 1324).
Fig. 2.
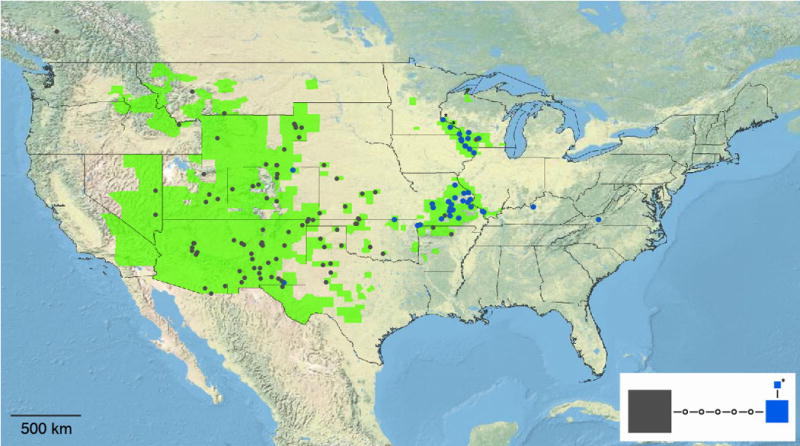
Plastid trnGR haplotype network (inset) and geographic distribution of the “western” (gray), “eastern” (blue), and “northern” (two MN individuals noted with asterisks) Myriopteris gracilis haplotypes.
The full Myriopteris alignment included 1,318 sites, with 235 sites removed. PartitionFinder identified the K80 and GTR+I+G models as most appropriate for the coding and non-coding portions of the trnGR region, respectively. The relaxed-clock/birth-death speciation tree analysis estimated the M. gracilis crown age at 2.26 mya (95% HPD: 0.72–4.16), and the crown age of the eastern + northern lineage at 0.67 mya (0.02–1.64) (Appendix S3). The trimmed Myriopteris alignment comprising one individual per species also included 1,318 sites, with 235 sites removed. PartitionFinder identified the K80 and general time-reversable with among-site gamma distribution (GTR+G) models as most appropriate for the coding and non-coding portions of the trnGR region. Post-burnin trees from the three BEAST runs identified a mean rate of 0.00127. The M. gracilis/M. lanosa/M. longifolia alignment included 1,158 sites, with 81 sites removed. PartitionFinder identified the K80 and GTR models as most appropriate for the coding and non-coding portions of the trnGR region. The relaxed-clock/coalescent tree prior analysis assuming the mean rate noted above estimated the M. gracilis crown age at 2.7 mya (0.74–5.02), and the crown age of the eastern + northern M. gracilis lineage at 0.67 mya (0.01–1.66).
The maximum clade credibility tree resulting from BEAST analysis of the full Myriopteris alignment (Appendix S3- Dryad Digital Repository: doi:10.5061/dryad.cs635) indicated that M. gracilis is monophyletic (1.0 Bayesian Posterior Probability- BPP). The slender lip fern was placed in a strongly supported clade (1.0 BPP) with M. lanosa and M. longipila, with M. lanosa and M. gracilis identified as sister taxa without support (0.42 BPP).
Genomic dataset
Both Illumina sequencing lanes produced >220 million useable barcoded reads (Elshire et al., 2011). Following tag identification and merging, 3,389,844 tags were identified. Following alignment, TASSEL identified 437,166 SNPs. Removal of individuals missing >80% of sites resulted in the retention of 73 individuals, and removing sites not present in all 73 individuals (0% missingness) resulted in a 17,581 site matrix. Of these, 16,480 (93.7%) sites were variable.
Three highly cohesive clusters of individuals are apparent in the plot of individual scores on the three most informative PCA axes (Fig. 3B). These same visually distinct groups were identified as highly ranked, statistically significant clusters by PCO-MC (Appendix S4) and received maximum support in likelihood/parsimony analyses (Fig. 3A). The most highly-ranked cluster (blue; stability = 90.72; Fig. 3B) comprised individuals from the Missouri Ozarks, the Driftless Zone (IA, IL, MN, WI), and the disjunct VA population (Fig. 4). All members of this genomically and morphologically distinctive (see below) blue cluster exhibited the eastern plastid haplotype or its rare northern variant (Fig. 2). In contrast, all but two of the remaining samples for which we have GBS data (Chatauqua Co., KS - Beck 1329; Weld Co., CO - Beck 1445) exhibited the western plastid haplotype (Fig. 2). The green cluster (six individuals; stability = 51.55; Fig. 3B) comprised individuals from the southern tier of the distribution (Fig. 4). Additional support for the green cluster was provided by trnGR indel variation not considered in the phylogenetic and haplotype analyses. Only eight of the 130 M. gracilis samples exhibited the “six A” character state at a poly-A region of the trnG intron: 5/6 green cluster individuals (the Chatauqua Co., KS sample exhibited the eastern haplotype) and three individuals for which GBS data were not available but which originate from the same geographic area as documented green cluster individuals (Wright Co., MO - Smith 4507; Lubbock Co., TX - Beck 1408; Baca Co., CO - Wickell 20). The red cluster (eight individuals; stability = 46.39; Fig. 3B) also comprised individuals from the southern tier of the M. gracilis range, particularly western TX and southern NM (Fig. 4). The next most stable cluster comprised all 45 remaining samples (stability = 26.80; Fig. 3B). These 45 samples originated from the western half of the M. gracilis range, although a specimen from Iowa Co., WI (Beck 1312) was a notable exception. Tree-based analyses establish that this large group is a conglomeration of multiple distinct clusters, with three pairs of samples also receiving maximum sister-group support in both likelihood and parsimony analyses. The first is a pair of samples from Sandoval (Wickell 26) and Sierra (Alexander 1431) Cos., NM, separated by 219 km (orange- Figs. 3, 4). The second is a pair of samples from Potter Co., TX (Beck 1381) and Union Co., NM (Wickell 24) separated by 167 km (purple- Figs. 3, 4). The third is a pair of samples from Guadalupe Co., NM (Wickell 41) and Hodgeman Co., KS (Wickell 02) separated by 532 km (teal- Figs. 3, 4). Although PCO-MC may not identify clusters of just two individuals, these three pairs were nevertheless each closely associated by PCA (Fig. 3B). These results and their relative geographic proximity strongly suggest these pairs represent additional distinct M. gracilis lineages. The remaining significant clusters identified by PCO-MC represent modified versions or combinations of those discussed above (Appendix S4).
Fig. 3.
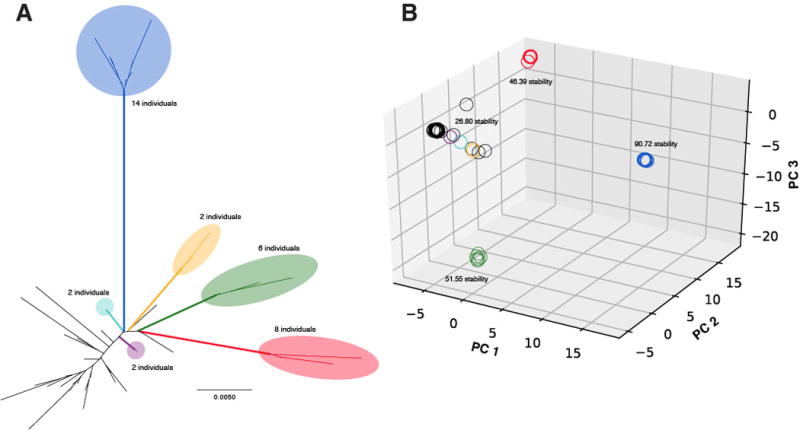
Results of tree-based and clustering analyses of 17,581 SNPs in 73 Myriopteris gracilis individuals. (A) Unrooted maximum likelihood phylogeny with clades receiving maximum support (100%) in both likelihood and parsimony analyses noted in bold colors. Colors correspond to lineages discussed throughout the text. (B) Plot of scores for each individual on the first three components obtained in a principal components analysis. Stability values derived from PCO-MC analysis are shown for four clusters.
Fig. 4.
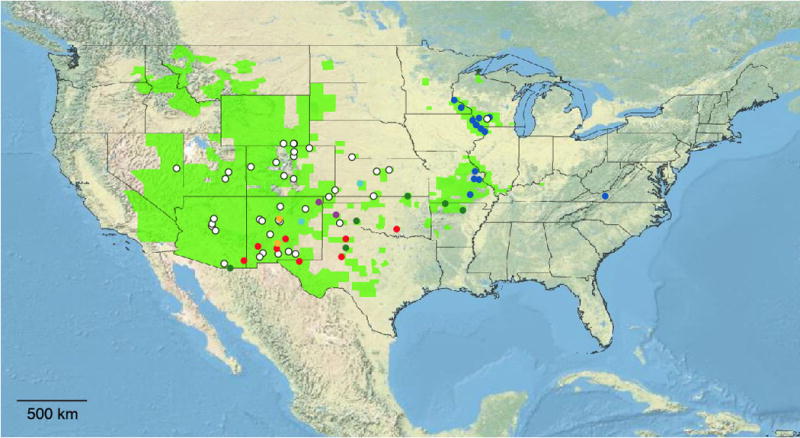
Geographic distribution of Myriopteris gracilis lineages identified by tree-based and clustering analyses of 17,581 SNPs in 73 individuals. Colors correspond to lineages outlined in Fig. 3, discussed in the text, and detailed in Appendix S1. Uncolored individuals in Fig. 3 are colored white for clarity.
Morphological dataset
Both rachis pubescence and pinna width were scorable on 67 of 73 specimens included in the full GBS analysis, including all 14 individuals assigned to the eastern lineage (blue; Figs. 3,4). Relative to the remaining material, these eastern individuals exhibited less pubescent rachises and significantly narrower pinnae (Fig. 5). Twelve of the 14 eastern-group individuals exhibited completely glabrous rachises (character state 1), with two individuals exhibiting widely scattered hairs (character state 2). Only three of the remaining 53 individuals exhibited completely glabrous rachises, all three of which were members of the green lineage. Eastern-group members exhibited significantly narrower pinna width (4.2 ± 0.7 mm) compared to western-group members (6.9 ± 1.5 mm) (Welch’s t-test: t = −9.692, df = 47.041, P = 8.62 × 10−13). The relatively narrow pinna collectively create an open, delicate appearance in leaves of eastern lineage individuals that is readily apparent (individuals N–U; Fig. 6). Notably, the Iowa Co., WI sample (Beck 1312) exhibiting both a western plastid and GBS profile displayed a western phenotype as well (arrow- Fig. 5).
Fig. 5.
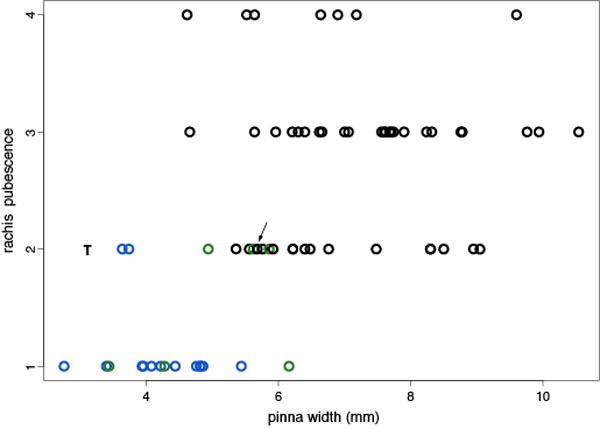
Bivariate plot of pinna width vs. rachis pubescence (1 = completely glabrous; 2 = exhibiting widely scattered hairs; 3 = generally hairy; 4 = densely hairy) for 67 of 73 Myriopteris gracilis specimens included in the full GBS analysis and an isotype specimen (T) for which only morphological data was assessed. Colors denote individuals assigned to the blue and green GBS clusters (see Figs. 3,4). The anomalous Iowa Co., WI sample is indicated with an arrow (see text).
Fig. 6.
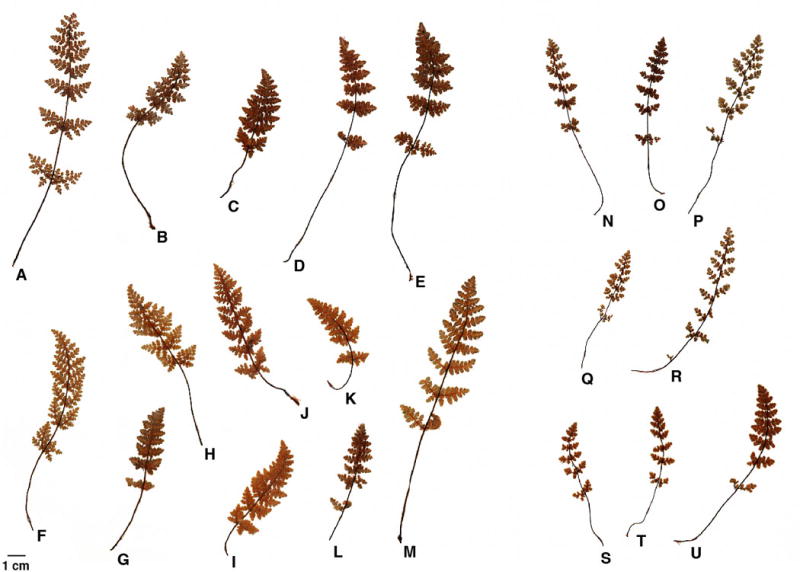
Example leaves illustrating the morphological distinction between the eastern lineage (N–U) and remaining Myriopteris gracilis material (A–M). Full details for all samples are presented in Appendix S1. (A) Wickell 56, Laramie Co., WY. (B) Beck 1442, Boulder Co., CO. (C) Wickell 44, Fremont Co., CO. (D) Beck 1445, Weld Co., CO. (E) Beck 1386, Ottowa Co., KS. (F) Alexander 1411, Yavapai Co., AZ. (G) Wickell 30, Cochise Co., AZ. (H) Wickell 37, Cibola Co., NM. (I) Alexander 1430, Luna Co., NM. (J) Wickell 41, Guadalupe Co., NM. (K) Alexander 1431, Sierra Co., NM. (L) Alexander 1452, Dona Aña Co., NM. (M) Beck 1435, Murray Co., OK. (N) Beck 1323, Winona Co., MN. (O) Beck 1319, Crawford Co., WI. (P) Beck 1317, Grant Co., WI. (Q) Beck 1327, Joe Daviess Co., IL. (R) Beck 1452, Jefferson Co., MO. (S) Wickell 14, Shannon Co., MO. (T) Wickell 5, Lincoln Co., MO. (U) Beck 1449, Pulaski Co., VA.
DISCUSSION
The absence of a slender lip fern sexual progenitor
The complete absence of sexuality among the 606 range-wide samples we examined suggests that the sexual diploid progenitor of M. gracilis is either extremely rare or no longer extant. Although our sampling along certain portions of the western edge of the distribution appears sparse (Fig. 1), the species is relatively uncommon in this region and previous attempts to identify sexual populations there have been unsuccessful (M. Windham, unpublished data). The apparent absence of sexual populations in M. gracilis is particularly conspicuous in light of numerous studies in which cryptic sexual populations have been discovered in species that were previously thought to be entirely asexual. Three prominent examples come from the “cheilanthoid” ferns, a monophyletic group of 400–500 species to which Myriopteris belongs (Windham et al., 2009). The smooth cliff-brake (Pellaea glabella Mett. ex Kuhn var. glabella), known from 23 states and two Canadian provinces, was long thought to be completely asexual until sexual populations were discovered in Missouri (Wagner et al., 1965). Two other Myriopteris species were, until quite recently, believed to be completely asexual. Grusz et al. (2009) reported the first discovery of rare sexual diploids of M. lindheimeri (Hook.) J. Sm., a species widely distributed from the southwest U.S. to central Mexico. Rare sexual populations of M. aurea (Poir.) Grusz & Windham have also been discovered. This species, ranging from the southwest U.S. to Argentina, was long thought to be uniformly asexual. However, application of the spore counting methodology used herein has led to the recent discovery of rare sexual populations in central Mexico (J. Beck, unpublished data). Non-plant examples include the fungus farming ant Mycoceperus smithii (Forel), in which sexual reproduction was inferred from the presence of sperm in females reproductive tracts despite the fact that the males of the species have yet to be identified (Rabeling et al., 2011). Ongoing cryptic sexual reproduction was also identified in 11 species of the gall-wasp genus Andricus (Hartig) by analysis of nuclear and mitochondrial allele frequencies (Stone et al., 2008).
Sexual progenitors are commonly found at lower latitudes relative to their asexual progeny (Lynch, 1984; Kearney, 2005; Hörandl, 2006), and the observation of increased M. gracilis lineage diversity near the southern limit of its U.S. range (AZ, NM and TX; Fig. 4) suggests that this region may have once harbored a diverse array of sexual populations. Future searches for rare sexual diploids should perhaps focus on the southernmost portion of the species’ range in southern NM, AZ, TX and the Mexican states of Chihuahua and Coahuila. Spore counts for this research were based on collections archived exclusively in U.S. herbaria; material archived in Mexican herbaria should be prioritized.
Two divergent lineages of the sexual ancestor
Two well-supported, geographically structured plastid lineages were observed in M. gracilis (Fig. 2), and the dating analysis estimated the divergence between these two groups at ca. 2.5 mya. This is a deep intraspecific divergence relative to those observed in other Myriopteris species. Multiple individuals of 15 species were included in the full dating analysis, and only Myriopteris lendigera J. Sm. (5.6 mya) and M. aurea (6.0 mya) exhibited older estimated crown ages (Appendix S3). Consistent with this relatively old divergence, a basic macro-morphological assessment established that the eastern slender lip fern lineage is phenotypically distinguishable from the more common western lineage (Figs. 5,6). We hypothesize that this divergence is a legacy of allopatry between eastern and western populations of the seemingly extinct sexual diploid progenitor of M. gracilis, as different asexual lineages arising in each region would then have received divergent plastomes. Although speculative, the distribution of the eastern lineage perhaps reflects range expansion during the Holocene Hypsithermal, a period of relative warm, dry conditions in North America centered around 7,000 years ago (Wright, 1976). These conditions brought an extension of prairie vegetation deep into the eastern deciduous forest (reviewed in Stuckey, 1981), increasing the habitability of the calcareous rock outcrops occupied by M. gracilis. Although the distinctiveness of the eastern and western lineages raises the question of formal taxonomic recognition, multiple factors run counter to this notion. If a new name were to be applied at either the specific or intraspecific rank, the original epithet would apply to the less common eastern taxon. An isotype specimen (Riehl 529, US; Grusz and Windham, 2013) is morphologically typical of the eastern “blue” lineage (Fig. 5), and a specimen assigned to the eastern lineage by both plastid and GBS datasets (Beck 1452) was collected ca. 15 km from the type location. Even if the eastern lineage is viewed as the result of a single origin, the remaining material comprises at least six lineages (Fig. 3). As an assemblage of evolutionarily independent lineages, recognizing the remaining western material as a single taxon would not be appropriate (de Queiroz, 2007).
Lineage diversity and the appearance of short-term success
The plastid data alone, which provide relatively limited insight into the recurrent formation of asexual lineages, indicate that M. gracilis represents a genetically diverse assemblage. The GBS data, presenting information from thousands of locations throughout the genome, provide a considerably more detailed picture of lineage diversity. Multiple genetic groups receive maximum support in phylogenetic analyses (Fig. 3A), many of which are evident in the three-dimensional PCA plot (Fig. 3B), with the larger groups identified as statistically significant and highly cohesive in multidimensional space by PCO-MC (Appendix S4). The genetic distinctiveness of these lineages, combined with morphological divergence and distributional trends in some cases, provide compelling evidence that M. gracilis comprises a diversity of multiple asexual lineages formed at different times, with each origin capturing and propagating a particular genotype of the sexual ancestor. If the unassigned individuals (uncolored in Fig. 3, white in Fig. 4) are conservatively viewed as a single cluster, a minimum of seven independent origins of M. gracilis can be inferred. This assessment is undoubtedly an underestimate of true lineage diversity, as both the six established lineages and the group of unassigned individuals exhibit additional variation. The lack of phylogenetic and multivariate clustering support for the group of unassigned individuals (Fig. 3A, Appendix S4) strongly suggests that this assemblage harbors additional lineages, as do certain cyto-nuclear patterns. Both unassigned (Beck 1445; Weld Co., CO) and green cluster individuals (Beck 1329; Chatauqua Co., KS) exhibited eastern plastid genomes (Fig. 2), suggesting additional origins from a sexual ancestor with these unique genomic characteristics.
With the discovery that the apomictic triploid fern Myriopteris gracilis comprises multiple genetic lineages, the species emerges as a prominent example of recurrent polyploid formation (Soltis et al., 2003) and lineage diversity in asexual taxa (Neiman et al., 2009). While our research shows once again that asexual taxa are capable of geographic success in the absence of a sexual progenitor, many questions regarding the evolutionary history and ecology of M. gracilis remain unanswered. Future research could include establishing the rate of lineage formation by estimating the relative age of all independent asexual lineages. This would require a rigorous, dated, intraspecific nuclear phylogeny, an effort that is complicated by the fact that M. gracilis is a polyploid derived from an extinct ancestor. The evolutionary history of individual M. gracilis nuclear genomes therefore departs considerably from a standard dichotomously branching tree, as the formation of an asexual lineage could potentially unite nuclear genomes from different portions of the sexual diploid phylogeny (Birky, 1996). Although a tree reconstructed from plastid sequences offers the opportunity to isolate and reconstruct the evolutionary history of the maternal portion of an asexual lineage’s genome, our single plastid locus results indicate that a whole-plastome sequencing approach is needed. Future efforts should also include evaluating phenotypic differentiation among independent M. gracilis lineages and the fit between location and genotype, a largely unexplored aspect of asexual evolution (Whitton et al., 2008; but see Stevens and Emery, 2015; Chambers and Emery, 2016).
Returning to the question of whether an obligate asexual is capable of short term success, the answer in the case of the slender lip fern is “yes, but…” Yes, the geographic distribution of M. gracilis comprises entirely asexual individuals, but this total range overstates the success of any of the recurrently formed lineages we identified. The occupation of large portions of the slender lip fern range by individual asexual lineages is nevertheless impressive. While restricted to the eastern portion of the range, the eastern (blue) lineage ranges from the Ozark Plateau north into the Driftless zone, with disjunct populations in KY and NC. Even if we only consider individuals for which we have GBS data, this range spans almost 1500 km from the northernmost population in Winona Co, MN (Beck 1323) to the easternmost collection in Pulaski Co., VA (Beck 1449). The green lineage is similarly impressive, documented from semi-xeric habitats in Cochise Co., AZ (Wickell 31) to mesic sites in the Arkansas Ozarks (Beck 1339, 1344), over 1700 km away (Fig 4). If either of these lineages indeed represent a single origin, they are perhaps what many researchers refer to as a “general purpose genotype,” capable of surviving in a wide range of conditions despite little genotypic variation (Lynch, 1984). The Iowa Co., WI specimen (Beck 1312) is also intriguing given that it is part of the largely western/southwestern unassigned group. As such, it likely shares a genome with samples from much drier sites. Ultimately however, while a number of its constituent lineages appear to have been quite successful when viewed in isolation, their success is diminished when placed in the context of M. gracilis’ total range. If this situation proves to be common among widespread asexual species, then our view of asexuality as an advantagous short-term strategy will require adjustment, perhaps with a shift in focus to the biogeography, ecology, and evolution of individual constituent lineages.
Supplementary Material
{kind=link}
Footnotes
This research was funded by the Wichita State University Department of Biological Sciences, an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences (P20 GM103418) from the National Institutes of Health, and by grants from the Kansas Academy of Science and the American Society of Plant Taxonomists. Research reported in this study was made possible in part by the services of the KU Genome Sequencing Core Laboratory. This lab is supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health under award number P20GM103638. The authors would like to thank Mary Liz Jameson for helpful insights, Patrick Alexander, Carl Rothfels, María Hilda Flores Olvera, and George Yatskievych for field collections, Gloria Caddell (Selman Living Laboratory) and Tara Littlefield (Kentucky State Nature Preserves Commission) for help in the field, Carl Rothfels and Fay-Wei Li for help with PURC, and Amanda Grusz for examining the isotype specimen. Thanks also to Amanda Grusz and two anonymous reviewers for helpful comments on a draft of this manuscript. Thanks to the Arizona and New Mexico Bureaus of Land Management; Kansas Department of Wildlife, Parks, and Tourism; Black Hills, Mark Twain, Ouachita, and Ozark National Forests; Minnesota Department of Natural Resources; Missouri Department of Conservation; Texas Parks and Wildlife Department; U.S. Forest Service Intermountain, Rocky Mountain, and Southwestern Regions; Wisconsin Department of Natural Resources; and the Virginia Department of Conservation and Recreation for collection permits. A special thanks to the curators of ARIZ, BRIT, COLO, CS, DUKE, JEPS, KANU, KSC, MO, NCU, NMC, OKLA, RENO, RM, UC, US, and UTC for permission to examine herbarium specimens.
LITERATURE CITED
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Beck JB, Allison JR, Pryer KM, Windham MD. Identifying multiple origins of polyploid taxa: a multilocus study of the hybrid cloak fern (Astrolepis integerrima; Pteridaceae) American Journal of Botany. 2012;99:1857–1865. doi: 10.3732/ajb.1200199. [DOI] [PubMed] [Google Scholar]
- Beck JB, Windham MD, Yatskievych G, Pryer KM. A diploids-first approach to species delimitation and interpreting polyploid evolution in the fern genus Astrolepis (Pteridaceae) Systematic Botany. 2010;35:223–234. [Google Scholar]
- Bell G. The Masterpiece of Nature. University of California Press; Berkeley, California, USA: 1982. [Google Scholar]
- Bierzychudek P. Patterns in plant parthenogenesis. Experientia. 1985;41:1255–1264. doi: 10.1007/978-3-0348-6273-8_9. [DOI] [PubMed] [Google Scholar]
- Birky CW. Heterozygosity, heteromorphy, and phylogenetic trees in asexual eukaryotes. Genetics. 1996;144:427–437. doi: 10.1093/genetics/144.1.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- Brunton D. The Wooly lip-fern (Cheilanthes feei Moore) in Alberta. Alberta Naturalist. 1979;9:121–125. [Google Scholar]
- Buffalo V. Scythe: A Bayesian adapter trimmer. 2015 Available at website GitHub repository: https://github.com/vsbuffalo/scythe.
- Chambers SM, Emery NC. Population differentiation and countergradient variation throughout the geographic range in the fern gametophyte Vittaria appalachiana. American Journal of Botany. 2016;103:86–98. doi: 10.3732/ajb.1500077. [DOI] [PubMed] [Google Scholar]
- Clark B. Earth Point: Tools for Google Earth. 2015 Available at: www.earthpoint.us.
- Clement M, Posada D, Crandal KA. TCS: a computer program to estimate gene genealogies. Molecular Ecology. 2000;9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- De Queiroz K. Species concepts and species delimitation. Systematic Biology. 2007;56:879–886. doi: 10.1080/10635150701701083. [DOI] [PubMed] [Google Scholar]
- Döpp W. Cytologische und genetische Untersuchungen innerhalb der Gattung Dryopteris. Planta. 1939;29:481. [Google Scholar]
- Drummond AJ, Suchard MA, Xie D D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond AJ, Bouckaert RR. Bayesian Evolutionary Analysis with BEAST 2. Cambridge University Press; Cambridge, UK: 2015. [Google Scholar]
- Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. A robust, simple genotyping by sequencing (GBS) approach for high diversity species. PLoS ONE. 2011;6:e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, Buckler ES. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE. 2014;9:e90346. doi: 10.1371/journal.pone.0090346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glesner RR, Tilman D. Sexuality and the components of environmental uncertainty: clues from geographic parthenogenesis in terrestrial animals. American Naturalist. 1978;112:659–673. [Google Scholar]
- Grusz AL. A current perspective on apomixis in ferns. Journal of Systematics and Evolution. 2016;54:656–665. [Google Scholar]
- Grusz AL, Windham MD. Toward a monophyletic Cheilanthes: the resurrection and recircumscription of Myriopteris (Pteridaceae) PhytoKeys. 2013;32:49–64. doi: 10.3897/phytokeys.32.6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grusz AL, Windham MD, Pryer KM. Deciphering the origins of apomictic polyploids in the Cheilanthes yavapensis complex (Pteridaceae) American Journal of Botany. 2009;96:1636–1645. doi: 10.3732/ajb.0900019. [DOI] [PubMed] [Google Scholar]
- Grusz AL, Windham MD, Yatskievych G, Huiet L, Gastony GJ, Pryer KM. Patterns of diversification in the xeric-adapted fern genus Myriopteris (Pteridaceae) Systematic Botany. 2014;39:698–714. [Google Scholar]
- Haufler CH, Pryer KM, Schuettpelz E, Sessa EB, Farrar DR, Moran R, Schneller JJ, et al. Sex and the single gametophyte; revising the homosporous vascular plant life cycle in the light of contemporary research. Bioscience. 2016;66:928–937. [Google Scholar]
- Hojsgaard D, Pellino M, Sharbel TF, Hörandl E. Resolving genome evolution patterns in asexual plants. In: Hörandl E, Appelhans MS, editors. Next Generation Sequencing in Plant Systematics. Koeltz Scientific Books; Königstein, Germany: 2015. pp. 119–153. [Google Scholar]
- Hörandl E. The complex causality of geographical parthenogenesis. New Phytologist. 2006;171:525–538. doi: 10.1111/j.1469-8137.2006.01769.x. [DOI] [PubMed] [Google Scholar]
- Hunter JD. Matplotlib: a 2D Graphics Environment. Computing in Science & Engineering. 2007;9:90–95. [Google Scholar]
- Janko K. Let us not be unfair to asexuals: Their ephemerality may be explained by neutral models without invoking any evolutionary constraints of asexuality. Evolution. 2014;68:569–576. doi: 10.1111/evo.12293. [DOI] [PubMed] [Google Scholar]
- Joshi NA, Fass JN. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files. 2011 Available at website GitHub repository: https://github.com/najoshi/sickle.
- Kartesz JT. North American Plant Atlas. Chapel Hill, N.C: 2015. The Biota of North America Program (BONAP) ( http://bonap.net/napa) [Google Scholar]
- Kearney M. Hybridization, glaciation and geographical parthenogenesis. Trends in Ecology & Evolution. 2005;20:495–502. doi: 10.1016/j.tree.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Lanfear R, Calcott B, Ho SY, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Molecular Biology and Evolution. 2012;29:1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundmark M, Saura A. Asexuality alone does not explain the success of clonal forms in insects with geographical parthenogenesis. Hereditas. 2006;143:23–32. doi: 10.1111/j.2006.0018-0661.01935.x. [DOI] [PubMed] [Google Scholar]
- Lynch M. Destabilizing hybridization, general purpose genotypes and geographic parthenogenesis. Quarterly Review of Biology. 1984;59:257–290. [Google Scholar]
- Manton I. Problems of Cytology and Evolution in the Pteridophyta. University Press; Cambridge, UK: 1950. [Google Scholar]
- Maynard Smith J. The Evolution of Sex. Cambridge University Press; Cambridge, UK: 1978. [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees; Proceedings of the Gateway Computing Environments Workshop (GCE); 14 Nov. 2010; New Orleans, Louisiana, USA. 2010. pp. 1–8. [Google Scholar]
- Neiman M, Meirmans S, Meirmans PG. What can asexual lineage age tell us about the maintenance of sex? Annals of the New York Academy of Sciences. 2009;1168:185–200. doi: 10.1111/j.1749-6632.2009.04572.x. [DOI] [PubMed] [Google Scholar]
- Otto SP, The evolutionary enigma of sex The American Naturalist. 2009;174:S1–S14. doi: 10.1086/599084. [DOI] [PubMed] [Google Scholar]
- Palmieri M. MS thesis. Southeast Missouri State University; Cape Girardeau, Missouri, USA: 2001. Presence and distribution of fungal symbiosis in Cheilanthes feei and Cheilanthes lanosa in southeastern Missouri and southern Illinois. [Google Scholar]
- Potts AJ, Hedderson TA, Grimm GW. Constructing phylogenies in the presence of intra-individual site polymorphisms (2ISPs) with a focus on the nuclear ribosomal cistron. Systematic Biology. 2014;63:1–16. doi: 10.1093/sysbio/syt052. [DOI] [PubMed] [Google Scholar]
- Rabeling C, Gonzales O, Schultz TR, Bacci M, Garcia MV, Verhaagh M, Mueller UG, et al. Cryptic sexual populations account for genetic diversity and ecological success in a widely distributed, asexual fungus-growing ant. Proceedings of the National Academy of Sciences. 2011;108:12366–12371. doi: 10.1073/pnas.1105467108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. FigTree v1.4. 2012 Available from http://tree.bio.ed.ac.uk/software/figtree/
- Rambaut A, Suchard MA, Xie D, Drummond AJ. Tracer v1.6. 2014 Available from http://beast.bio.ed.ac.uk/Tracer.
- Reeves PA, Richards CM. Accurate inference of subtle population structure (and other genetic discontinuities) using principal coordinates. PLoS ONE. 2009;4:e4269. doi: 10.1371/journal.pone.0004269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitzel AM, Herrera S, Layden MJ, Martindale MQ, Shank TM. Going where traditional markers have not gone before: utility of and promise for RAD sequencing in marine invertebrate phylogeography and population genomics. Molecular Ecology. 2013;22:2953–2970. doi: 10.1111/mec.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross L, Hardy NB, Okusu A, Normark BB. Large population size predicts the distribution of asexuality in scale insects. Evolution. 2013;67:196–206. doi: 10.1111/j.1558-5646.2012.01784.x. [DOI] [PubMed] [Google Scholar]
- Rothfels CJ, Sigel EM, Windham MD. Cheilanthes feei T. Moore (Pteridaceae) and Dryopteris erythrosora (D.C. Eaton) Kunze (Dryopteridaceae) new for the flora of North Carolina. American Fern Journal. 2012a;102:184–186. [Google Scholar]
- Rothfels CJ, Larsson A, Kuo LY, Korall P, Chiou WL, Pryer KM. Overcoming deep roots, fast rates, and short internodes to resolve the ancient rapid radiation of eupolypod II ferns. Systematic Biology. 2012b;61:490–509. doi: 10.1093/sysbio/sys001. [DOI] [PubMed] [Google Scholar]
- Rothfels CJ, Pryer KM, Li F-Wei. Next-generation polyploid phylogenetics: rapid resolution of hybrid polyploid complexes using PacBio single-molecule sequencing. New Phytologist. 2017;213:413–429. doi: 10.1111/nph.14111. [DOI] [PubMed] [Google Scholar]
- RStudio Team. RStudio: Integrated Development for R. RStudio, Inc; Boston, MA: 2015. [Google Scholar]
- Schwander T, Crespi BJ. Twigs on the tree of life? Neutral and selective models for integrating macroevolutionary patterns with microevolutionary processes in the analysis of asexuality. Molecular Ecology. 2009;18:28–42. doi: 10.1111/j.1365-294X.2008.03992.x. [DOI] [PubMed] [Google Scholar]
- Schuettpelz E, Pryer KM. Evidence for a Cenozoic radiation of ferns in an angiosperm dominated canopy. Proceedings of the National Academy of Sciences. 2009;106:11200–11205. doi: 10.1073/pnas.0811136106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettpelz E, Grusz AL, Windham MD, Pryer KM. The utility of nuclear gapCp in resolving polyploid fern origins. Systematic Botany. 2008;33:621–629. [Google Scholar]
- Sigel EM, Windham MD, Haufler CH, Pryer KM. Phylogeny, divergence time estimates, and phylogeography of the diploid species of the Polypodium vulgare complex (Polypodiaceae) Systematic Botany. 2014;39:1042–1055. [Google Scholar]
- Silvertown J. The evolutionary maintenance of sexual reproduction: evidence from the ecological distribution of asexual reproduction in clonal plants. International Journal of Plant Sciences. 2008;169:157–168. [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones STM, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Research. 2009;19:1117–1123. doi: 10.1101/gr.089532.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis DE, Soltis PS, Tate JA. Advances in the study of polyploidy since Plant Speciation. New Phytologist. 2003;161:173–191. [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens SM, Emery NC. Dispersal limitation and population differentiation in performance beyond a northern range limit in an asexually reproducing fern. Diversity and Distributions. 2015;21:1242–1253. [Google Scholar]
- Stone GN, Atkinson RJ, Rokas A, Aldrey JLN, Melika G, Acs Z, McVean GA, et al. Evidence for widespread cryptic sexual generations in apparently purely asexual Andricus gallwasps. Molecular Ecology. 2008;17:652–665. doi: 10.1111/j.1365-294X.2007.03573.x. [DOI] [PubMed] [Google Scholar]
- Stuckey RL. Origin and development of the concept of the Prairie Peninsula. In: Stuckey RL, Reese KJ, editors. Proceedings of the Sixth North American Prairie Conference, The Prairie Peninsula-in the “shadow” of Transeau. The Ohio State University; Columbus, Ohio, USA: 1981. pp. 4–23. [Google Scholar]
- Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (*and other methods) Sinauer Associates; Sunderland MA: 2002. [Google Scholar]
- Tilquin A, Kokko H. What does the geography of parthenogenesis teach us about sex? Philosophical Transactions of the Royal Society B. 2016;371:20150538. doi: 10.1098/rstb.2015.0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk PJ. Apomixis: basics for non-botanists. In: Schön I, Martens K, van Dijk P, editors. Lost Sex; the Evolutionary Biology of Parthenogenesis. Springer; New York, New York, USA: 2009. pp. 47–62. [Google Scholar]
- Wagner. Biosystematics and evolutionary noise. Taxon. 1970;19:146–151. [Google Scholar]
- Wagner WH, Farrar DR, Chen KL. A new sexual form of Pellaea glabella var. glabella from Missouri. American Fern Journal. 1965;4:171–178. [Google Scholar]
- Walker TG. The cytogenetics of ferns. In: Dyer AF, editor. The experimental biology of ferns. Academic Press; London, UK: 1979. pp. 87–132. [Google Scholar]
- Whitton J, Sears CJ, Baack EJ, Otto SP. The dynamic nature of apomixis in the angiosperms. International Journal of Plant Sciences. 2008;169:169–182. [Google Scholar]
- Williams GC. Sex and Evolution. Princeton University Press; Princeton, New Jersey, USA: 1975. [Google Scholar]
- Windham MD, Rabe EW. Cheilanthes. In: Flora of North America Editorial Committee, editor. Flora of North America. Vol. 2. Oxford University Press; New York, New York, USA: 1993. pp. 152–169. [Google Scholar]
- Windham MD, Yatskievych G. Chromosome studies of cheilanthoid ferns (Pteridaceae, Cheilanthoideae) from the western United States and Mexico. American Journal of Botany. 2003;90:1788–1800. doi: 10.3732/ajb.90.12.1788. [DOI] [PubMed] [Google Scholar]
- Windham MD, Huiet L, Schuettpelz E, Grusz AL, Rothfels C, Beck J, Yatskievych G, Pryer KM. Using plastid and nuclear DNA sequences to redraw generic boundaries and demystify species complexes in cheilanthoid ferns. American Fern Journal. 2009;99:128–132. [Google Scholar]
- Wright HE. The dynamic nature of holocene vegetation, a problem in paleoclimatology, biogeography, and stratigraphic nomenclature. Quaternary Research. 1976;6:581–596. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.