

Abstract
The last two decades have seen a surge in antimalarial drug development with product development partnerships taking a leading role. Resistance of Plasmodium falciparum to the artemisinin derivatives, piperaquine and mefloquine in Southeast Asia means new antimalarials are needed with some urgency. There are at least 13 agents in clinical development. Most of these are blood schizonticides for the treatment of uncomplicated falciparum malaria, under evaluation either singly or as part of two-drug combinations. Leading candidates progressing through the pipeline are artefenomel–ferroquine and lumefantrine-KAF156, both in Phase 2b. Treatment of severe malaria continues to rely on two parenteral drugs with ancient forebears: artesunate and quinine, with sevuparin being evaluated as an adjuvant therapy. Tafenoquine is under review by stringent regulatory authorities for approval as a single-dose treatment for Plasmodium vivax relapse prevention. This represents an advance over standard 14-day primaquine regimens; however, the risk of acute haemolytic anaemia in patients with glucose-6-phosphate dehydrogenase deficiency remains. For disease prevention, several of the newer agents show potential but are unlikely to be recommended for use in the main target groups of pregnant women and young children for some years. Latest predictions are that the malaria burden will continue to be high in the coming decades. This fact, coupled with the repeated loss of antimalarials to resistance, indicates that new antimalarials will be needed for years to come. Failure of the artemisinin-based combinations in Southeast Asia has stimulated a reappraisal of current approaches to combination therapy for malaria with incorporation of three or more drugs in a single treatment under consideration.
Key Points
| Product development partnerships have revived antimalarial drug development, and a variety of target candidate and product profiles have been defined to support control and elimination goals |
| New agents (arterolane, cipargamin, KAF156) on the horizon show potential to replace failing artemisinin combination therapies as part of novel combinations |
| The loss of front-line therapies to resistance has stimulated a reappraisal of the current approach to combination therapy for malaria, with consideration of a switch from dual to triple drug combinations |
Introduction
Malaria is a protozoan parasitic disease transmitted by the female Anopheles mosquito, which infects around half a billion people in tropical and sub-tropical countries each year [1]. Six species of Plasmodium cause human disease regularly: P. falciparum, P. vivax, P. ovale curtisi, P. ovale wallikeri, P. malariae and P. knowlesi. P. falciparum is responsible for almost 90% of all infections and for most of the 445,000 deaths that occur annually; most are in African children. P. vivax and P. ovale are relapsing malarias with a dormant hypnozoite stage in the liver which requires targeted therapy to prevent relapse, and P. knowlesi is a predominantly zoonotic infection with macaques acting as the natural hosts. The main strategies to control malaria globally are vector control with long-lasting insecticide-treated bed nets, early diagnosis and treatment with artemisinin-based combination therapies (ACTs), and chemoprevention in pregnant women and young children. After decades of research, a malaria vaccine (RTS,S/AS01) received a positive opinion from the European Medicines Agency under Article 58 in 2015; however, it is only moderately efficacious and has an uncertain future [2]. Its introduction is unlikely to lessen the demand for antimalarial drugs.
The use of ACTs to treat malaria dates back to the early 1990s. At that time the situation in Southeast Asia was bleak; chloroquine, sulfadoxine-pyrimethamine (SP) and mefloquine had been deployed sequentially and fallen to resistance. In 1979, Chinese scientists had published the chemical structure of qinghaosu or artemisinin, a potent antimalarial derived from the sweet wormwood plant Artemisia annua [3, 4]. White proposed partnering the fast-acting, highly efficacious, but rapidly eliminated artemisinin derivatives with more slowly eliminated drugs as short-course (3-day) highly effective combination treatments, with the aim of improving cure rates and slowing the development of resistance [5]. ACTs were adopted fairly quickly in Southeast Asia with some produced locally, e.g. Vietnam has produced various dihydroartemisinin (DHA)–piperaquine combinations since 1995.
Meanwhile chloroquine- and pyrimethamine-resistant strains of falciparum malaria had spread to Africa leading to an increase in child mortality [6–8]. This crisis renewed the impetus to bring malaria under control, and greater financial resources were made available. An important part of the response was the recognition that new drugs were needed, and that an innovative approach was required to stimulate their development. New product development partnerships (PDPs) were created, such as Medicines for Malaria Venture (MMV, 1999) and the Drugs for Neglected Diseases Initiative (2003). The PDPs harnessed the expertise of the pharmaceutical industry and malaria academics, with financial support coming from a variety of (mainly public and philanthropic) sources. They were given a head start by focusing on the fruits of the labours of Chinese scientists and pharma [9]. By 2006, ACTs had become the recommended treatments for falciparum malaria worldwide [10]. The first ACT produced to recognised international standards of good manufacturing practice (GMP) was artemether–lumefantrine (Coartem®; Novartis), approved by the US FDA in April 2009 [11]. Artemether–lumefantrine, DHA–piperaquine, artesunate–amodiaquine, artesunate–mefloquine and artesunate–sulfadoxine–pyrimethamine are currently the most used combinations. Also available are pyronaridine–artesunate, artemisinin–naphthoquine, and arterolane–piperaquine. Arterolane is a newer synthetic peroxide developed in India resembling the artemisinin derivatives. P. knowlesi malaria is treated in the same way as falciparum malaria [12]. For uncomplicated disease caused by Plasmodium vivax, ovale or malariae chloroquine remains the first line treatment in most parts of the world; however, there are increasing reports of resistance in Plasmodium vivax [13]. In Indonesia where failure rates are high, the ACT, DHA–piperaquine, has replaced chloroquine for treatment. Severe malaria continues to be treated by either parenteral artesunate (the drug of choice), or quinine or quinidine (USA only) [14, 15]. A number of adjuvant therapies have been evaluated over the years but have not been shown to improve outcome [16].
Malaria case numbers and deaths have more than halved over the last 15 years and the goal of global elimination is being targeted again. At the same time, a new threat has emerged in Southeast Asia in the form of artemisinin resistance. Evidence that Plasmodium falciparum resistant to artemisinin has emerged has accrued steadily since 2008 [17–20]. Contributing factors may have been the continued availability of artesunate monotherapy in the region for many years or falsified or substandard drugs. Artemisinin resistance manifests clinically as delayed parasite clearance and was defined phenotypically using a parasite clearance half-life of ≥ 5 h or a high day 3 parasite positivity rate until molecular markers for resistance were reported in 2014: mutations in the kelch gene on chromosome 13 of the parasite [21]. Complete non-response to artemisinin treatment has not been described to date. The blunting of the early parasiticidal effect of the artemisinin derivatives leaves a larger residuum of parasites for the partner drugs to clear. Inevitably resistance to these drugs has now emerged in areas where artemisinin resistance had taken hold and ACTs are failing in Cambodia, Vietnam and Thailand [22–25] The mismatch of elimination kinetics between the artemisinin derivatives and the drugs it is partnered with was often considered a risk for development of resistance, although to date, resistance to the rapidly eliminated more potent drug (artemisinin) has become apparent first.
In 2018, the situation in Southeast Asia is serious. In Cambodia, treatment has switched back to artesunate–mefloquine from failing DHA–piperaquine, which had replaced failing artesunate–mefloquine in 2008. Pyronaridine–artesunate will be introduced as an alternative treatment option in areas of drug resistance this year. Small numbers of artemisinin-resistant parasites have been found in India and the Americas [26, 27], while in Africa the news is more reassuring so far with no evidence that resistance has become established [28]. However, vigilance is needed, including for the possibility of kelch-independent reduced susceptibility. Triple artemisinin-based combinations, comprising a standard dual ACT with another slowly eliminated antimalarial are being evaluated as a possible holding measure until new agents are available (NCT02453308).
On this backdrop, the development strategy for antimalarial drugs has evolved. Early on, ambitious development goals of single-dose curative (including radical cure) combination treatments were emphasised. There has since been a shift to defining a broader portfolio of target product and candidate profiles suited to different goals of control and elimination, with less emphasis on single-exposure therapy. These include molecules for case management, prevention, transmission blocking and relapse prevention, described in detail in recent reviews [29, 30]. “Resistance-proofing” future compounds in both the selection process and by considering combinations of more than two molecules in new products is a priority.
The process of identifying new antimalarials, dose-finding and evaluation has also evolved over the last 10 years. Phenotypic screening still dominates with target-based methods employing molecular or genome scanning used increasingly [31–34]. There has been a collaborative approach to drug discovery and development with initiatives such as the Malaria Box, an open access resource containing 400 compounds with antimalarial activity made available free of charge until December 2015 [35]. Newer approaches to dose finding include determining the minimum inhibitory concentration of drugs by tracking sub-microscopic parasite densities using ultrasensitive PCR in human challenge models or malaria patients [36, 37].
Chemoprevention for malaria is used in vulnerable groups in endemic areas and in travellers. Strategies targeting pregnant women and young children include intermittent preventive therapy (IPT) with SP for pregnant women and infants, and seasonal malaria chemoprevention (SMC) using SP-amodiaquine in children aged < 5 years living in the Sahel. Administration of a complete treatment course of antimalarials in both IPT and SMC ensures that anyone already harbouring parasites with few or no symptoms receives an effective treatment. Mefloquine and DHA–piperaquine have been evaluated as alternatives for IPT in pregnancy in the face of worsening SP resistance [38–41]. Antimalarials for IPT or SMC ideally have a prolonged protective effect against infection allowing dosing to be adapted to typical antenatal clinic attendance or expanded programme for immunisation schedules.
For travellers, compounds with a shorter protective efficacy, including those requiring daily administration are acceptable. Possession of causal prophylactic activity is advantageous since such agents protect against infection and can be stopped soon after leaving an endemic area. Antimalarials most commonly prescribed to travellers are atovaquone–proguanil, doxycycline and mefloquine, although highly publicised neuropsychiatric side effects following mefloquine make it unpopular among both travellers and prescribers.
Here we review the antimalarials in clinical development and comment on their potential, with an emphasis on those that could succeed failing ACTs for treatment. The prospects for new preventive therapies and agents in the preclinical and discovery phase are also summarised.
Search Strategy
A two-stage approach was used. First a search of recent reviews of new antimalarials and new antimalarials not covered by these was performed in PubMed: (((“new antimalarial*”[Title/Abstract]) AND review [Publication Type]) AND (“2012/10/01”[Date-Publication]: “3000”[Date-Publication])) AND (“Antimalarials”[MAJR]) AND (“2016/11/01”[Date-Publication]: “3000”[Date-Publication]). The global portfolio of antimalarial medicines on the MMV website was also consulted [42]. Each antimalarial found using this approach was then searched for individually by name(s) in PubMed and Embase databases, for example, ((KAE609 OR KAE 609 OR KAE-609 OR NITD609 OR NITD 609 OR NITD-609 OR cipargamin) AND malaria) and on the ClinicalTrials.gov website (https://www.clinicaltrials.gov/).
Drugs in Clinical Development
There are around 13 new antimalarial drugs in clinical development, nine of which are in Phase 2 [31]. The majority are blood schizonticides intended for the treatment of uncomplicated P. falciparum (Fig. 1). The chances of successful progress through the antimalarial development pipeline from Phase 2 to registration have been estimated to range from 34% (2a) to 60% (2b) [30].
Fig. 1.
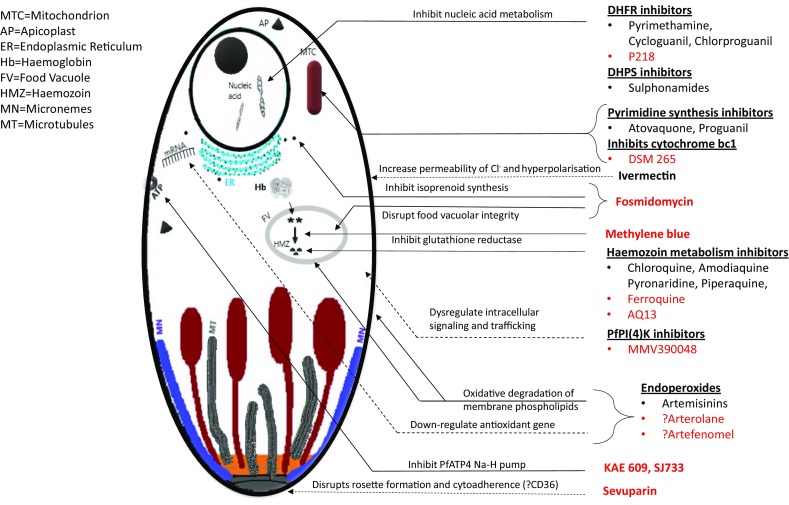
Schematic representation of intra-erythrocytic trophozoite showing sites of action of newer antimalarials. Agents in red are still in development
New Drugs in Clinical Development for Treatment
An ideal antimalarial treatment would be a potent inhibitor of parasite multiplication, short-course or single-dose, well tolerated in pregnant women and young children, have a good safety profile, be affordable and have a low propensity for resistance to develop. An expanded list of criteria used to assess antimalarials is shown in Table 1 with illustrative examples from drugs in use. Several new agents have been studied in Phase 2a before being paired with partner drugs for evaluation as a combination product (Table 2). The positive experience of using ACTs has led to a preference for at least one partner in a new combination to reduce the parasite biomass rapidly. Encouragingly, this is the case with the ozonides (e.g. OZ439), imidazolopiperazines (KAF 156) and the spiroindolone cipargamin (KAE 609), all potent inhibitors of parasite multiplication (Fig. 2).
Table 1.
Criteria used to assess antimalarials
| Individual antimalarial drugs | Examples |
|---|---|
| Anti-parasitic activity | |
| Stage-specificity, e.g. blood schizonticide (treatment), gametocytocide or sporontocide (transmission-blocking), hypnozoiticide (relapse prevention), hepatic schizonticide (causal prophylaxis) | The artemisinin derivatives have the broadest stage-specificity of action of all registered antimalarials (trophozoites, including young rings, gametocytes, with the exception of Stage V) Only the 8-aminoquinolines (primaquine, tafenoquine) are active against hypnozoites for relapse prevention in vivax or ovale malaria |
| Pharmacokinetics | |
| Speed of action (including dependence on co-factors, e.g. for absorption) | Lumefantrine AUC is the principal determinant of cure following artemether–lumefantrine treatment (absorption is enhanced by coadministration with fat) |
| Speed of elimination | Slowly eliminated drugs (e.g. piperaquine) have the added advantage of a longer post-treatment prophylactic effect and hence fewer episodes of malaria in high-transmission areas [115] |
| Pharmacodynamics | |
| Parasiticidal effect: relates to asexual stage-specific activity | The most potent antimalarials have the greatest inhibitory effect on parasite multiplication. The average parasite biomass in an adult with uncomplicated falciparum malaria is > 1012. Artesunate reduces the biomass by ~ 104 per asexual life-cycle (48 h) in sensitive infections. Thus, if used alone ≥ 6-day treatment (3 cycles) must be given to have the best chance of cure [5] |
| Safety/toxicity | |
| Pregnant women and children | ACTs were contraindicated in the first trimester of pregnancy due to concerns of embryotoxicity in animals and a lack of safety data in humans until recently |
| Repeated dosing | Patients in high transmission areas may have multiple episodes of malaria per year. Repeated dosing of artesunate-pyronaridine was not recommended initially due to safety concerns (signal of hepatotoxicity). This caution has since been lifted |
| Propensity for resistance to develop | Atovaquone–proguanil is extremely vulnerable to resistance development due to rapid selection of cytochrome b mutations |
| Cost | Artemether–lumefantrine costs US$0.38–1.3 per treatment [116] |
| Antimalarial combinations | |
|---|---|
| Formulation | |
| Co-formulations, tablet burden | All leading ACTs now exist as fixed-dose combinations with the exception of AS-SP |
| Paediatric formulations | Most leading ACTs have paediatric dosage forms |
| Posology | |
| Dose number and frequency | Most antimalarials are given as 3-day treatments. Artemether lumefantrine has the highest dose frequency (6 doses) |
| Matched pharmacokinetics | |
| Elimination half-lives | For the ACTs in use there is a mismatch of the elimination kinetics of artemisinin derivatives and partner drugs, which leaves the slowly eliminated drug unprotected |
| Drug–drug interactions | Triple ACTs under evaluation exploit inverse resistance selection properties of different partner drug pairings, e.g. amodiaquine and lumefantrine |
| Global recommendations to add a single low dose of primaquine to ACTs in areas targeting elimination necessitate study of potential drug-drug interactions with any new treatment |
Table 2.
New agents in clinical development or under review by stringent regulatory authorities
| Name and development partners | Type/target | Activity spectrum | First in human | In vivo or in vitro efficacy data | Safety | Vision for development |
|---|---|---|---|---|---|---|
| Under review by stringent regulatory authorities | ||||||
| Tafenoquine GSK, MMV, US Army | 3-phenoxy-substituted 8-aminoquinoline, i.e. synthetic analogue of primaquine | PV blood and tissue schizonticide, gametocytocidal | FIH study in HAVs reported in 1998 defined long half-life (14 days) and good tolerability [117] | Relapse-free efficacy at 6 months of chloroquine (CQ) + 300 mg tafenoquine 89.2% (77–95%) compared to 37.5% (23–52) for CQ alone [118] | Acute haemolytic anaemia in G6PD deficiency, elevated creatinine, methaemoglobinaemia | Single-dose relapse prevention for PV (given in combination with blood schizonticide) |
| Phase 2 | ||||||
| Artefenomel (formerly OZ439) MMV (+ ferroquine) |
Synthetic endoperoxide; several theories on mechanism of action [119] | Blood schizonticide for falciparum and vivax | Study of artefenomel in HAVs demonstrated a terminal half-life of 25–30 h; exposure increased by co-administration with food [120]. Evaluation in human induced blood-stage malaria model predicted an MIC of 4.1 ng/mL [121] FQ FIH data in HAVs were not published in a primary report [122] |
Artefenomel-piperaquine, PCt1/2 (min–max) in Vietnam was 6.1 (1.1–12.7) vs 3.5 (1.2–7.7) h in Africa and D-28 ACPR were 66.2% (95% CI 54.6–76.6) vs 74.5% (95% CI 7.81–79.7) [49] | Dose independent QTc prolongation, mild transaminitis [45, 122, 123] | Single dose combination treatment [49] (NCT02497612) |
| KAF156 MMV, Novartis (+ lumefantrine solid dispersion formulation) |
Imidazolopiperazine; unknown mechanism of action | Multi-stage (liver, blood and transmission-blocking) [124, 125] | KAF 156 FIH study in male HAVs published in 2014 with PK [126] | Median (IQR) PCt1/2 3.5 h (3.4–3.8) multiple-dose and 3.4 (3.2–4.1) single dose. Single-dose D-28 ACPR 67% [50] | Post-treatment mild transaminitis. Asymptomatic bradycardia [50] | Single dose multi-stage treatment (with lumefantrine) |
| Cipargamin (formerly) KAE609 MMV, Novartis |
Spiroindolone PfATP4 inhibitor [64, 127] |
Blood schizonticide for falciparum and vivax | Single- and multiple-dose cohorts studied. Dose-related gastrointestinal and genitourinary adverse events reported [128] | Median (IQR) PCt1/2 0.9 h (0.78–1.07) in PF [63]. D28 ACPR 50% at 30 mg, 60% at 20 mg, 29% at 10 mg, 14% at 15 mg stat dosing [129]. In vivo MIC estimated as 0.126 ng/mL [37] | Hepatotoxicity concerns-dose-escalation safety study ongoing (NCT03334747) | To combine with longer-acting partner drug not prone to PfATP4 mutation |
| Fosmidomycin (+ piperaquine) JOMAA |
Inhibitor of 1-deoxy-d-xylulose 5-phosphate reductoisomerase in isoprenoid biosynthesis pathway | Uncomplicated PF blood schizonticide | PK study of parenteral and oral fosmidomycin reported in 1982 [62] | PCT 49 h in small study of 5 days monotherapy and D14 cure rate of 89% [130]. With piperaquine D28 efficacy 100% [60] | Mild post-treatment transaminitis [130] | Combination therapy in areas with no piperaquine resistance |
| DSM265 Takeda, MMV, UT Southwestern |
Dihydroorotate dehydrogenase inhibitor | Uncomplicated PF schizonticide, liver stage activity; no gametocytocidal activity; no data in PV | Favourable early safety, tolerability, pharmacokinetic, and activity data published in HAVs and human challenge study in 2017 [66] | Parasite reduction ratio at 48 h in the human challenge study measured 1.55 (95% CI 1.42–1.67), which was lower than that following a 10 mg/kg dose of mefloquine (2.34) | Mild, e.g. headache [66] | Prevention (travellers’ market) and partner drug in combination therapy |
| AQ-13 Immtech and Tulane University |
Modified 4-aminoquinoline | Blood schizonticide for falciparum and vivax | HAV study published in 2007 showed a similar tolerability and PK profile to chloroquine [70] | Mean PCT 47.3 h (43.5–51.1) vs 32.5 (28.0–37.0) for artemether–lumefantrine (AL): by day 42, 0/28 vs 2/33 recrudescences in AL arm [69] | No grade 2–4 adverse events [69] | Partner drug in combination therapy |
| Methylene Blue University of Heidelberg |
Phenothiazine derivative. Prevents haem polymerisation by inhibiting P. falciparum glutathione reductase | PF schizonticide and potent gametocytocidal agent (male and female Stage V gametocytes) | First description of methylene blue to treat malaria was in 1891 [131] | D28 ACPR 72–84% in semi-immune adults [132]; 95% in combination with amodiaquine [133] | Increased vomiting when combined with artesunate–amodiaquine [82] | Part of a triple combination with ACTs + transmission blocking |
| Sevuparin (DF02) Dilaforette-Karolinska Institute |
Anti-adhesive polysaccharide derived from heparin with eliminated antithrombin binding site [87] | Blocks merozoite invasion and sequestration [87] | Well tolerated in HAVs [87] | ≥ 100 μg/mL inhibits cytoadherence [86]. No significant difference in parasite clearance between sevuparin-atovaquone–proguanil (AP) vs AP alone [87] | No adverse event leading to study withdrawal. No bleeding AEs [87] | Adjunctive treatment for severe malaria |
| MMV 390048 MMV and University of Cape Town |
Aminopyridine PfPI (4) K inhibitor [71] |
Blood schizonticide inhibits gametocytogenesis and oocyst formation (preclinical) [71] | FIH (NCT02230579; NCT02554799), PK (NCT02783820) and human challenge studies (NCT02281344) are complete (data not published) | Phase 2a study (NCT02880241) recruitment suspended for interim data analysis | Not published | Part of single-dose treatment, transmission- blocking, chemoprevention [71, 134] |
| Phase 1 | ||||||
| P218 MMV, Janssen (Biotec Thailand) |
PF-dihydrofolate reductase (DHFR) inhibitor | Blood schizonticide but inactive across all gametocyte stages [135] | Safety, tolerability and PK study in HAVs completed; not published (NCT02885506) | In vitro IC50 (SD) for quadruple DHFR mutant is 56 nM (20) compared to > 100,000 for pyrimethamine [136] | – | Chemoprevention |
| CDRI 97/78 Ipca |
Trioxane | Blood schizonticide | Well-tolerated in HAVs with half-life of 11.85 ± 1.94 h [137] | – | AEs were few and not severe [137] | Part of a combination therapy |
| M5717 (formerly) DDD107498 MMV, Merck, University of Dundee |
Quinoline-4-carboxamide inhibits P. falciparum translation elongation factor-2 required for protein synthesis [72] | Multi-stage (liver, blood and transmission blocking) | FIH study of single ascending dose, multiple ascending dose and malaria challenge model in healthy subjects is recruiting (NCT03261401) | More potent than artesunate in ex vivo study: median EC50 0.81 nM (range 0.29–3.29 nM) [72] | – | Single-dose treatment costing US$1 |
| SJ733 MMV, University of Kentucky, Eisai |
Dihydroisoquinoline inhibiting PfATP4 | Blood schizonticide for falciparum and vivax, inhibits gametocytogenesis [138, 139] | Dose-escalation study in HAVs (NCT02661373) is due to complete recruitment in July 2018. A human challenge (early induced blood stage malaria infection) study is complete (NCT02867059) | EC50 range 10–60 nM. Effective dose and exposure (AUCED90) superior to artesunate in animal models [139] | No toxicity in mouse model with a dose ~ 43-fold higher than effective dose [139] | Part of single dose treatment |
| ACT-451840 Actelion Pharmaceuticals [140] |
Phenylalanine-based compound; unknown mechanism of action | Blood schizonticide | Single ascending-dose study in HAVs. Exposure increased with food. Half-life ~ 34 h [141] | Minimal parasiticidal concentration was 1.5 ng/mL [141] | No adverse event leading to withdrawal [141] | Combination therapy, e.g. with lumefantrine or mefloquine [140] |
ACPR adequate clinical and parasitological response, AE adverse event, ALT alanine aminotransferase, FIH first in human, HAV healthy adult volunteer, MMV Medicines for Malaria Venture, PCT parasite clearance time, PF Plasmodium falciparum, PCt1/2 parasite clearance half-life, PK pharmacokinetic, PV Plasmodium vivax
Fig. 2.

Graph representing comparative parasite clearance rates after treatment with different antimalarial classes.
Adapted from White NJ [150] with permission
Antimalarials with a synthetic peroxide scaffold, e.g. arterolane, artefenomel, were prioritised for development initially because the advantageous properties of the artemisinin derivatives were replicated (rapid onset, strong parasiticidal effect), but with modifications to remove some of the disadvantages (reliance on agricultural supply, short duration of action, reproductive toxicity in animals) [43]. The establishment of artemisinin resistance raises the concern of cross-resistance due to chemical similarities between the two groups of compounds. By contrast, cipargamin and KAF 156 are structurally unrelated to the artemisinin derivatives.
Artefenomel–ferroquine is a combination of a fast- and long-acting synthetic ozonide, artefenomel (previously OZ439), which has an elimination half-life of 46–62 h [44, 45], and ferroquine (FQ), a 4-aminoquinoline. The goal is to create a single-dose curative treatment, and a multicentre dose-finding Phase 2b study (NCT02497612) is underway. An advantage of this product is that neither of the constituent drugs has been deployed as monotherapy previously. FQ retains activity against chloroquine- and piperaquine-resistant parasites in vitro and has a long elimination half-life of 16 days. It is only moderately efficacious as monotherapy but when combined with artesunate (daily dose of 4 or 6 mg/kg FQ plus artesunate 4 mg/kg for 3 days) the polymerase chain reaction (PCR)-corrected efficacy at 28 days for the treatment of uncomplicated falciparum malaria was 99% (95% CI 93–100) [46]. The potential for cross resistance of artemisinin-resistant parasites to artefenomel has been investigated. Evidence from in vitro ring-stage survival assays showed cross resistance only for parasites carrying the kelch13 I543T mutation, and not the more well-known C580Y mutation [47]. In a clinical study close to the Thailand-Myanmar border in 2015, median parasite clearance following treatment of 19 patients with kelch13 mutant parasites (including C580Y) compared to wild type was 5.5 vs 4.4 h; p = 0.34 [48]. Artefenomel was originally paired with piperaquine, and a Phase 2 study found parasite clearance times were longer in patients with parasites carrying mutations in kelch13= [49]. More clinical data are needed to resolve this question; however, this is challenging to provide since artemisinin-resistant malaria in Southeast Asia is now typically concentrated in remote areas making conducting Phase 3 trials of new drugs in these populations less feasible than previously.
Lumefantrine-KAF156 combines an aryl-amino alcohol, already in widespread use in the artemether–lumefantrine (LUM) combination, with the highly potent imidazolopiperazine KAF156, which has multistage activity and an elimination half-life of 48.7 ± 7.9 h [50]. Regimens comprising different single daily doses given for one, two or three days are being evaluated in a Phase 2b study in West Africa (NCT03167242). A new formulation of lumefantrine, LUM-SDF (Lumefantrine Solid Dispersion Formulation) that can be given once daily is being studied in this trial [51]. This has an advance over the artemether–lumefantrine co-formulation in widespread use, which is administered twice daily, since lumefantrine absorption has been shown to be fat-dependent and dose-limited [52]. The prospects for LUM-KAF156 in areas where artemether–lumefantrine has been used for a long time, or in areas of multidrug resistance are unclear [53, 54]. Piperaquine was assessed as a potential partner for KAF156 initially, and a drug-drug interaction study in healthy volunteers was performed with reassuring findings; however, development of this combination is not being pursued currently [55].
Fosmidomycin–piperaquine Fosmidomycin was developed as an antibacterial drug in the 1980s. This inhibitor of isoprenoid biosynthesis has a good safety profile but has shown variable efficacy both as monotherapy or when partnered with clindamycin [56–58]. Most recently, it has been partnered with piperaquine, a bisquinoline developed in the 1960s, which already forms part of a leading ACT in combination with dihydroartemisinin [59]. A small Phase 2b trial in Ghanaian patients of all ages showed high efficacy of fosmidomycin–piperaquine [60]. Of note, the dose of piperaquine is 16 mg/kg bodyweight, which has been demonstrated to be sub-optimal in the standard DHA–piperaquine ACT in children less than 5 years of age [61]. This is likely to be of relevance for the combination with fosmidomycin, a less potent drug than DHA with an elimination half-life of around 2 h [62]. Unfortunately, piperaquine resistance has now been reported from Cambodia, Thailand and Vietnam, effectively ruling this combination out as a successor for the failing ACTs in Southeast Asia [25].
A feature of the combinations in Phase 2 is that they all contain one partner which may have compromised efficacy in areas of multidrug resistance. This is a potential setback for the new treatments and may stimulate new combinations of the more potent drug classes with partners not in widespread use such as pyronaridine and methylene blue.
Cipargamin, a spiroindolone and potent, long-acting (elimination half-life 18.5 ± 4.7 h [63, 64]) blood schizonticide is still under evaluation as a single agent. A dose-escalation safety study is recruiting in Mali (NCT03334747) with a special focus on hepatotoxicity after signals in earlier studies [63]. If cipargamin is shown to be well-tolerated, it has potential to become part of a single-dose treatment. A Phase 1 drug-drug interaction study of cipargamin and piperaquine showed no increase in exposure to either drug and no safety concerns [65].
DSM-265 is a long-acting (elimination half-life 86–118 h in experimental malaria infection) dihydroorotate dehydrogenase inhibitor with blood and liver stage activity in Phase 2 development for treatment and prevention. It is a less potent inhibitor of parasite multiplication than some of the newer classes, thus is likely to be partnered with a more rapidly effective agent [66, 67]. In vitro studies suggest a relatively low barrier to resistance selection, so measures to protect this drug, such as matching with a partner with similar elimination kinetics and only deploying as part of a fixed-dose combination will be important [68].
AQ-13 is a substituted 4-aminoquinoline, which has already been in clinical development for more than 10 years. A small proof-of-concept study reported in 2017 showed it to be non-inferior to artemether–lumefantrine in symptomatic adult males in Mali [69]. In terms of activity and potency it is likely to be similar to ferroquine. The mean (95% CI) elimination half-life was reported as 3.9 (2.4–5.3) days in malaria patients in the Mali study [69] compared to an earlier finding of 14.3 days [6.2–39.3] in healthy adult volunteers [70].
MMV 390048 is an aminopyridine Plasmodium phosphatidylinositol 4-kinase inhibitor currently in Phase 2a (NCT02880241). This is a multistage antimalarial with potential for chemoprevention [71]. Another treatment prospect that has moved into clinical development recently is DDD 107498, also known as M5717, an inhibitor of peptide elongation factor 2. This is a multiple-stage antimalarial with activity against blood stages, pre-erythrocytic stages, and male and female mature gametocytes [72, 73].
New Drugs in Clinical Development for Prevention
There is a paucity of new agents in development for chemoprevention alone. DSM-265 shows potential as a prophylactic agent for travellers, although doses studied so far in a human challenge model have not conferred seven days of protection from malaria infection, which would be required for weekly dosing [74].
Pregnant women and infants are typically excluded from studies of new antimalarials, despite bearing the brunt of most malaria-associated morbidity and mortality. As a result, they are the last to receive them since it takes time for sufficient efficacy and safety data to accumulate post-registration. Ideally, drugs used for prevention should be different to those deployed for treatment, but currently this is not the case and adequate data on treatment of malaria in pregnancy with a new agent is required before it is considered acceptable to assess use for prevention. SP continued to be used for prevention after it had been abandoned as a treatment in some countries since it retains some protective effect in the absence of high-grade resistance. Antimalarial under-dosing in pregnant women and young children has been recognised belatedly for several drugs, e.g. DHA–piperaquine, SP, artemether–lumefantrine, and this is a risk for the new agents, none of which have yet been evaluated in pregnancy [75–77].
New Drugs in Clinical Development for Relapse Prevention (Plasmodium vivax and Plasmodium ovale)
Like primaquine, tafenoquine is an 8-aminoquinoline active against asexual stages of P. vivax, but more importantly against hypnozoites which cause relapse. This long-acting drug (elimination half-life 14–19 days) is being developed for single-dose relapse prevention for vivax malaria and a new drug application was submitted to US FDA and the Australian Therapeutic Goods Administration in late 2017 [78]. The development process has been slow and painstaking, necessitated by the known potential for haematological toxicity (acute haemolytic anaemia) in persons with glucose-6-phosphate dehydrogenase (G6PD) deficiency, an X-linked enzymopathy found in malaria-endemic areas. No clinically significant PK-PD interactions or safety concerns were observed when chloroquine and tafenoquine were co-administered in healthy adult volunteers [79]. Tafenoquine has the potential to revolutionise radical therapy of the relapsing malarias by enhancing patient adherence compared to the standard 14 days of primaquine and may be a potent tool to accelerate vivax malaria elimination; however, it will need to be deployed with concurrent reliable point-of-care G6PD activity testing to screen out individuals at risk. Differences in metabolism of primaquine with a resulting impact on efficacy for relapse prevention have been observed in association with allelic variation in the cytochrome 2D6 gene [80]. Preliminary investigation has indicated the same may not be true for tafenoquine, but more evidence is needed [81].
New Drugs in Clinical Development for Transmission-Blocking
Reducing malaria transmission substantially will accelerate malaria elimination. Drugs preventing malaria transmission target gametocytes, sporogony or the mosquito vector by reducing its survival (endectocides). Single low-dose (0.25 mg/kg) primaquine is now recommended alongside antimalarial treatment in endemic countries targeting elimination for its activity against mature gametocytes; thus, the potential for drug-drug interactions with new blood schizonticides needs to be evaluated. Very few new agents are in development specifically for transmission-blocking, although some have multistage activity, e.g. KAF 156. Methylene blue is an old parasiticidal agent with blood stage activity and the added benefit of activity against mature male and female P. falciparum gametocytes. It is being developed by the University of Heidelberg in combination with ACTs as a strategy to protect against artemisinin resistance emergence and reduce transmission [82].
Re-purposing of Antimicrobial Drugs for Malaria Prevention or Treatment
Several antibiotics have weak-to-moderate antimalarial activity, e.g. doxycycline, clindamycin, azithromycin, and cotrimoxazole. Cotrimoxazole is given to patients with HIV to protect against Pneumocystis jirovecii pneumonia and has been shown to reduce malaria infections, and is a possible candidate for use as prevention in HIV-uninfected pregnant women or children [83]. Azithromycin is amenable to once-daily dosing and has a good safety profile; therefore it could theoretically be combined as part of a short-course once-daily combination therapy or be used for prevention. Drawbacks include selection for resistance in other common important pathogens, e.g. Salmonella enterica serovar Typhi, Streptococcus pneumoniae. In the 1970s, the discovery of ivermectin, a common anti-parasitic agent used to treat animals, led to the Nobel Prize in Medicine being awarded to William C. Campbell and Satoshi Ōmura in 2015. The prize was shared with Tu Youyou in recognition of the discovery of qinghaosu (artemisinin) to treat malaria. Ivermectin has also been shown to reduce malaria transmission by reducing mosquito survival [84]. It has other uses for human disease, e.g. mass treatment for lymphatic filariasis, thus has the potential to be used as a tool in integrated disease-control programmes [85].
New Adjunctive Therapies in Development for the Treatment of Severe Malaria
The search for adjunctive therapies capable of improving the outcome from severe malaria continues after serial alternatives have failed to show any benefit, or in some cases, caused harm [16]. Sevuparin is a polysaccharide heparin analogue, which retains the anti-adhesive effects of heparin without the antithrombin properties and has been shown to block merozoite invasion, cytoadherence and rosetting [86, 87]. Studies in patients with severe malaria are planned.
Drugs in Pre-clinical Development
An up-to-date analysis has estimated that molecules in pre-clinical development have an 8% chance of becoming a registered product [30]. Agents in active development are shown in Table 3. MMV 253 (previously AZ13721412) is a very long-acting Plasmodium ATPase inhibitor in early preclinical development, with the goal of being part of a single-dose radical cure [88, 89]. JPC-3210 is another promising long-acting drug shown to be active against multidrug resistant P. falciparum in vitro [90, 91].
Table 3.
Agents in preclinical development
| Name/ development partners | Type/target | Activity spectrum | Other characteristics | Vision for development |
|---|---|---|---|---|
| MMV 253 (previously AZ13721412)/MMV and Zydus Cadila [142] | Triaminopyrimidine, Plasmodium ATPase inhibitor | Blood schizonticide | Long acting | Part of a single-dose radical cure |
| AN 13762/MMV, University of California, San Francisco [143] | Benzoxaborole, mechanism of action unknown | Blood schizonticide | Reassuring pre-clinical toxicology | Part of a single-dose radical cure, prevention |
| JPC-3210/MMV and Zydus Cadila [90] | Aminomethylphenol | Blood schizonticide | Long acting | Treatment and prevention |
| UCT 943 (previously MMV 642943)/MMV, H3D, University of Cape Town | PfPI(4)K inhibitor [144] | Multi-stage, falciparum and vivax | – | Back-up for MMV048 [71] (part of a single-dose radical cure, transmission blocking, prevention) |
| NPC1161B/University of Mississippi [93, 145] | 8-Aminoquinoline (single enantiomer) | Multi-stage | Superior radical curative activity to primaquine in animal models [146] | Vivax malaria relapse prevention |
| SC83288/University of Heidelberg [92] | Amicarbalide derivative | Blood schizonticide | Rapid action (log parasite reduction ratio ~ 3 in a mouse model) | Severe malaria treatment |
| SAR121 (previously MMV688533)/Sanofi, MMV | Unknown mechanism of action | Blood schizonticide | Long acting | Part of a single-dose radical cure |
| DM1157(PL 69)/DesignMedix | “Reversed chloroquine”, similar mechanism of action | Blood schizonticide | No published data (similar to chloroquine) | Part of a single-dose radical cure [147] |
SC83288 is the only drug in preclinical development for severe malaria. This amicarbalide derivative is fast acting and adapted for parenteral administration. Preliminary findings in a murine model have demonstrated a three-log parasite reduction per asexual life cycle, which is approximately 10-fold lower than that observed with parenteral artesunate treatment in humans [92].
In late preclinical development for relapse prevention is another 8-aminoquinoline, NPC1161B, developed by the University of Mississippi [93]. The development plan is to see whether this single enantiomer drug has a more favourable haematological toxicity profile than tafenoquine in Phase 1.
Antimalarial Drug Discovery
Drug discovery for malaria is in a healthy state, with numerous compounds under investigation following successful screening programmes. As evidenced by the development of the artemisinin derivatives, it is possible to register highly successful antimalarials without fully understanding the drug target or mechanism of action. Phytochemical screening is still being pursued to identify new antimalarials. From the target-based approaches, a number of Plasmodium proteases have been singled out; these perform critical functions in the parasite life cycle [94]. There are different families such as the falcipain cysteine proteases, aminopeptidases, and plasmepsins (aspartic proteases). Plasmepsin V inhibitors have been proposed as powerful new agents, and recent elucidation of the function of plasmepsins IX and X has revealed them to be two new promising additional targets for potential multistage antimalarials [95–97]. Another target that has garnered a lot of interest is Na+-ATPase 4, the site of action of the potent cipargamin and SJ557733, both in clinical development, and of 21A092, a pyrazoleamide in the discovery phase [98]. Pathways involved in haem detoxification are also under study [99]. Work continues on developing parasite-selective inhibitors of phenylalanyl-tRNA synthetase, histone deacetylase, hypoxanthine–guanine–(xanthine) phosphoribosyltransferase and N-myristoyltransferase [100–103]. Another avenue of research and development is the creation of hybrid molecules, i.e. combining two synergistic agents with different targets as a way of increasing the barrier to resistance [104, 105].
As a general rule, available libraries are biased in favour of compounds with blood-stage activity but there has been progress in the discovery of agents capable of preventing transmission through dedicated screening programmes such as that of the GSK Tres Cantos Antimalarial Set (TCAMS), which identified 98 out of 13,533 molecules with activity against trophozoites and stage V gametocytes and prioritized 56 compounds [106]. Torin 2, an agent with multistage activity, including gametocytes, identified in a different screening programme, has been selected for lead optimisation by the National Center for Advancing Translational Sciences (NCATS) programme of development of malaria transmission-blocking drugs [107, 108]. Detailed studies of mature P. falciparum gametocytes in human infections have discovered that females tend to outnumber males by a ratio of ~four to one and that they differ in their susceptibility to antimalarial drugs, with males tending to be more sensitive [109, 110]. This knowledge may be exploited in the development of new gametocytocidal drugs but requires new methods of assessment to distinguish males from females [111, 112]. To date, the assessment of transmission blocking in clinical trials of new drugs has relied on gametocyte quantitation in peripheral blood by microscopic examination, which is inadequate.
Discovery of new drugs for relapse prevention is hampered by the challenge of finding suitable pre-clinical models to screen for activity against hypnozoites but there has been progress, for example, with the development of an in vitro liver stage drug assay [113, 114]. A novel class of non-8-aminoquinoline compounds, the PI4K inhibitors e.g. KAI407 and KDU691, has been shown to kill early P. cynomolgi dormant liver stages in vitro [113, 114].
Conclusion
The malaria drug development pipeline, at present, is in a healthier state than for many years; however, there are no novel compounds ready to be deployed in areas where existing treatments are failing (Fig. 3). Of the newer agents, some classes possess multistage activity and are highly potent in terms of their ability to inhibit parasite multiplication (Fig. 1). While there has been encouraging progress towards malaria elimination in some countries, e.g. Sri Lanka was certified as malaria free in 2016, this is not universal, and recent reports suggest progress has stalled [1]. This makes it highly likely that there will be a continued need for new drugs for many years to come.
Fig. 3.
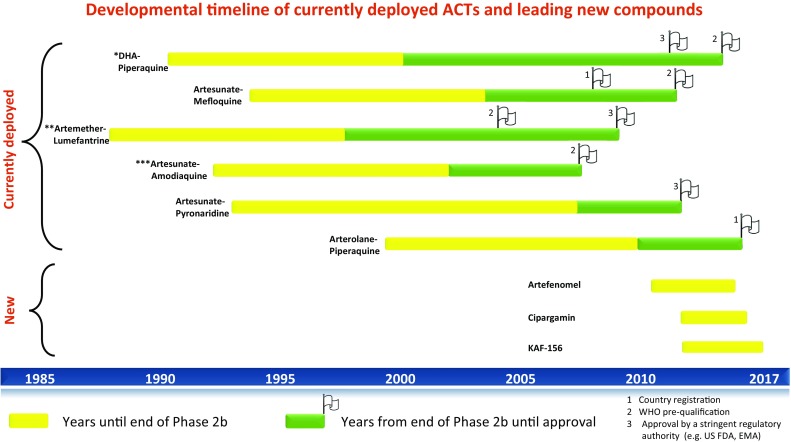
Developmental time-line of currently deployed ACTs versus leading compounds. Information sourced from [11, 31, 148, 149]. Single asterisk: Dihydroartemisinin–piperaquine (WHO pre-qualification refers to Eurartesim® by sigma tau in 2015); approved by EMA in 2011. Double asterisk: Artemether–lumefantrine (WHO pre-qualification for Coartem® by Novartis in 2004, products by Ipca and Cipla in 2009; others have followed since then). Triple asterisk: Artesunate–amodiaquine (WHO pre-qualification for Sanofi’s ASAQ Winthrop® in 2008, Ipca and Guilin products in 2012, Ajanta in 2013 and Cipla in 2014)
Looking to the future, a variety of target product profiles tailored to control and elimination goals in different populations have been defined, but the highest priority is still to ensure a continued supply of effective blood schizonticides and to maximise their useful therapeutic life. Single exposure treatments are desirable but not essential. Continued investment in drug discovery will help to sustain the current pipeline.
Prevention for groups at highest risk is likely to rely on the same classes of antimalarials as used for treatment, given the difficulties of developing drugs for use in pregnant women and children. There are calls to accelerate reproductive toxicology studies by bringing them further forward in the development process [31].
For relapse prevention there is good news, with tafenoquine closer to registration. However, the lack of options safe for use in populations where G6PD deficiency is prevalent is an impediment to the elimination of vivax malaria [34]. Recent progress in the development of methods to screen for hypnozoiticidal and transmission-blocking compounds may lead to more agents being identified in the coming years.
The failure of the ACTs in Southeast Asia has led to a re-evaluation of combination therapy for malaria. Rather than only using triple combinations of existing drugs as a salvage strategy to try to improve cure rates in areas where resistance has already taken hold, there is an argument for introducing them in countries with no evidence of resistance, sooner rather than later, as well as ensuring all new combinations developed are composed of three or more drugs. The drawbacks of this approach include increased development time, increased costs and increased risks of toxicity; thus, any ambition to produce a novel triple combination that is efficacious against all strains of malaria in time to replace currently failing ACTs may have to be sacrificed in the interests of expediency. However, this could be the goal for future generations of novel antimalarial combination therapies.
Acknowledgements
Thanks to Olaf Mueller, Larry Walker, Philip Rosenthal, John Haselden and Andrea Ruecker for updates and/or advice, and to the anonymous reviewers for constructive criticism. The Myanmar Oxford Clinical Research Unit is part of the MORU Tropical Health Network funded by the Wellcome Trust.
Compliance with Ethical Standards
Conflict of interest
APP was an investigator on trials of artefenomel sponsored by Medicines for Malaria Venture and KAF156 and cipargamin sponsored by Novartis. EAA was a co-investigator on studies of DHA–piperaquine funded by Medicines for Malaria Venture and of artesunate–mefloquine funded by Drugs for Neglected Disease Initiative.
References
- 1.World Health Organization . World malaria report. Geneva: World Health Organization; 2017. [Google Scholar]
- 2.World Health Organization Malaria vaccine: WHO position paper, January 2016—recommendations. Vaccine. 2017 doi: 10.1016/j.vaccine.2016.10.070. [DOI] [PubMed] [Google Scholar]
- 3.Antimalaria studies on Qinghaosu. Chin Med J. 1979;92(12):811–6. [PubMed]
- 4.White NJ, Hien TT, Nosten FH. A brief history of Qinghaosu. Trends Parasitol. 2015;31(12):607–610. doi: 10.1016/j.pt.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White N. Antimalarial drug resistance and combination chemotherapy. Philos Trans R Soc Lond Ser B Biol Sci. 1999;354(1384):739–749. doi: 10.1098/rstb.1999.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snow RW, Trape JF, Marsh K. The past, present and future of childhood malaria mortality in Africa. Trends Parasitol. 2001;17(12):593–597. doi: 10.1016/s1471-4922(01)02031-1. [DOI] [PubMed] [Google Scholar]
- 7.Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6(4):861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roper C, Pearce R, Nair S, Sharp B, Nosten F, Anderson T. Intercontinental spread of pyrimethamine-resistant malaria. Science (New York, NY) 2004;305(5687):1124. doi: 10.1126/science.1098876. [DOI] [PubMed] [Google Scholar]
- 9.Maxmen A. Busting the billion-dollar myth: how to slash the cost of drug development. Nature. 2016;536(7617):388–390. doi: 10.1038/536388a. [DOI] [PubMed] [Google Scholar]
- 10.World Health Organization . Guidelines for the treatment of malaria. 3. Geneva: World Health Organization; 2015. [PubMed] [Google Scholar]
- 11.Premji ZG. Coartem: the journey to the clinic. Malar J. 2009;8(Suppl 1):S3. doi: 10.1186/1475-2875-8-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barber BE, Grigg MJ, William T, Yeo TW, Anstey NM. The treatment of Plasmodium knowlesi malaria. Trends Parasitol. 2017;33(3):242–253. doi: 10.1016/j.pt.2016.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Price RN, von Seidlein L, Valecha N, Nosten F, Baird JK, White NJ. Global extent of chloroquine-resistant Plasmodium vivax: a systematic review and meta-analysis. Lancet Infect Dis. 2014;14(10):982–991. doi: 10.1016/S1473-3099(14)70855-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dondorp AM, Fanello CI, Hendriksen IC, Gomes E, Seni A, Chhaganlal KD, et al. Artesunate versus quinine in the treatment of severe falciparum malaria in African children (AQUAMAT): an open-label, randomised trial. Lancet (Lond, Engl) 2010;376(9753):1647–1657. doi: 10.1016/S0140-6736(10)61924-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dondorp A, Nosten F, Stepniewska K, Day N, White N, South East Asian Quinine Artesunate Malaria Trial Group Artesunate versus quinine for treatment of severe falciparum malaria: a randomised trial. Lancet (Lond, Engl) 2005;366(9487):717–725. doi: 10.1016/S0140-6736(05)67176-0. [DOI] [PubMed] [Google Scholar]
- 16.John CC, Kutamba E, Mugarura K, Opoka RO. Adjunctive therapy for cerebral malaria and other severe forms of Plasmodium falciparum malaria. Expert Rev Anti Infect Ther. 2010;8(9):997–1008. doi: 10.1586/eri.10.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM, et al. Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med. 2008;359(24):2619–2620. doi: 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- 18.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, et al. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009;361(5):455–467. doi: 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashley EA, Dhorda M, Fairhurst RM, Amaratunga C, Lim P, Suon S, et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2014;371(5):411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miotto O, Amato R, Ashley EA, MacInnis B, Almagro-Garcia J, Amaratunga C, et al. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet. 2015;47(3):226–234. doi: 10.1038/ng.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505(7481):50–55. doi: 10.1038/nature12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thanh NV, Thuy-Nhien N, Tuyen NT, Tong NT, Nha-Ca NT, Dong LT, et al. Rapid decline in the susceptibility of Plasmodium falciparum to dihydroartemisinin–piperaquine in the south of Vietnam. Malar J. 2017;16(1):27. doi: 10.1186/s12936-017-1680-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phyo AP, Ashley EA, Anderson TJC, Bozdech Z, Carrara VI, Sriprawat K, et al. Declining efficacy of artemisinin combination therapy against P. Falciparum malaria on the Thai–Myanmar border (2003–2013): the role of parasite genetic factors. Clin Infect Dis Off Publ Infect Dis Soc Am. 2016;63(6):784–791. doi: 10.1093/cid/ciw388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amato R, Lim P, Miotto O, Amaratunga C, Dek D, Pearson RD, et al. Genetic markers associated with dihydroartemisinin-piperaquine failure in Plasmodium falciparum malaria in Cambodia: a genotype-phenotype association study. Lancet Infect Dis. 2017;17(2):164–173. doi: 10.1016/S1473-3099(16)30409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Witkowski B, Duru V, Khim N, Ross LS, Saintpierre B, Beghain J, et al. A surrogate marker of piperaquine-resistant Plasmodium falciparum malaria: a phenotype-genotype association study. Lancet Infect Dis. 2017;17(2):174–183. doi: 10.1016/S1473-3099(16)30415-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chenet SM, Akinyi Okoth S, Huber CS, Chandrabose J, Lucchi NW, Talundzic E, et al. Independent emergence of the Plasmodium falciparum Kelch propeller domain mutant allele C580Y in Guyana. J Infect Dis. 2016;213(9):1472–1475. doi: 10.1093/infdis/jiv752. [DOI] [PubMed] [Google Scholar]
- 27.Mishra N, Bharti RS, Mallick P, Singh OP, Srivastava B, Rana R, et al. Emerging polymorphisms in falciparum Kelch 13 gene in Northeastern region of India. Malar J. 2016;15(1):583. doi: 10.1186/s12936-016-1636-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menard D, Khim N, Beghain J, Adegnika AA, Shafiul-Alam M, Amodu O, et al. A worldwide map of Plasmodium falciparum K13-propeller polymorphisms. N Engl J Med. 2016;374(25):2453–2464. doi: 10.1056/NEJMoa1513137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burrows JN, van Huijsduijnen RH, Mohrle JJ, Oeuvray C, Wells TN. Designing the next generation of medicines for malaria control and eradication. Malar J. 2013;12:187. doi: 10.1186/1475-2875-12-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burrows JN, Duparc S, Gutteridge WE, Hooft van Huijsduijnen R, Kaszubska W, Macintyre F, et al. New developments in anti-malarial target candidate and product profiles. Malar J. 2017;16(1):26. doi: 10.1186/s12936-016-1675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wells TN, Hooft van Huijsduijnen R, Van Voorhis WC. Malaria medicines: a glass half full? Nat Rev Drug Discov. 2015;14(6):424–442. doi: 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- 32.Bushell E, Gomes AR, Sanderson T, Anar B, Girling G, Herd C, et al. Functional profiling of a plasmodium genome reveals an abundance of essential genes. Cell. 2017;170(2):260e8–272e8. doi: 10.1016/j.cell.2017.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hovlid ML, Winzeler EA. Phenotypic screens in antimalarial drug discovery. Trends Parasitol. 2016;32(9):697–707. doi: 10.1016/j.pt.2016.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cowell AN, Istvan ES, Lukens AK, Gomez-Lorenzo MG, Vanaerschot M, Sakata-Kato T, et al. Mapping the malaria parasite druggable genome by using in vitro evolution and chemogenomics. Science (New York, NY) 2018;359(6372):191–199. doi: 10.1126/science.aan4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wells TN, Willis P, Burrows JN, Hooft van Huijsduijnen R. Open data in drug discovery and development: lessons from malaria. Nat Rev Drug Discov. 2016;15(10):661–662. doi: 10.1038/nrd.2016.154. [DOI] [PubMed] [Google Scholar]
- 36.McCarthy JS, Marquart L, Sekuloski S, Trenholme K, Elliott S, Griffin P, et al. Linking murine and human Plasmodium falciparum challenge models in a translational path for antimalarial drug development. Antimicrob Agents Chemother. 2016;60(6):3669–3675. doi: 10.1128/AAC.02883-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hien TT, White NJ, Thuy-Nhien NT, Hoa NT, Thuan PD, Tarning J, et al. Estimation of the in vivo MIC of cipargamin in uncomplicated Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2017;61(2):e01940-16. doi: 10.1128/AAC.01940-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tagbor H, Cairns M, Bojang K, Coulibaly SO, Kayentao K, Williams J, et al. A non-inferiority, individually randomized trial of intermittent screening and treatment versus intermittent preventive treatment in the control of malaria in pregnancy. PloS One. 2015;10(8):e0132247. doi: 10.1371/journal.pone.0132247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radeva-Petrova D, Kayentao K, ter Kuile FO, Sinclair D, Garner P. Drugs for preventing malaria in pregnant women in endemic areas: any drug regimen versus placebo or no treatment. Cochrane Database Syst Rev. 2014;(10):CD000169. 10.1002/14651858.cd000169.pub3. [DOI] [PMC free article] [PubMed]
- 40.Desai M, Gutman J, L’Lanziva A, Otieno K, Juma E, Kariuki S, et al. Intermittent screening and treatment or intermittent preventive treatment with dihydroartemisinin–piperaquine versus intermittent preventive treatment with sulfadoxine–pyrimethamine for the control of malaria during pregnancy in western Kenya: an open-label, three-group, randomised controlled superiority trial. Lancet (Lond, Engl) 2015;386(10012):2507–2519. doi: 10.1016/S0140-6736(15)00310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Permala J, Tarning J, Nosten F, White NJ, Karlsson MO, Bergstrand M. Prediction of improved antimalarial chemoprevention with weekly dosing of dihydroartemisinin–piperaquine. Antimicrob Agents Chemother. 2017;61(5):e02491-16. doi: 10.1128/AAC.02491-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Medicines for Malaria Venture. Global portfolio of antimalarial medicines 2017. https://www.mmv.org/research-development/mmv-supported-projects. Accessed 19 Jan 2018.
- 43.Clark RL. Improved safety margin for embryotoxicity in rats for the new endoperoxide artefenomel (OZ439) as compared to artesunate. Antimicrob Agents Chemother. 2017 doi: 10.1002/bdr2.1170. [DOI] [PubMed] [Google Scholar]
- 44.Charman SA, Arbe-Barnes S, Bathurst IC, Brun R, Campbell M, Charman WN, et al. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci USA. 2011;108(11):4400–4405. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phyo AP, Jittamala P, Nosten FH, Pukrittayakamee S, Imwong M, White NJ, et al. Antimalarial activity of artefenomel (OZ439), a novel synthetic antimalarial endoperoxide, in patients with Plasmodium falciparum and Plasmodium vivax malaria: an open-label phase 2 trial. Lancet Infect Dis. 2015 doi: 10.1016/S1473-3099(15)00320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Held J, Supan C, Salazar CL, Tinto H, Bonkian LN, Nahum A, et al. Ferroquine and artesunate in African adults and children with Plasmodium falciparum malaria: a phase 2, multicentre, randomised, double-blind, dose-ranging, non-inferiority study. Lancet Infect Dis. 2015;15(12):1409–1419. doi: 10.1016/S1473-3099(15)00079-1. [DOI] [PubMed] [Google Scholar]
- 47.Straimer J, Gnadig NF, Stokes BH, Ehrenberger M, Crane AA, Fidock DA. Plasmodium falciparum K13 mutations differentially impact ozonide susceptibility and parasite fitness in vitro. mBio. 2017;8(2):e00172-17. doi: 10.1128/mBio.00172-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Phyo AP, Jittamala P, Nosten FH, Pukrittayakamee S, Imwong M, White NJ, et al. Antimalarial activity of artefenomel (OZ439), a novel synthetic antimalarial endoperoxide, in patients with Plasmodium falciparum and Plasmodium vivax malaria: an open-label phase 2 trial. Lancet Infect Dis. 2016;16(1):61–69. doi: 10.1016/S1473-3099(15)00320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Macintyre F, Adoke Y, Tiono AB, Duong TT, Mombo-Ngoma G, Bouyou-Akotet M, et al. A randomised, double-blind clinical phase II trial of the efficacy, safety, tolerability and pharmacokinetics of a single dose combination treatment with artefenomel and piperaquine in adults and children with uncomplicated Plasmodium falciparum malaria. BMC Med. 2017;15(1):181. doi: 10.1186/s12916-017-0940-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.White N, Duong TT, Uthaisil C, Nosten F, Phyo AP, Hanboonkunupakarn B, et al. Antimalarial activity of KAF156 in falciparum and vivax malaria. N Engl J Med. 2016;375(12):1152–60. doi: 10.1056/NEJMoa1602250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jain JP, Leong FJ, Chen L, Kalluri S, Koradia V, Stein DS, et al. Bioavailability of lumefantrine is significantly enhanced with a novel formulation approach, an outcome from a randomized, open-label pharmacokinetic study in healthy volunteers. Antimicrob Agents Chemother. 2017;61(9):e00868-17. doi: 10.1128/AAC.00868-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashley EA, Stepniewska K, Lindegardh N, McGready R, Annerberg A, Hutagalung R, et al. Pharmacokinetic study of artemether–lumefantrine given once daily for the treatment of uncomplicated multidrug-resistant falciparum malaria. Trop Med Int Health. 2007;12(2):201–208. doi: 10.1111/j.1365-3156.2006.01785.x. [DOI] [PubMed] [Google Scholar]
- 53.Denis MB, Tsuyuoka R, Lim P, Lindegardh N, Yi P, Top SN, et al. Efficacy of artemether–lumefantrine for the treatment of uncomplicated falciparum malaria in northwest Cambodia. Trop Med Int Health. 2006;11(12):1800–1807. doi: 10.1111/j.1365-3156.2006.01739.x. [DOI] [PubMed] [Google Scholar]
- 54.Sondo P, Derra K, Diallo Nakanabo S, Tarnagda Z, Kazienga A, Zampa O, et al. Artesunate–amodiaquine and artemether–lumefantrine therapies and selection of Pfcrt and Pfmdr1 Alleles in Nanoro, Burkina Faso. PloS One. 2016;11(3):e0151565. doi: 10.1371/journal.pone.0151565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leong FJ, Jain JP, Feng Y, Goswami B, Stein DS. A phase 1 evaluation of the pharmacokinetic/pharmacodynamic interaction of the anti-malarial agents KAF156 and piperaquine. Malar J. 2018;17(1):7. doi: 10.1186/s12936-017-2162-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oyakhirome S, Issifou S, Pongratz P, Barondi F, Ramharter M, Kun JF, et al. Randomized controlled trial of fosmidomycin–clindamycin versus sulfadoxine–pyrimethamine in the treatment of Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2007;51(5):1869–1871. doi: 10.1128/AAC.01448-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fernandes JF, Lell B, Agnandji ST, Obiang RM, Bassat Q, Kremsner PG, et al. Fosmidomycin as an antimalarial drug: a meta-analysis of clinical trials. Future Microbiol. 2015;10(8):1375–1390. doi: 10.2217/FMB.15.60. [DOI] [PubMed] [Google Scholar]
- 58.Borrmann S, Lundgren I, Oyakhirome S, Impouma B, Matsiegui PB, Adegnika AA, et al. Fosmidomycin plus clindamycin for treatment of pediatric patients aged 1 to 14 years with Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2006;50(8):2713–2718. doi: 10.1128/AAC.00392-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Davis TM, Hung TY, Sim IK, Karunajeewa HA, Ilett KF. Piperaquine: a resurgent antimalarial drug. Drugs. 2005;65(1):75–87. doi: 10.2165/00003495-200565010-00004. [DOI] [PubMed] [Google Scholar]
- 60.Mombo-Ngoma G, Remppis J, Sievers M, Zoleko Manego R, Endamne L, Kabwende L, et al. Efficacy and safety of fosmidomycin–piperaquine as non-artemisinin-based combination therapy for uncomplicated falciparum malaria—a single-arm, age-de-escalation proof of concept study in Gabon. Clin Infect Dis Off Publ Infect Dis Soc Am. 2017 doi: 10.1093/cid/cix1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.WorldWide Antimalarial Resistance Network DPSG The effect of dosing regimens on the antimalarial efficacy of dihydroartemisinin–piperaquine: a pooled analysis of individual patient data. PLoS Med. 2013;10(12):e1001564. doi: 10.1371/journal.pmed.1001564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murakawa T, Sakamoto H, Fukada S, Konishi T, Nishida M. Pharmacokinetics of fosmidomycin, a new phosphonic acid antibiotic. Antimicrob Agents Chemother. 1982;21(2):224–230. doi: 10.1128/aac.21.2.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.White NJ, Pukrittayakamee S, Phyo AP, Rueangweerayut R, Nosten F, Jittamala P, et al. Spiroindolone KAE609 for falciparum and vivax malaria. N Engl J Med. 2014;371(5):403–410. doi: 10.1056/NEJMoa1315860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rottmann M, McNamara C, Yeung BKS, Lee MCS, Zou B, Russell B, et al. Spiroindolones, a potent compound class for the treatment of malaria. Science (New York, NY) 2010;329(5996):1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stein DS, Jain JP, Kangas M, Lefevre G, Machineni S, Griffin P, et al. Open-label, single-dose, parallel-group study in healthy volunteers to determine the drug-drug interaction potential between KAE609 (cipargamin) and piperaquine. Antimicrob Agents Chemother. 2015;59(6):3493–3500. doi: 10.1128/AAC.00340-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCarthy JS, Lotharius J, Ruckle T, Chalon S, Phillips MA, Elliott S, et al. Safety, tolerability, pharmacokinetics, and activity of the novel long-acting antimalarial DSM265: a two-part first-in-human phase 1a/1b randomised study. Lancet Infect Dis. 2017;17(6):626–635. doi: 10.1016/S1473-3099(17)30171-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McCarthy JS, Griffin PM, Sekuloski S, Elliott SM, Marquart L, Klein K, et al. A dose ranging clinical trial to evaluate the pharmacokinetics and pharmacodynamics of the combination of DSM265 and OZ439 in a Plasmodium falciparum induced blood stage malaria system. Am J Trop Med Hyg. 2015;93(4 Supplement):367–368. [Google Scholar]
- 68.Phillips MA, Lotharius J, Marsh K, White J, Dayan A, White KL, et al. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Transl Med. 2015;7(296):aaa6645. doi: 10.1126/scitranslmed.aaa6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Koita OA, Sangare L, Miller HD, Sissako A, Coulibaly M, Thompson TA, et al. AQ-13, an investigational antimalarial, versus artemether plus lumefantrine for the treatment of uncomplicated Plasmodium falciparum malaria: a randomised, phase 2, non-inferiority clinical trial. Lancet Infect Dis. 2017;17(12):1266–1275. doi: 10.1016/S1473-3099(17)30365-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mzayek F, Deng H, Mather FJ, Wasilevich EC, Liu H, Hadi CM, et al. Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clin Trials. 2007;2(1):e6. doi: 10.1371/journal.pctr.0020006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Paquet T, Le Manach C, Cabrera DG, Younis Y, Henrich PP, Abraham TS, et al. Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4-kinase. Sci Transl Med. 2017;9(387):eaad9735. doi: 10.1126/scitranslmed.aad9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baragana B, Hallyburton I, Lee MC, Norcross NR, Grimaldi R, Otto TD, et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 2015;522(7556):315–320. doi: 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.ClinicalTrials.gov. First-in-human trial of single ascending dose, multiple ascending dose and malaria challenge model in healthy subjects NCT03261401. 2017. https://clinicaltrials.gov/ct2/show/study/NCT03261401?view=results. Accessed 14 Jan 2018.
- 74.Sulyok M, Ruckle T, Roth A, Murbeth RE, Chalon S, Kerr N, et al. DSM265 for Plasmodium falciparum chemoprophylaxis: a randomised, double blinded, phase 1 trial with controlled human malaria infection. Lancet Infect Dis. 2017;17(6):636–644. doi: 10.1016/S1473-3099(17)30139-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hoglund RM, Workman L, Edstein MD, Thanh NX, Quang NN, Zongo I, et al. Population pharmacokinetic properties of piperaquine in falciparum malaria: an individual participant data meta-analysis. PLoS Med. 2017;14(1):e1002212. doi: 10.1371/journal.pmed.1002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barnes KI, Little F, Smith PJ, Evans A, Watkins WM, White NJ. Sulfadoxine–pyrimethamine pharmacokinetics in malaria: pediatric dosing implications. Clin Pharmacol Ther. 2006;80(6):582–596. doi: 10.1016/j.clpt.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 77.Tarning J, McGready R, Lindegardh N, Ashley EA, Pimanpanarak M, Kamanikom B, et al. Population pharmacokinetics of lumefantrine in pregnant women treated with artemether–lumefantrine for uncomplicated Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2009;53(9):3837–3846. doi: 10.1128/AAC.00195-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.GSK . GSK submits US regulatory application for single-dose tafenoquine for Plasmodium vivax malaria. London: GSK; 2017. [Google Scholar]
- 79.Miller AK, Harrell E, Ye L, Baptiste-Brown S, Kleim JP, Ohrt C, et al. Pharmacokinetic interactions and safety evaluations of coadministered tafenoquine and chloroquine in healthy subjects. Br J Clin Pharmacol. 2013;76(6):858–867. doi: 10.1111/bcp.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vuong C, Xie LH, Potter BM, Zhang J, Zhang P, Duan D, et al. Differential cytochrome P450 2D metabolism alters tafenoquine pharmacokinetics. Antimicrob Agents Chemother. 2015;59(7):3864–3869. doi: 10.1128/AAC.00343-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.St Jean PL, Xue Z, Carter N, Koh GCKW, Duparc S, Taylor M, et al. Tafenoquine treatment of Plasmodium vivax malaria: suggestive evidence that CYP2D6 reduced metabolism is not associated with relapse in the phase 2b DETECTIVE trial. Malar J. 2016;15(1):97. doi: 10.1186/s12936-016-1145-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Coulibaly B, Pritsch M, Bountogo M, Meissner PE, Nebie E, Klose C, et al. Efficacy and safety of triple combination therapy with artesunate–amodiaquine–methylene blue for falciparum malaria in children: a randomized controlled trial in Burkina Faso. J Infect Dis. 2015;211(5):689–697. doi: 10.1093/infdis/jiu540. [DOI] [PubMed] [Google Scholar]
- 83.Mbeye NM, ter Kuile FO, Davies MA, Phiri KS, Egger M, Wandeler G, et al. Cotrimoxazole prophylactic treatment prevents malaria in children in sub-Saharan Africa: systematic review and meta-analysis. Trop Med Int Health. 2014;19(9):1057–1067. doi: 10.1111/tmi.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ouedraogo AL, Bastiaens GJ, Tiono AB, Guelbeogo WM, Kobylinski KC, Ouedraogo A, et al. Efficacy and safety of the mosquitocidal drug ivermectin to prevent malaria transmission after treatment: a double-blind, randomized, clinical trial. Clin Infect Dis Off Publ Infect Dis Soc Am. 2015;60(3):357–365. doi: 10.1093/cid/ciu797. [DOI] [PubMed] [Google Scholar]
- 85.Alout H, Foy BD. Ivermectin: a complimentary weapon against the spread of malaria? Expert Rev Anti Infect Ther. 2017;15(3):231–240. doi: 10.1080/14787210.2017.1271713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saiwaew S, Sritabal J, Piaraksa N, Keayarsa S, Ruengweerayut R, Utaisin C, et al. Effects of sevuparin on rosette formation and cytoadherence of Plasmodium falciparum infected erythrocytes. PloS One. 2017;12(3):e0172718. doi: 10.1371/journal.pone.0172718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Leitgeb AM, Charunwatthana P, Rueangveerayut R, Uthaisin C, Silamut K, Chotivanich K, et al. Inhibition of merozoite invasion and transient de-sequestration by sevuparin in humans with Plasmodium falciparum malaria. PloS One. 2017;12(12):e0188754. doi: 10.1371/journal.pone.0188754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tekwani BL, Walker LA. 8-Aminoquinolines: future role as antiprotozoal drugs. Curr Opin Infect Dis. 2006;19(6):623–631. doi: 10.1097/QCO.0b013e328010b848. [DOI] [PubMed] [Google Scholar]
- 89.Fonteilles-Drabek S, Reddy D, Wells TN. Managing intellectual property to develop medicines for the world’s poorest. Nat Rev Drug Discov. 2017;16(4):223–224. doi: 10.1038/nrd.2017.24. [DOI] [PubMed] [Google Scholar]
- 90.Chavchich M, Birrell GW, Ager AL, MacKenzie DO, Heffernan GD, Schiehser GA, et al. Lead selection of a new aminomethylphenol, JPC-3210, for malaria treatment and prevention. Antimicrob Agents Chemother. 2016;60(5):3115–3118. doi: 10.1128/AAC.03066-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Birrell GW, Heffernan GD, Schiehser GA, Anderson J, Ager AL, Morales P, et al. Characterization of the preclinical pharmacology of the new 2-aminomethylphenol, JPC-3210, for malaria treatment and prevention. Antimicrob Agents Chemother. 2018 doi: 10.1128/AAC.01335-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pegoraro S, Duffey M, Otto TD, Wang Y, Rosemann R, Baumgartner R, et al. SC83288 is a clinical development candidate for the treatment of severe malaria. Nat Commun. 2017;8:14193. doi: 10.1038/ncomms14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nanayakkara NP, Ager AL, Jr, Bartlett MS, Yardley V, Croft SL, Khan IA, et al. Antiparasitic activities and toxicities of individual enantiomers of the 8-aminoquinoline 8-[(4-amino-1-methylbutyl)amino]-6-methoxy-4-methyl-5-[3,4-dichlorophenoxy]quinoline succinate. Antimicrob Agents Chemother. 2008;52(6):2130–2137. doi: 10.1128/AAC.00645-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Deu E. Proteases as antimalarial targets: strategies for genetic, chemical, and therapeutic validation. FEBS J. 2017;284(16):2604–2628. doi: 10.1111/febs.14130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pino P, Caldelari R, Mukherjee B, Vahokoski J, Klages N, Maco B, et al. A multistage antimalarial targets the plasmepsins IX and X essential for invasion and egress. Science (New York, NY) 2017;358(6362):522–528. doi: 10.1126/science.aaf8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nasamu AS, Glushakova S, Russo I, Vaupel B, Oksman A, Kim AS, et al. Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion. Science (New York, NY) 2017;358(6362):518–522. doi: 10.1126/science.aan1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hodder AN, Sleebs BE, Czabotar PE, Gazdik M, Xu Y, O’Neill MT, et al. Structural basis for plasmepsin V inhibition that blocks export of malaria proteins to human erythrocytes. Nat Struct Mol Biol. 2015;22(8):590–596. doi: 10.1038/nsmb.3061. [DOI] [PubMed] [Google Scholar]
- 98.Jimenez-Diaz MB, Ebert D, Salinas Y, Pradhan A, Lehane AM, Myrand-Lapierre ME, et al. (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc Natl Acad Sci USA. 2014;111(50):E5455–E5462. doi: 10.1073/pnas.1414221111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gupta P, Mehrotra S, Sharma A, Chugh M, Pandey R, Kaushik A, et al. Exploring heme and hemoglobin binding regions of plasmodium heme detoxification protein for new antimalarial discovery. J Med Chem. 2017;60(20):8298–8308. doi: 10.1021/acs.jmedchem.7b00089. [DOI] [PubMed] [Google Scholar]
- 100.Andrews KT, Tran TN, Fairlie DP. Towards histone deacetylase inhibitors as new antimalarial drugs. Curr Pharm Des. 2012;18(24):3467–3479. [PubMed] [Google Scholar]
- 101.Keough DT, Rejman D, Pohl R, Zbornikova E, Hockova D, Croll T, et al. Design of Plasmodium vivax hypoxanthine-guanine phosphoribosyltransferase inhibitors as potential antimalarial therapeutics. ACS Chem Biol. 2017 doi: 10.1021/acschembio.7b00916. [DOI] [PubMed] [Google Scholar]
- 102.Wright MH, Clough B, Rackham MD, Rangachari K, Brannigan JA, Grainger M, et al. Validation of N-myristoyltransferase as an antimalarial drug target using an integrated chemical biology approach. Nat Chem. 2014;6(2):112–121. doi: 10.1038/nchem.1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kato N, Comer E, Sakata-Kato T, Sharma A, Sharma M, Maetani M, et al. Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature. 2016;538(7625):344–349. doi: 10.1038/nature19804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Agarwal D, Gupta RD, Awasthi SK. Are antimalarial hybrid molecules a close reality or a distant dream? Antimicrob Agents Chemother. 2017;61(5):e00249-17. doi: 10.1128/AAC.00249-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Reddy PL, Khan SI, Ponnan P, Tripathi M, Rawat DS. Design, synthesis and evaluation of 4-aminoquinoline-purine hybrids as potential antiplasmodial agents. Eur J Med Chem. 2017;126:675–686. doi: 10.1016/j.ejmech.2016.11.057. [DOI] [PubMed] [Google Scholar]
- 106.Almela MJ, Lozano S, Lelievre J, Colmenarejo G, Coteron JM, Rodrigues J, et al. A new set of chemical starting points with Plasmodium falciparum transmission-blocking potential for antimalarial drug discovery. PloS One. 2015;10(8):e0135139. doi: 10.1371/journal.pone.0135139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hanson KK, Ressurreicao AS, Buchholz K, Prudencio M, Herman-Ornelas JD, Rebelo M, et al. Torins are potent antimalarials that block replenishment of Plasmodium liver stage parasitophorous vacuole membrane proteins. Proc Natl Acad Sci USA. 2013;110(30):E2838–E2847. doi: 10.1073/pnas.1306097110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.National Center for Advancing Translational Sciences NIoH. Development of malaria transmission-blocking drugs. 2017. https://ncats.nih.gov/trnd/projects/active/malaria-transmission-blocking-drugs. Accessed 16 Jan 2018.
- 109.Delves MJ, Ruecker A, Straschil U, Lelievre J, Marques S, Lopez-Barragan MJ, et al. Male and female Plasmodium falciparum mature gametocytes show different responses to antimalarial drugs. Antimicrob Agents Chemother. 2013;57(7):3268–3274. doi: 10.1128/AAC.00325-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Delves MJ. Plasmodium cell biology should inform strategies used in the development of antimalarial transmission-blocking drugs. Future Med Chem. 2012;4(18):2251–2263. doi: 10.4155/fmc.12.182. [DOI] [PubMed] [Google Scholar]
- 111.Stone W, Sawa P, Lanke K, Rijpma S, Oriango R, Nyaurah M, et al. A molecular assay to quantify male and female Plasmodium falciparum gametocytes: results from 2 randomized controlled trials using primaquine for gametocyte clearance. J Med Chem. 2017;216(4):457–467. doi: 10.1093/infdis/jix237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ruecker A, Mathias DK, Straschil U, Churcher TS, Dinglasan RR, Leroy D, et al. A male and female gametocyte functional viability assay to identify biologically relevant malaria transmission-blocking drugs. Antimicrob Agents Chemother. 2014;58(12):7292–7302. doi: 10.1128/AAC.03666-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zeeman AM, van Amsterdam SM, McNamara CW, Voorberg-van der Wel A, Klooster EJ, van den Berg A, et al. KAI407, a potent non-8-aminoquinoline compound that kills Plasmodium cynomolgi early dormant liver stage parasites in vitro. Antimicrob Agents Chemother. 2014;58(3):1586–1595. doi: 10.1128/AAC.01927-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Campo B, Vandal O, Wesche DL, Burrows JN. Killing the hypnozoite—drug discovery approaches to prevent relapse in Plasmodium vivax. Pathog Glob Health. 2015;109(3):107–122. doi: 10.1179/2047773215Y.0000000013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yeka A, Dorsey G, Kamya MR, Talisuna A, Lugemwa M, Rwakimari JB, et al. Artemether–lumefantrine versus dihydroartemisinin–piperaquine for treating uncomplicated malaria: a randomized trial to guide policy in Uganda. PloS One. 2008;3(6):e2390. doi: 10.1371/journal.pone.0002390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.World Health Organization . Global supply of artemether–lumefantrine before, during, and after the Memorandum of Understanding between WHO and Novartis. Geneva: World Health Organization; 2011. [Google Scholar]
- 117.Brueckner RP, Lasseter KC, Lin ET, Schuster BG. First-time-in-humans safety and pharmacokinetics of WR 238605, a new antimalarial. Am J Trop Med Hyg. 1998;58(5):645–649. doi: 10.4269/ajtmh.1998.58.645. [DOI] [PubMed] [Google Scholar]
- 118.Llanos-Cuentas A, Lacerda MV, Rueangweerayut R, Krudsood S, Gupta SK, Kochar SK, et al. Tafenoquine plus chloroquine for the treatment and relapse prevention of Plasmodium vivax malaria (DETECTIVE): a multicentre, double-blind, randomised, phase 2b dose-selection study. Lancet. 2014;383(9922):1049–1058. doi: 10.1016/S0140-6736(13)62568-4. [DOI] [PubMed] [Google Scholar]
- 119.Jefford CW. Synthetic peroxides as potent antimalarials. News and views. Curr Top Med Chem. 2012;12(5):373–399. doi: 10.2174/156802612799362940. [DOI] [PubMed] [Google Scholar]
- 120.Moehrle JJ, Duparc S, Siethoff C, van Giersbergen PL, Craft JC, Arbe-Barnes S, et al. First-in-man safety and pharmacokinetics of synthetic ozonide OZ439 demonstrates an improved exposure profile relative to other peroxide antimalarials. Br J Clin Pharmacol. 2013;75(2):524–537. doi: 10.1111/j.1365-2125.2012.04368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.McCarthy JS, Baker M, O’Rourke P, Marquart L, Griffin P, Hooft van Huijsduijnen R, et al. Efficacy of OZ439 (artefenomel) against early Plasmodium falciparum blood-stage malaria infection in healthy volunteers. J Antimicrob Chemother. 2016;71(9):2620–2627. doi: 10.1093/jac/dkw174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mombo-Ngoma G, Supan C, Dal-Bianco MP, Missinou MA, Matsiegui PB, Ospina Salazar CL, et al. Phase I randomized dose-ascending placebo-controlled trials of ferroquine—a candidate anti-malarial drug—in adults with asymptomatic Plasmodium falciparum infection. Malar J. 2011;10:53. doi: 10.1186/1475-2875-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Darpo B, Ferber G, Siegl P, Laurijssens B, Macintyre F, Toovey S, et al. Evaluation of the QT effect of a combination of piperaquine and a novel anti-malarial drug candidate OZ439, for the treatment of uncomplicated malaria. Br J Clin Pharmacol. 2015;80(4):706–715. doi: 10.1111/bcp.12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kuhen KL, Chatterjee AK, Rottmann M, Gagaring K, Borboa R, Buenviaje J, et al. KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment, and prevention of disease transmission. Antimicrob Agents Chemother. 2014;58(9):5060–5067. doi: 10.1128/AAC.02727-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Meister S, Plouffe DM, Kuhen KL, Bonamy GMC, Wu T, Barnes SW, et al. Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science (New York, NY) 2011;334(6061):1372–1377. doi: 10.1126/science.1211936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Leong FJ, Zhao R, Zeng S, Magnusson B, Diagana TT, Pertel P. A first-in-human randomized, double-blind, placebo-controlled, single- and multiple-ascending oral dose study of novel Imidazolopiperazine KAF156 to assess its safety, tolerability, and pharmacokinetics in healthy adult volunteers. Antimicrob Agents Chemother. 2014;58(11):6437–6443. doi: 10.1128/AAC.03478-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Spillman NJ, Allen RJ, McNamara CW, Yeung BK, Winzeler EA, Diagana TT, et al. Na(+) regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe. 2013;13(2):227–237. doi: 10.1016/j.chom.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Leong FJ, Li R, Jain JP, Lefevre G, Magnusson B, Diagana TT, et al. A first-in-human randomized, double-blind, placebo-controlled, single- and multiple-ascending oral dose study of novel antimalarial Spiroindolone KAE609 (Cipargamin) to assess its safety, tolerability, and pharmacokinetics in healthy adult volunteers. Antimicrob Agents Chemother. 2014;58(10):6209–6214. doi: 10.1128/AAC.03393-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Tran HT, White NJ, Nguyen T-NT, Nhu HT, Phung TD, Tarning J, et al. Estimation of the in-vivo minimum inhibitory concentration of cipargamin in uncomplicated Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2016;61:e01940-16. doi: 10.1128/AAC.01940-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Missinou MA, Borrmann S, Schindler A, Issifou S, Adegnika AA, Matsiegui P-B, et al. Fosmidomycin for malaria. Lancet. 2002;360(9349):1941–1942. doi: 10.1016/S0140-6736(02)11860-5. [DOI] [PubMed] [Google Scholar]
- 131.Guttmann P, Ehrlich P. Ueber die Wirkung des Methylenblau bei Malaria. Berliner Klinische Wochenschrift. 1891;39:953–956. [Google Scholar]
- 132.Bountogo M, Zoungrana A, Coulibaly B, Klose C, Mansmann U, Mockenhaupt FP, et al. Efficacy of methylene blue monotherapy in semi-immune adults with uncomplicated falciparum malaria: a controlled trial in Burkina Faso. Trop Med Int Health. 2010;15(6):713–717. doi: 10.1111/j.1365-3156.2010.02526.x. [DOI] [PubMed] [Google Scholar]
- 133.Zoungrana A, Coulibaly B, Sié A, Walter-Sack I, Mockenhaupt FP, Kouyaté B, et al. Safety and efficacy of methylene blue combined with artesunate or amodiaquine for uncomplicated falciparum malaria: a randomized controlled trial from Burkina Faso. PloS One. 2008;3(2):e1630. doi: 10.1371/journal.pone.0001630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Paquet T, Chibale K, Street L, Younis Y, Cabrera GD, Le Manach C, et al. Discovery of the 2-aminopyridine MMV390048 as an anti-malarial with the potential to be a component in a single dose cure. Basic Clin Pharmacol Toxicol. 2014;115:226. [Google Scholar]
- 135.Plouffe David M, Wree M, Du Alan Y, Meister S, Li F, Patra K, et al. High-throughput assay and discovery of small molecules that interrupt malaria transmission. Cell Host Microbe. 2016;19(1):114–126. doi: 10.1016/j.chom.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yuthavong Y, Tarnchompoo B, Vilaivan T, Chitnumsub P, Kamchonwongpaisan S, Charman SA, et al. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc Natl Acad Sci USA. 2012;109(42):16823–16828. doi: 10.1073/pnas.1204556109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shafiq N, Rajagopalan S, Kushwaha HN, Mittal N, Chandurkar N, Bhalla A, et al. Single ascending dose safety and pharmacokinetics of CDRI-97/78: first-in-human study of a novel antimalarial drug. Malar Res Treat. 2014;2014:372521. doi: 10.1155/2014/372521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Spillman NJ, Kirk K. The malaria parasite cation ATPase PfATP4 and its role in the mechanism of action of a new arsenal of antimalarial drugs. Int J Parasitol Drugs Drug Resist. 2015;5(3):149–162. doi: 10.1016/j.ijpddr.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jiménez-Díaz MB, Ebert D, Salinas Y, Pradhan A, Lehane AM, Myrand-Lapierre M-E, et al. (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc Natl Acad Sci. 2014;111(50):E5455–E5462. doi: 10.1073/pnas.1414221111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Boss C, Aissaoui H, Amaral N, Bauer A, Bazire S, Binkert C, et al. Discovery and characterization of ACT-451840: an antimalarial drug with a novel mechanism of action. ChemMedChem. 2016;11(18):1995–2014. doi: 10.1002/cmdc.201600298. [DOI] [PubMed] [Google Scholar]
- 141.Bruderer S, Hurst N, de Kanter R, Miraval T, Pfeifer T, Donazzolo Y, et al. First-in-humans study of the safety, tolerability, and pharmacokinetics of ACT-451840, a new chemical entity with antimalarial activity. Antimicrob Agents Chemother. 2015;59(2):935–942. doi: 10.1128/AAC.04125-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hameed PS, Solapure S, Patil V, Henrich PP, Magistrado PA, Bharath S, et al. Triaminopyrimidine is a fast-killing and long-acting antimalarial clinical candidate. Nat Commun. 2015;6:6715. doi: 10.1038/ncomms7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sindhe KMV, Sonoiki E, Guo D, Gut J, Legac J, Tran Y, et al. A new benzoxaborole with an apparent novel mechanism of action against Plasmodium falciparum. In: American Society of tropical medicine and hygiene 65th annual meeting, Atlanta, Georgia. 2016.
- 144.McNamara CW, Lee MC, Lim CS, Lim SH, Roland J, Simon O, et al. Targeting Plasmodium PI(4)K to eliminate malaria. Nature. 2013;504(7479):248–253. doi: 10.1038/nature12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Delves M, Plouffe D, Scheurer C, Meister S, Wittlin S, Winzeler EA, et al. The activities of current antimalarial drugs on the life cycle stages of Plasmodium: a comparative study with human and rodent parasites. PLoS Med. 2012;9(2):e1001169. doi: 10.1371/journal.pmed.1001169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.LaMontagne MP, Blumbergs P, Strube RE. Antimalarials. 14. 5-(aryloxy)-4-methylprimaquine analogues. A highly effective series of blood and tissue schizonticidal agents. J Med Chem. 1982;25(9):1094–1097. doi: 10.1021/jm00351a017. [DOI] [PubMed] [Google Scholar]
- 147.Peyton DH. Reversed chloroquine molecules as a strategy to overcome resistance in malaria. Curr Top Med Chem. 2012;12(5):400–407. doi: 10.2174/156802612799362968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ubben D, Poll EM. MMV in partnership: the Eurartesim® experience. Malar J. 2013;12:211. doi: 10.1186/1475-2875-12-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Wells S, Diap G, Kiechel JR. The story of artesunate–mefloquine (ASMQ), innovative partnerships in drug development: case study. Malar J. 2013;12:68. doi: 10.1186/1475-2875-12-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.White NJ. Assessment of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrob Agents Chemother. 1997;41(7):1413–1422. doi: 10.1128/aac.41.7.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]