

Abstract
Alpha-synuclein (aS) forms toxic intermediates ranging from small oligomers and protofibrils to large amyloid fibrils. Understanding the time course of aS fibril formation and the role played by its regions is critical for therapeutic intervention. Here, we used pulsed hydrogen-deuterium-exchange and mass spectrometry (HDX-MS) for the first time to probe kinetic intermediates of the full aS aggregation in vitro, achieving kinetic snapshots containing spatially resolved protein information about critical stages. Monitoring the resultant mass shifts show distinct binomial abundances for two main exchange profiles, one that represents a fast-exchanging, solvent-accessible species and another with a more protected nature. We show using a series of proteolytic peptides from the full protein that self-association is most pronounced in the NAC region and less so for either termini. The N-terminus, however, shows a minor protected population at mid- and late times, whereas the C-terminus shows predominantly unimodal HDX, indicating that these regions are devoid of any large conformational rearrangements. Focusing on the hydrophobic core, we confirmed and modeled the different isotopic distributions and calculated their relative fractions to discern their individual contributions. The data fitting reports respective t1/2 values, which are nearly identical and do not depend on location. We followed the aggregation by complementary transmission electron microscopy to observe the morphology of aggregates and circular dichroism to assess changes in secondary structure. Our results provide a detailed picture of aS aggregation in vitro, and demonstrate that HDX-MS offers unique spatially resolved, co-existing kinetic intermediates in solution. This new platform is suitable for testing promising inhibitors of aS aggregation.
Keywords: Pulsed hydrogen-deuterium exchange, mass spectrometry, bimodal HDX profiles, amyloids, aggregation kinetics, kinetic modeling
Graphical Abstract
Introduction
A key feature of Parkinson’s disease is the formation and deposition of amyloid fibrils comprised of alpha-synuclein (aS). In the most prevalent, sporadic form of PD, the major constituent of Lewy bodies and Lewy neurites is aggregated aS.1, 2, 3 Under physiological conditions, aS is expressed most abundantly at synapses and performs multiple functions in the cell.4, 5 In the cellular environment, aS can undergo a wide array of post-translational modifications, importantly N-terminal acetylation, that modulate and influence its biological and pathological properties.5, 6, 7, 8 Although aS, a 140 residue protein, is natively unstructured, it has a high propensity to misfold owing to changes in its local chemical or cellular environment, and to form neurotoxic oligomers, protofibrils, and insoluble β-sheets containing fibrillary structures.9
Numerous biochemical and biophysical studies in vitro have focused on understanding the causative events triggering its structural transitions, and on defining the conformational rearrangements leading to its self-assembly.10, 11, 12, 13, 14 Although circular dichroism (CD), Fourier Transfer Infrared spectroscopy (FTIR), Electron Paramagnetic Resonance (EPR), small angle X-ray scattering (SAXS), transmission electron microscopy (TEM), atomic force microscopy (AFM), super-resolution microscopy, among others, provide opportunities to interrogate these structures on a global, full-protein level,15 they provide little regional information. High resolution X-ray crystallography, cryo-EM, and NMR can give atomic-level resolution typically at several mg/mL protein concentration, but they are challenged when investigating IPDs that present polymorphism and/or a complex ensemble with heterogeneous and/or dynamic, transient conformational properties. Recent developments in solid-state NMR have enabled acquisition of a high resolution structure of aS fibrils with unprecedented detail,16 showing a topology similar to a Greek key. This approach reports on the end-stage or mature form but not on intermediate states. Thioflavin-T (ThT) fluorescence and single-molecule fluorescence measure kinetic changes by using spectral properties of fluorophores15 but do not yield any regional or peptide-level information.
As a complementary analytical tool, HDX-MS can address stability, folding and misfolding, protein-protein interactions, and ligand binding, affording structural information for the full protein, providing insights at the near-residue level.17, 18 With regard to aS, continuous HDX studies provide information about the solvent accessibility and H-bonding of key intermediates ranging from the monomeric protein to the fully formed late stage fibrils19 including a subset of diverse, mutant oligomers20 or oligomers formed from the wild type protein.21 By using a variable, narrow deuteration time window with respect to aggregation, stable or metastable structures can be followed. Continuous HDX, however, is compromised by the slow aggregation of aS where the kinetics of HDX are fast and outpace the aggregation whereas for fast aggregation (e.g., that of Aβ42), the kinetics are convolved with those of aggregation.
Thus, another avenue is needed to capture the regional-dependent, self-assembly of amyloid proteins as a function of aggregation time. To this end, we implemented pulsed HDX of aS to monitor structural transitions as a function of time under standard in vitro aggregation conditions (pH, concentration, agitation). The merits of this approach, developed previously in our laboratory, are exemplified in studies of other amyloidogenic proteins such as Aβ4222, CsgE 23 and CsgA.24
To examine the events of aS aggregation, we monitored its time-dependent aggregation and found remarkably high HDX protection in the central, non-amyloid-β-component (NAC) and nearby regions, pinpointing the residues driving prominent aggregation. We observed that the N and C-termini do not gain significant protection and, thus, are only moderately affected or involved in the misfolding. Moreover, we observed two populations for the NAC region, one rapidly and another slowly exchanging, as reflected by HDX bimodal distributions whose relative intensities shift as the aggregation progresses. The occurrence of these binomial distributions is not unique to aS, and seems to be a characteristic signature of amyloidogenic proteins undergoing conformational rearrangements.19, 20, 21, 23, 25
Here, we describe the different aggregation stages of aS as a function of time and provide regional information of the critical residues involved in amyloid formation. In so doing, we demonstrate again the utility of pulsed HDX-MS as a platform for following amyloid formation.
Results and Discussion
aS encompasses three main structural domains: the aliphatic, N-terminal terminal domain (residues 1-60) that binds to lipids and proteins, the aggregation-prone, hydrophobic Non-Amyloid-β-Component (NAC) region (residues 61-95), and the highly acidic, proline-rich C-terminal domain (residues 96-140),26 as displayed in Figure 1.
Figure 1.

The three main structural domains of wild type aS. The alipathic N- terminus (1–60, brown) is followed by the central, hydrophobic core (61–95, orange) with putatively a high propensity for aggregation, and the acidic C-terminus (96–140, blue). The corresponding amino acid sequence is shown below the image from residue 1 to 140.
Its aggregation depends on a number of factors including, for example, familial point mutations,27 concentration, pH, buffer conditions, temperature, the presence or absence of co-factors, crowding effects, and post-translational modifications.28 Amyloid formation is a multi-step process, involving nucleation, growth, and elongation, and in some instances is followed by underlying secondary processes including fragmentation, as discussed in a recent review.15 Although considerable knowledge has been accumulated on the aggregation kinetics of aS, novel methods are actively sought to address the specifics of its aggregation landscape, and evaluate the effects of potential inhibitors (e.g., small molecules and drug candidates) that can halt or re-direct the aggregation pathway toward non-cytotoxic species.
Pulsed HDX has shown promise in probing alternate conformational features in the aggregation of Aβ4222 and various permutations of curli.23, 24 To examine the events of misfolding and to address which peptic residues play critical roles in the aggregation of aS, we chose to apply pulsed HDX here by collecting samples at various points during in vitro aS aggregation (see Methods section) performed at 80 μM protein concentration, neutral pH, 37 °C with continuous agitation by a 2 mm glass stir bead, starting from a solution containing monomerized aS. To report on self-assembly, we submitted samples to a short, 1 min pulse of D2O, quenched the HDX by adding 3 M urea with 1% TFA, decreasing the pH 2.5 to dissociate to monomers, and then enzymatically digested by porcine pepsin to generate peptides, which were desalted and eluted by reversed-phase HPLC for MS detection.
Pulsed HDX-MS pattern of aS indicates that aggregation primarily takes place in the NAC
Given the disordered and dynamic nature of aS, one expects high and rapid deuterium exchange even at short times, indicating high solvent accessibility and minimal H bonding.29 When aS self-assembles, however, the solvent-accessibility of exchangeable amide residues decreases as a measure of misfolding and aggregation, and concomitantly the residues become less dynamic. This change in solvent accessibility is revealed in spatial detail over both the entire sequence and the time course of aggregation. This encouraged us to obtain a detailed map of aS aggregation that can provide fresh insights about the structural determinants governing aS aggregation. As expected, at the starting time points, aS is very open, showing little protection and high solvent accessibility all across its sequence. There is some moderate residual structure for particular segments or protein regions, as revealed by initial uptakes of < 85%. This is in line with another observation that at neutral pH, aS displays a smaller Stokes-radius than expected for a disordered protein, as determined by SEC, pointing to some residual structure. The HDX result is also consistent with NMR diffusion and radius of gyration (Rg) measurements obtained by a Guinier approximation of SAXS data.10 Native mass spectrometry measurements also describe the structural plasticity of aS with multiple conformers that can be modulated by changes in pH30, or by various aggregation conditions.31 Moreover, nano-electrospray ionization (nano-ESI) mass spectrometry showed that other co-factors, such as metals and alcohols can influence the conformational equilibrium of alternate conformers32.
We choose for presentation one representative peptic peptide from each structural domain to illustrate the regional behavior of the full protein during aggregation. For example, although the N-terminus undergoes considerable HDX in 1 min pulse, typical of a disordered region, the HDX remains nearly constant for the entire aggregation time course. The isotopic distribution of peptide 5–17 (+1), is unimodal up to 31 h, and thereafter, it displays moderate bimodality for ≥ 53 h, suggesting that misfolding eventually propagates onto this region (Figure 2A; the full time series for HDX of this region of the protein is shown in Figure S2 in the Supporting Information). The relative contribution of the more protected population remains below 0.15. Previously, both Mysling et al.21 and Paslawski et al.,20 using continuous HDX, assigned the N-terminal region in aS oligomers to be protected. Solid-state NMR of aS fibrils indicates rigidity on the N-terminus from residue 22 onward.33
Figure 2.

Mass spectra of peptide 5–17 (+1; MKGLSKAKEGVVA) indicating mass shifts at various time points of the aggregation reaction as compared to an undeuterated control at the top left (blue spectrum). The isotopic envelopes exist as binomial distributions with a very small, protected second component ≥ 53 h. B. Mass spectra of peptide 95–113 (+2; VKKDQLGKNEEGAPQEGIL) at various time points of the aggregation as compared to the undeuterated control at the top left (blue spectra). The isotopic envelopes remain predominantly unimodal.
The C-terminus remains solvent-exposed throughout the entire aggregation time, as exhibited by the mass-shifted, predominantly unimodal envelopes indicating extensive HDX, as illustrated by a representative peptide, 95–113 (+2) (Figure 2B; the complete time series of this peptide is shown in Figure S3 in the Supporting Information). This is in agreement with NMR results showing that at least 35 residues on the C-terminal remain flexible and disordered for mature aS fibrils.33, 34
In contrast, our data for region 55–76 demonstrate that aS misfolds following a lag phase where initiating steps of nucleation give rise to larger conformational transitions and cause changes in hydrogen bonding reflected by the resultant HDX pattern (Figure 3; the full time series for HDX of this region of the protein is shown in Figure S4 in the Supporting Information). This 55–76 region includes the boundary of the N-terminus and a part of the central, hydrophobic domain. We found multimodal distributions for this region suggestive of oligomerization and self-assembly, whereby a population develops containing amide hydrogens that exchange with deuterium less rapidly than the initially appearing, rapidly exchanging population. This indicates that there are at least two classes of protein molecules in solution: one whose residues become buried (or tightly H bonded) during aggregation and a second that has higher solvent exposure and exchanges faster. The latter group is referred to as species 1, whereas the fast-exchanging population is denoted as species 2. Although we cannot rule out multiple conformers in each of these two distinct populations, we can clearly resolve two pools of exchange profiles.
Figure 3.
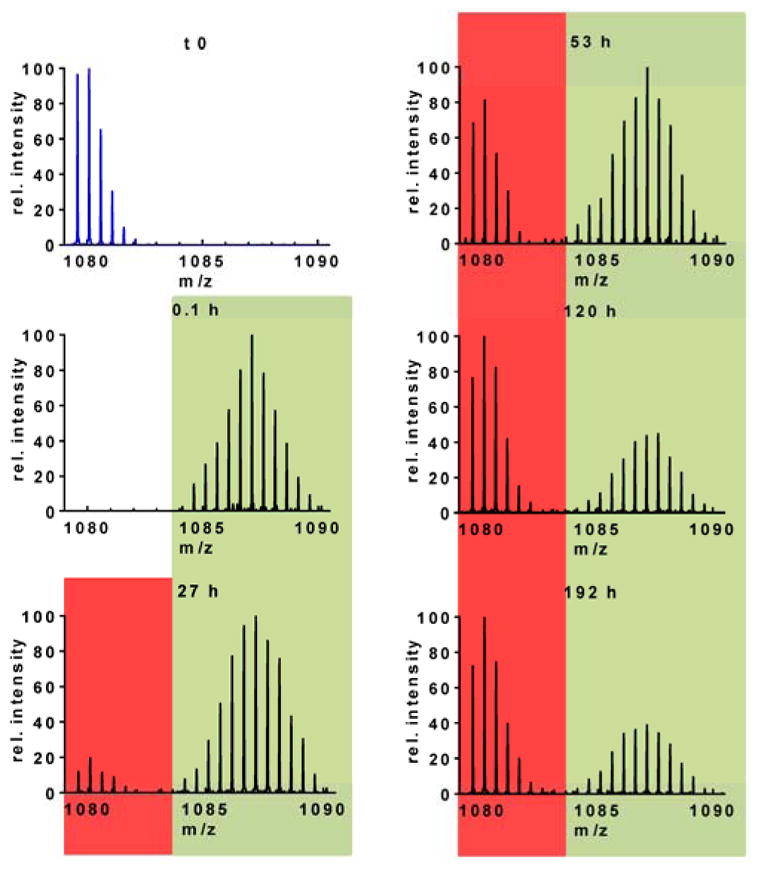
Mass spectra during HDX covering peptide 55–76 (+2; VAEKTKEQVTNVGGAVVTGVTA) indicating mass shifts at various time points of the aggregation reaction as compared to the undeuterated control at the top left (blue spectrum). The isotopic envelopes exhibit a binomial distribution shaded with both red (species 1) and green (species 2). The protected, slow exchanging population is shaded by red bars and denoted as species 1, whereas the fast exchanging, initial population that gradually begins to deplete in abundance is shaded in green and referred to as species 2.
Kinetics of aS aggregation involving the hydrophobic region
HDX shows that the aggregation is driven by the hydrophobic NAC region, in agreement with previous findings by complementary biophysical methods.35, 36, 37 Supporting our conclusion is the previous continuous HDX of aggregated fibrils that show a dramatic reduction in solvent accessibility in the region of 39–97.19 Only subsequent studies focusing on the oligomeric intermediates, however, describe binomial distributions.20, 21
This exchange outcome is similar to that of an EX1 regime, where alternate protein states can be detected based on their deuterium content whose intrinsic exchange rate is greater than that of the closing rate of protein refolding, revealing kinetic information. The-on exchange of aS, however, is attributed to two different species with alternate structural contents undergoing an EX2 exchange.38 In an EX2 mechanism, backbone amides of proteins are typically exposed for a short time and exchange with the heavier isotope gradually over the exchange time, where respective mass shifts of unimodal isotopic clusters inform about solvent accessibility. In the current scenario, short, 1 min pulses of deuteration were applied, to different aggregation samples comprised of alternate conformational states that developed in solution as a function of time. The reaction mixture for labeling consisted of mixed species, some of which were open with exposed amides exchanging rapidly, and some of which were folded yielding a slower exchange. These mixed states contributed to a differential exchange, yielding an EX1-like pattern with multinomial distributions.
We observed that the relative abundance of the more protected, solvent-shielded population increases as the aggregation process accelerates, and gives rise to a sigmoidal curve as seen in Figure 4.
Figure 4.
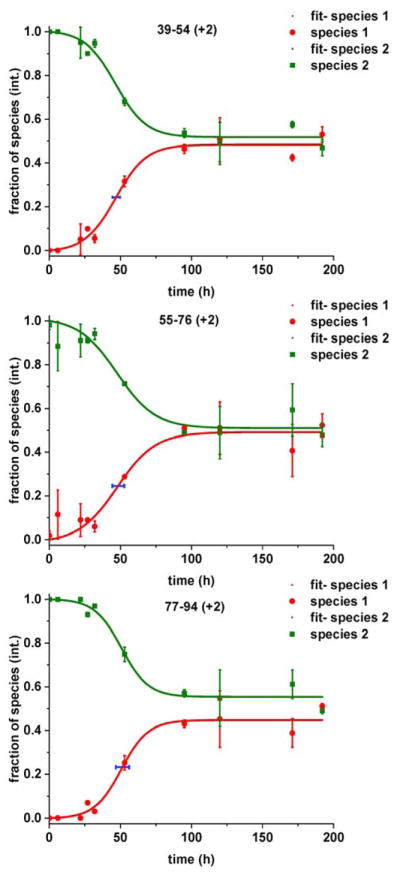
Changes in the fractional species 1 or 2 of selected peptic peptides that reside nearby or in the NAC region of aS, taken from the bimodal distributions seen in HDX. Species 1 (red) represents the fraction of the slow exchanging population, whereas species 2 (green) represents the fraction of fast exchanging, solvent-accessible population. The horizontal, blue error bar represents the standard deviation of the respective t1/2 values.
The protection appears at various times for different regions of the protein (e.g., at 6 h for peptide 55–76 (+2) and 22 h for 77–89 (+1) (Figure S5 in the Supporting Information) or at 27 h for 39–54 (+2). To resolve the relative contribution of each of the two main populations detected in our data, we semi-automatically analyzed data for each peptic peptide, by using MathCAD, verifying and fitting the isotopic distribution of the respective isotopic clusters at a given time point. We calculated their average relative abundances compared to the total of both species and expressed it as a fractional species. We did this analysis while considering carefully the chemical composition, potential isoforms, and contaminants and inspecting the peptides at all time points.
We are able to show the fractional species for three peptic peptides located in this region (and other peptides located also near or in the NAC region are shown in Figure S6 in the Supporting Information), all starting with a unimodal isotopic pattern consistent with high solvent accessibility, denoted by species 2 (green). This species gives way to another population of species 1 (red), which is the aggregated, more protected counterpart. We performed a number of measurements at a later date (at 32 h, 120 h and 171 h), to ensure reproducibility, found good agreement, and added these data to the existing number of replicates. We were able to achieve full coverage of aS; the pulsed HDX of those peptides that did not change during the aggregation are given in Figure S7 in the Supporting Information.
The t1/2 for maximum fraction of species 1 reached during aggregation
To characterize better the kinetics of each peptic peptides displaying protection, we took the fraction of protected species 1 and kinetically modeled these data. Our model is based on the Finke-Watzky two-stage model (FW-2), previously applied to aS by Morris et al.39 The model assumes the conversion of a natively disordered protein (A) to an aggregation-competent state (B) by a slow nucleation process followed by a fast, autocatalytic surface growth. We implemented a minor modification that allows for reversion of species B to A as described in the methods section. We determined the maximum fractional species 1 of peptic peptides nearby and in the NAC region (Figure S8 A and Table S1 in the Supporting Information). The maximum fractional species 1’s intensity was derived from the fit, and it varies between 0.43 ± 0.06 and 0.64 ± 0.12. Two-tailed, p-value analysis did not reveal statistically significant differences among the peptic peptides discussed here.
The time taken to reach half of the maximum intensity (t1/2) in a ThT assay is often used to describe the kinetics of amyloidogenic systems.40 We adopted this criterion and extended it to characterize the propensity for aggregation as a function of protein region (Figure S8 B and Table S2 in the Supporting Information) as a reporter for aggregation propensity. Note that the standard deviation is that for the fitted t1/2 in response to the uncertainty represented by the experimental data as calculated by bootstrapping (see Methods). The t1/2 values are the shortest for regions 39–54 (+2), 55–76 (+2) and 77–94 (+2), less than 50 h for the first two peptides and around 51 h for the latter. Although we were not able to observe statistically significant differences among the peptic peptides described here because the coarseness of our measurements does not allow resolution of nuances in their respective t1/2 values. The peptic peptides shown here afford medium resolution but are not able to yield residue-level information about the structural determinants of aS that are pivotal in governing aggregation.
Complementary measurements of aggregation
To follow the morphology of fibril formation, we obtained an electron micrograph at each time point analyzed by HDX-MS. We observed small, spherical oligomers before 22 h, and thereafter protofibrillar and larger fibrillar species (Figure S9 in the Supporting Information). The presence of macroscopic larger aggregates or fibrillar forms is largely consistent with the appearance of binomial distribution in time detected by our HDX analyses.
To assess transitions in the secondary structure, we performed circular dichroism (CD) at the same dilution that we used for pulsed labeling. The CD measurements revealed a major transition of aS from a natively unstructured spectrum with a minimum near 200 nm to a more ordered secondary structure at 22 h and thereafter (Figure S10 in the Supporting Information). This transition coincides with the appearance of bimodal distributions for certain NAC-region located peptides observed by HDX-MS.
Conclusions
Much has been uncovered about the role of aS in neurodegeneration by combined efforts gleaned from cell biology, neuroscience, biochemistry, protein biophysics and structural biology.41 Nevertheless, additional approaches that do not disrupt the natively unstructured form, require small amounts of material, and provide both kinetic and spatially resolved information are needed. To fulfill in part this need, we implemented pulsed HDX-MS to study the self-assembly of aS in vitro and thereby provide evidence for alternate populations of aS species that report on its aggregation as a function of time. We highlight the existence of two main populations that are dynamic and change in their relative abundance during the course of the aggregation.
We demonstrate that the aggregation is centered in the core region of aS, whereas the N-termini’s contribution to aggregation is moderate, and the C-terminus remains solvent-accessible throughout the aggregation. We show a number of peptic peptides in the central, NAC region, and through curve analysis, we establish their respective t1/2 times.
Moreover, we can classify the peptides according to their HDX-MS data into four categories (peptides that show no change, very weak protection at late time points, moderate gain in protection at mid and/or late time points, and high protection) as highlighted in Figure 5, where we overlay our peptides on a recently proposed solid-state NMR fibril structure, validated by TEM and X-ray fiber diffraction.16 It is important to note that structural polymorphism of aS fibrils has been documented before, describing at least two types of aS fibrils, straight and twisted, formed almost under identical conditions.33 This model emphasizes a characteristic aS fibril structure with parallel, in-register β-sheets arising from hydrophobic residues with diverse structural entities, encompassing a salt bridge (between E46-K80), a key glutamine ladder (residue Q79), steric zippers (e.g., V49, V77 and V82) and packing of small amino acids (A, G and S) primarily in the turn regions. Residues 55-62 are ascertained as disordered in contrast to our findings, whereas I88, A91 and F94 form the inside of the Greek key, alongside the neighboring hydrophobic residues. Heise et al.,33 relying on solid-state NMR, described two less structured turns or loop regions in between β-strands in the central core region, spanning residues 66-68 and 83-86.
Figure 5.

Solid-state NMR structure of aS fibrils resembling a Greek-key topology taken from Tuttle et al. (PDB: 2n0a) colored with the HDX exchange pattern, where blue represents no change, orange shows a moderate gain in protection at the mid or late time points of aggregation, yellow shows very weak protection at the late stages of aggregation and red shows substantial protection developing during the course of aggregation.
The exact boundaries of aS fibrils structural core are somewhat unclear; different arrangements were reported in the literature. Using quenched HDX-NMR, Vilar et al.34 placed the protected core between residues 35-96, harboring five β-strands. Moreover, solid-state NMR conducted by the same authors revealed β-sheet secondary structure between residues 30-110.34 The solid-state NMR structure considered here16 indicates a β-strand boundary starting from residue 38, which is part of our peptic peptide 5–38 (+3), and shows some evidence of multinomial distributions reflective of aggregation (Figure S11 in the Supporting Information). Nevertheless, the adjacent peptide 39–54 (+2), and those discussed above, show a much more apparent bimodality as a function of time. Unfortunately, we do not have a representative peptide spanning residues 95-96 that demarks the outside boundary of the β-sheet (arrowhead pointing beyond amino acid 94 in Figure 5), but these residues may be relevant structural determinants of our aS samples, and are not seen simply because pepsin cleavage favors other sites. Peptide 95–113 (+2) is a large peptide, in which the contribution of these two structure ordering residues can be diluted by the disordered, solvent-accessible constituents.
Thus, pulsed HDX-MS opens a kinetic dimension to the solid-state NMR architecture, illuminating the evolution of protection patterns prior to a fibrillar state across a long time course for aggregation. TEM imaging and CD support structural rearrangements ≥ 22 h, as detected by mass spectrometry, but they are not capable of providing spatially resolved information. Pulsed HDX-MS is also well-suited for the study of aS aggregation, and possibly that of other amyloidogenic proteins. Moreover, the method development paves the way for further investigations of aS inhibitors and small molecules as potential therapeutic targets, and these studies are underway.
Materials and Methods
All reagents and chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA) unless otherwise stated.
Protein expression and purification of wild type aS
WT aS protein expression was performed in E. coli BL21 (DE3) Gold. Bacteria was grown in Luria Broth (LB) and induced for expression at OD600 0.5 with 1 mM Isopropyl β-D-1-thiogalactopyranose (IPTG) that was further incubated at 30 °C overnight. Cells were harvested by centrifugation at 4,000 rpm at 4 °C for 30 min in a Sorvall centrifuge (SLC-6000 rotor). The pellet was frozen, then thawed, and lysed mechanically in 10 mM Tris-HCl with 1 mM EDTA, pH 8.0 (lysis buffer), containing 1 mM phenylmethylsulfonyl fluoride (PMSF) and 2 μL benzonase. The lysate was boiled at 100 °C for 20 min. Ammonium sulfate (361 mg/mL) was added to the supernatant and stirred at 4 °C for 15 min. After centrifugation at 4,000 rpm at 4 °C for 30 min (Eppendorf 5810R), the pellet was re-suspended in 25 mM Tris, pH 7.7 purification buffer. After re-suspension, the excess ammonium sulfate was removed by buffer exchanging the pellet in a Spectra/Por 3.5 kDa MWCO (VWR) dialysis tubing in 25 mM Tris buffer, pH 7.7 overnight with continuous stirring. The crude lysate solution was filtered through a 0.22 μM syringe filter and loaded onto a 6 mL ResourceTM Q column (GE HealthCare). The column was equilibrated with 5 column volumes (CV) of 25 mM Tris purification buffer (pH 7.7), then with 5 CV of 25 mM Tris and 1 M NaCl, pH 7.7 elution buffer, and finally with 5 CV of purification buffer. The aS was eluted with a linear salt gradient from 0% to 100% buffer. SDS-PAGE analysis of ResourceTM Q fractions was performed, Coomassie stained, and fractions enriched for protein were pooled. Fractions containing pure aS were collected, and aS was dialyzed extensively at 4 °C in 3 L 10 mM ammonium bicarbonate buffer, pH 7.3, with the buffer being changed at least three times. Aliquots were made of the purified protein and lyophilized for long term storage at −20 °C. The protein concentration was measured by UV-Vis spectrophotometry (Nanodrop Onec, Thermo Scientific, Madison, WI) at 280 nm using the extinction coefficient of 5960 M−1 cm−1. The purity of protein was verified by denaturing mass spectrometry (Figure S1 in the Supporting Information) obtained in positive-ion mode on a Bruker MaXis 4G mass spectrometer fitted with an ESI source, at 400 V capillary voltage, 1 bar nebulizer gas, 8 L/min dry gas, 200 °C dry temperature using a 9.5 min linear gradient.
Aggregation assay
To ensure a seedless, monomeric starting material, lyophilized aS was dissolved in 10 mM NaOH, sonicated for 15 min, and filtered through a 50 kDa MWCO Vivaspin filter unit (Millipore) for 15 min at 14,000 rpm on a benchtop centrifuge (Eppendorf 5418, Hamburg, Germany). The flow-through was collected, and UV-Vis spectrophotometry was used to determine the concentration of aS at neutral pH. The aggregation assay was set up by diluting aS to a 80 μM final concentration into 100 mM sodium phosphate, 10 mM NaCl, and the pH was adjusted to pH 7.4. 60 μL of the solution was pipetted into low binding Eppendorf tubes containing a 2 mm glass stir bead in replicates and agitated at 37 °C, at continuous shaking at 300 rpm on an Eppendorf Thermomixer 5350 benchtop shaker (Hauppauge, NY). Vials were sealed with parafilm to minimize any evaporation. Samples were collected at 0 h, 0.1 h, 6 h, 22 h, 27 h, 32 h, 53 h, 95 h, 120 h, 171 h, 192 h, snap-frozen, and stored at −80 °C.
HDX-MS
Pulsed deuterium labeling was performed by adding 1 μL 80 μM aS solution taken at various time points of the assay, 4 μL 1X aggregation buffer to 20 μL of D2O buffer for 1 min at 21 °C. Ice–cold reducing quench buffer, 3 M urea with 1% trifluoroacetic acid (TFA), pH 2.5 was added to the solution at 1:1 (v/v) to quench the HDX reaction. Online pepsin digestion and HPLC separation were performed as previously described. 42 Briefly, protein samples were digested over a custom–packed online pepsin column. The resulting peptides were trapped and desalted on a C8 trap column (Zorbax XDB, Agilent Inc., Santa Clara, CA). Separation of the peptide mixtures was carried out on a 1.9 μM reversed–phase C18 column (Hypersil Gold Thermo Fisher Scientific, Waltham, MA) with a 9.5 min linear gradient. All the experiments were performed in duplicate (n = 2), unless otherwise stated.
MS analysis of the eluted peptides was conducted on a LTQ-FTICR instrument (Thermo Scientific, Santa Clara, CA) using a positive–ion electrospray ionization (ESI) mode. The ESI spray voltage was set to 5 kV, the temperature to 275 °C, the capillary voltage to 38 V, the sheath gas flow rate to 10 L/min and nebulizer gas flow rate to 8 L/min, respectively.
HDX–MS Data Analysis
Identification of peptic peptides was achieved by submitting them to collision–induced dissociation (CID) fragmentation on a LTQ-FTICR (Thermo Scientific, Santa Clara, CA) instrument. Product–ion spectra were then submitted to MassMatrix (version 2.4.2)43 for identification, and manually inspected. All data were processed by HDX Workbench (Scripps Inst., Florida) to determine differential deuterium uptakes by using the centroid of isotopic profiles, with D2O dilution taken into account. For envelopes with multimodal distributions, a customized program implemented in MathCAD (v14, Parametric Technology Corp., MA) was used to fit the HDX isotopic envelope for each aS peptide with two or three binomial distributions as appropriate. Isotopic clusters were confirmed by using the chemical composition of a peptide and calculating the probabilities of deuterium uptake of each given peptide, where the first two amides and prolines were excluded. After cluster selection, relative intensities were used for modeling to determine fractional species. Fractional species were expressed as follows:
For peptides with unimodal distribution, the deuterium uptake percentage was acquired using HDX Workbench (Scripps Institute, CA) based on using Eq. 1:
| (1) |
where mHDX and mt0 are the centroid mass of the deuterated and non–deuterated peptides, respectively, 0.8 corresponds to the final D2O content of the buffer system, and (N − 2) represents the number of the amide hydrogens that are exchangeable, assuming first two residues retain no deuterium. Curve fitting and all data plots were performed with Prism 6 (GraphPad, La Jolla, CA). HDX pattern was overlaid on the solid-state NMR structure (PDB: 2n0a)16 using the UCSF Chimera software package.44
Kinetic Modeling
The Finke-Watzky (FW)-polymerization model (a two-step mechanism of slow, continuous aggregation) was applied to aS aggregation39, 45 with a minor alteration. The first step entails conversion to an aggregation competent species , and as a deviation from the model, we allow for a minor back-conversion step , which is almost negligible. The second main step is that denotes a fast, autocatalytic surface growth.
Experimental fractional species were fit with this model to afford the maximum fractional species 1 and the t1/2, where the latter is defined as the time it is taken to reach half maximum intensity. The nonlinear least-squares fit of the kinetic model to the species fraction at nine times was performed by the Minimize function in MathCAD (v14, Parametric Technology Corp., MA). The uncertainty in the parameters that result from measured uncertainty in the data was obtained by computing the statistics from 1024 trials of a “bootstrap” like procedure. For each trial, an independent nine-coordinate random Gaussian zero-mean unit-variance vector was computed. Each random vector coordinate was rescaled so that its population standard deviation would match that of the corresponding time-point species fraction and then added to the mean species fraction. The resulting simulated kinetic curve was fitted to produce the resulting parameters for that trial. The population standard deviation for a parameter was calculated from the 1024 trial results. The significance of the difference in a parameter between two kinetic curves was estimated by compiling a 101 bin histogram of all combinations of trial differences (10242 differences). The fraction of those trials that exceeded in distance the absolute value of the mean difference from the difference mean value was taken as the two-tailed empirical p-value. This was judged to work because the histogram distributions are broader than the mean differences.
Transmission electron microscopy
Carbon films on 200-mesh copper grids (Ted Pella) were loaded with 10 μL of aS aggregation products for 30 s, followed by two rounds of washing with 10 μL deionized H2O for 30 s. A volume of 10 μL of uranyl acetate stain was loaded onto the film for 30 s. Excess liquid was wicked off using a Chem Wipe, the grids were air-dried, and viewed using a FEI Tecnai G2 Spirit microscope (Thermo Fisher Scientific, Hillsboro, Oregon).
Circular dichroism
Circular dichroism spectra were obtained on a J-715 instrument (JASCO Corp, Tokyo, Japan) at scanning speed: 50 nm/min, in continuous mode, with a response time: of 1 s, bandwidth of 1 nm, accumulation of 5 spectra, using a cuvette with a path length of 0.02 cm in 100 mM sodium phosphate with 10 mM NaCl, at 8 μM protein concentration. Ellipticity was expressed as (degcm X 106)/(path length (cm) X conc. (μM) X number of peptide bonds).
Supplementary Material
Acknowledgments
We would like to thank Professor Jan Bieschke for providing materials for and access to recombinant protein expression and purification. We thank Professor Carl Frieden for access to circular dichroism. We would like to acknowledge the Nano Research Facility at Washington University in St Louis for access to TEM. We thank the American Parkinson’s Disease Association and the NIH (Grant P41GM103422) for funding.
Footnotes
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- 1.Nussbaum RL. Genetics of synucleinopathies. Cold Spring Harb Perspect Med. 2017 doi: 10.1101/cshperspect.a024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ozansoy M, Basak AN. The central theme of parkinson’s disease: Alpha-synuclein. Mol Neurobiol. 2013;47:460–465. doi: 10.1007/s12035-012-8369-3. [DOI] [PubMed] [Google Scholar]
- 3.Al-Hilaly YK, Biasetti L, Blakeman BJ, Pollack SJ, Zibaee S, Abdul-Sada A, Thorpe JR, Xue WF, Serpell LC. The involvement of dityrosine crosslinking in alpha-synuclein assembly and deposition in Lewy bodies in Parkinson’s disease. Sci Rep. 2016;6:39171. doi: 10.1038/srep39171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Uversky VN. Alpha-synuclein misfolding and neurodegenerative diseases. Curr Protein Pept Sci. 2008;9:507–540. doi: 10.2174/138920308785915218. [DOI] [PubMed] [Google Scholar]
- 5.Theillet FX, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D, Selenko P. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature. 2016;530:45–50. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 6.Beyer K, Ariza A. Alpha-synuclein posttranslational modification and alternative splicing as a trigger for neurodegeneration. Mol Neurobiol. 2013;47:509–524. doi: 10.1007/s12035-012-8330-5. [DOI] [PubMed] [Google Scholar]
- 7.Oueslati A, Fournier M, Lashuel HA. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: Implications for parkinson’s disease pathogenesis and therapies. Prog Brain Res. 2010;183:115–145. doi: 10.1016/S0079-6123(10)83007-9. [DOI] [PubMed] [Google Scholar]
- 8.Mason RJ, Paskins AR, Dalton CF, Smith DP. Copper binding and subsequent aggregation of alpha-synuclein are modulated by N-terminal acetylation and ablated by the H50Q missense mutation. Biochemistry. 2016;55:4737–4741. doi: 10.1021/acs.biochem.6b00708. [DOI] [PubMed] [Google Scholar]
- 9.Stroo E, Koopman M, Nollen EAA, Mata-Cabana A. Cellular regulation of amyloid formation in aging and disease. Front Neurosci. 2017;11:64. doi: 10.3389/fnins.2017.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uversky VN, Eliezer D. Biophysics of Parkinson’s disease: Structure and aggregation of alpha-synuclein. Curr Protein Pept Sci. 2009;10:483–499. doi: 10.2174/138920309789351921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sidhu A, Segers-Nolten I, Subramaniam V. Conformational compatibility is essential for heterologous aggregation of alpha-synuclein. ACS Chem Neurosci. 2016;7:719–727. doi: 10.1021/acschemneuro.5b00322. [DOI] [PubMed] [Google Scholar]
- 12.Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J Am Chem Soc. 2005;127:476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 13.Munishkina LA, Fink AL, Uversky VN. Accelerated fibrillation of alpha-synuclein induced by the combined action of macromolecular crowding and factors inducing partial folding. Curr Alzheimer Res. 2009;6:252–260. doi: 10.2174/156720509788486491. [DOI] [PubMed] [Google Scholar]
- 14.Flagmeier P, Meisl G, Vendruscolo M, Knowles TPJ, Dobson CM, Buell AK, Galvagnion C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of a-synuclein aggregation. Proc Natl Acad Sci U S A. 2016;113:10328–10333. doi: 10.1073/pnas.1604645113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buell AK, Dobson CM, Knowles TPJ. The physical chemistry of the amyloid phenomenon: Thermodynamics and kinetics of filamentous protein aggregation. Essays Biochem. 2014;56:11–39. doi: 10.1042/bse0560011. [DOI] [PubMed] [Google Scholar]
- 16.Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VMY, George JM, Rienstra CM. Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol. 2016;23(5):409–415. doi: 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chalmers MJ, Busby SA, Pascal BD, West GM, Griffin PR. Differential hydrogen/deuterium mass spectrometry analysis of protein-ligand interactions. Expert Rev Proteomics. 2011;8:43–59. doi: 10.1586/epr.10.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Konermann L, Pan J, Liu Y. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc Rev. 2011;40:1224–1234. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- 19.Del Mar C, Greenbaum EA, Mayne L, Englander SW, Woods VL., Jr Structure and properties of alpha-synuclein and other amyloids determined at the amino acid level. Proc Natl Acad Sci U S A. 2005;102:15477–15482. doi: 10.1073/pnas.0507405102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paslawski W, Mysling S, Thomsen K, Jorgensen TJ, Otzen DE. Co-existence of two different alpha-synuclein oligomers with different core structures determined by hydrogen/deuterium exchange mass spectrometry. Angew Chem Int Ed Engl. 2014;53:7560–7563. doi: 10.1002/anie.201400491. [DOI] [PubMed] [Google Scholar]
- 21.Mysling S, Betzer C, Jensen PH, Jorgensen TJ. Characterizing the dynamics of alpha-synuclein oligomers using hydrogen/deuterium exchange monitored by mass spectrometry. Biochemistry. 2013;52:9097–9103. doi: 10.1021/bi4009193. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Rempel DL, Zhang J, Sharma AK, Mirica LM, Gross ML. Pulsed hydrogen-deuterium exchange mass spectrometry probes conformational changes in amyloid beta (Aβ) peptide aggregation. Proc Natl Acad Sci U S A. 2013;110:14604–14609. doi: 10.1073/pnas.1309175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, Shu Q, Rempel DL, Frieden C, Gross ML. Continuous and pulsed hydrogen-deuterium exchange and mass spectrometry characterize CsgE oligomerization. Biochemistry. 2015;54:6475–6481. doi: 10.1021/acs.biochem.5b00871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, Shu Q, Rempel DL, Frieden C, Gross ML. Understanding curli amyloid-protein aggregation by hydrogen–deuterium exchange and mass spectrometry. International Journal of Mass Spectrometry. 2016;420:16–23. doi: 10.1016/j.ijms.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh J, Sabareesan AT, Mathew MK, Udgaonkar JB. Development of the structural core and of conformational heterogeneity during the conversion of oligomers of the mouse prion protein to worm-like amyloid fibrils. J Mol Biol. 2012;423:217–231. doi: 10.1016/j.jmb.2012.06.040. [DOI] [PubMed] [Google Scholar]
- 26.Breydo L, Wu JW, Uversky VN. Alpha-synuclein misfolding and Parkinson’s disease. Biochim Biophys Acta. 2012;1822:261–285. doi: 10.1016/j.bbadis.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Fujioka S, Ogaki K, Tacik PM, Uitti RJ, Ross OA, Wszolek ZK. Update on novel familial forms of Parkinson’s disease and multiple system atrophy. Parkinsonism Relat Disord. 2014;20(Suppl 1):S29–34. doi: 10.1016/S1353-8020(13)70010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uversky VN. Amyloidogenesis of natively unfolded proteins. Curr Alzheimer Res. 2008;5:260–287. doi: 10.2174/156720508784533312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goswami D, Devarakonda S, Chalmers MJ, Pascal BD, Spiegelman BM, Griffin PR. Time window expansion for HDX analysis of an intrinsically disordered protein. J Am Soc Mass Spectrom. 2013;24:1584–1592. doi: 10.1007/s13361-013-0669-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bernstein SL, Liu D, Wyttenbach T, Bowers MT, Lee JC, Gray HB, Winkler JR. Alpha-synuclein: Stable compact and extended monomeric structures and pH dependence of dimer formation. J Am Soc Mass Spectrom. 2004;15:1435–1443. doi: 10.1016/j.jasms.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 31.Phillips AS, Gomes AF, Kalapothakis JM, Gillam JE, Gasparavicius J, Gozzo FC, Kunath T, MacPhee C, Barran PE. Conformational dynamics of alpha-synuclein: Insights from mass spectrometry. Analyst. 2015;140:3070–3081. doi: 10.1039/c4an02306d. [DOI] [PubMed] [Google Scholar]
- 32.Natalello A, Benetti F, Doglia SM, Legname G, Grandori R. Compact conformations of alpha-synuclein induced by alcohols and copper. Proteins. 2011;79:611–621. doi: 10.1002/prot.22909. [DOI] [PubMed] [Google Scholar]
- 33.Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Molecular-level secondary structure, polymorphism, and dynamics of full-length alpha-synuclein fibrils studied by solid-state NMR. Proc Natl Acad Sci U S A. 2005;102:15871–15876. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vilar M, Chou HT, Luhrs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, Riek R. The fold of alpha-synuclein fibrils. Proc Natl Acad Sci U S A. 2008;105:8637–8642. doi: 10.1073/pnas.0712179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen M, Margittai M, Chen J, Langen R. Investigation of alpha-synuclein fibril structure by site-directed spin labeling. J Biol Chem. 2007;282:24970–24979. doi: 10.1074/jbc.M700368200. [DOI] [PubMed] [Google Scholar]
- 36.Forloni G, Bertani I, Calella AM, Thaler F, Invernizzi R. Alpha-synuclein and parkinson’s disease: Selective neurodegenerative effect of alpha-synuclein fragment on dopaminergic neurons in vitro and in vivo. Ann Neurol. 2000;47:632–640. [PubMed] [Google Scholar]
- 37.Uversky VN, Li J, Souillac P, Millett IS, Doniach S, Jakes R, Goedert M, Fink AL. Biophysical properties of the synucleins and their propensities to fibrillate: Inhibition of alpha-synuclein assembly by beta- and gamma-synucleins. J Biol Chem. 2002;277:11970–11978. doi: 10.1074/jbc.M109541200. [DOI] [PubMed] [Google Scholar]
- 38.Ferraro DM, Lazo N, Robertson AD. EX1 hydrogen exchange and protein folding. Biochemistry. 2004;43:587–594. doi: 10.1021/bi035943y. [DOI] [PubMed] [Google Scholar]
- 39.Morris AM, Finke RG. Alpha-synuclein aggregation variable temperature and variable pH kinetic data: A re-analysis using the Finke-Watzky 2-step model of nucleation and autocatalytic growth. Biophys Chem. 2009;140:9–15. doi: 10.1016/j.bpc.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Arosio P, Knowles TPJ, Linse S. On the lag phase in amyloid fibril formation. Phys Chem Phys. 2015;17:7606–7618. doi: 10.1039/c4cp05563b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: From structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J, Rodnin MV, Ladokhin AS, Gross ML. Hydrogen-deuterium exchange and mass spectrometry reveal the pH-dependent conformational changes of diphtheria toxin T domain. Biochemistry. 2014;53:6849–6856. doi: 10.1021/bi500893y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu H, Freitas MA. MassMatrix: A database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics. 2009;9:1548–1555. doi: 10.1002/pmic.200700322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 45.Morris AM, Watzky MA, Agar JN, Finke RG. Fitting neurological protein aggregation kinetic data via a 2-step, minimal/”Ockham’s razor” model: The Finke-Watzky mechanism of nucleation followed by autocatalytic surface growth. Biochemistry. 2008;47:2413–2427. doi: 10.1021/bi701899y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.