

ABSTRACT
Staphylococcus aureus is a leading cause of device-associated biofilm infections, which represent a serious health care concern based on their chronicity and antibiotic resistance. We previously reported that S. aureus biofilms preferentially recruit myeloid-derived suppressor cells (MDSCs), which promote monocyte and macrophage anti-inflammatory properties. This is associated with increased myeloid arginase-1 (Arg-1) expression, which has been linked to anti-inflammatory and profibrotic activities that are observed during S. aureus biofilm infections. To determine whether MDSCs and macrophages utilize Arg-1 to promote biofilm infection, Arg-1 was deleted in myeloid cells by use of Tie-2Cre mice. Despite Arg-1 expression in biofilm-associated myeloid cells, bacterial burdens and leukocyte infiltrates were similar between wild-type (WT) and Arg-1fl/fl;Tie-2Cre conditional knockout (KO) mice from days 3 to 14 postinfection in both orthopedic implant and catheter-associated biofilm models. However, inducible nitric oxide synthase (iNOS) expression was dramatically elevated in biofilm-associated MDSCs from Arg-1fl/fl;Tie-2Cre animals, suggesting a potential Arg-1-independent compensatory mechanism for MDSC-mediated immunomodulation. Treatment of Arg-1fl/fl;Tie-2Cre mice with the iNOS inhibitor N6-(1-iminoethyl)-l-lysine (l-NIL) had no effect on biofilm burdens or immune infiltrates, whereas treatment of WT mice with the Arg-1/ornithine decarboxylase inhibitor difluoromethylornithine (DFMO) increased bacterial titers, but only in the surrounding soft tissues, which possess attributes of a planktonic environment. A role for myeloid-derived Arg-1 in regulating planktonic infection was confirmed using a subcutaneous abscess model, in which S. aureus burdens were significantly increased in Arg-1fl/fl;Tie-2Cre mice compared to those in WT mice. Collectively, these results indicate that the effects of myeloid Arg-1 are context dependent and are manifest during planktonic but not biofilm infection.
KEYWORDS: arginase-1, biofilm, S. aureus, macrophage, myeloid-derived suppressor cell
INTRODUCTION
Staphylococcus aureus is a leading cause of community-acquired and nosocomial infections in humans (1–7), and the presence of foreign materials increases infection risk due to the ability of S. aureus to form biofilms (3, 8, 9). Biofilms are heterogeneous bacterial communities that are difficult to eradicate because of their chronicity and recalcitrance to antibiotic therapy (3, 8, 9). In addition, previous work from our laboratory has shown that S. aureus biofilms actively skew the host immune response toward an anti-inflammatory phenotype that is characterized by the preferential recruitment of myeloid-derived suppressor cells (MDSCs) and anti-inflammatory monocytes and macrophages, whereas neutrophils and T cell infiltrates are less abundant (10–15). MDSCs recruited to the site of biofilm infection express interleukin-10 (IL-10), which is one contributing mechanism of immune suppression (12). In addition, biofilm-associated MDSCs and macrophages upregulate arginase-1 (Arg-1) expression, suggesting that Arg-1 may further potentiate the anti-inflammatory and profibrotic cellular responses that are favored during S. aureus biofilm infections (10, 16, 17).
Arg-1 catalyzes the hydrolysis of arginine to ornithine and urea (18). During an immune response, myeloid-derived Arg-1 is an important enzyme involved in the regulation of arginine availability (18–22), and previous studies have demonstrated that myeloid-derived Arg-1 can inhibit proinflammatory immune responses (18, 23–27). Arginine is a substrate for both Arg-1 and inducible nitric oxide synthase (iNOS), which are characteristic markers of anti- and proinflammatory macrophages, respectively (24, 28–30). In myeloid cells, ornithine generation by Arg-1 is used for proline or polyamine synthesis through ornithine decarboxylase (ODC), which promotes fibrosis by collagen formation (18). A previous study demonstrated the importance of host-derived polyamines for S. aureus clearance and wound healing in a subcutaneous (s.c.) abscess model that represents planktonic infection (31); however, a role for Arg-1 action in the context of biofilms has not yet been explored. Biofilm infections are often encompassed by a fibrotic response, which coincides with robust Arg-1 expression (10, 15, 16). Arg-1 activity also inhibits NO-mediated killing of pathogens by macrophages, since arginine is the primary substrate for iNOS (28–30). Furthermore, extracellular arginine depletion leads to reduced CD3ζ chain expression and the inability to augment cyclin D3 and cdk4, leading to T cell hyporesponsiveness and cell cycle arrest, respectively (19–23, 26, 32). However, the effects of MDSC Arg-1 on inhibition of T cell proliferation may be context dependent, as some studies have not demonstrated a role for the enzyme (33, 34). Previous work reported that myeloid-derived Arg-1 expression has a pathological role in several cancers, Alzheimer's disease, and bacterial and helminth infections, establishing it as a key enzyme in dictating disease pathogenesis and immune outcomes (18, 24, 35, 36). Therefore, elevated Arg-1 expression by MDSCs and/or macrophages may participate in preventing an effective immune response to S. aureus biofilm infection through the depletion of extracellular arginine and suppression of proinflammatory responses.
The objective of the present study was to examine the functional role of myeloid-derived Arg-1 in promoting device-associated S. aureus biofilm infection. Using conditional knockout (KO) mice to delete Arg-1 in myeloid cells (Arg-1fl/fl;Tie-2Cre mice) (24, 37), we established that Arg-1 expression was significantly reduced in myeloid cells in vitro and in MDSCs and macrophages isolated from S. aureus biofilm infections ex vivo. Utilizing two device-associated S. aureus biofilm infection models, we found that bacterial burdens were not dramatically different between wild-type (WT) and Arg-1fl/fl;Tie-2Cre myeloid conditional KO mice. Further investigation showed that MDSCs from Arg-1fl/fl;Tie-2Cre conditional KO animals had upregulated iNOS expression, suggesting a possible compensatory mechanism for arginine depletion and immunomodulation. However, treatment of Arg-1fl/fl;Tie-2Cre mice with a small-molecule inhibitor of iNOS did not affect S. aureus biofilm burdens or leukocyte infiltrates. Additive effects were also not observed when mice were treated with a small-molecule inhibitor of NADPH oxidase. However, in the orthopedic implant model, treatment of WT mice with the arginase and ODC inhibitor difluoromethylornithine (DFMO) increased bacterial titers, but only in the surrounding soft tissue, which possesses some attributes of a planktonic environment, not in the joint and femur, which model biofilm growth. Indeed, a role for myeloid-derived Arg-1 in regulating planktonic infection was confirmed using a subcutaneous abscess model in which S. aureus burdens were significantly increased in Arg-1fl/fl;Tie-2Cre mice compared to those in WT animals. Collectively, these results demonstrate that myeloid-derived Arg-1 controls S. aureus growth during planktonic but not biofilm growth in vivo. By extension, the lack of Arg-1 involvement during biofilm formation represents another immune evasion mechanism leading to infection persistence.
RESULTS
Arginase-1 expression and enzyme activity are significantly reduced in myeloid cells from Arg-1 conditional KO mice.
Arginase-1 is a marker of anti-inflammatory macrophages and inhibits inflammatory responses through depletion of available arginine stores (18, 22, 24, 35, 38, 39). Our laboratory previously reported increased Arg-1 expression in MDSCs, monocytes, and macrophages infiltrating tissues surrounding an S. aureus biofilm infection (10, 12–14, 16). Although MDSC depletion improved biofilm clearance by enhancing monocyte proinflammatory activity (12, 14), the mechanism of action and whether Arg-1 was involved remained unclear. This was an important issue to address, since Arg-1 activity has been implicated in several types of cancers, Alzheimer's disease, and infection (18, 24, 35, 36). In the current study, we used two mouse models of device-associated S. aureus biofilm infection to determine the role of myeloid-derived Arg-1 in shaping leukocyte infiltrates and promoting biofilm persistence. Prior reports demonstrated that LysM-driven expression of Cre recombinase is inefficient at flox-mediated Arg-1 deletion (24, 40). This was confirmed in the current study, where significant Arg-1 activity remained in Arg-1fl/fl;LysMCre macrophages compared to that in wild-type cells (see Fig. S1 in the supplemental material). As an alternative approach, we utilized Arg-1fl/fl;Tie-2Cre mice, which have been shown to efficiently delete Arg-1 from myeloid cells (24). We first confirmed efficient Arg-1 deletion in myeloid cells from Arg-1fl/fl;Tie-2Cre conditional KO mice. Both Arg-1 expression and enzyme activity were significantly increased in wild-type Arg-1fl/fl;Tie-2null macrophages in response to IL-4 (Fig. 1A and B). In contrast, bone marrow-derived macrophages from Arg-1fl/fl;Tie-2Cre mice displayed significant decreases in Arg-1 expression and activity relative to those in Arg-1fl/fl;Tie-2null control cells, both at baseline and following IL-4 treatment, confirming effective gene deletion (Fig. 1A and B). Arginase activity in Arg-1fl/fl;Tie-2Cre macrophages may result from mitochondrial Arg-2 expression that can also catalyze the substrate used for the arginase assay (41).
FIG 1.
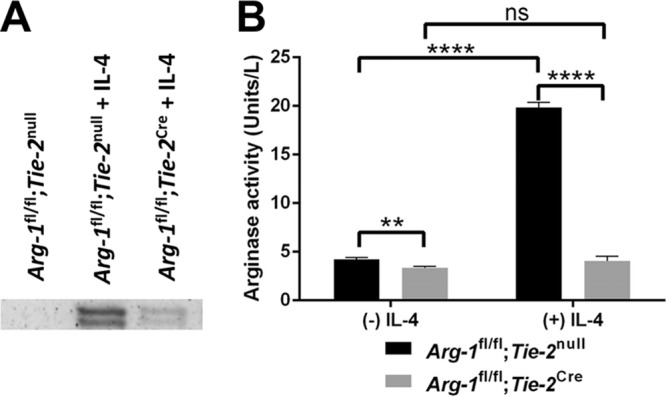
Arginase-1 expression and enzymatic activity are significantly reduced in Arg-1fl/fl;Tie-2Cre bone marrow-derived macrophages. Bone marrow-derived macrophages were left unstimulated or treated with rmIL-4 (10 ng/ml) for 24 h, whereupon whole-cell protein extracts (20 μg) were used for Western blotting (A) and arginase activity assays (B). Arginase activity in whole-cell extracts was measured by the amount of enzyme that converted 1.0 μmol of l-arginine to ornithine and urea per minute. ns, not significant; **, P < 0.01; ****, P < 0.0001 (one-way ANOVA with Bonferroni's multiple-comparison test).
Myeloid-derived arginase-1 does not dramatically affect S. aureus device-associated biofilm infection.
Based on our in vitro studies demonstrating that Arg-1 activity was significantly reduced in Arg-1fl/fl;Tie-2Cre conditional KO macrophages, we next examined the role of Arg-1 in the establishment and persistence of S. aureus biofilms by using two infection models. The first was an orthopedic implant-associated biofilm infection model, which our laboratory has shown is dominated by MDSCs that express Arg-1 and monocytes polarized toward an anti-inflammatory state (12–14). No significant differences in biofilm burdens or leukocyte infiltrates were observed between Arg-1fl/fl;Tie-2Cre conditional KO and Arg-1fl/fl;Tie-2null mice over the 2-week infection period (Fig. 2). We next examined a mouse model of catheter-associated biofilm infection, which is typified by more robust macrophage infiltrates, fibrotic responses, and Arg-1 expression than those in the orthopedic implant model (10, 15). Although MDSC infiltrates and bacterial burdens were significantly increased in catheter-associated tissues of Arg-1fl/fl;Tie-2Cre conditional KO mice at day 10 postinfection (Fig. 3), this did not dramatically alter biofilm growth at later time points (days 14 to 28) (data not shown). No significant differences in arginase activity were observed in either tissue or femur homogenates from WT and Arg-1fl/fl;Tie-2Cre mice in the orthopedic implant model (Fig. S2). This is likely because fibroblasts and other stromal cells outnumber myeloid cells in these compartments and contribute to the arginase pool at the site of infection. In addition, we did not observe any dramatic alterations in tissue integrity and collagen content as determined by hematoxylin and eosin (H&E) and trichrome staining, respectively (data not shown). As a quantitative means to assess collagen formation, a hydroxyproline assay was performed. Although hydroxyproline levels were elevated at day 3 postinfection in Arg-1fl/fl;Tie-2Cre mice in the catheter-associated infection model (Fig. S3), this was not observed at later time points, which confirmed the similarities in collagen levels between the two strains. Collectively, these results indicate that although Arg-1 expression is increased at the site of S. aureus biofilm infection, the enzyme itself in myeloid cells is not critical for regulating biofilm growth.
FIG 2.

Arginase-1 expression in myeloid cells does not influence S. aureus orthopedic implant infection. Titanium orthopedic implants were placed in the femurs of Arg-1fl/fl;Tie-2null (WT) and Arg-1fl/fl;Tie-2Cre mice and inoculated with 103 CFU S. aureus LAC. (A to C) Tissues surrounding infected implants, femurs, and knee joints were collected at the indicated intervals postinfection to quantify bacterial burdens. (D to F) Infiltrating leukocyte populations in implant-associated tissues were evaluated by flow cytometry. (D) MDSCs; (E) monocytes; (F) macrophages. Results are representative of two independent experiments.
FIG 3.

Arginase-1 in myeloid cells does not dramatically affect S. aureus catheter-associated biofilm infection. Catheter-associated infections were established in Arg-1fl/fl;Tie-2null (WT) and Arg-1fl/fl;Tie-2Cre mice following challenge with 103 CFU of S. aureus LAC. Animals were sacrificed at the indicated intervals postinfection, whereupon bacterial burdens in the surrounding tissue (A) and catheter (B) were determined and infiltrating leukocyte populations evaluated by flow cytometry (C to E). Results are representative of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (unpaired 2-tailed Student's t test).
iNOS expression is increased in biofilm-associated MDSCs from Arg-1fl/fl;Tie-2Cre conditional KO mice.
To establish that the overall lack of differences between Arg-1fl/fl;Tie-2Cre myeloid conditional KO and Arg-1fl/fl;Tie-2null mice in both the catheter and orthopedic implant infection models was not due to inefficient Arg-1 deletion in vivo, Arg-1 expression was examined in myeloid cells recovered from the site of infection. Both MDSCs (CD45+ Ly6Ghigh Ly6C+) and monocytes (CD45+ Ly6G− Ly6C+) were isolated by fluorescence-activated cell sorter (FACS) analysis from Arg-1fl/fl;Tie-2Cre and Arg-1fl/fl;Tie-2null control mice at days 7 and 14 postinfection, whereupon Arg-1 expression and activity were assessed by reverse transcription-quantitative PCR (RT-qPCR) and enzymatic assays, respectively (Fig. 4). As expected, Arg-1 expression and activity were dramatically reduced in macrophages and MDSCs from Arg-1fl/fl;Tie-2Cre conditional KO mice compared to those in cells from Arg-1fl/fl;Tie-2null controls in the catheter-associated infection model at days 7 and 14 (Fig. 4A). Because myeloid conditional Arg-1 deletion was verified in this model, this analysis was not repeated with the orthopedic implant biofilm model. Interestingly, MDSCs from Arg-1fl/fl;Tie-2Cre myeloid conditional KO mice showed a dramatic increase in iNOS expression (Fig. 4A). Since prior studies from our laboratory revealed a critical role for MDSCs in attenuating monocyte/macrophage proinflammatory activity and promoting biofilm persistence (12, 14), this suggested that a potential compensatory mechanism for arginine depletion may exist via upregulation of iNOS by MDSCs in the absence of Arg-1.
FIG 4.
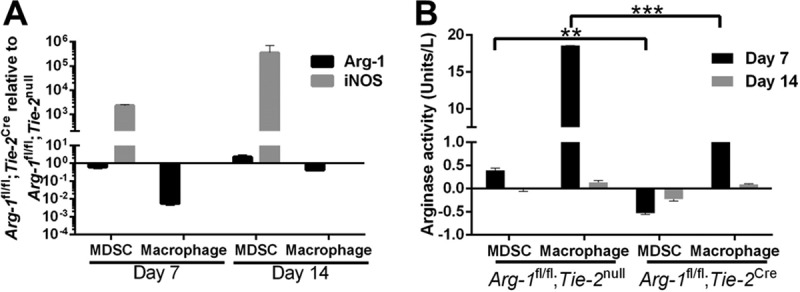
MDSCs and macrophages from Arg-1fl/fl;Tie-2Cre mice express limited arginase-1 in vivo. Catheter-associated infections were established in Arg-1fl/fl;Tie-2null (WT) and Arg-1fl/fl;Tie-2Cre mice following challenge with 103 CFU of S. aureus LAC. Animals were sacrificed at days 7 and 14 postinfection, whereupon MDSCs (CD45+ Ly6Ghigh Ly6C+) and monocytes (CD45+ Ly6G− Ly6C+) were isolated by FACS analysis for RT-qPCR quantification of Arg-1 and iNOS expression (A) and arginase activity assay (B). Results are representative of two independent experiments. **, P < 0.01; ***, P < 0.001 (unpaired 2-tailed Student's t test).
iNOS or ROS do not compensate for arginase-1 deficiency in myeloid cells.
Given the observed increase in iNOS expression in MDSCs from Arg-1 myeloid conditional KO mice, we treated Arg-1fl/fl;Tie-2Cre animals with N6-(1-iminoethyl)-l-lysine (l-NIL), a small-molecule inhibitor with selectivity for iNOS (cytokine inducible) over neuronal NOS (nNOS) and endothelial NOS (eNOS) (42). Both Arg-1fl/fl;Tie-2Cre myeloid conditional KO and Arg-1fl/fl;Tie-2null control mice received daily administration of l-NIL or vehicle in the S. aureus orthopedic implant model. No significant differences in bacterial burdens from the knee, surrounding soft tissue, femur, or leukocyte infiltrates were observed with l-NIL treatment (Fig. 5A to C and data not shown), suggesting that the observed increase in iNOS expression by MDSCs does not compensate for reduced myeloid Arg-1. We next examined the potential role of NADPH oxidase as a redundant pathway for Arg-1 in modulating biofilm persistence, since reactive oxygen species (ROS) production by MDSCs has also been implicated in modulating their suppressive activity (43, 44). Arg-1fl/fl;Tie-2Cre myeloid conditional KO and Arg-1fl/fl;Tie-2null control mice were treated with the small-molecule NADPH oxidase inhibitor apocynin, which also revealed no role in limiting the establishment or persistence of orthopedic implant-associated biofilm infection (Fig. 5A to C).
FIG 5.

iNOS or reactive oxygen species do not compensate for arginase-1 deficiency in myeloid cells. S. aureus orthopedic implant infections were established in Arg-1fl/fl;Tie-2null (WT) or Arg-1fl/fl;Tie-2Cre mice (A to C) or in C57BL/6 mice (D to F), whereupon animals received daily treatments of apocynin (6 mg/kg) (A to C), l-NIL (40 mg/kg) (A to C), or DFMO (50 mg/kg) (D to F) beginning 5 h prior to infection. Bacterial burdens in surrounding soft tissues (A and D), the femur (B and E), and the knee joint (C and F) were quantified at the indicated time points postinfection. *, P < 0.05; ***, P < 0.001 (unpaired 2-tailed Student's t test).
Inhibition of polyamine synthesis promotes bacterial growth in tissues surrounding biofilm infections.
Although our results suggested that myeloid Arg-1 deletion had minimal effects on biofilm development, we treated WT mice with difluoromethylornithine (DFMO), a small-molecule inhibitor of both arginase and ornithine decarboxylase (ODC), in the orthopedic implant infection model to examine the potential redundancy of ODC during S. aureus biofilm infection. Interestingly, bacterial burdens were increased at days 7 and 10 postinfection with DFMO treatment but were manifest only in the soft tissue surrounding the biofilm infection site (Fig. 5D). This finding agrees with an earlier report showing that polyamines are critical for antistaphylococcal responses and resolution of skin abscesses following DFMO treatment (31). Importantly, our study advances this finding by identifying myeloid cells as a critical source of Arg-1 during subcutaneous S. aureus abscess formation, since bacterial burdens were significantly increased in abscesses from Arg-1fl/fl;Tie-2Cre mice compared to those from WT mice (Fig. 6A). No gross differences in abscess size or dermatonecrosis were observed between Arg-1fl/fl;Tie-2Cre myeloid conditional KO and Arg-1fl/fl;Tie-2null control mice (Fig. 6B and data not shown). We did not explore the effects of DFMO on MDSC function, since our data demonstrated that myeloid-derived Arg-1 does not play a major role during S. aureus biofilm formation. Collectively, these results demonstrate that myeloid-derived Arg-1 controls S. aureus growth during planktonic but not biofilm growth in vivo.
FIG 6.
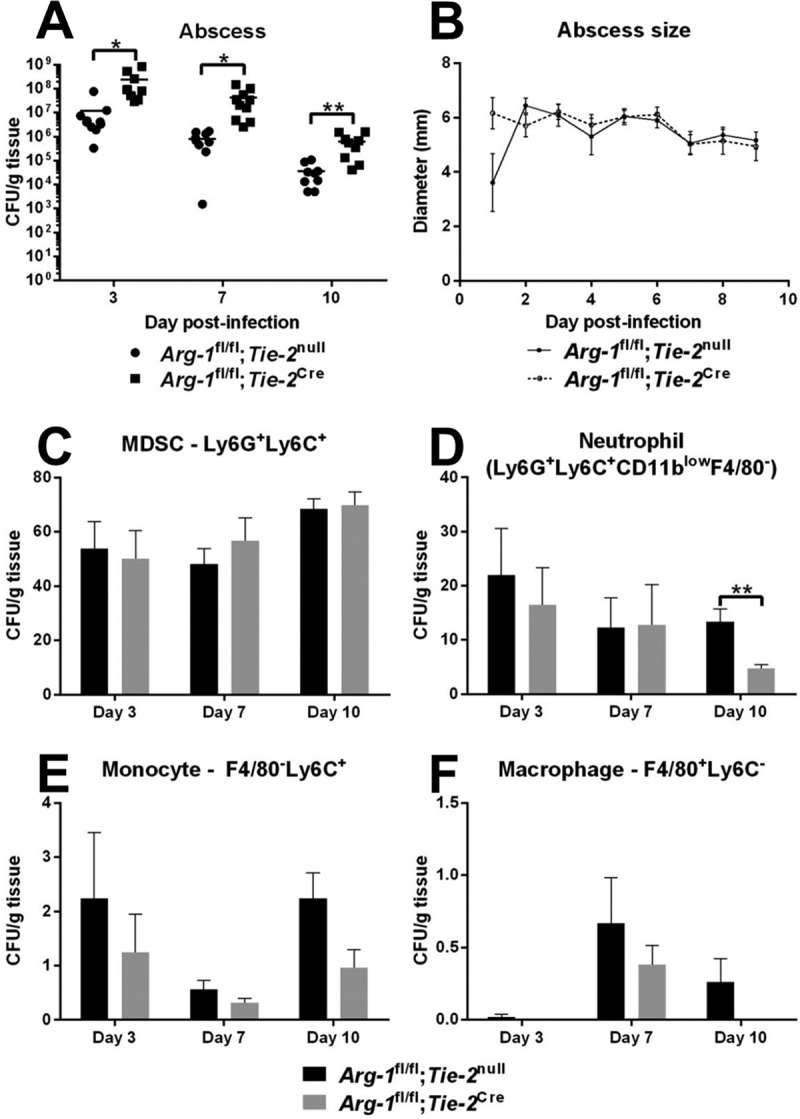
Myeloid-derived arginase-1 contributes to S. aureus clearance during subcutaneous abscess infection. Subcutaneous abscesses were established in Arg-1fl/fl;Tie-2null (WT) and Arg-1fl/fl;Tie-2Cre mice following inoculation with 107 CFU S. aureus LAC. Animals were sacrificed at days 3, 7, and 10 postinfection, whereupon abscess-associated bacterial burdens (A) and leukocyte infiltrates (C to F) were measured. (B) Abscess sizes were monitored by use of calipers throughout the course of infection. Results are representative of two independent experiments. *, P < 0.05; **, P < 0.01 (unpaired 2-tailed Student's t test).
DISCUSSION
Our laboratory has demonstrated that MDSCs are preferentially recruited to S. aureus biofilm infections, where they inhibit monocyte proinflammatory activity to promote infection persistence (10, 12–16). MDSCs are the primary source of IL-10 during biofilm infection, and prior studies have established that antibody-mediated depletion of MDSCs and IL-10 deficiency both result in reduced S. aureus biofilm burdens (10, 12, 14, 16). However, bacteria still remain in implant-associated tissues in both of these scenarios, indicating the existence of additional immunosuppressive mechanisms. Previous reports have shown that MDSCs and macrophages inhibit innate and adaptive immune responses through extracellular arginine depletion by Arg-1 (23, 26, 45). Our recent studies revealed that Arg-1 expression is increased in MDSCs during S. aureus orthopedic implant biofilm infection and that macrophages surrounding biofilm-infected catheters dramatically upregulate Arg-1 (10–12, 14). Furthermore, host polyamine synthesis is important for controlling S. aureus growth in nonbiofilm infection models, such as models of sepsis and subcutaneous abscesses (31). Arg-1 also plays a critical role in models of cancer, Alzheimer's disease, and infection because of its immunosuppressive properties (18, 24, 35, 36). Based on these observations, we sought to determine whether myeloid-derived Arg-1 facilitates S. aureus biofilm persistence, which may represent a viable treatment target since the enzyme is currently being explored as a therapeutic target in Alzheimer's disease (35).
Despite efficient depletion of myeloid-derived arginase activity from Arg-1fl/fl;Tie-2Cre mice, minimal differences in bacterial burdens or leukocyte infiltrates were observed in two distinct S. aureus device-associated biofilm infection models. MDSCs are the primary leukocyte infiltrate in both orthopedic implant and catheter-associated biofilm infections, whereas fewer T cells and neutrophils are present. However, one important distinction is that macrophage infiltrates are more abundant in catheter-associated infections than in the orthopedic implant model (10, 12–14). The finding that leukocyte infiltrates were similar in Arg-1fl/fl;Tie-2Cre myeloid conditional KO and Arg-1fl/fl;Tie-2null control mice in both models supports previous reports that Arg-1 activity regulates cellular activation rather than differential leukocyte recruitment (18, 23–25, 27). However, cellular activation status was not examined in the current study, since no dramatic differences in biofilm titers were observed.
Interestingly, a marked increase in iNOS expression was observed in MDSCs from Arg-1fl/fl;Tie-2Cre myeloid conditional KO animals both 7 and 14 days following infection. A previous study reported a similar phenomenon, where deletion of macrophage Arg-1 potentiated NO production by increasing arginine availability (24). To determine whether this increase in iNOS expression could compensate for myeloid-targeted Arg-1 depletion, Arg-1fl/fl;Tie-2Cre mice were treated with l-NIL, a small-molecule inhibitor of iNOS. l-NIL had no effect on biofilm burdens or leukocyte infiltrates in Arg-1fl/fl;Tie-2Cre myeloid conditional KO mice, indicating that increased iNOS expression is not a significant compensatory mechanism for promoting biofilm persistence. In addition, unpublished data from our laboratory, using iNOS KO mice, demonstrated that iNOS does not play a role in S. aureus biofilm infections. In contrast, McInnes et al. reported increased bacterial burdens and mortality during S. aureus septic arthritis in iNOS KO mice (46), identifying a distinction between biofilm and planktonic infections. In addition, the current study demonstrates that NADPH does not have an impact on biofilm growth, since apocynin treatment had no effect on S. aureus burdens. This differs from the results of a recent study of NADPH oxidase-deficient mice by Sun et al., which revealed a critical role for ROS in controlling S. aureus pneumonia (47). Therefore, our study has revealed novel distinctions between S. aureus biofilm and planktonic infections, since iNOS and NADPH oxidase do not affect biofilm formation but do regulate host immunity to planktonic infection.
As another potential point of redundancy, we examined whether ODC contributes to S. aureus persistence during biofilm infection, since a prior report revealed a critical role for polyamine synthesis in controlling S. aureus growth in a mouse skin abscess model (31). Interestingly, treatment of WT animals with the arginase/ODC inhibitor DFMO increased bacterial burdens in the soft tissue surrounding infected orthopedic implants at days 7 and 10 postinfection, whereas no changes in bacterial titers were observed in the knee joint, femur, or implant. The selectivity of DFMO action in the tissue is reminiscent of our findings with the catheter-associated infection model, in which S. aureus burdens in Arg-1fl/fl;Tie-2Cre conditional KO mice at day 10 postinfection were elevated in the surrounding soft tissues, which possess some attributes of a planktonic environment since bacteria have dispersed from the catheter surface. To further demonstrate the importance of Arg-1 in the context of planktonic infection, we took advantage of a subcutaneous abscess model in which a role for polyamines in controlling bacterial growth was previously shown (31). Importantly, our study advances this finding by identifying myeloid cells as a critical source of Arg-1 during subcutaneous S. aureus abscess formation, since bacterial burdens were significantly increased in abscesses from Arg-1fl/fl;Tie-2Cre mice compared to those in WT mice. Collectively, these results demonstrate that myeloid-derived Arg-1 activity and polyamine synthesis control S. aureus growth during planktonic but not biofilm growth in vivo.
The complete repertoire of mechanisms used by MDSCs to inhibit immune function and promote S. aureus biofilm establishment and persistence is still unknown. Previous studies from our laboratory implicated roles for IL-12 and IL-10 in the indirect recruitment and immunosuppressive function of MDSCs, respectively (12, 13). In cancer models, myeloid-derived Arg-1 has been shown to provide two complementary functions. First, depletion of extracellular arginine stores modulates the immune response by decreasing NO production, synthesis of arginine-containing proteins, and T cell activation (18, 29, 30, 48). Second, arginine metabolism provides precursor molecules for collagen and polyamine synthesis, which leads to fibrotic tissue deposition, vascular remodeling, and direct antibacterial actions (23, 28–31, 39). Although prior work suggested that myeloid Arg-1 might be important for promoting S. aureus biofilm infection (12–14), the present study demonstrates that myeloid Arg-1 does not directly influence the immune response to S. aureus biofilm infections or induce a compensatory increase in arginine flux toward reactive nitrogen intermediate (RNI)/ROS production. However, these observations do not provide a specific mechanism by which this occurs. Namely, they do not rule out potential effects of Arg-1 on arginine bioavailability to support S. aureus growth and fitness in vivo. For example, previous studies demonstrated that S. aureus growth and virulence are dependent on exogenous arginine or arginine biosynthesis via proline (49). Alternatively, S. aureus biofilm induction of Arg-1 expression in nonmyeloid cells, such as fibroblasts or other mesenchymal cells, may override any potential contribution of myeloid-derived Arg-1 activity during S. aureus biofilm infection. This would require crossing of an Arg-1fl/fl line with a Cre line directed by the type I collagen promoter Col1a2 to target gene deletion in fibroblasts (50). Furthermore, mitochondrial Arg-2 released from dead or dying cells may also contribute to the overall pool of arginase enzyme activity (41). Additional studies are needed to address these possibilities; however, the present study demonstrates that myeloid-derived Arg-1 has minimal effects on S. aureus biofilm growth but instead plays a unique role in controlling S. aureus planktonic infections.
MATERIALS AND METHODS
Mice.
Male and female C57BL/6 mice (8 weeks of age) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Arg-1fl/fl;Tie-2Cre and Arg-1fl/fl;LysMCre mice were generated by crossing Arg-1fl/fl animals with Tie-2Cre and LysMCre animals, respectively, both on a C57BL/6 background (The Jackson Laboratory), with Arg-1fl/fl littermates used as WT controls. This study was conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals (51). The animal protocol was approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center.
Generation of bone marrow-derived macrophages.
Macrophages were expanded from the bone marrow of Arg-1fl/fl;Tie-2Cre conditional KO, Arg-1fl/fl;LysMCre, and Arg-1fl/fl;Tie-2null control mice by flushing long bones with sterile RPMI 1640 serum-free medium and culturing cells in RPMI 1640 medium containing 10% fetal bovine serum (FBS) (Atlanta Biologicals, Atlanta, GA), penicillin-streptomycin-amphotericin B (Fungizone), and 5% conditioned medium from L929 fibroblasts as a source of macrophage colony-stimulating factor (M-CSF) (11, 52). FACS analysis revealed that >99% of cells were macrophages, based on CD11b and F4/80 staining, after a 6-day culture period (data not shown). At in vitro day 6, macrophages were harvested, replated, and treated with 10 ng/ml recombinant mouse IL-4 (rmIL-4) (BioLegend, San Diego, CA) for 24 h, whereupon cellular extracts were prepared for Western blotting and Arg-1 activity assays. Western blotting was performed as previously described (53), with an anti-Arg-1 antibody (goat anti-mouse; 1:200) (sc-271430; Santa Cruz Biotechnology) followed by a rabbit anti-goat IgG conjugated to horseradish peroxidase (HRP) (Abcam, Cambridge, MA). Arginase activity in macrophages was measured using an arginase activity assay kit (Sigma-Aldrich, St. Louis, MO).
Mouse models of device-associated S. aureus biofilm infection.
Mouse models of S. aureus catheter-associated and orthopedic implant biofilm infections were performed as previously described (10–13, 15, 17). Briefly, for both device-associated models, mice were anesthetized with ketamine-xylazine (100 mg/kg of body weight-5 mg/kg of body weight), and the surgical site was disinfected with povidone-iodine. The health status of mice was regularly monitored throughout the course of infection, and all mice exhibited normal ambulation and no discernible pain behaviors. S. aureus catheter-associated biofilm infections were performed as previously described (10, 15, 16). Briefly, a 1-cm subcutaneous incision was made in the flank, and a blunt probe was used to create a pocket for insertion of a 1-cm sterile 16-gauge Teflon-coated intravenous (i.v.) catheter (Excel International, St. Petersburg, FL). The incision was sealed using Vetbond tissue adhesive (3M, St. Paul, MN), whereupon a total of 103 CFU of S. aureus USA300 LAC (54) in 20 μl 1× phosphate-buffered saline (PBS) was injected directly into the catheter lumen to establish biofilm formation.
To model infectious complications that can arise in patients following arthroplasty, a mouse model of S. aureus orthopedic implant infection was used as previously described (12–14, 17, 55). Briefly, after a surgical plane of ketamine-xylazine anesthesia was achieved, a medial incision was created through the quadriceps, with lateral displacement to access the distal femur. A burr hole was made in the femoral intercondylar notch through the intramedullary canal, using a 26-gauge needle, whereupon a precut 0.8-cm orthopedic-grade Kirschner wire (0.6-mm diameter) (Nitinol [nickel-titanium]; Custom Wire Technologies, Port Washington, WI) was inserted into the intramedullary canal, leaving ∼1 mm protruding into the joint space. The exposed wire surface was inoculated with 103 CFU of S. aureus USA300 LAC in 2 μl of 1× PBS. Following closure of the surgical site with absorbable nylon sutures, animals received s.c. buprenorphine (Buprenex) (0.1 mg/kg; Reckitt Benckiser, Hull, United Kingdom) for the first 24 h after surgery for pain relief. Detailed time course studies were initiated for the catheter-associated model, with mice sacrificed on days 3, 5, 7, and 10 postinfection. Subsequent studies were conducted with a more limited set of time points in the orthopedic infection model, with mice analyzed at days 3, 7, and 14 postinfection.
Mouse model of S. aureus abscess infection.
S. aureus subcutaneous abscesses were induced as previously described (31, 49). Briefly, Arg-1fl/fl;Tie-2Cre conditional KO and Arg-1fl/fl;Tie-2null control mice were anesthetized with ketamine-xylazine (100 mg/kg-5 mg/kg), the flank was shaved using clippers, and the surgical site was disinfected with povidone-iodine. Mice received subcutaneous injections of S. aureus (107 CFU in 20 μl PBS), and abscess sizes were measured daily by use of calipers throughout the course of infection. Mice were euthanized at days 3, 7, and 10 postinfection to quantify bacterial burdens and leukocyte infiltrates.
Recovery of infected tissues for S. aureus enumeration.
The soft tissue surrounding catheter-associated infections was collected, weighed, and dissociated using the blunt end of a 3.0-ml syringe in 500 μl of PBS supplemented with a protease inhibitor cocktail tablet (Roche Diagnostics, Indianapolis, IN). Catheters were placed in 500 μl of PBS for sonication to dissociate bacteria from the catheter surface. For the orthopedic implant infection model, the titanium implant, femur, knee joint, and surrounding soft tissue were collected by first removing the skin, whereupon the tissue ventral to the patellar tendon was excised, weighed, and disrupted using the blunt end of a 3.0-ml syringe in 500 μl of PBS supplemented with a protease inhibitor cocktail tablet. The remaining muscle and tendons were removed and excluded from analysis. The knee joint and femur were separated and homogenized individually by use of a hand-held homogenizer for 30 s. The titanium implant was carefully removed and vortexed in 200 μl PBS to dislodge biofilm-associated bacteria. Subcutaneous abscesses were collected, weighed, and dissociated using the blunt end of a 3.0-ml syringe in 500 μl of PBS. S. aureus titers were quantified using tryptic soy agar (TSA) plates supplemented with 5% sheep blood (Remel, Lenexa, KS) and are expressed as CFU per milliliter for catheters/titanium implants or CFU per gram of wet tissue weight.
Flow cytometry.
To characterize leukocyte infiltrates associated with soft tissues surrounding the infected knee, catheter, or abscess, homogenates were passed through a 35-μm filter (BD Falcon, BD Biosciences). The cellular filtrate was washed with PBS supplemented with 2% heat-inactivated FBS and collected by centrifugation (300 × g, 5 min), whereupon red blood cells (RBCs) were lysed using RBC lysis buffer (BioLegend, San Diego, CA). Cells were washed and resuspended in PBS containing 2% FBS and then incubated with TruStain fcX (BioLegend, San Diego, CA) to minimize nonspecific antibody binding. Cells were then stained with Live/Dead Fixable Blue dead cell stain (Invitrogen, Eugene, OR), CD45-allophycocyanin (CD45-APC), Ly6G-phycoerythrin (Ly6G-PE), Ly6C-peridinin chlorophyll protein (PerCP)-Cy5.5, and F4/80-PE-Cy7 (BioLegend, San Diego, CA). An aliquot of pooled cells was stained with isotype-matched control Abs to assess the degree of nonspecific staining per treatment group. For individual samples, 10,000 to 100,000 events were analyzed using BD FACSDiva software, with cell populations expressed as percentages of total viable CD45+ leukocytes.
Hydroxyproline assay.
To determine the relative collagen content in infected tissues, hydroxyproline assays were performed using a hydroxyproline assay kit (Sigma-Aldrich). Briefly, samples were prepared by homogenization of 10 mg of infected tissue in 100 μl of water, transferred to a pressure-tight vial with 100 μl of 12 M hydrochloric acid, and hydrolyzed at 120°C for 3 h. A total of 10 to 50 μl per sample was transferred to a 96-well plate and evaporated at 60°C. Following the addition of reaction buffer, the plate was read at 570 nm by use of an iMark microplate absorbance reader (Bio-Rad, Hercules, CA).
Statistics.
Significant differences between experimental groups were determined by an unpaired two-tailed Student t test or one-way analysis of variance (ANOVA) with Bonferroni's multiple-comparison test, using GraphPad Prism 6 (GraphPad Software, La Jolla, CA). For all analyses, P values of <0.05 were considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the NIH National Institute of Allergy and Infectious Diseases (NIAID) under grant 2P01AI083211 (project 4 to T.K.).
We thank Rachel Fallet for managing the mouse colony and Craig Semerad, Victoria Smith, and Samantha Wall of the UNMC Flow Cytometry Core Facility for assistance with FACS analysis.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00206-18.
REFERENCES
- 1.Fowler VG Jr, Olsen MK, Corey GR, Woods CW, Cabell CH, Reller LB, Cheng AC, Dudley T, Oddone EZ. 2003. Clinical identifiers of complicated Staphylococcus aureus bacteremia. Arch Intern Med 163:2066–2072. doi: 10.1001/archinte.163.17.2066. [DOI] [PubMed] [Google Scholar]
- 2.Morgenstern M, Erichsen C, Hackl S, Mily J, Militz M, Friederichs J, Hungerer S, Buhren V, Moriarty TF, Post V, Richards RG, Kates SL. 2016. Antibiotic resistance of commensal Staphylococcus aureus and coagulase-negative staphylococci in an international cohort of surgeons: a prospective point-prevalence study. PLoS One 11:e0148437. doi: 10.1371/journal.pone.0148437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naber CK. 2009. Staphylococcus aureus bacteremia: epidemiology, pathophysiology, and management strategies. Clin Infect Dis 48(Suppl 4):S231–S237. doi: 10.1086/598189. [DOI] [PubMed] [Google Scholar]
- 4.Walls RJ, Roche SJ, O'Rourke A, McCabe JP. 2008. Surgical site infection with methicillin-resistant Staphylococcus aureus after primary total hip replacement. J Bone Joint Surg Br 90:292–298. doi: 10.1302/0301-620X.90B3.20155. [DOI] [PubMed] [Google Scholar]
- 5.Self WH, Wunderink RG, Williams DJ, Zhu Y, Anderson EJ, Balk RA, Fakhran SS, Chappell JD, Casimir G, Courtney DM, Trabue C, Waterer GW, Bramley A, Magill S, Jain S, Edwards KM, Grijalva CG. 2016. Staphylococcus aureus community-acquired pneumonia: prevalence, clinical characteristics, and outcomes. Clin Infect Dis 63:300–309. doi: 10.1093/cid/ciw300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tande AJ, Osmon DR, Greenwood-Quaintance KE, Mabry TM, Hanssen AD, Patel R. 2014. Clinical characteristics and outcomes of prosthetic joint infection caused by small colony variant staphylococci. mBio 5:e01910-. doi: 10.1128/mBio.01910-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tande AJ, Patel R. 2014. Prosthetic joint infection. Clin Microbiol Rev 27:302–345. doi: 10.1128/CMR.00111-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guggenbichler JP, Assadian O, Boeswald M, Kramer A. 2011. Incidence and clinical implication of nosocomial infections associated with implantable biomaterials—catheters, ventilator-associated pneumonia, urinary tract infections. GMS Krankenhhyg Interdiszip 6:Doc18. doi: 10.3205/dgkh000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klein E, Smith DL, Laxminarayan R. 2007. Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999–2005. Emerg Infect Dis 13:1840–1846. doi: 10.3201/eid1312.070629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanke ML, Heim CE, Angle A, Sanderson SD, Kielian T. 2013. Targeting macrophage activation for the prevention and treatment of Staphylococcus aureus biofilm infections. J Immunol 190:2159–2168. doi: 10.4049/jimmunol.1202348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanke ML, Kielian T. 2012. Deciphering mechanisms of staphylococcal biofilm evasion of host immunity. Front Cell Infect Microbiol 2:62. doi: 10.3389/fcimb.2012.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heim CE, Vidlak D, Kielian T. 2015. Interleukin-10 production by myeloid-derived suppressor cells contributes to bacterial persistence during Staphylococcus aureus orthopedic biofilm infection. J Leukoc Biol 98:1003–1013. doi: 10.1189/jlb.4VMA0315-125RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heim CE, Vidlak D, Scherr TD, Hartman CW, Garvin KL, Kielian T. 2015. IL-12 promotes myeloid-derived suppressor cell recruitment and bacterial persistence during Staphylococcus aureus orthopedic implant infection. J Immunol 194:3861–3872. doi: 10.4049/jimmunol.1402689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heim CE, Vidlak D, Scherr TD, Kozel JA, Holzapfel M, Muirhead DE, Kielian T. 2014. Myeloid-derived suppressor cells contribute to Staphylococcus aureus orthopedic biofilm infection. J Immunol 192:3778–3792. doi: 10.4049/jimmunol.1303408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thurlow LR, Hanke ML, Fritz T, Angle A, Aldrich A, Williams SH, Engebretsen IL, Bayles KW, Horswill AR, Kielian T. 2011. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J Immunol 186:6585–6596. doi: 10.4049/jimmunol.1002794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanke ML, Angle A, Kielian T. 2012. MyD88-dependent signaling influences fibrosis and alternative macrophage activation during Staphylococcus aureus biofilm infection. PLoS One 7:e42476. doi: 10.1371/journal.pone.0042476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heim CE, Hanke ML, Kielian T. 2014. A mouse model of Staphylococcus catheter-associated biofilm infection. Methods Mol Biol 1106:183–191. doi: 10.1007/978-1-62703-736-5_17. [DOI] [PubMed] [Google Scholar]
- 18.Munder M. 2009. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol 158:638–651. doi: 10.1111/j.1476-5381.2009.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makarenkova VP, Bansal V, Matta BM, Perez LA, Ochoa JB. 2006. CD11b+/Gr-1+ myeloid suppressor cells cause T cell dysfunction after traumatic stress. J Immunol 176:2085–2094. doi: 10.4049/jimmunol.176.4.2085. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez PC, Quiceno DG, Ochoa AC. 2007. l-Arginine availability regulates T-lymphocyte cell-cycle progression. Blood 109:1568–1573. doi: 10.1182/blood-2006-06-031856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, Ochoa JB, Ochoa AC. 2003. l-Arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol 171:1232–1239. doi: 10.4049/jimmunol.171.3.1232. [DOI] [PubMed] [Google Scholar]
- 22.Zhu X, Pribis JP, Rodriguez PC, Morris SM Jr, Vodovotz Y, Billiar TR, Ochoa JB. 2014. The central role of arginine catabolism in T-cell dysfunction and increased susceptibility to infection after physical injury. Ann Surg 259:171–178. doi: 10.1097/SLA.0b013e31828611f8. [DOI] [PubMed] [Google Scholar]
- 23.Bronte V, Serafini P, De Santo C, Marigo I, Tosello V, Mazzoni A, Segal DM, Staib C, Lowel M, Sutter G, Colombo MP, Zanovello P. 2003. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol 170:270–278. doi: 10.4049/jimmunol.170.1.270. [DOI] [PubMed] [Google Scholar]
- 24.El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, Basaraba RJ, Konig T, Schleicher U, Koo MS, Kaplan G, Fitzgerald KA, Tuomanen EI, Orme IM, Kanneganti TD, Bogdan C, Wynn TA, Murray PJ. 2008. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol 9:1399–1406. doi: 10.1038/ni.1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Modolell M, Choi BS, Ryan RO, Hancock M, Titus RG, Abebe T, Hailu A, Muller I, Rogers ME, Bangham CR, Munder M, Kropf P. 2009. Local suppression of T cell responses by arginase-induced l-arginine depletion in nonhealing leishmaniasis. PLoS Negl Trop Dis 3:e480. doi: 10.1371/journal.pntd.0000480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, Antonia S, Ochoa JB, Ochoa AC. 2004. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 64:5839–5849. doi: 10.1158/0008-5472.CAN-04-0465. [DOI] [PubMed] [Google Scholar]
- 27.Wijnands KA, Hoeksema MA, Meesters DM, van den Akker NM, Molin DG, Briede JJ, Ghosh M, Kohler SE, van Zandvoort MA, de Winther MP, Buurman WA, Lamers WH, Poeze M. 2014. Arginase-1 deficiency regulates arginine concentrations and NOS2-mediated NO production during endotoxemia. PLoS One 9:e86135. doi: 10.1371/journal.pone.0086135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaur U, Roberts SC, Dalvi RP, Corraliza I, Ullman B, Wilson ME. 2007. An effect of parasite-encoded arginase on the outcome of murine cutaneous leishmaniasis. J Immunol 179:8446–8453. doi: 10.4049/jimmunol.179.12.8446. [DOI] [PubMed] [Google Scholar]
- 29.Maarsingh H, Leusink J, Bos IS, Zaagsma J, Meurs H. 2006. Arginase strongly impairs neuronal nitric oxide-mediated airway smooth muscle relaxation in allergic asthma. Respir Res 7:6. doi: 10.1186/1465-9921-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meurs H, McKay S, Maarsingh H, Hamer MA, Macic L, Molendijk N, Zaagsma J. 2002. Increased arginase activity underlies allergen-induced deficiency of cNOS-derived nitric oxide and airway hyperresponsiveness. Br J Pharmacol 136:391–398. doi: 10.1038/sj.bjp.0704725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thurlow LR, Joshi GS, Clark JR, Spontak JS, Neely CJ, Maile R, Richardson AR. 2013. Functional modularity of the arginine catabolic mobile element contributes to the success of USA300 methicillin-resistant Staphylococcus aureus. Cell Host Microbe 13:100–107. doi: 10.1016/j.chom.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zabaleta J, McGee DJ, Zea AH, Hernandez CP, Rodriguez PC, Sierra RA, Correa P, Ochoa AC. 2004. Helicobacter pylori arginase inhibits T cell proliferation and reduces the expression of the TCR zeta-chain (CD3zeta). J Immunol 173:586–593. doi: 10.4049/jimmunol.173.1.586. [DOI] [PubMed] [Google Scholar]
- 33.Bian Z, Abdelaal AM, Shi L, Liang H, Xiong L, Kidder K, Venkataramani M, Culpepper C, Zen K, Liu Y. 28 February 2018. Arginase-1 is neither constitutively expressed in nor required for myeloid-derived suppressor cell-mediated inhibition of T-cell proliferation. Eur J Immunol doi: 10.1002/eji.201747355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deshane J, Zmijewski JW, Luther R, Gaggar A, Deshane R, Lai JF, Xu X, Spell M, Estell K, Weaver CT, Abraham E, Schwiebert LM, Chaplin DD. 2011. Free radical-producing myeloid-derived regulatory cells: potent activators and suppressors of lung inflammation and airway hyperresponsiveness. Mucosal Immunol 4:503–518. doi: 10.1038/mi.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kan MJ, Lee JE, Wilson JG, Everhart AL, Brown CM, Hoofnagle AN, Jansen M, Vitek MP, Gunn MD, Colton CA. 2015. Arginine deprivation and immune suppression in a mouse model of Alzheimer's disease. J Neurosci 35:5969–5982. doi: 10.1523/JNEUROSCI.4668-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knippenberg S, Brumshagen C, Aschenbrenner F, Welte T, Maus UA. 2015. Arginase 1 activity worsens lung-protective immunity against Streptococcus pneumoniae infection. Eur J Immunol 45:1716–1726. doi: 10.1002/eji.201445419. [DOI] [PubMed] [Google Scholar]
- 37.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. 2001. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol 230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 38.Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, Sanchez-Pino D, Hernandez C, Wyczechowska DD, Ochoa AC, Rodriguez PC. 2015. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res 75:275–283. doi: 10.1158/0008-5472.CAN-14-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. 2017. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol 8:289. doi: 10.3389/fimmu.2017.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murray PJ, Wynn TA. 2011. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol 89:557–563. doi: 10.1189/jlb.0710409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi S, Park C, Ahn M, Lee JH, Shin T. 2012. Immunohistochemical study of arginase 1 and 2 in various tissues of rats. Acta Histochem 114:487–494. doi: 10.1016/j.acthis.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 42.Grant SK, Green BG, Stiffey-Wilusz J, Durette PL, Shah SK, Kozarich JW. 1998. Structural requirements for human inducible nitric oxide synthase substrates and substrate analogue inhibitors. Biochemistry 37:4174–4180. doi: 10.1021/bi972481d. [DOI] [PubMed] [Google Scholar]
- 43.Gabrilovich DI, Nagaraj S. 2009. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. 2012. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kusmartsev S, Gabrilovich DI. 2003. Inhibition of myeloid cell differentiation in cancer: the role of reactive oxygen species. J Leukoc Biol 74:186–196. doi: 10.1189/jlb.0103010. [DOI] [PubMed] [Google Scholar]
- 46.McInnes IB, Leung B, Wei XQ, Gemmell CC, Liew FY. 1998. Septic arthritis following Staphylococcus aureus infection in mice lacking inducible nitric oxide synthase. J Immunol 160:308–315. [PubMed] [Google Scholar]
- 47.Sun K, Yajjala VK, Bauer C, Talmon GA, Fischer KJ, Kielian T, Metzger DW. 2016. Nox2-derived oxidative stress results in inefficacy of antibiotics against post-influenza S. aureus pneumonia. J Exp Med 213:1851–1864. doi: 10.1084/jem.20150514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, Rodriguez PC, Sica A, Umansky V, Vonderheide RH, Gabrilovich DI. 2016. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 7:12150. doi: 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nuxoll AS, Halouska SM, Sadykov MR, Hanke ML, Bayles KW, Kielian T, Powers R, Fey PD. 2012. CcpA regulates arginine biosynthesis in Staphylococcus aureus through repression of proline catabolism. PLoS Pathog 8:e1003033. doi: 10.1371/journal.ppat.1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng B, Zhang Z, Black CM, de Crombrugghe B, Denton CP. 2002. Ligand-dependent genetic recombination in fibroblasts: a potentially powerful technique for investigating gene function in fibrosis. Am J Pathol 160:1609–1617. doi: 10.1016/S0002-9440(10)61108-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 52.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. 2009. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci 29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aldrich A, Kielian T. 2011. Central nervous system fibrosis is associated with fibrocyte-like infiltrates. Am J Pathol 179:2952–2962. doi: 10.1016/j.ajpath.2011.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.DeLeo FR, Otto M, Kreiswirth BN, Chambers HF. 2010. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 375:1557–1568. doi: 10.1016/S0140-6736(09)61999-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scherr TD, Lindgren KE, Schaeffer CR, Hanke ML, Hartman CW, Kielian T. 2014. Mouse model of post-arthroplasty Staphylococcus epidermidis joint infection. Methods Mol Biol 1106:173–181. doi: 10.1007/978-1-62703-736-5_16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.