

ABSTRACT
Salmonella enterica, a common cause of diarrhea, has to colonize the gut lumen to elicit disease. In the gut, the pathogen encounters a vast array of environmental stresses that cause perturbations in the bacterial envelope. The CpxRA two-component system monitors envelope perturbations and responds by altering the bacterial gene expression profile. This allows Salmonella to survive under such harmful conditions. Therefore, CpxRA activation is likely to contribute to Salmonella gut infection. However, the role of the CpxRA-mediated envelope stress response in Salmonella-induced diarrhea is unclear. Here, we show that CpxRA is dispensable for the induction of colitis by S. enterica serovar Typhimurium, whereas it is required for gut colonization. We prove that CpxRA is expressed during gut infection and that the presence of antimicrobial peptides in growth media activates the expression of CpxRA-regulated genes. In addition, we demonstrate that a S. Typhimurium strain lacking the cpxRA gene is able to cause colitis but is unable to continuously colonize the gut. Finally, we show that CpxRA-dependent gut colonization requires the host gut inflammatory response, while DegP, a CpxRA-regulated protease, is dispensable. Our findings reveal that the CpxRA-mediated envelope stress response plays a crucial role in Salmonella gut infection, suggesting that CpxRA might be a promising therapeutic target for infectious diarrhea.
KEYWORDS: CpxRA, Salmonella, colitis, diarrhea, gut colonization
INTRODUCTION
Fine-tuned gene expression is necessary for bacterial survival in diverse environments. To achieve appropriate regulation, bacteria must sense changes in their environment and adapt to them. Therefore, bacteria have developed a number of sophisticated signal transduction systems called two-component systems (1). CpxRA is a two-component signal transduction system present in Gram-negative bacteria. It consists of the inner membrane-bound histidine sensor kinase CpxA and the cytoplasmic response regulator CpxR (2). CpxA senses environmental stimuli that cause envelope stress and responds through autophosphorylation at a conserved histidine residue, followed by the transfer of the phosphate to CpxR at a conserved aspartate residue (3). Phosphorylated CpxR alters the transcriptional profile of a large number of genes, alleviating envelope stress and allowing bacteria to adapt to changes in the environment (3). Periplasmic CpxP participates in the CpxRA two-component system response, acting as a negative-feedback regulator. Specifically, CpxP suppresses the CpxRA response by maintaining CpxA in an unphosphorylated state under noninducible conditions (4, 5). Under inducible conditions, phosphorylated CpxR upregulates the expression of cpxP, which avoids the overexpression of the CpxRA two-component system (5).
CpxRA activation is also involved in the response of bacteria to misfolded proteins in the periplasmic space and the inner membrane. Phosphorylated CpxR increases the expression of degP, which encodes a periplasmic protease that degrades misfolded proteins, allowing bacteria to cope with misfolded-protein-related envelope stress (6–8). Besides degP, phosphorylated CpxR also alters the transcriptional activity of numerous genes involved in protein folding, peptidoglycan degradation, efflux, and membrane respiration (9–11). In this way, Gram-negative bacteria adapt to various types of envelope stress, surviving under harmful environmental conditions.
Host mucosal immunity has a deleterious effect on enteropathogenic bacteria. For instance, epithelial cells produce antimicrobial peptides that can damage the bacterial membrane, leading to envelope stress. Therefore, CpxRA activation is thought to play a crucial role during infection by enteropathogenic bacteria. Several studies on the role of CpxRA in the pathogenesis of enteropathogenic bacteria have been reported to date, with conflicting results. For instance, in Citrobacter rodentium, enteropathogenic Escherichia coli (EPEC), and Shigella spp., CpxRA has been found to be involved in pathogenesis (12) and the expression of virulence factors (13–16) and virulence regulators (17, 18). However, other studies suggest that CpxRA activation inhibits virulence-associated phenotypes in EPEC and Yersinia spp. (15, 19, 20). It is likely that the final effect of CpxRA on bacterial infection depends on the fine-tuned control of gene expression by CpxRA.
The Gram-negative bacterium Salmonella enterica, in particular Salmonella enterica serovar Typhimurium, causes self-limiting diarrhea in humans, calves, and primates. CpxRA activation has been found to negatively affect the pathogenesis of S. Typhimurium in a mouse model of infection, where the classical mouse model for typhoid fever in humans was used (21). In the mouse, the normal gut microbiota prevents intestinal colonization upon oral infection with S. Typhimurium, which leads to a typhoid-like infection rather than gastrointestinal disease as in humans. Therefore, the role of CpxRA in S. Typhimurium gut infection remains unknown. Here, we have used the streptomycin mouse model of infection to study the role of CpxRA in Salmonella-induced colitis and gut infection. In this model, oral treatment of mice with streptomycin transitionally reduces the normal gut microbiota, allowing the colonization of the gut by S. Typhimurium and the induction of gut inflammation (22, 23). Using this model, we show that CpxRA activation contributes to S. Typhimurium gut infection, especially gut colonization. Our findings suggest that CpxRA might be a novel therapeutic target for infectious diarrhea inflicted by enteropathogenic bacteria.
RESULTS
In vivo expression of cpxRA and CpxRA-regulated genes during S. Typhimurium gut infection.
To study the role of the CpxRA two-component system in gut infection by S. Typhimurium, we first examined the expression of cpxR, cpxA, and the CpxRA-regulated genes cpxP and degP in feces during S. Typhimurium intestinal infection. Streptomycin-pretreated C57BL/6 mice were infected with S. Typhimurium wild-type strain SL1344 by oral gavage (5 × 107 CFU). On days 1 and 3 postinfection (p.i.), fecal pellets were harvested, and total RNA was extracted. Quantitative PCR (qPCR) on cDNA samples prepared from feces demonstrated that all four genes were expressed in the gut on days 1 and 3 p.i. (Fig. 1). Furthermore, the cpxP expression level tended to be increased on day 3 p.i. compared with that on day 1 p.i. (Fig. 1). This result suggests that CpxRA is expressed in the feces during intestinal infection, since the expression of cpxP occurs during CpxRA activation (4, 5). As expected, expression of the Salmonella genes rpoD, cpxR, cpxA, cpxP, and degP was not detected in uninfected mice, confirming that the above-described results were specific for S. Typhimurium. Therefore, the data imply the possibility that the CpxRA two-component system is expressed during S. Typhimurium intestinal infection.
FIG 1.
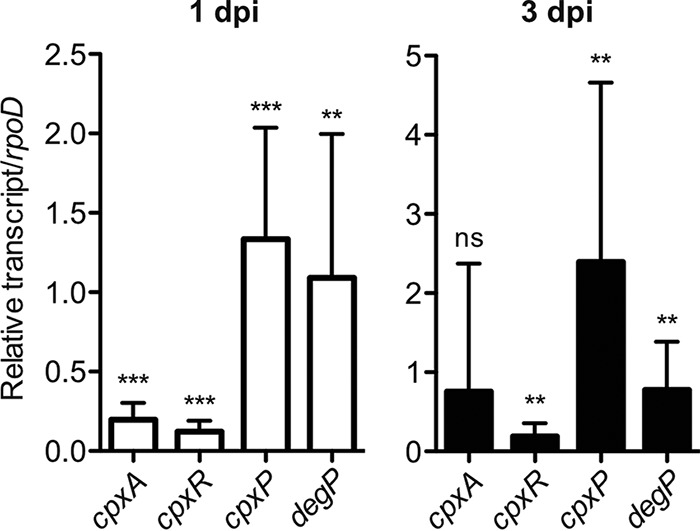
In vivo expression of cpxRA and CpxRA-regulated genes. C57BL/6 mice were pretreated with streptomycin (25 mg per mouse 24 h before infection) and left uninfected or infected orally with 5 × 107 CFU S. Typhimurium wild-type strain SL1344 for 3 days. mRNA levels of cpxRA and cpxRA-regulated genes (cpxP and degP) in the gut were measured by reverse transcription-qPCR. Data were normalized to rpoD expression levels (n = 12). dpi, day(s) postinfection. Bars represent means ± standard deviations. ns, not significant (P ≥ 0.05); **, P < 0.01; ***, P < 0.001 (as determined by a one-sample t test).
Antimicrobial peptides activate CpxRA.
Some environmental stimuli have been found to activate CpxRA in E. coli (6, 24–29), including antimicrobial peptides that are constitutively expressed in the gut lumen (28). Therefore, we decided to study whether the S. Typhimurium CpxRA signal transduction system also responds to antimicrobial peptides, stimuli that could be found during gut colonization. To this end, strains T429 and T443, containing degP::lacZ and cpxP::lacZ chromosomal transcriptional fusions, respectively, were generated in S. Typhimurium wild-type strain SH100, an ATCC 14028 derivative harboring spontaneous resistance to nalidixic acid (30). Moreover, an in-frame cpxRA deletion (ΔcpxRA::kan) was introduced into T429 by phage transduction with a P22 phage lysate prepared from the T192 strain (S. Typhimurium SL1344 ΔcpxRA::kan), yielding strain T442. The expression levels of degP and cpxP were measured in T429 and T443 in the presence or absence of the antimicrobial peptide polymyxin B by using a β-galactosidase assay. As shown in Fig. 2A, higher levels of β-galactosidase activity were observed in the presence of polymyxin B, while the deletion of the cpxRA genes (T442) resulted in the reduced expression of degP, even in the presence of polymyxin B (Fig. 2B). Complementation of the cpxRA genes in T442 restored the induction of degP by polymyxin B (Fig. 2B). These results suggest that an in vitro environmental stimulus usually found in the gut lumen, such as the presence of antimicrobial peptides, activates S. Typhimurium CpxRA.
FIG 2.
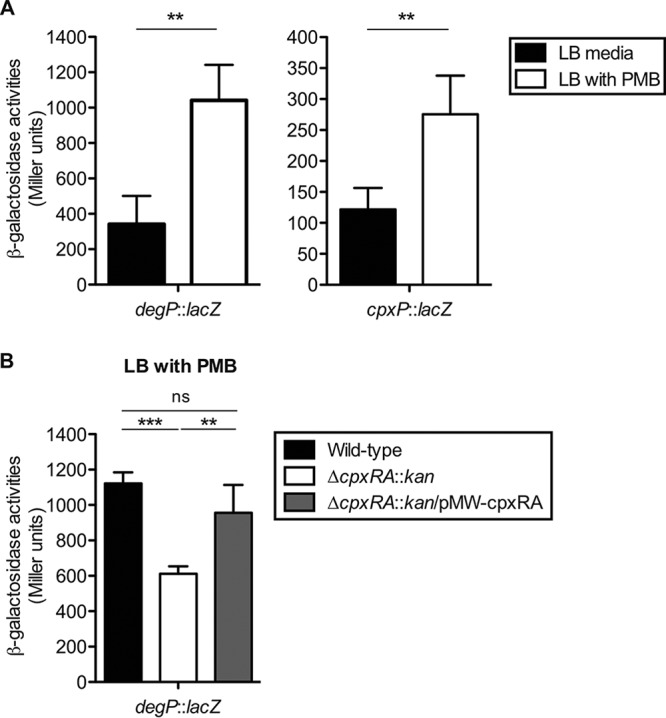
CpxRA is activated by antimicrobial peptides. (A) S. Typhimurium wild-type strains were grown in LB medium or LB medium with 1 μg/ml polymyxin B (PMB) to the stationary growth phase (A600 of ≥1.0), and β-galactosidase activities (Miller units) were measured. (B) The S. Typhimurium wild-type strain, the ΔcpxRA::kan mutant strain, or the mutant strain harboring pMW-cpxRA, which contains a chromosomal degP::lacZ fusion, was grown in LB medium with 1 μg/ml polymyxin B to the stationary growth phase (A600, ≥1.0), and β-galactosidase activities (Miller units) were measured. Error bars represent the standard deviations of the means from at least five independent experiments. ns, not significant (P ≥ 0.05); **, P < 0.01; ***, P < 0.001 (as determined by a Mann-Whitney U test).
CpxRA is dispensable for induction of colitis.
To examine whether the CpxRA two-component system is required for the induction of colitis by S. Typhimurium, the pathogenicities of the wild-type strain and a cpxRA deletion mutant were compared by using the streptomycin mouse model. Specifically, two groups of five streptomycin-pretreated C57BL/6 mice were infected with 5 × 107 CFU of S. Typhimurium SL1344 (wild type) or T192 (ΔcpxRA::kan) by oral gavage. On day 2 p.i., the bacterial load in feces was enumerated by plating serial dilutions onto selective medium. The bacterial loads were high for both S. Typhimurium strains, and no significant differences were observed (Fig. 3A). Similarly, histopathological analysis of the cecal mucosa revealed inflammation in the gut of both groups of mice (Fig. 3B and C). Measurement of fecal lipocalin-2, an inflammatory marker, suggested that intestinal inflammation is induced after infection (Fig. 3D). These results suggest that either CpxRA is not required for S. Typhimurium enteropathy on day 2 p.i. or its function is redundant.
FIG 3.
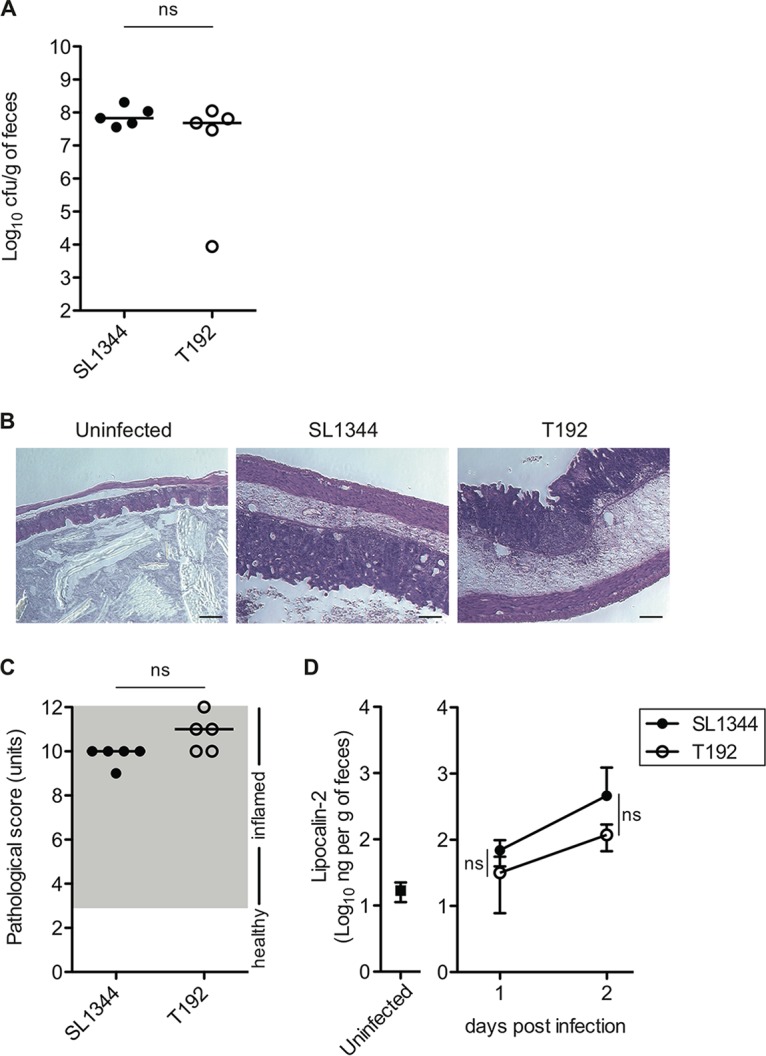
CpxRA is dispensable for induction of S. Typhimurium colitis on day 2 postinfection. Streptomycin-treated C57BL/6 mice (n = 5) were infected intragastrically for 2 days with 5 × 107 CFU S. Typhimurium wild-type strain SL1344 or ΔcpxRA::kan mutant strain T192. (A) S. Typhimurium loads in feces on day 2 postinfection. (B) Representative H&E-stained cecal sections (magnification, ×100). Bars, 100 μm. (C) Cecal pathological scores for H&E-stained cecal tissue sections. (D) Fecal lipocalin-2 levels monitored by an ELISA. Data points represent means ± standard deviations. For panels A and C, bars indicate the medians. ns, not significant (P ≥ 0.05) (as determined by a Mann-Whitney U test).
CpxRA is involved in gut colonization.
Next, we investigated whether CpxRA contributes to S. Typhimurium gut colonization using a competitive index (CI) assay. Streptomycin-pretreated C57BL/6 mice were infected with a 1:1 mixture of the SL1344 and ΔcpxRA::kan (T192) strains via oral gavage (5 × 107 CFU). On days 1 and 4 p.i., the bacterial load in feces was analyzed. On day 4 p.i., the mice were sacrificed, and bacterial loads in the cecum lumen, mesenteric lymph nodes (mLN), and spleen were monitored. The bacterial loads in feces were not significantly different on day 1 p.i. (CI, 1.65) (Fig. 4A). In contrast, on day 4 p.i., cpxRA mutant strain T192 displayed a competitive-colonization defect in the gut (CI, 12.59) (Fig. 4A) as well as in the cecum lumen, mLN, and spleen (Fig. 4B to D). Fecal lipocalin-2 measurements suggested that the mice developed gut inflammation (Fig. 4E). These results indicate that CpxRA is required by S. Typhimurium for gut colonization during acute colitis and for further systemic infection.
FIG 4.

CpxRA is required for S. Typhimurium gut colonization on day 4 postinfection in mixed infections. Streptomycin-treated C57BL/6 mice (n = 5) were infected intragastrically for 4 days with a 1:1 mixture (total of 5 × 107 CFU) of S. Typhimurium wild-type (wt) strain SL1344 and ΔcpxRA::kan mutant strain T192. (A to D) S. Typhimurium loads in feces (A), cecal contents (B), mLN (C), and spleen (D) were determined by selective plating, and competitive-infection indices (CIs) were subsequently determined on days 1 and 4 p.i. dpi, day(s) postinfection. (E) Fecal lipocalin-2 levels were determined by an ELISA. Data points represent means ± standard deviations. For panels A to D, horizontal bars indicate the medians. ns, not significant (P ≥ 0.05); *, P < 0.05; **, P < 0.01 (as determined by a Mann-Whitney U test).
CpxRA contributes to gut colonization in a TTSS-2-independent manner.
To clarify whether CpxRA contributes to sustained gut colonization, we used attenuated S. Typhimurium strain T145, a deletion mutant of the ssaV gene encoding a component of type III secretion system 2 (TTSS-2). Infection with the S. Typhimurium TTSS-2 gene mutant strains allows the evaluation of sustained gut colonization by S. Typhimurium in the streptomycin-treated mouse colitis model because the attenuated S. Typhimurium strain cannot spread to systemic sites, resulting in local gut infection (31, 32). Furthermore, the use of this strain also allows the evaluation of whether CpxRA-dependent colonization requires the TTSS-2 gene, since CpxRA activation has been shown to repress TTSS-2 gene expression (33). Streptomycin-pretreated C57BL/6 mice were coinfected with T145 (ΔssaV::cat) and T198 (ΔssaV ΔcpxRA::kan) by oral gavage (5 × 107 CFU). S. Typhimurium loads in feces were monitored for 8 days. On day 1 p.i., both strains displayed comparable abilities to colonize the gut (CI, 1) (Fig. 5A). However, by day 4 p.i., S. Typhimurium strain T198 exhibited a substantial colonization defect (CI, 30) (Fig. 5A) that extended to days 6 and 8 p.i. (Fig. 5A). The colonization defect was also observed in the cecum and mLN on day 8 p.i. (Fig. 5B and C). Measurement of fecal lipocalin-2 levels revealed that the mice developed gut inflammation, especially on day 4 p.i., when the inflammation levels were maximal (Fig. 5D). These results indicate that CpxRA contributes to sustained gut colonization by S. Typhimurium and that CpxRA-dependent colonization does not involve TTSS-2.
FIG 5.

CpxRA is required for sustained S. Typhimurium gut colonization in mixed infection. Streptomycin-treated C57BL/6 mice (n = 9) were infected for 8 days with a 1:1 mixture (total of 5 × 107 CFU intragastrically) of the S. Typhimurium ΔssaV::cat (T145) and ΔssaV ΔcpxRA::kan (T198) strains. (A) CIs of S. Typhimurium loads in feces on days 1, 4, 6, and 8 p.i. (B) CIs of S. Typhimurium loads in cecal contents on day 8 p.i. (C) CIs of S. Typhimurium loads in mLN on day 8 p.i. dpi, day(s) postinfection. (D) Fecal lipocalin-2 levels were monitored by an ELISA. Data points represent means ± standard deviations. For panels A to C, bars indicate the medians. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (as determined by a Mann-Whitney U test).
DegP is dispensable for gut colonization.
We next investigated whether impaired colonization by the cpxRA mutant is due to a defect in the degradation of misfolded proteins in the periplasmic space. To this end, we constructed a S. Typhimurium mutant strain lacking the CpxRA-regulated periplasmic protease DegP and performed a competitive-infection assay with wild-type strain SL1344. Streptomycin-pretreated C57BL/6 mice were infected with a 1:1 mixture of SL1344 and the ΔdegP::cat strain (TM1739) by oral gavage (5 × 107 CFU). For 4 days, the bacterial loads in feces were analyzed. In addition, on day 4 p.i., mice were sacrificed, and the bacterial loads in the cecum lumen were determined. The bacterial loads of the two S. Typhimurium strains in feces were not significantly different on days 1, 2, 3, and 4 p.i. (Fig. 6A). Similarly, no significant differences in the S. Typhimurium loads in the cecum were observed on day 4 p.i. (Fig. 6B). These results suggest that DegP is dispensable for gut colonization in a competitive-infection assay, and therefore it, is not involved in CpxRA-dependent gut colonization.
FIG 6.

DegP is not involved in S. Typhimurium gut colonization in mixed infection. Streptomycin-treated C57BL/6 mice (n = 5) were infected intragastrically for 4 days with a 1:1 mixture (total of 5 × 107 CFU) of S. Typhimurium wild-type strain SL1344 and ΔdegP::cat mutant strain TM1739. (A) CIs of S. Typhimurium loads in feces on days 1, 2, 3, and 4 p.i. (B) CIs of S. Typhimurium loads in cecal contents at 4 days postinfection (dpi). Bars indicate the medians. ns, not significant (P ≥ 0.05) (as determined by a Mann-Whitney U test).
CpxRA-dependent gut colonization requires the TTSS-1 gene and the TTSS-1-inducible host inflammatory response.
TTSS-1-triggered gut inflammation promotes S. Typhimurium gut colonization (34, 35). Thus, we investigated whether CpxRA-dependent gut colonization requires TTSS-1-inducible gut inflammation. To this end, we started by constructing a competitive-infection assay with an avirulent S. Typhimurium TTSS-1 TTSS-2 double mutant strain (T249 [ΔinvG ΔssaV::cat]) that does not induce an inflammatory response during gut infection. The isogenic S. Typhimurium mutant strain colonizes the gut but remains silent, as it does not have two key virulence factors and cannot elicit mucosal inflammation (34, 35). Therefore, we tested whether T249 is incapable of eliciting gut inflammation in the streptomycin mouse model. Streptomycin-pretreated C57BL/6 mice were infected with 5 × 107 CFU of S. Typhimurium T249 by oral gavage, and S. Typhimurium loads in feces were monitored for 3 days. Colonization levels of T249 in feces remained high on days 1 to 3 p.i., whereas histopathological analysis of the cecal mucosa and fecal lipocalin-2 measurements suggested that the mice did not develop gut inflammation (Fig. 7A to C). Thus, we confirmed that T249 is an avirulent S. Typhimurium mutant strain that colonizes the gut but cannot elicit gut inflammation.
FIG 7.

CpxRA-dependent gut colonization requires a host inflammatory response. (A to C) Streptomycin-treated C57BL/6 mice (n = 5) were infected for 3 days with 5 × 107 CFU of the S. Typhimurium ΔinvG ΔssaV::cat strain (T249) via gavage. (A) S. Typhimurium loads in feces on days 1, 2, and 3 postinfection. (B) Cecal pathological scores for H&E-stained cecal tissue sections. (C) Fecal lipocalin-2 levels monitored by an ELISA. (D and E) Streptomycin-treated C57BL/6 mice (n = 5) were infected for 4 days with a 1:1 mixture (total of 5 × 107 CFU intragastrically) of the S. Typhimurium ΔinvG ΔssaV::cat (T249) and ΔinvG ΔssaV ΔcpxRA::kan (T441) strains. (D) CIs of S. Typhimurium loads in feces on days 1, 2, 3, and 4 p.i. and in cecal contents on day 4 p.i. (E) Fecal lipocalin-2 levels on days 1 and 4 p.i. were determined by an ELISA. For panels A, C, and E, data points represent means ± standard deviations. For panels B and D, bars indicate the medians. ns, not significant (P ≥ 0.05); *, P < 0.05 (as determined by a Mann-Whitney U test).
Streptomycin-pretreated C57BL/6 mice were infected with a 1:1 mixture of strains T249 and T441 (ΔinvG ΔssaV ΔcpxRA::kan) by oral gavage (5 × 107 CFU), and the S. Typhimurium loads in feces were monitored for 4 days. On day 4 p.i., mice were sacrificed, and the bacterial loads in the cecum lumen were determined. As shown in Fig. 7D, the bacterial loads of the two S. Typhimurium strains in feces were not significantly different on days 1, 3, and 4 p.i., nor were the colonization levels in the cecum on day 4 p.i. (Fig. 7D). Gut inflammation was verified by measuring fecal lipocalin-2 levels (Fig. 7E), indicating that mice with mixed infection did not develop gut inflammation. These results suggest that CpxRA activation is beneficial for S. Typhimurium gut colonization when there is a TTSS-1-inducible host inflammatory response.
DISCUSSION
Enteric pathogens encounter a broad range of stimuli in the gut, including high osmolarity and the presence of antimicrobial peptides. These stresses cause perturbations in the bacterial cell envelope, and bacteria try to adapt by activating two-component systems, such as CpxRA, that respond to envelope stresses. Therefore, the aim of this work was to study the role of CpxRA in a mouse model of colitis. We show that cpxRA and the CpxRA-regulated genes degP and cpxP are expressed in the feces during intestinal infection and in vitro in the presence of antimicrobial peptides. These data suggest that CpxRA is activated under these conditions, probably as the result of bacterial envelope perturbations caused by the antimicrobial peptides and other stresses found in the gut. Furthermore, we show that CpxRA is not involved in the induction of colitis but is required for gut colonization; that is, the CpxRA-mediated envelope stress response allows S. Typhimurium to colonize the gut.
Some environmental stimuli known to activate CpxRA in E. coli include the overexpression of the outer membrane lipoprotein NlpE (6, 24), misfolded P pilin subunits (25), high concentrations of external copper (29), high osmolarity (27, 36), antimicrobial cationic polyethylenimine (26), and antimicrobial peptides (28). The osmolarity of the gut lumen is quite high (equivalent to 0.3 M NaCl or higher), and antimicrobial peptides, such as α-defensin, are dominantly present (37, 38). Therefore, it is likely that, in the gut, S. Typhimurium senses these environmental stimuli, which leads to the activation of CpxRA. Our findings showing that cpxRA and the CpxRA-regulated genes are induced in the presence of antimicrobial peptides support this hypothesis.
Antimicrobial peptides are known to activate the S. Typhimurium PhoPQ two-component system by the direct binding of the peptides to the PhoQ sensor (39). Although our results show that S. Typhimurium CpxRA also responds to antimicrobial peptides, a direct interaction between the CpxA sensor and the peptides has not been reported so far. Therefore, it is possible that, unlike PhoQ, CpxA senses envelope perturbations caused by the antimicrobial peptides. Interestingly, CpxRA activation has been shown to mediate resistance to antimicrobial peptides in both E. coli and S. Typhimurium (28, 40), and resistance to α-helical antimicrobial peptides is required, at least in part, for S. Typhimurium gut colonization (41). Therefore, an impaired resistance to antimicrobial peptides might account for the reduced competitive fitness of the S. Typhimurium cpxRA mutant in the gut.
Other scenarios could explain the involvement of CpxRA in S. Typhimurium gut colonization. For instance, CpxRA-regulated motility may affect gut colonization. It has been demonstrated that CpxRA activation in E. coli has a negative effect on bacterial motility (42, 43), whereas mutations of the CpxRA-regulated dsbA and degP genes result in decreased motility in EPEC (42, 44). Like E. coli, the S. Typhimurium dsbA mutant displays a motility defect (30). Since motility allows S. Typhimurium to exploit mucosal inflammation and contributes to sustained gut colonization (45, 46), it is possible that the loss of the CpxRA-mediated tuning of motility could result in impaired gut colonization. Alternatively, a derepression of certain respiratory complexes in the absence of CpxRA could result in reduced gut colonization. In that sense, it was recently shown that CpxRA regulates the expression of certain respiratory complexes in EPEC, whose derepression is toxic for the bacteria during envelope stresses (11). Indeed, dysbiosis-derived oxygen facilitates the growth of S. Typhimurium in the gut lumen through cytochrome bd-II oxidase-dependent aerobic respiration (47). Therefore, CpxRA-regulated aerobic respiration might be essential for the growth of S. Typhimurium in the gut. Our results showing that DegP is dispensable for S. Typhimurium gut colonization imply that the periplasmic accumulation of misfolded toxic proteins is unlikely to be the cause of impaired colonization by the cpxRA mutant, opening the door to other hypotheses, such as the involvement of aerobic respiration. Deciphering the mechanism underlying CpxRA-regulated aerobic respiration in S. Typhimurium and its involvement in gut colonization deserves further investigation.
The colonization defect of the S. Typhimurium cpxRA mutant depends on the host inflammatory response. In general, gut inflammation is a protective response of the host against infection by enteric pathogens. For example, the levels of gut luminal mucin, secreted from goblet cells, increase massively during gut inflammation, resulting in the formation of a robust mucosal barrier that inhibits the access of pathogens to intestinal epithelial cells. However, flagellum-mediated motility allows S. Typhimurium to localize to and use the mucosal components as high-energy nutrients for enhanced growth (45, 46). In addition, inflammation provides S. Typhimurium with a respiratory electron acceptor that members of the resident microbiota are unable to utilize, thereby allowing S. Typhimurium to outcompete the resident commensal bacteria in the gut (34, 48, 49). It remains unclear how host inflammation causes the impaired-colonization phenotype of the S. Typhimurium cpxRA mutant in the gut. However, it is possible that decreased resistance to antimicrobial peptides, such as defensin, and reduced motility contribute, at least in part, to it. In any case, our results allow us to conclude that S. Typhimurium CpxRA-dependent gut colonization benefits from pathogen-exploited inflammation.
In summary, environmental stresses in the gut cause perturbations in the S. Typhimurium envelope. This leads to CpxRA expression, which is essential for gut colonization by S. Typhimurium, similarly to what was previously reported for C. rodentium, a model for EPEC and enterohemorrhagic E. coli (EHEC) infection (12). Therefore, CpxRA might be a promising therapeutic target for gut infections by enteric pathogens.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Bacterial strains and plasmids used in this study are listed in Table 1. S. Typhimurium strain SL1344 was mouse virulent and used for mouse infection experiments. S. Typhimurium strain SH100, an ATCC 14028 derivative, was used for β-galactosidase assays. Bacterial strains were grown in LB medium containing the appropriate antibiotic(s). For mouse infection experiments, bacteria were grown in LB medium supplemented with 0.3 M NaCl and the appropriate antibiotic(s) for 13 h at 37°C under mild aeration (160 rpm) and subcultured at a 1:20 dilution rate for 4 h under the same conditions except that LB medium was used without supplementation with antibiotics.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or description | Reference or source |
|---|---|---|
| Salmonella enterica serovar Typhimurium strains | ||
| SL1344 | Wild-type S. Typhimurium; hisG | 54 |
| T192 | SL1344 ΔcpxAR::kan | This study |
| SH100 | Wild-type S. Typhimurium; ATCC 14028 derivative | 30 |
| T429 | SH100 degP::lacZ | This study |
| T442 | SH100 ΔcpxAR::kan degP::lacZ | This study |
| T446 | T442 harboring pMW-cpxRA | This study |
| T443 | SH100 cpxP::lacZ fusion | This study |
| T145 | SL1344 ΔssaV::cat | 41 |
| T198 | SL1344 ΔssaV ΔcpxAR::kan | This study |
| TM1739 | SL1344 ΔdegP::cat | This study |
| T249 | SL1344 ΔinvG ΔssaV::cat | This study |
| T441 | SL1344 ΔinvG ΔssaV ΔcpxAR::kan | This study |
| Plasmids | ||
| pLD-lacZΩ | Integrational plasmid with a promoterless lacZ gene | 30 |
| pLD-degPZ | degP::lacZ transcriptional fusion in pLD-lacZΩ | This study |
| pLD-cpxPZ | cpxP::lacZ transcriptional fusion in pLD-lacZΩ | This study |
| pMW118 | Low-copy-no. expression vector | NipponGene |
| pMW-cpxRA | pMW118 containing the cpxRA gene from SL1344 | This study |
Construction of S. Typhimurium gene deletion mutants.
S. Typhimurium strains harboring chromosomal in-frame deletions were created by using the lambda red homologous-recombination system (50). Primers used for the construction of S. Typhimurium mutants are listed in Table 2.
TABLE 2.
List of primers used in this study
| Primer | Sequence (5′–3′) | Usage(s) |
|---|---|---|
| qrpoD-FW | CGATCTTATCACCGGCTTTGT | qPCR |
| qrpoD-RV | TTCTTCATCTTCGTCTTCGTCATC | qPCR |
| qcpxR-FW | AGCAGCAGAGCAGCGACAA | qPCR |
| qcpxR-RV | CAGCAAATAGAGCAGGGTGAA | qPCR |
| qcpxA-FW | TCGGCTTCTCGGTGGATAAA | qPCR |
| qcpxA-RV | GCGATAGAACGGACGGAAGA | qPCR |
| qcpxP-FW | AGCGTAGCGCGCAAAATC | qPCR |
| qcpxP-RV | CTGTTCGTGCCTTGCCTGT | qPCR |
| qdegP-FW | CGTCGTCACCAACAACCAC | qPCR |
| qdegP-RV | TTTGCCCACCACTTTAGCATC | qPCR |
| cpxA-2 | CGAGATAAAAAATCGGCCTGCATTCGCAGGCCGATGGTTTGTGTAGGCTGGAGCTGCTTC | Mutant |
| cpxR-1 | CGCCTGATGACGTAATTTCTGCCTCGGAGGTACGTAAACACATATGAATATCCTCCTTAG | Mutant |
| STM4057-FW | AATGGCCCGACGGTCGCCCGTTTGC | Mutant |
| cpxP-RV | CATAACAGCAGCGGTAACTTTGCGC | Mutant |
| degP-Pro-SalI | AAAGTCGACATCCGGATGTAGAACAGCTTG | Plasmid, mutant |
| degP-Rev-BamHI | AAAGGATCCAGTGCACTCATTGCTAATGTG | Plasmid, mutant |
| ProcpxP-FW-SalI | AAAGTCGACCGGCGCAAAATAGCCCTGAT | Plasmid |
| cpxP-RV-BamHI | AAAGGATCCCACCGGGGTGCCAGTTATCG | Plasmid |
| cpxR-FW-HindIII | GGGAAGCTTTCAACGAGAGACAGTTTACG | Plasmid |
| cpxA-RV-SalI | AAAGTCGACCGACGGCGAGATAAAAAATC | Plasmid |
| invG-red-FW | GCGGAAATTATCAAATATTATTCAATTGGCAGACAAATGAGTGTAGGCTGGAGCTGCTTC | Mutant |
| invG-red-RV | TTCTGGAAAATGAAATACCGGAGGTTGAGCCAGGAATCATCATATGAATATCCTCCTTAG | Mutant |
| invG-FW | CAGCAAATTATTACGCCTTC | Mutant |
| invG-RV | AGGACTAAATCACTGGGGTC | Mutant |
| degP-red-FW | ACAGCAATTTTGCGTTACCTGTTAATCGAGATTGAAACACGTGTAGGCTGGAGCTGCTTC | Mutant |
| degP-red-RV | GCAAATAAATAGAACTATCACCACGCTGAATATTCAGCGCCATATGAATATCCTCCTTAG | Mutant |
Construction of S. Typhimurium strains with a chromosomal transcriptional lacZ fusion.
The DNA fragments containing the degP promoter region were amplified by PCR using primer set degP-Pro-SalI and degP-Rev-BamHI or ProcpxP-FW-SalI and cpxP-RV-BamHI (Table 2). The PCR products digested with SalI and BamHI were ligated into the same sites of pLD-lacZΩ containing a promoterless lacZ gene (30), yielding pLD-degPZ and pLD-cpxPZ. The resulting plasmid was transferred from E. coli SM10λpir to S. Typhimurium strain SH100 by conjugation, and subsequently, the cpxRA::kan allele from T192 was transduced via the P22 phage.
Construction of a complementary plasmid.
The complementary plasmid pMW-cpxRA was constructed by using DNA fragments containing the cpxRA gene generated by PCR with primers cpxR-FW-HindIII and cpxA-RV-SalI (Table 2), and S. Typhimurium strain SL1344 chromosomal DNA as the template, which were digested with HindIII and SalI and then ligated between the same sites of pMW118.
Ethical statement.
The use of mice for infection experiments was reviewed and approved by the Kitasato University Institutional Animal Care and Use Committee (permit numbers A13-6, J96-1, and J13-1).
Mouse infection experiments.
All mice used in this study were of the C57BL/6 background and maintained at the institute of experiments of animals at the School of Pharmacy, Kitasato University, or purchased from Japan SLC. Infection experiments were performed as described previously (51). Pretreatment with 25 mg streptomycin by gavage was performed, and 24 h later, mice were infected with 5 × 107 CFU S. Typhimurium strains by the same route. To determine S. Typhimurium population sizes, fecal pellets, cecal contents, mesenteric lymph nodes (mLN), and spleens were freshly collected and homogenized in sterile phosphate-buffered saline (PBS) containing 0.5% Tergitol for differential plating onto MacConkey agar plates (Nissui Pharmaceutical) supplemented with the appropriate antibiotics (50 μg/ml streptomycin, 50 μg/ml kanamycin, and 10 μg/ml chloramphenicol). The competitive index was calculated by the division of the population sizes of S. Typhimurium strains by those of their derivative mutants. Parts of cecal tissue were fixed in 4% formaldehyde (Mildform; Wako Pure Chemical Industries, Ltd.) and embedded in paraffin. Cryosections were prepared, air dried, and then stained with hematoxylin and eosin (H&E). To determine the degree of inflammation, the pathological score was monitored, as previously described (51), evaluating submucosal edema, polymorphonuclear leukocyte infiltration, goblet cell numbers, and epithelial damage, resulting in maximum scores of 13.
Reverse transcription-quantitative PCR for quantifying bacterial gene expression levels in the gut.
Total RNA from murine feces was isolated by using the RNeasy powermicrobiome kit (Qiagen). Five hundred micrograms of total RNA was used for reverse transcription using TaqMan reverse transcription reagents (ThermoFisher Scientific). qPCR was conducted with a CFX96 real-time PCR detection system (Bio-Rad), using SsoAdvanced universal SYBR green supermix (Bio-Rad) or Kapa SYBR fast qPCR master mix (Kapa Biosystems). Relative transcript levels were normalized to the values for the rpoD gene and calculated by using the 2−ΔCT method (52).
β-Galactosidase assay.
β-Galactosidase activities of reporter gene fusions were determined according to standard procedures (53). Briefly, bacterial reporter strains were grown overnight in LB medium, diluted 1:100, and subcultured for 2.5 h in the same medium. For inducing conditions, polymyxin B (1 μg/ml; Wako) was supplemented after 2 h of subculturing, and the culture was then incubated for a further 30 min. One hundred microliters of the bacterial culture was added to 900 μl of Z buffer (60 mM Na2HPO4·7H2O, 40 mM NaH2PO4·H2O, 10 mM KCl, 1 mM MgSO4·7H2O, 50 mM β-mercaptoethanol), and 20 μl of 0.1% (wt/vol) SDS and 40 μl of chloroform were mixed well. After incubation for 5 min at 28°C, the reaction was started by the addition of 200 μl of o-nitrophenyl-β-d-galactopyranoside (ONPG; Wako) in 0.1 M potassium phosphate buffer (pH 7) (4 mg/ml) and developed at 28°C. The reactions were stopped by the addition of 500 μl of 1 M Na2CO3, and the optical densities at 420 nm were measured. Miller units were calculated as described previously (53).
Quantification of fecal lipcalin-2.
Fecal pellets were homogenized and diluted in PBS. The resulting serial dilutions were analyzed by an enzyme-linked immunosorbent assay (ELISA) using a mouse lipocalin-2/NGAL detection kit (R&D), according to the manufacturer's instructions.
Statistical analysis.
Statistical tests were performed by using GraphPad Prism (version 5) for Mac OS X (GraphPad Software). Statistical significance (P < 0.05) was determined by a Mann-Whitney U test or Student's t test. In the in vivo expression experiment, a one-sample t test was employed, as the theoretical mean value was zero.
ACKNOWLEDGMENTS
We thank Haruka Koga, Taiga Watanabe, Tsuyoshi Otake, Nao Nakamura, and Riku Hirota for technical support.
This work was supported in part by JSPS KAKENHI grants JP16K08783 (to T.M.) and JP15K08470 (to T.H.). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Hoch JA. 2000. Two-component and phosphorelay signal transduction. Curr Opin Microbiol 3:165–170. doi: 10.1016/S1369-5274(00)00070-9. [DOI] [PubMed] [Google Scholar]
- 2.Macritchie DM, Raivio TL. 29 July 2009, posting date Envelope stress responses. EcoSal Plus 2009 doi: 10.1128/ecosalplus.5.4.7. [DOI] [PubMed] [Google Scholar]
- 3.Raivio TL, Silhavy TJ. 1997. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J Bacteriol 179:7724–7733. doi: 10.1128/jb.179.24.7724-7733.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Danese PN, Silhavy TJ. 1998. CpxP, a stress-combative member of the Cpx regulon. J Bacteriol 180:831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raivio TL, Popkin DL, Silhavy TJ. 1999. The Cpx envelope stress response is controlled by amplification and feedback inhibition. J Bacteriol 181:5263–5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danese PN, Snyder WB, Cosma CL, Davis LJ, Silhavy TJ. 1995. The Cpx two-component signal transduction pathway of Escherichia coli regulates transcription of the gene specifying the stress-inducible periplasmic protease, DegP. Genes Dev 9:387–398. doi: 10.1101/gad.9.4.387. [DOI] [PubMed] [Google Scholar]
- 7.Lipinska B, Fayet O, Baird L, Georgopoulos C. 1989. Identification, characterization, and mapping of the Escherichia coli htrA gene, whose product is essential for bacterial growth only at elevated temperatures. J Bacteriol 171:1574–1584. doi: 10.1128/jb.171.3.1574-1584.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strauch KL, Johnson K, Beckwith J. 1989. Characterization of degP, a gene required for proteolysis in the cell envelope and essential for growth of Escherichia coli at high temperature. J Bacteriol 171:2689–2696. doi: 10.1128/jb.171.5.2689-2696.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Price NL, Raivio TL. 2009. Characterization of the Cpx regulon in Escherichia coli strain MC4100. J Bacteriol 191:1798–1815. doi: 10.1128/JB.00798-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raivio TL. 2014. Everything old is new again: an update on current research on the Cpx envelope stress response. Biochim Biophys Acta 1843:1529–1541. doi: 10.1016/j.bbamcr.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 11.Guest RL, Wang J, Wong JL, Raivio TL. 2017. A bacterial stress response regulates respiratory protein complexes to control envelope stress adaptation. J Bacteriol 199:e00153-17. doi: 10.1128/JB.00153-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomassin JL, Giannakopoulou N, Zhu L, Gross J, Salmon K, Leclerc JM, Daigle F, Le Moual H, Gruenheid S. 2015. The CpxRA two-component system is essential for Citrobacter rodentium virulence. Infect Immun 83:1919–1928. doi: 10.1128/IAI.00194-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang HZ, Donnenberg MS. 1996. DsbA is required for stability of the type IV pilin of enteropathogenic Escherichia coli. Mol Microbiol 21:787–797. doi: 10.1046/j.1365-2958.1996.431403.x. [DOI] [PubMed] [Google Scholar]
- 14.Nevesinjac AZ, Raivio TL. 2005. The Cpx envelope stress response affects expression of the type IV bundle-forming pili of enteropathogenic Escherichia coli. J Bacteriol 187:672–686. doi: 10.1128/JB.187.2.672-686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vogt SL, Nevesinjac AZ, Humphries RM, Donnenberg MS, Armstrong GD, Raivio TL. 2010. The Cpx envelope stress response both facilitates and inhibits elaboration of the enteropathogenic Escherichia coli bundle-forming pilus. Mol Microbiol 76:1095–1110. doi: 10.1111/j.1365-2958.2010.07145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humphries RM, Griener TP, Vogt SL, Mulvey GL, Raivio T, Donnenberg MS, Kitov PI, Surette M, Armstrong GD. 2010. N-Acetyllactosamine-induced retraction of bundle-forming pili regulates virulence-associated gene expression in enteropathogenic Escherichia coli. Mol Microbiol 76:1111–1126. doi: 10.1111/j.1365-2958.2010.07192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakayama S, Watanabe H. 1998. Identification of cpxR as a positive regulator essential for expression of the Shigella sonnei virF gene. J Bacteriol 180:3522–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitobe J, Arakawa E, Watanabe H. 2005. A sensor of the two-component system CpxA affects expression of the type III secretion system through posttranscriptional processing of InvE. J Bacteriol 187:107–113. doi: 10.1128/JB.187.1.107-113.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Macritchie DM, Ward JD, Nevesinjac AZ, Raivio TL. 2008. Activation of the Cpx envelope stress response down-regulates expression of several locus of enterocyte effacement-encoded genes in enteropathogenic Escherichia coli. Infect Immun 76:1465–1475. doi: 10.1128/IAI.01265-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carlsson KE, Liu J, Edqvist PJ, Francis MS. 2007. Influence of the Cpx extracytoplasmic-stress-responsive pathway on Yersinia sp.-eukaryotic cell contact. Infect Immun 75:4386–4399. doi: 10.1128/IAI.01450-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Humphreys S, Rowley G, Stevenson A, Anjum MF, Woodward MJ, Gilbert S, Kormanec J, Roberts M. 2004. Role of the two-component regulator CpxAR in the virulence of Salmonella enterica serotype Typhimurium. Infect Immun 72:4654–4661. doi: 10.1128/IAI.72.8.4654-4661.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaiser P, Diard M, Stecher B, Hardt WD. 2012. The streptomycin mouse model for Salmonella diarrhea: functional analysis of the microbiota, the pathogen's virulence factors, and the host's mucosal immune response. Immunol Rev 245:56–83. doi: 10.1111/j.1600-065X.2011.01070.x. [DOI] [PubMed] [Google Scholar]
- 23.Wotzka SY, Nguyen BD, Hardt WD. 2017. Salmonella Typhimurium diarrhea reveals basic principles of enteropathogen infection and disease-promoted DNA exchange. Cell Host Microbe 21:443–454. doi: 10.1016/j.chom.2017.03.009. [DOI] [PubMed] [Google Scholar]
- 24.Snyder WB, Davis LJ, Danese PN, Cosma CL, Silhavy TJ. 1995. Overproduction of NlpE, a new outer membrane lipoprotein, suppresses the toxicity of periplasmic LacZ by activation of the Cpx signal transduction pathway. J Bacteriol 177:4216–4223. doi: 10.1128/jb.177.15.4216-4223.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones CH, Danese PN, Pinkner JS, Silhavy TJ, Hultgren SJ. 1997. The chaperone-assisted membrane release and folding pathway is sensed by two signal transduction systems. EMBO J 16:6394–6406. doi: 10.1093/emboj/16.21.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lander BA, Checchi KD, Koplin SA, Smith VF, Domanski TL, Isaac DD, Lin S. 2012. Extracytoplasmic stress responses induced by antimicrobial cationic polyethylenimines. Curr Microbiol 65:488–492. doi: 10.1007/s00284-012-0182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jubelin G, Vianney A, Beloin C, Ghigo JM, Lazzaroni JC, Lejeune P, Dorel C. 2005. CpxR/OmpR interplay regulates curli gene expression in response to osmolarity in Escherichia coli. J Bacteriol 187:2038–2049. doi: 10.1128/JB.187.6.2038-2049.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Audrain B, Ferrieres L, Zairi A, Soubigou G, Dobson C, Coppee JY, Beloin C, Ghigo JM. 2013. Induction of the Cpx envelope stress pathway contributes to Escherichia coli tolerance to antimicrobial peptides. Appl Environ Microbiol 79:7770–7779. doi: 10.1128/AEM.02593-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamamoto K, Ishihama A. 2005. Transcriptional response of Escherichia coli to external copper. Mol Microbiol 56:215–227. doi: 10.1111/j.1365-2958.2005.04532.x. [DOI] [PubMed] [Google Scholar]
- 30.Miki T, Okada N, Danbara H. 2004. Two periplasmic disulfide oxidoreductases, DsbA and SrgA, target outer membrane protein SpiA, a component of the Salmonella pathogenicity island 2 type III secretion system. J Biol Chem 279:34631–34642. doi: 10.1074/jbc.M402760200. [DOI] [PubMed] [Google Scholar]
- 31.Endt K, Stecher B, Chaffron S, Slack E, Tchitchek N, Benecke A, Van Maele L, Sirard JC, Mueller AJ, Heikenwalder M, Macpherson AJ, Strugnell R, von Mering C, Hardt WD. 2010. The microbiota mediates pathogen clearance from the gut lumen after non-typhoidal Salmonella diarrhea. PLoS Pathog 6:e1001097. doi: 10.1371/journal.ppat.1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miki T, Goto R, Fujimoto M, Okada N, Hardt WD. 2017. The bactericidal lectin regIIIbeta prolongs gut colonization and enteropathy in the streptomycin mouse model for Salmonella diarrhea. Cell Host Microbe 21:195–207. doi: 10.1016/j.chom.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 33.De la Cruz MA, Perez-Morales D, Palacios IJ, Fernandez-Mora M, Calva E, Bustamante VH. 2015. The two-component system CpxR/A represses the expression of Salmonella virulence genes by affecting the stability of the transcriptional regulator HilD. Front Microbiol 6:807. doi: 10.3389/fmicb.2015.00807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, Dougan G, von Mering C, Hardt WD. 2007. Salmonella enterica serovar Typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol 5:2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hapfelmeier S, Stecher B, Barthel M, Kremer M, Muller AJ, Heikenwalder M, Stallmach T, Hensel M, Pfeffer K, Akira S, Hardt WD. 2005. The Salmonella pathogenicity island (SPI)-2 and SPI-1 type III secretion systems allow Salmonella serovar Typhimurium to trigger colitis via MyD88-dependent and MyD88-independent mechanisms. J Immunol 174:1675–1685. doi: 10.4049/jimmunol.174.3.1675. [DOI] [PubMed] [Google Scholar]
- 36.Prigent-Combaret C, Brombacher E, Vidal O, Ambert A, Lejeune P, Landini P, Dorel C. 2001. Complex regulatory network controls initial adhesion and biofilm formation in Escherichia coli via regulation of the csgD gene. J Bacteriol 183:7213–7223. doi: 10.1128/JB.183.24.7213-7223.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gupta S, Chowdhury R. 1997. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect Immun 65:1131–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salzman NH, Underwood MA, Bevins CL. 2007. Paneth cells, defensins, and the commensal microbiota: a hypothesis on intimate interplay at the intestinal mucosa. Semin Immunol 19:70–83. doi: 10.1016/j.smim.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 39.Bader MW, Sanowar S, Daley ME, Schneider AR, Cho U, Xu W, Klevit RE, Le Moual H, Miller SI. 2005. Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell 122:461–472. doi: 10.1016/j.cell.2005.05.030. [DOI] [PubMed] [Google Scholar]
- 40.Weatherspoon-Griffin N, Zhao G, Kong W, Kong Y, Morigen, Andrews-Polymenis H, McClelland M, Shi Y. 2011. The CpxR/CpxA two-component system up-regulates two Tat-dependent peptidoglycan amidases to confer bacterial resistance to antimicrobial peptide. J Biol Chem 286:5529–5539. doi: 10.1074/jbc.M110.200352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goto R, Miki T, Nakamura N, Fujimoto M, Okada N. 2017. Salmonella Typhimurium PagP- and UgtL-dependent resistance to antimicrobial peptides contributes to the gut colonization. PLoS One 12:e0190095. doi: 10.1371/journal.pone.0190095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacRitchie DM, Acosta N, Raivio TL. 2012. DegP is involved in Cpx-mediated posttranscriptional regulation of the type III secretion apparatus in enteropathogenic Escherichia coli. Infect Immun 80:1766–1772. doi: 10.1128/IAI.05679-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.De Wulf P, Kwon O, Lin EC. 1999. The CpxRA signal transduction system of Escherichia coli: growth-related autoactivation and control of unanticipated target operons. J Bacteriol 181:6772–6778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miki T, Okada N, Kim Y, Abe A, Danbara H. 2008. DsbA directs efficient expression of outer membrane secretin EscC of the enteropathogenic Escherichia coli type III secretion apparatus. Microb Pathog 44:151–158. doi: 10.1016/j.micpath.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 45.Stecher B, Barthel M, Schlumberger MC, Haberli L, Rabsch W, Kremer M, Hardt WD. 2008. Motility allows S. Typhimurium to benefit from the mucosal defence. Cell Microbiol 10:1166–1180. doi: 10.1111/j.1462-5822.2008.01118.x. [DOI] [PubMed] [Google Scholar]
- 46.Stecher B, Hapfelmeier S, Muller C, Kremer M, Stallmach T, Hardt WD. 2004. Flagella and chemotaxis are required for efficient induction of Salmonella enterica serovar Typhimurium colitis in streptomycin-pretreated mice. Infect Immun 72:4138–4150. doi: 10.1128/IAI.72.7.4138-4150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rivera-Chavez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu G, Velazquez EM, Lebrilla CB, Winter SE, Baumler AJ. 2016. Depletion of butyrate-producing clostridia from the gut microbiota drives an aerobic luminal expansion of Salmonella. Cell Host Microbe 19:443–454. doi: 10.1016/j.chom.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, Roth JR, Baumler AJ. 2010. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467:426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Winter SE, Baumler AJ. 2014. Dysbiosis in the inflamed intestine: chance favors the prepared microbe. Gut Microbes 5:71–73. doi: 10.4161/gmic.27129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barthel M, Hapfelmeier S, Quintanilla-Martinez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Russmann H, Hardt WD. 2003. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun 71:2839–2858. doi: 10.1128/IAI.71.5.2839-2858.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 53.Miller JH. 1992. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 54.Hoiseth SK, Stocker BA. 1981. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291:238–239. doi: 10.1038/291238a0. [DOI] [PubMed] [Google Scholar]