

ABSTRACT
Severe bacterial (pneumococcal) infections are commonly associated with influenza and are significant contributors to the excess morbidity and mortality of influenza. Disruption of lung tissue integrity during influenza participates in bacterial pulmonary colonization and dissemination out of the lungs. Interleukin-22 (IL-22) has gained considerable interest in anti-inflammatory and anti-infection immunotherapy over the last decade. In the current study, we investigated the effect of exogenous IL-22 delivery on the outcome of pneumococcal superinfection postinfluenza. Our data show that exogenous treatment of influenza virus-infected mice with recombinant IL-22 reduces bacterial dissemination out of the lungs but is without effect on pulmonary bacterial burden. Reduced systemic bacterial dissemination was linked to reinforced pulmonary barrier functions, as revealed by total protein measurement in the bronchoalveolar fluids, intratracheal fluorescein isothiocyanate-dextran tracking, and histological approaches. We describe an IL-22-specific gene signature in the lung tissue of influenza A virus (IAV)-infected (and naive) mice that might explain the observed effects. Indeed, exogenous IL-22 modulates the gene expression profile in a way that suggests reinforcement of tissue integrity. Our results open the way to alternative approaches for limiting postinfluenza bacterial superinfection, particularly, systemic bacterial invasion.
KEYWORDS: epithelial barrier, influenza, interleukin 22, superinfection
INTRODUCTION
Despite the application of vaccination programs and antiviral drugs, influenza A virus (IAV) infection is one of the most important causes of respiratory tract diseases and is responsible for widespread morbidity and mortality every winter (H1N1 and H3N2 strains) (1). Severe bacterial, including pneumococcal, infections are commonly associated with influenza and are significant contributors to the excess morbidity and mortality of influenza during epidemics and pandemics (2, 3). Mechanisms leading to bacterial superinfection include denudation of the epithelial cell surface (through neuraminidase activity) and disruption of the lung barrier integrity, both of which are effects due to the virus itself. This physical alteration promotes bacterial adherence and systemic invasion (1, 2, 4, 5). On the other hand, dysfunction of pulmonary innate effector cells greatly contributes to bacterial superinfection (for reviews, see references 1–3 and 6–8). Today's treatments of secondary bacterial infections are still not effective enough. Moreover, antibiotic resistance is a major issue. Hence, there is an urgent need for novel therapies.
Interleukin-22 (IL-22) is emerging as an important, though versatile, cytokine in many physiological and pathological situations. This IL-10-related cytokine acts on nonhematopoietic cells at barrier surfaces, particularly on epithelial cells (9–12). Experimental models and clinical studies have revealed that IL-22 can be protective or proinflammatory depending on the tissue and the context in which it is expressed (10, 13–18). In the intestine, IL-22 maintains epithelial barrier integrity, controls the dissemination of commensal bacteria, and protects against pathogenic intruders (19–25). These effects are mediated by induction of genes related to epithelial cell regeneration and proliferation, antimicrobial peptides (Reg3β and Reg3γ), chemotactic factors, and mucins. Through these properties, IL-22 protects against inflammatory intestine disorders (21). In the pulmonary compartment, IL-22 protects against acute and chronic airway inflammation, including ventilator-induced lung injury and lung fibrosis, whereas its role in asthma is more ambiguous (26–31). IL-22 plays a role in pulmonary mucosal defenses against intracellular and extracellular bacteria, including Klebsiella pneumoniae, Chlamydia muridarum, Staphylococcus aureus, Haemophilus influenzae, Streptococcus pneumoniae, and Mycobacterium tuberculosis (32–44). The positive role of IL-22 in respiratory fungal infections has also been reported (45, 46). Fewer studies have been devoted to decipher the function of IL-22 during respiratory viral infections, and most of them focused on IAV infection (47).
During IAV infection, IL-22 is rapidly produced in the lung tissue by natural killer (NK) cells, nonconventional T cells (γδ T cells and natural killer T [NKT] cells), and type 3 innate lymphoid cells (48–51). We along with others have shown that endogenous IL-22 is protective (reduced pneumonia) during H1N1 and H3N2 influenza virus infections (49, 50, 52). In these systems, IL-22 might protect against epithelial damages caused by virus replication although underlining mechanisms are still elusive (49). IL-22 might also participate in airway epithelial regeneration and barrier repair (50, 52). Importantly, production of endogenous IL-22 during influenza virus infection leads to reduced susceptibility to secondary bacterial (pneumococcal) infection (51). In the current study, we examined the effect of exogenous IL-22 supplementation in the control of secondary bacterial infection postinfluenza. Our data show that IL-22 supplementation during influenza fails to hamper bacterial development in the lung compartment but reduces systemic bacterial dissemination. Our study identified for the first time an IL-22-specific gene signature in lung tissue that might explain the observed effects.
RESULTS
Exogenous IL-22 induces a classical gene signature in the liver but not in the lungs.
We first tested the in vivo biological activity of IL-22-Fc, a fusion protein with a prolonged half-life in vivo (53). To this end, the expression of genes known to be targeted by IL-22 (19–25, 41, 42) was analyzed by quantitative reverse transcription-PCR (RT-PCR). Compared to treatment with the IgG control, intraperitoneal (i.p.) inoculation of IL-22-Fc induced upregulation of Saa3, Reg3b, and C3 transcripts in the liver at 24 h postadministration (Fig. 1A, left panel). In marked contrast, intranasal (i.n.) administration of IL-22-Fc failed to induce gene expression in the liver, even at 72 h postadministration (Fig. 1A, left panel; also data not shown). This can be partly explained by the low concentration of IL-22-Fc in the serum at 24 h post-intranasal administration (Fig. 1A, right panel). We next turned to analyze the effect of IL-22-Fc on gene expression in the pulmonary compartment. Relative to treatment with the IgG control, and regardless of the mode of inoculation and the time point analyzed (including 8 h), IL-22-Fc failed to upregulate mRNAs encoding Saa3, Reg3b, C3 as well as other potential targets such as Reg3g, S100a9, and S100a8 (Fig. 1B, left panel; also not shown). Of note, IL-22-Fc was still detected in the bronchoalveolar lavage (BAL) fluids at 24 h and, to a lesser extent, at 72 h after i.n. inoculation (Fig. 1B, right panel). Collectively, our data indicate that IL-22-Fc is biologically active but fails to induce a classical IL-22 gene signature in the lung tissue.
FIG 1.

Effect of IL-22 treatment on gene expression in the liver and in the lungs. (A) Mice were i.p. treated with 100 μg of IL-22-Fc or isotype control. After 24 h, liver RNAs were extracted, and gene expression was analyzed by quantitative RT-PCR. The concentration of IL-22 in the blood was measured by enzyme-linked immunosorbent assay at 24 h and 72 h after inoculation. (B) Mice were i.n. treated with 100 μg of IL-22-Fc or an isotype control. Lung RNAs were extracted at 24 h after treatment. IL-22 was measured by enzyme-linked immunosorbent assay in the BAL fluids at 24 h and 72 h postinoculation. Data are expressed as fold increase over average gene expression in isotype-treated mice and represent the means ± standard deviations. One representative experiment out of two (n = 3 to 6), except for panel A (n = 8, two pooled experiments), is shown. *, P < 0.05; **, P < 0.01 (Mann-Whitney t test for data in the left panels and a Kruskal-Wallis one-way ANOVA for data in the right panels).
Exogenous IL-22 induces gene expression in the lung tissue suggestive of enhanced barrier functions.
To decipher the impact of exogenous IL-22 on pulmonary gene expression, we performed a transcriptomic analysis of the whole lung. Relative to expression levels in the IgG-treated control, 431 genes were differentially expressed in response to IL-22-Fc at 8 h postadministration, including 273 upregulated genes and 158 downregulated genes (P < 0.05; fold change, >2) (Fig. 2A). At 48 h posttreatment, the proportion of upregulated genes was less important (101 versus 294 downregulated genes) (see Fig. S1A in the supplemental material). Gene ontology (GO) analysis revealed six families of genes whose expression levels are generally down-modulated at 8 h by IL-22-Fc, most of them having metabolic (including lipid and amino acid) functions (Fig. 2B, left panel). Of interest was the identification of several families (mostly upregulated genes) involved in cellular functions, including growth and organization/maintenance (Fig. 2B, right). Similar modulated pathways were observed at 48 h posttreatment (Fig. S1B). A heat map of selected genes (8 h), on a total of 20, representative for the category cellular growth and proliferation is shown in Fig. 2C. Some genes are described to display transcriptional activity or chromatin binding, including Kdm6a, Bcl11b, Arid3c, Nlrp1b, Lhx5, Myt1, Pax1, and Cbx3 (Fig. 2C). In this family, genes involved in epithelial cell proliferation were detected, including Egfr, Egr4, Bcl11b, Flg2 (also involved in epithelial barrier), and Foxn1 (epithelial differentiation) (Fig. 2C, right, and Table 1). Within the cell morphology family, members belonging to the adhesion proteins, including Cd9 (tetraspanin), Cdh9 (cadherin), Cntn4 (contactin), Angptl1, (angiopoietin), pappa2, (pappalysin), and Cib1 (calmyrin) and to the extracellular matrix-associated proteins, such as Col12a1 (collagen), Col24a1, Thsd7b (thrombospondin), and Adamts3 (disintegrin/metallopeptidase) were identified (Fig. 2). This gene regulation suggests that IL-22 treatment could result in increased adherence strength of the epithelial structure and consequently could participate in maintaining tissue integrity. At 8 h, a small number of immune genes were also found to be upregulated by IL-22, including Nlrp1b, IL-21, Cxcl9, Tnfrsf19, Gzmk, Cd4, Ptcra, and Skint4 (Table 1 and Fig. S1C). Surprisingly, no antimicrobial peptide genes were significantly upregulated upon IL-22-Fc stimulation. Of note, flow cytometry analysis revealed that, at the dose used, IL-22 did not modulate the number and frequency of immune cells in the lung tissue, including macrophages, dendritic cells, neutrophils, nonconventional T cells (γδ and NKT cells), and conventional B/T lymphocytes (Fig. 2E; also data not shown). Collectively, this unbiased analysis demonstrates that, upon IL-22 stimulation, the lung modulates its gene expression profile in a way that suggests reinforcement of barrier functions.
FIG 2.

Transcriptomic signature of IL-22 in the lungs of naive mice. (A) Total RNAs from Ig-treated mice or IL-22-Fc-treated mice (5 μg/animal) were extracted from the lungs at 8 h poststimulation. Hierarchical clustering diagrams showing individual replicates are represented. Normalized intensity values (IL-22-Fc condition compared to IgG condition) are depicted in blue (downregulated) or red (upregulated) (P < 0.05 and fold change of >2). The magnitude of the regulation is illustrated by the intensity of the color. Hierarchical clustering representation indicates a minimal effect of the isotype control in the lung tissue (data not shown). (B) Gene ontological analysis. Significantly enriched pathways are represented as a bar plot (IPA). For panels A and B, data from three mice are shown. (C) Heat maps of genes that were differentially regulated by IL-22 treatment and that belong to the cellular growth and proliferation family (GO:0008283) and, within this family, belonging to the epithelial growth and proliferation subfamily (GO:0050673). Representative genes are noted on the right. Each column represents data from one individual mouse, with three mice per group. (D) Heat maps of genes represented in the families of adhesion proteins (GO:0022610) and extracellular-matrix (ECM)-associated proteins (GO:0031012), both belonging to the morphology family. (E) Naive mice were treated with IL-22-Fc or IgG (5 μg/animal), and 8 h later, cells from whole lungs were analyzed by flow cytometry. The mean number ± standard deviation of alveolar macrophages (CD45+ Siglec F+ CD11blow) and neutrophils (CD11b+ Ly6G+ Siglec F−) are depicted (n = 8; two independent experiments).
TABLE 1.
Genes belonging to the epithelial growth and proliferation family and the immune response family that are altered by IL-22 in naive micea
| GO family and gene symbol | Fold increase | Name | Function(s) | Role(s) in the lung |
|---|---|---|---|---|
| Epithelial growth and proliferation (GO:0050673) | ||||
| Egfr | 2.7 | Epidermal growth factor receptor | Differentiation, barrier functions | Repair of damaged alveolar epithelium, mucin production |
| Egr4 | 3.6 | Early growth response 4 | Differentiation, barrier functions | Unknown |
| Bcl11b | 2.6 | B cell leukemia lymphoma 11B | T-cell differentiation and proliferation | Unknown |
| Flg2 | 5.8 | Filaggrin family member 2 (S100 fused-type protein) | Epithelial homeostasis, barrier functions, epithelial differentiation | Unknown |
| Foxn1 | 3 | Forkhead box N1 | Antimicrobial activity | Unknown |
| Immune response (GO:0006955) | ||||
| Nlrp1b | 2.4 | NLR family, pyrin domain-containing 1B | Inflammasome, host defense | Unknown |
| Il21 | 2 | Interleukin-21 | Immune regulation, host defense | Control IL-17 production |
| Cxcl9 | 2 | Chemokine (C-X-C motif) ligand 9 | CXCR3 ligand | Inflammation, defense |
| Tnfrsfl9 | 4.5 | Tumor necrosis factor receptor superfamily, member 19 | Development, apoptosis | Unknown |
| Gzmk | 3.1 | Granzyme K | Cell lysis | Defense (?) |
| Cd4 | 2.3 | CD4 antigen | T cell marker | Defense |
| Ptcra | 2.9 | Pre-T cell receptor α-type chain precursor | TCR component | Unknown |
| Skint4 | 2.6 | Selection and upkeep of intraepithelial T cells 4 | T cell maturation (?) | Unknown |
Gene expression was assessed at 8 h. Uncertain functions/roles are indicated with a question mark.
Treatment with IL-22 during influenza virus infection results in enhanced barrier functions.
We next analyzed the effect of IL-22-Fc supplementation on gene expression in the context of IAV infection. To this end, mice were i.n. treated with IL-22-Fc or the isotype control 7 days after IAV infection, the peak of the inflammatory response. Compared to expression levels in the IgG1-treated control, 1,231 genes were differently regulated (P < 0.05; fold change, >2), of which 1,038 genes were upregulated and 193 genes were downregulated upon IL-22 treatment (8 h) (Fig. S2A). This number contrasted with the 431 regulated genes under noninfected conditions, indicating that IL-22 acts in concert with mediators produced during infection. As observed in naive animals, the dominant pathways promoted by IL-22 during IAV infection relate to metabolism and cellular (growth, maintenance, and repair) functions (Fig. 3A). Of note, the intensity of the response was drastically increased in comparison to the response under naive (mock) conditions. The most highly significant family found to be modulated by IL-22 was the group of cellular growth and proliferation genes, which included a total of 114 genes (Fig. S2B and data not shown). Of particular interest, several genes within the family of epithelial cell proliferation genes were upregulated upon IL-22 treatment. This includes growth factors or receptor for growth factors (Fgf9, Fgfr4, Gdf5, MstI, Ptn, Bmp10, and Bmpr2) and genes relevant in lung development, structure, and angiogenesis, such as Lgr5, Efna2, Sema5a, Sox5, Vash1, Kif3c, and Col25a1 (Fig. 3B and Table 2). Moreover, IL-22 treatment triggered the expression of genes involved in physical barrier (e.g., apical junction complex and components of the desmosome) and repair/regeneration functions, the so-called tissue integrity family (Fig. 3C and Table 3). This includes cell-cell adhesion molecules such as claudin and cadherin (Cldn24, Pcdh15, Cdh16, and dsg1c) and molecules participating in extracellular matrix components such as papilin (Papln) and reelin (Reln). Other genes with potentially important functions in lung barrier were identified, including desmoyokin (Ahnak), follistatin like-1 (Fstl1), involucrin (Ivl), otogelin (Otog), thymosin β10 (Tmsb10), parkin 2 (Park2), chloride channel calcium-activated 4a (Clca4a), and spectrin beta (Sptbn1). The modulated expression of these genes was confirmed by quantitative RT-PCR (Fig. S2C and data not shown). These data suggest that treatment with IL-22 might restrict loss of epithelial barrier functions due to IAV. In agreement with this hypothesis, IL-22 treatment reduced total protein concentration in the BAL fluids of infected animals (Fig. 3D). To further demonstrate improved pulmonary barrier properties following IL-22 treatment, diffusion of fluorescein isothiocyanate (FITC)-dextran across the pulmonary barrier was measured. As revealed in Fig. 3E, IL-22 treatment lowered the translocation of FITC-dextran from airspace to plasma. The positive effect of exogenous IL-22 was confirmed by histological examinations of lung sections (Fig. 4). Indeed, a significant reduction of airway inflammation (pneumonia) was noticed in IL-22-treated IAV-infected animals relative to the level in IgG-treated controls (Fig. 4A, top panels, and B). In IL-22-treated mice, alveolitis and bronchiolitis (in tendency) were less pronounced (Fig. 4A, middle and bottom panels, and B). In addition, epithelia surrounding the bronchi from IL-22-treated mice were less damaged. Signs of severe injury, characterized by augmented loss of intercellular cohesion and denuded epithelium, were observed in IAV-infected mice and were less pronounced after IL-22 supplementation (Fig. 4A, bottom panels). Together, these results indicate that supplementation of IL-22 during influenza virus infection reduces lung pathology and enhances pulmonary barrier functions.
FIG 3.

Effect of IL-22 supplementation on pulmonary gene expression and barrier functions. (A to C) Transcriptomic signature of IL-22 in the lungs of IAV-infected mice. Mice were i.n. infected with 500 PFU of the H1N1 pandemic IAV strain. At 7 days postinfection, mice were treated with PBS, the isotype control (5 μg/animal), or IL-22-Fc (5 μg/animal). Lung samples were collected 8 h later for further transcriptomic analysis. Ontological analysis is shown in panel A. Significantly enriched pathways are represented as a bar plot (IPA). Data from four mice are shown. In panels B and C, heat maps show genes that were differentially regulated by IL-22 treatment and that belong to the epithelial growth and proliferation family (GO:0050673) and to the tissue integrity family. Representative genes are noted on the right. Each column represents data from one individual mouse, with four mice per group. (D) Protein concentrations in the BAL fluids of IAV-infected mice treated with IL-22-Fc or IgG. Data represent the means ± standard deviations (n = 10 to 12; three pooled experiments). (E) IAV-infected mice, untreated or treated with IL-22, were inoculated with FITC-dextran. One hour later, FITC-dextran was quantified in the blood. Data represent the means ± standard deviations (n = 8; two pooled experiments). *, P < 0.05; ***, P < 0.001 (Kruskal-Wallis one-way ANOVA).
TABLE 2.
Genes of the epithelial growth and proliferation family (GO:0050673) altered by IL-22 in IAV-infected micea
| Gene symbol | Fold increase | Nameb | Function(s) | Role(s) in the lungc |
|---|---|---|---|---|
| Fgf9 | 2.2 | Fibroblast growth factor 9 | Cell differentiation, antiapoptotic | Development, organogenesis, antifibrotic, repair |
| Fgfr4 | 2.4 | Fibroblast growth factor receptor 4 | Epithelial-mesenchymal transition | Alveolar development, repair (?) |
| Gdf5 | 3.2 | Growth differentiation factor 5 (TGF-β family) | Cell differentiation, angiogenesis, antiapoptotic | Unknown |
| Ptn | 7.5 | Pleiotrophin (related to midkine) | Cell differentiation, angiogenesis | Fetal development |
| Bmp10 | 3.4 | Bone morphogenetic protein 10 (TGF-β family) | Endothelial homeostasis, angiogenesis | Fetal development (?) |
| Bmpr2 | 7 | Bone morphogenetic protein receptor 2 (TGF-β family) | Tissue remodeling | Arterial hypertension |
| Lgr5 | 7.7 | Leucine-rich repeat-containing G protein-coupled receptor 5 | Marker of adult stem cells (epithelial tissues) | Morphological structure, alveoli and bronchi |
| Efna2 | 8.3 | Ephrin A2 | Angiogenesis, vascular permeability | Lung inflammation |
| Sema5a | 2.2 | Sema domain 7, thrombospondin repeats, TM domain, short cytoplasmic domain 5A | Development biology, axonal guidance | Prognostic biomarker for lung cancer |
| Sox5 | 3.4 | Sex determining region Y-box 5 | Epithelial-mesenchymal transition | Development |
| Vash1 | 3.4 | Vasohibin 1 | Inhibits angiogenesis | Reduces bronchiolitis |
| Kif3c | 4.4 | Kinesin family member 3C | Microtubule dynamic, ciliogenesis | Epithelial-mesenchymal transition |
| Col25a1 | 5 | Collagen type 25α1 | Basement membrane, structure (brain) | Unknown |
Gene expression was assessed at 8 h.
TM, transmembrane.
Uncertain roles are indicated with a question mark.
TABLE 3.
Genes of the tissue integrity family altered by IL-22 in IAV-infected micea
| Gene symbol | Fold increase | Name | Function(s)b | Role in the lung |
|---|---|---|---|---|
| Cldn24 | 17.8 | Claudine 24 | Tight junction component, epithelial barrier integrity | Unknown |
| Cdh16 | 2 | Cadherin 16 | Tissue development, cell-cell adhesion | Unknown |
| Pcdh15 | 20.3 | Protocadherin 15 | Cell-cell adhesion | Unknown |
| Dsglc | 5 | Desmoglein 1 gamma (cadherin-like) | Cell-cell junction, desmosome | Dysregulated expression in cancer |
| Papln | 15.3 | Papilin | ECM protein, basement membrane | Unknown |
| Reln | 5.5 | Reelin | ECM protein barrier, healing remodeling | Unknown |
| Ahnak | 5.3 | Desmoyokin | Cell-cell junction, desmosome | Tumor suppressor (?) |
| Flst1 | 4.9 | Follistatin like-1 | Tight junctions, transepithelial resistance | Alveolar epithelial differentiation |
| Ivl | 2.2 | Involucrin | Keratinocyte differentiation, epidermal barrier | Dysregulated expression (cancer) |
| Otog | 11.4 | Otogelin | Fibrillar network, mucus-like protective layer | Unknown |
| Tmsb10 | 21.1 | Thymosin β1 | Cytoskeleton, angiogenesis, reparation | Protection against damage (?) |
| Park2 | 14.7 | Parkin2 | E3 ubiquitin ligase, mitophagy | Regulate inflammation and fibrosis |
| Clca4a | 5 | Chloride channel calcium activated 4A | Intestinal crypt epithelial cells | Unknown |
| Sptbn1 | 10.1 | Spectrin beta | Membrane cytoskeleton, gap junction (?) | Unknown |
Gene expression was assessed at 8 h. Uncertain functions/roles are indicated with a question mark.
ECM, extracellular matrix.
FIG 4.

Histological analysis of lung sections from IAV-infected (at 7 days postinfection) mice untreated or treated with IL-22. (A) Representative lung sections stained with hematoxylin and eosin are shown. Alveolar lesions (middle row) and perivascular and peribronchic infiltrates combined with lesions of alveolitis (bottom row) are shown. Arrows indicate denuded epithelia. Bv, blood vessel, Br, bronchiole. (B) Sections were scored blindly for levels of immunopathology. Data represent the means ± standard deviations (n = 5 mice/group). *, P < 0.05; **, P < 0.01 (Mann-Whitney t test).
Exogenous IL-22 alters immune gene expression and prevents the recruitment of inflammatory monocytes in the lungs of IAV-infected mice.
We next turned to analyze the effects of IL-22 supplementation on immune gene expression during influenza virus infection. Gene expression profiling revealed that IL-22 modulates genes associated with cell adhesion, leukocyte migration, and immune response (Fig. 5A and B). With respect to the last category, interferon (IFN) type I receptor (Ifnar1) and IFN type I-stimulated genes, including Ifna11 and Ifitm10, were upregulated (Fig. 5B and Table 4). Although these genes might play a role in virus clearance, IL-22 supplementation failed to reduce the viral load in the lungs (Fig. 5C). In parallel, Lif, C9, Nlrp9a, Sema5a, Cd300lf, Skint4, Skint1, and IL22ra1 transcripts were upregulated in response to IL-22 (Fig. 5B and Table 4). With respect to IL22ra1, this indicates an autocrine loop in line with the presence of STAT-3 binding sites in the proximal promoter region of IL-22R1. Some antibacterial innate immune genes were also induced by IL-22, including defensins (Defa1 and Defb14), the two members of the emerging cytokine family midkine/pleiotrophin (Mdk and Ptn), and the pentraxin family member Crp (Fig. 5B and Table 4). Influenza virus infection is associated with a recruitment of inflammatory monocytes, neutrophils, and NK cells. Of interest, flow cytometry analysis indicated that while IL-22 did not impact the recruitment of neutrophils and NK cells, it significantly lowered that of inflammatory monocytes, a population known to promote lung (epithelial) damage during influenza virus infection (54–56) (Fig. 5D). IAV infection reduces the number of alveolar macrophages and conventional dendritic cells, a process that can lead to secondary bacterial infections (57, 58; also submitted for publication). IL-22 supplementation did not have an impact on the number of alveolar macrophages and conventional dendritic cells (Fig. 5E). Of note, IL-22 had no effect on the frequency/number of nonconventional T cells (NKT and γδ T cells) and conventional B/T lymphocytes (data not shown). Collectively, IL-22 treatment during influenza alters immune gene expression and reduces the number of inflammatory monocytes in the lungs, a population that exacerbates bacterial superinfection (56).
FIG 5.

(A) Heat maps of genes belonging to cell adhesion (GO:0007155) and leukocyte migration (GO:0050900) families. (B) Heat map of genes belonging to the immune response family (GO:0006955). (C) Determination of the viral load after IL-22 treatment. IAV M1 mRNA levels were measured by quantitative RT-PCR. Data are expressed as cycle threshold (CT) values. The dashed line represents the detection threshold (n = 6). (D) IAV-infected mice were treated at 7 days postinfection with IL-22-Fc (5 μg/animal) or the isotype control. Eight hours later, cells from whole lungs were analyzed by flow cytometry. The mean number ± standard deviation of inflammatory monocytes (IM) (CD45+ Siglec F− Ly6G− Ly6C+ CD11b+ CCR2+), neutrophils (CD45+ CD11b+ Ly6G+ Siglec F−), and NK cells (CD45+ NKp46+ TCRβ−; TCRβ is T cell receptor beta) are depicted. (E) The mean numbers ± standard deviations of alveolar macrophages (CD45+ Siglec F+ CD11blow) and conventional dendritic cells (cDC) (CD45+ CD11chigh MHC class II+ Siglec F− CD64−; MHC is major histocompatibility complex) are depicted. (D and E) As a control, the number of immune cells from mock-treated mice are indicated (n = 8; two pooled experiments). The statistical analysis between mock-treated and IAV-infected mice is not depicted. *, P < 0.05 (Kruskal-Wallis one-way ANOVA).
TABLE 4.
Genes of the immune response family (GO:0006955) altered by IL-22 in IAV-infected micea
| Gene symbol | Fold increase | Nameb | Function(s) | Role in the lung |
|---|---|---|---|---|
| Ifnar1 | 3 | Interferon (type 1) receptor | Immune stimulation | Defense (virus) |
| Ifna11 | 8.2 | Interferon alpha 11 | Immune stimulation | Defense (virus) |
| Ifitm10 | 4.3 | Interferon-induced TM protein 10 | Immune stimulation (virus) | Unknown |
| Cxcl2 | 2 | Chemokine (C-X-C motif) ligand 2 | Cell recruitment | Defense wound healing |
| Lif | 8.2 | Leukemia inhibitory factor (IL-6 member) | Immune regulation, differentiation of myeloid cells | Protection during (viral) pneumonia |
| C9 | 2.5 | Complement component 9 | Bacterial lysis, phagocytosis, cell recruitment | Defense |
| Nlrp9a | 7.2 | NLR family, pyrin domain containing 9A | Inflammasome, defense | Unknown |
| Sema5a | 2.2 | Semaphorin 5A | Cellular immune response | Unknown |
| Cd3001f | 6.7 | CD300 antigen like family member F | Efferocytosis (?), dendritic cell functions (?) | Unknown |
| Skint4 | 74.6 | Selection and upkeep of intraepithelial T cell 4 | T cell maturation (?) | Unknown |
| Skint1 | 8.6 | Selection and upkeep of intraepithelial T cell 1 | Epidermal T cell selection | Unknown |
| defa1 | 3 | Defensin α1 | Defense | Unknown (?) |
| defb14 | 2 | Defensin β14 | Defense | Unknown |
| Mdk | 3 | Midkine | Defense | Development of pulmonary fibrosis |
| Ptn | 7.6 | Pleiotrophin | Defense | Dysregulated expression (cancer) |
| Crp | 2.1 | C-reactive protein (pentraxin-related) | Defense | Protection during infection |
Gene expression was assessed at 8 h. Uncertain functions/roles are indicated with a question mark.
TM, transmembrane.
IL-22 inoculation reduces systemic bacterial dissemination in superinfected animals.
Impaired lung (barrier) integrity and a defective innate immune response are the two major causes leading to bacterial superinfection postinfluenza (1–4, 6–8). We have previously shown that endogenous production of IL-22 during influenza virus infection (H3N2) is important to control secondary bacterial outgrowth in the lungs (51). As Fig. 6A (left panel) shows, this is also the case after infection with H1N1 IAV. Indeed, compared to superinfected Il22−/− mice, IL-22-proficient animals had a decreased bacterial load in the lung compartment. In contrast, superinfected IL-22-proficient mice had an identical number of bacteria in the spleen (Fig. 6A, right). As our current data show that IL-22 reinforces lung barrier properties, we hypothesized that supplementation of IL-22 in the context of IAV infection could reduce the extent of secondary bacterial infection. Intranasal inoculation of IL-22-Fc before S. pneumoniae challenge failed to reduce the number of bacteria in the lung tissue (Fig. 6B, left panel). Of interest, IL-22 supplementation led to a significant decrease (∼10-fold less) of bacteria in the spleen in doubly infected mice (Fig. 6B, right panel). Collectively, treatment with IL-22 just before pneumococcal challenge significantly reduces systemic invasion in superinfected mice, an effect that probably relies on improved lung integrity.
FIG 6.
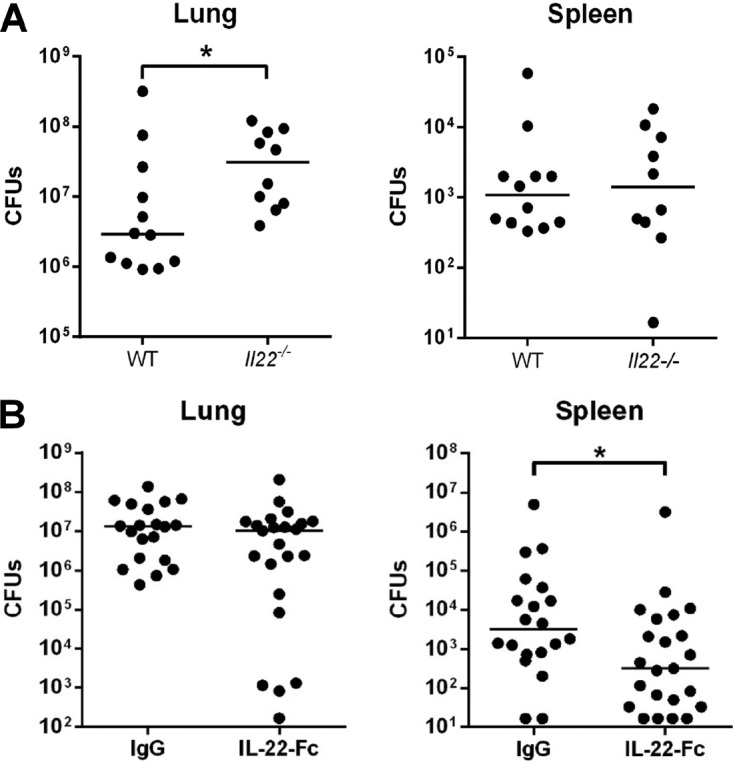
Effect of endogenous and exogenous IL-22 in local and systemic bacterial loads in the context of influenza. (A) Role of endogenous IL-22 on secondary bacterial infection postinfluenza. Wild type (WT) or Il22−/− mice were infected with 500 PFU of the H1N1 pandemic IAV strain. Seven days later, IAV-infected mice were challenged with S. pneumoniae (1 × 103 CFU). (B) Effect of IL-22 supplementation on bacterial superinfection postinfluenza. At day 7 post-IAV infection, mice were i.n. inoculated with 5 μg of IL-22-Fc or an isotype control. After 16 h, mice were challenged with S. pneumoniae. (A and B) Doubly infected mice were sacrificed 30 h after S. pneumoniae challenge, and the number of CFU was determined in the lungs and spleens. Total CFU counts in each tissue are represented. The solid line corresponds to the median values. Results from a pool of two independent experiments (n = 10 to 12 mice) (A) or three independent experiments (n = 20 to 23 mice) (B) are shown. *, P < 0.05 (Mann-Whitney t test).
DISCUSSION
A major cause of respiratory failure during IAV infection is damage to the epithelial barrier, including the epithelial-endothelial barrier of the pulmonary alveolus (7). Interleukin-22 has recently gained significant attention as a protective agent in diseases driven by epithelial injury, particularly in the skin and intestine (10, 12, 17, 59). In the lung tissue, its role is more diverse, and its effects on pulmonary disease outcomes seems to depend, in part, on the synthesis of other factors, including IL-17 (14). In the context of influenza virus and secondary bacterial infection, recent data suggest that endogenous IL-22 is beneficial (48–52). In view of the medical and economic burden of influenza, we investigated in the current study the effect of exogenous IL-22 on the outcome of secondary bacterial (pneumococcal) infection postinfluenza.
To our surprise, the molecular signature induced by IL-22 supplementation in the lung compartment is still elusive. Microarray analysis on whole lungs was determined in naive animals and in animals infected 7 days before with IAV. We chose this time point as it corresponds to the peak of susceptibility to bacterial infection. Pathway analysis of the IL-22-specific genes revealed that the top pathways modulated in naive animals are related to metabolism (downregulation) and cellular functions (upregulation), including growth, morphology, organization, and maintenance. Analysis of modulated genes indicated a reinforcement of the lung (epithelial) barrier functions under naive conditions. Under resting conditions, the IL-22 receptor (IL-22R1 subunit) mainly locates on tracheobronchial epithelial cells and, to a lesser extent, on airway epithelial cells (large and small airways, but not parenchyma) (52). Of interest, recent data indicate that the IL-22 receptor is also expressed by (microvascular) endothelial cells, especially at alveolar sites (60, 61). Although it has been suggested that neutrophil proteolytic enzymes might cleave the IL-22 receptor (62), its expression appears to increase during the course of influenza (at least at 21 days postinfection) to localize at the sites of parenchymal lung remodeling/repair (repairing alveoli) (52). Moreover, our published data indicate that expression of IL-22 binding protein, a potent inhibitor of IL-22 biological activity, decreases during influenza (51). Together, these findings indicate that IL-22 immunotherapy might (positively) impact the outcome of influenza virus pathogenesis. IL-22 supplementation induces the modulation of a myriad of genes during influenza virus infection (at 7 days postinfection), thus confirming the functionality of the IL-22 receptor. The majority of genes (>84%) identified were upregulated. Among them were genes implicated in epithelial functions, including epithelial growth/differentiation (e.g., fibroblast growth factors and transforming growth factor β [TGF-β] family members) and in epithelial-endothelial barrier properties (claudin, cadherin [like], follistatin, involucrin, and reelin). IAV is known to strongly damage the apical junction complex of epithelial cells, which are critical to regulate paracellular permeability and to ensure barrier integrity. Of interest, the expression of several genes encoding factors involved in cell-cell junction (tight junctions and desmosomal plaques) and in tissue repair/protection (growth factors, vasohibin 1, thymosin, and parkin 2) was induced by IL-22. This analysis thus identifies a plethora of novel targets of IL-22 that might be important in lung tissue protection. In the future, it will be interesting to study the functions of these factors (some of which have unknown functions in the lungs) (Tables 2 and 3) in pathologies driven by epithelial injury, including severe influenza. Our unbiased analysis suggests that, in the lung, exogenous IL-22 might reinforce physical barriers. In line with this, IL-22 treatment during influenza enhanced the pulmonary epithelial-endothelial barrier function, as assessed by a decrease in the total protein concentration in BAL fluids of treated animals. Supplementation of IL-22 also lowered the translocation of FITC-dextran across the pulmonary barrier. Finally, analysis of lung sections revealed reduced pulmonary injury and a clear improvement of the epithelial layer in mice treated with IL-22. Disruption of the pulmonary barrier contributes to systemic bacterial superinfection in the context of IAV infection. In line with the above findings, exogenous IL-22 hampered (∼10-fold less) the dissemination of the bacteria out of the lungs. Under naive conditions, to cause invasive disease, pneumococci must attach and translocate across the epithelium, pass through the extracellular matrix, and then enter the bloodstream by crossing the endothelium. In the context of prior influenza, bacterial dissemination out of the lungs is greatly facilitated and depends on multiple factors, including exposition of new attachment sites for the bacteria (due to epithelial cell surface denudation) and disruption of the alveolar capillary barrier integrity. The beneficial role for exogenous IL-22 in bacterial dissemination might be due to its direct restricting effect on barrier damage due to IAV replication. It might also arise from reduced recruitment of inflammatory monocytes, a cell population noted to have negative effects on epithelial damage and secondary bacterial infections (54–56). Finally, exogenous IL-22 might enhance barrier functions by promoting repair and regeneration (reepithelialization). It will be interesting in the future to investigate the mode of action of IL-22 in these settings.
Although IL-22 supplementation modulates (albeit moderately) the expression of innate immune genes, it had no impact on the local antibacterial immunity in the context of prior influenza virus infection. This is in contrast to the endogenous function of IL-22, which reduced local bacterial loads without affecting bacterial dissemination (51) (Fig. 6A). One may question why, in our setting (i.n. inoculation), IL-22 failed to reduce pneumococcal load in the lungs of superinfected animals. Two main reasons may explain this finding. First, recent data indicate that IL-22 signaling in the liver (but not in the lungs) is essential to control pneumococcal load in mice singly infected with S. pneumoniae (41). Endogenous (as well as exogenous) IL-22 induces the expression of complement C3 in the liver, a hub protein for activation of complement pathways. The IL-22 signaling in the liver ultimately leads to potentiate C3 opsonization on bacterial surfaces (probably via neutrophils), thus reducing pulmonary bacterial load. Our data (Fig. 1 and data not shown) show that i.n. delivery of IL-22 failed to promote C3 expression in the liver (as assessed by PCR analysis), which may explain the lack of protective effect on pulmonary bacterial load in superinfected mice. The second reason derives from the fact that influenza virus can alter the function of effector cells involved in the control of bacterial infection, including macrophages and neutrophils (58, 63–66).
Excessive inflammation triggered by secondary bacterial infection is considered to be the key contributor of the morbidity and mortality associated with bacterial superinfection postinfluenza. In line with this, and despite the positive effect of exogenous IL-22 on reduced bacterial systemic dissemination, IL-22 treatment failed to ameliorate the morbidity and mortality indexes of superinfected animals (data not shown). On the other hand, we have previously demonstrated that the reduced bacterial load in IL-22-proficient mice, relative to that in IL-22-deficient mice, is associated with a better survival outcome (51). Hence, endogenous IL-22 restricts bacterial replication in the lungs, thus improving the overall survival rate after superinfection, while exogenous IL-22 limits bacterial dissemination without a beneficial effect on survival. Therefore, the clinical relevance of our finding is still uncertain, and a combination of treatments, combining IL-22 with conventional antibacterial drugs (i.e., antibiotics), may increase therapeutic effectiveness.
To sum up, our study describes for the first time an IL-22-mediated gene expression signature in the lungs under resting and inflammatory conditions and opens new avenues of potential mechanisms through which IL-22 might mediate positive effects on tissue integrity in the context of acute (lung) inflammation. This study also demonstrates that IL-22 supplementation lowers bacterial dissemination out of the lungs in the context of influenza, an effect that may stem from reinforced lung barrier functions. Whether IL-22 may be used in combined therapy to control bacterial superinfection postinfluenza will be worth future study.
MATERIALS AND METHODS
Ethics statement.
All animal work conformed with the Lille Pasteur Institute's animal care and use guidelines and was approved by the regional investigational review board (Comité d'Ethique en Expérimentation Animale Nord Pas-de-Calais, reference AF 16/20090) and the French Ministry for Research (Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche, references CEAA75 and 00357.03).
Animals.
Eight- to 10-week-old male C57BL/6J mice were purchased from Janvier (Le Genest-St-Isle, France). Interleukin-22-deficient mice have been described previously (67). For IAV and S. pneumoniae infection, mice were maintained in a biosafety level 2 facility in the Animal Resource Center at the Lille Pasteur Institute (Lille, France).
Infection with IAV and S. pneumoniae.
For infection with IAV, 50 μl of phosphate-buffered saline (PBS) alone (mock) or containing 500 PFU of the H1N1 pandemic IAV strain (a kind gift from M Rosa-Calatrava, Centre International de Recherche en Infectiologie, Lyon, France) was intranasally (i.n.) administered to anesthetized mice. Superinfection was performed as follows. Mice were infected with IAV, and 7 days later, mice were i.n. inoculated with 1 × 103 CFU of S. pneumoniae serotype 1 (clinical isolate E1586). Viable bacteria in the lungs and spleen were counted 30 h after the S. pneumoniae challenge by plating serial 10-fold dilutions of lung or spleen homogenates onto blood agar plates. The plates were incubated at 37°C overnight, and CFU were counted 24 h later. Determination of viral load was performed by quantitative RT-PCR on lung RNA for viral matrix protein (M1), as described previously (68).
Treatment with IL-22.
Recombinant mouse fusion protein IL-22-Fc was from Genentech (South San Francisco, CA) (a kind gift from W. Ouyang) (53), and the isotype control (IgG1) was from Bio X Cell (West Lebanon, NH). For the functional analysis, 100 μg of IL-22-Fc or the isotype control was i.n. or intraperitoneally (i.p.) administered to anesthetized (noninfected) mice. For the microarray analysis, 5 μg of IL-22-Fc or isotype control was i.n. administered to noninfected mice or IAV-infected mice (7 days postinfection). To study the effects of IL-22 on bacterial superinfection, IAV-infected mice were i.n. treated with IL-22 (5 μg), and 16 h later they were infected with S. pneumoniae.
Assessment of the lung pathology.
For histopathologic examination, lungs were fixed by inflation and immersion in PBS–3.2% paraformaldehyde and embedded in paraffin. To evaluate airway inflammation, we subjected fixed lung slices (5-μm sections) to hematoxylin and eosin staining (68). Three evenly distributed sections per lung were microscopically evaluated by a certified pathologist. The dissemination and the nature of pathological alterations were scored using specified lung inflammation parameters. The lung histopathological score (pneumonia) was expressed as the sum of the following three parameters, graded on a scale of 0 (absent) to 4 (severe): (i) alveolitis, characterized by thickness of the alveolar wall, alveolar protein exudate, and inflammatory cells; (ii) bronchiolitis, characterized by bronchial necrosis, desquamation, and denudation; (iii) vascularitis, characterized by inflammatory cells in the media/intima and media fibrinoid necrosis.
Assessment of gene expression by quantitative RT-PCR.
Total RNAs from whole lungs and livers were extracted. The cDNAs were analyzed using quantitative RT-PCRs as described previously (51). Specific primers (listed in Table 5) were designed using Primer Express software (Applied Biosystems, Villebon sur Yvette, France). Data were normalized against expression of the gapdh gene and are expressed as a fold increase over the mean gene expression level in isotype-treated mice (68).
TABLE 5.
Quantitative RT-PCR primers used in this study
| Target | Forward primer | Reverse primer |
|---|---|---|
| Gapdh | 5′-GCAAAGTGGAGATTGTTGCCA-3′ | 5′-GCCTTGACTGTGCCGTTGA-3′ |
| Saa3 | 5′-GATGACTTTAGCAGCCCAGGC-3′ | 5′-GCTGGTCAAGGGTCTAGAGAC-3′ |
| Reg3b | 5′-GAATATACCCTCCGCACGCA-3′ | 5′-GGTCATGGAGCCCAATCCAA-3′ |
| C3 | 5′-GGCTGAAACACCTGATCGTGA-3′ | 5′-TGTTCGGTCTGGTCCAGGTAGT-3′ |
| S100a9 | 5′-GCAGCATAACCACCATCATCG-3′ | 5′-TGTGCTTCCACCATTTGTCTGA-3′ |
| S100a8 | 5′-CACCATGCCCTCTACAAGAATG-3′ | 5′-CACCATCGCAAGGAACTCC-3′ |
| Gdf5 | 5′-GAGTAACAGAGAGCCTGTGAAG-3′ | 5′-GCAGGAATCTTCAGAAGGTGA-3′ |
| Ptn | 5′-GCTGCCTTCCTGGCATTGAT-3′ | 5′-GCACACACTCCACTGCCATTCT-3′ |
| Bmp10 | 5′-TGACCCTTTGCTGGTTGTGT-3′ | 5′-ATCGGGCCCACTGAAGAAAG-3′ |
| Lgr5 | 5′-GTCGTCCTTCCCTGTGACTG-3′ | 5′-CTGTAAGGCTCGGTTCCCTG-3′ |
| Efna2 | 5′-TCCCTGACATTGCTGGTGAC-3′ | 5′-CCCCTGGATCTCCGGTATTC-3′ |
| Cldn24 | 5′-CTTCAGAACGGCCATGCAAT-3′ | 5′-CCATCTCGTTCAGCTCCAAGT-3′ |
| Pcdh15 | 5′-CGGCCAAGTTCACTACAGCC-3′ | 5′-TGCCACGACAACCAGTTCAT-3′ |
| Dsg1c | 5′-GAGTGCTCCCTAGCATTGGT-3′ | 5′-AATCGCCCCAAGCTTAAAATTC-3′ |
| Park2 | 5′-ACCCACAGAAAACCACAGCA-3′ | 5′-CCAAAGGTGCACCCTCATCT-3′ |
| IAV (M1) | 5′-AAGAACAATCCTGTCACCTCTGA-3′ | 5′-CAAAGCGTCTACGCTGCAGTCC-3′ |
Transcriptional analysis.
Lung transcriptional profiling was performed using Agilent's SurePrint G3 mouse gene expression microarray kit (version 2; 8 by 60,000 spots) (Agilent Technologies, Santa Clara, CA). A single-color design was used to provide two types of comparisons: (i)IgG1-treated mice versus IL-22-Fc-treated mice, and (ii) PBS-treated infected mice versus IgG1-treated infected mice versus IL-22-Fc-treated infected mice. Cy3-labeled cRNAs were prepared from 100 ng of total RNA using a one-color Low Input Quick Amp labeling kit (Agilent Technologies). Specific activities and cRNA yields were determined using a NanoDrop ND-1000 instrument (Thermo Fisher Scientific). For each sample, 600 ng of Cy3-labeled cRNA (specific activity of >9 pmol of Cy3/μg of cRNA) was fragmented at 60°C for 30 min. cRNA was hybridized to the microarrays for 17 h at 65°C in a rotating hybridization oven as previously described (69). After hybridization, microarrays were washed and then dried immediately. After being washed, the slides were scanned using a G2565CA scanner system (Agilent Technologies), with a resolution of 3 μm and a 20-bit dynamic range. The resulting TIFF images were analyzed with Feature Extraction software, version 10.7.3.1 (Agilent Technologies), using a GE1_107_Sep09 protocol. MIAME (minimum information about microarray experiment)-compliant data were deposited in ArrayExpress at EMBL. Identification of differentially expressed genes and functional investigations were done using GeneSpring software (version 14.8; Agilent Technologies). Differentially expressed genes were identified using a moderated t test, and a P value cutoff of 5% was applied. A fold change cutoff of >2 was then added to select the differentially expressed genes between the control and treated/infected conditions. Hierarchical clustering analysis was performed to analyze genes that were differentially expressed during infection and upon IL-22 treatment (similarity measure, Euclidean; linkage rules, Wards). For further analysis, data files were uploaded into the Ingenuity Pathways Analysis (IPA) software (Ingenuity Systems). Right-tailed Fisher's exact test was used to calculate a P value for the probability that each biological function and disease assigned to that data set is due to chance alone.
Analysis of pulmonary immune cells by flow cytometry.
Lung mononuclear cells from naive mice or IAV-infected mice, untreated or treated with IL-22-Fc, were labeled for dead cells. To identify immune cells, lung mononuclear cells were labeled with appropriate dilutions of conjugated antibodies exactly as described in Barthelemy et al. (57). The data were acquired on an LSR Fortessa cytometer (Becton Dickinson Biosciences, Rungis, France) running FACSDiva software and then analyzed with FlowJo software.
Measurement of pulmonary permeability.
Bronchoalveolar lavage (BAL) fluids were recovered from mock-treated and IAV-infected mice (7 days postinfection). Total protein was quantified in the BAL fluids by standard procedures (Bio-Rad Laboratories). Fluorescein isothiocyanate (FITC)-dextran (4,000 molecular weight; Sigma-Aldrich, St. Louis, MO) was dissolved in sterile saline and was introduced to the airspace via oropharyngeal aspiration (60 μl, 5 mg/kg) (7 days postinfection). After 1 h, mice were euthanized, and blood was collected by cardiac puncture. FITC-dextran in plasma was quantified using a Tecan (Männedorf, Switzerland) plate reader and a 485-nm filter.
Statistical analyses.
Results are expressed as the means ± standard deviations. A Mann-Whitney unpaired t test was used to compare two groups, unless otherwise specified. Comparisons of more than two groups were performed with a nonparametric Kruskal-Wallis one-way analysis of variance (ANOVA), followed by Dunn's posttest (using Prism software, version 6; GraphPad). The threshold for statistical significance was set to a P value of <0.05.
Accession number(s).
MIAME-compliant microarray data were deposited in the EMBL ArrayExpress database (https://www.ebi.ac.uk/arrayexpress/) under accession number E-MTAB-6044.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the Institut National de la Santé et de la Recherche Médicale, the Centre national de la Recherche Scientifique, the University of Lille, and the Pasteur Institute of Lille and Inserm Transfert (CoPoC Flu22). A.B. and V.S. were recipients of a doctoral fellowship from the Ministère de l'Education Nationale de la Recherche et Technique. C.F. is supported by the Institut National de la Santé et de la Recherche Médicale, R.L.G. is supported by the Institut National de la recherche Agronomique, and F.T. is supported by the Centre National de la Recherche Scientifique.
We thank the Animal Resource Center at the Lille Pasteur Institute and the BICeL flow cytometry core facility. We thank M. Moroldo (Centre de Ressources Biologiques pour la Génomique des Animaux Domestiques et d'Intérêt Economique, Institut National de la Recherche Agronomique, Jouy-en-Josas, France) for lung transcriptional profilings. We acknowledge J. C. Renauld and L. Dumoutier (Ludwig Institute, Bruxels, Belgium) for the gift of Il22−/− mice, W. Ouyang (Genentech, San Francisco, CA) for the gift of IL-22-Fc, and M. Rosa Calatrava (Centre International de Recherche en Infectiologie, Lyon, France) for the gift of H1N1p.
We declare that we have no competing financial, professional, or personal interests.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00706-17.
REFERENCES
- 1.Morens DM, Taubenberger JK, Fauci AS. 2008. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis 198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McCullers JA. 2006. Insights into the interaction between influenza virus and pneumococcus. Clin Microbiol Rev 19:571–582. doi: 10.1128/CMR.00058-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Snelgrove RJ, Godlee A, Hussell T. 2011. Airway immune homeostasis and implications for influenza-induced inflammation. Trends Immunol 32:328–334. doi: 10.1016/j.it.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 4.Pittet LA, Hall-Stoodley L, Rutkowski MR, Harmsen AG. 2010. Influenza virus infection decreases tracheal mucociliary velocity and clearance of Streptococcus pneumoniae. Am J Respir Cell Mol Biol 42:450–460. doi: 10.1165/rcmb.2007-0417OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li N, Ren A, Wang X, Fan X, Zhao Y, Gao GF, Cleary P, Wang B. 2015. Influenza viral neuraminidase primes bacterial coinfection through TGF-β-mediated expression of host cell receptors. Proc Natl Acad Sci U S A 112:238–243. doi: 10.1073/pnas.1414422112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Sluijs KF, van der Poll T, Lutter R, Juffermans NP, Schultz MJ. 2010. Bench-to-bedside review: bacterial pneumonia with influenza—pathogenesis and clinical implications. Crit Care 14:219. doi: 10.1186/cc8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Short KR, Kroeze EJBV, Fouchier RAM, Kuiken T. 2014. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis 14:57–69. doi: 10.1016/S1473-3099(13)70286-X. [DOI] [PubMed] [Google Scholar]
- 8.Metzger DW, Sun K. 2013. Immune dysfunction and bacterial coinfections following influenza. J Immunol 191:2047–2052. doi: 10.4049/jimmunol.1301152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dumoutier L, Renauld JC. 2002. Viral and cellular interleukin-10 (IL-10)-related cytokines: from structures to functions. Eur Cytokine Netw 13:5–15. [PubMed] [Google Scholar]
- 10.Sonnenberg GF, Fouser LA, Artis D. 2011. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol 12:383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- 11.Eidenschenk C, Rutz S, Liesenfeld O, Ouyang W. 2014. Role of IL-22 in microbial host defense. Curr Top Microbiol Immunol 380:213–236. doi: 10.1007/978-3-662-43492-5_10. [DOI] [PubMed] [Google Scholar]
- 12.Dudakov JA, Hanash AM, van den Brink MR. 2015. Interleukin-22: immunobiology and pathology. Annu Rev Immunol 33:747–785. doi: 10.1146/annurev-immunol-032414-112123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. 2004. IL-22 increases the innate immunity of tissues. Immunity 21:241–254. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 14.Eyerich S, Eyerich K, Cavani A, Schmidt-Weber C. 2010. IL-17 and IL-22: siblings, not twins. Trends Immunol 31:354–361. doi: 10.1016/j.it.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Witte E, Witte K, Warszawska K, Sabat R, Wolk K. 2010. Interleukin-22: a cytokine produced by T, NK and NKT cell subsets, with importance in the innate immune defense and tissue protection. Cytokine Growth Factor Rev 21:365–379. doi: 10.1016/j.cytogfr.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 16.Wolk K, Witte E, Witte K, Warszawska K, Sabat R. 2010. Biology of interleukin-22. Semin Immunopathol 32:17–31. doi: 10.1007/s00281-009-0188-x. [DOI] [PubMed] [Google Scholar]
- 17.Sabat R, Ouyang W, Wolk K. 2014. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat Rev Drug Discov 13:21–38. doi: 10.1038/nrd4176. [DOI] [PubMed] [Google Scholar]
- 18.Mühl H, Scheiermann P, Bachmann M, Härdle L, Heinrichs A, Pfeilschifter J. 2013. IL-22 in tissue-protective therapy. Br J Pharmacol 169:761–771. doi: 10.1111/bph.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W. 2008. Interleukin-22 Mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 20.Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirch S, Weigmann B, Wirtz S, Ouyang W, Neurath MF, Beker C. 2009. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 206:1465–1472. doi: 10.1084/jem.20082683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. 2008. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest 118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, Shibata N, Grunberg S, Sinha R, Zahm AM, Tardif MR, Sathaliyawala T, Kubota M, Farber DL, Collman RG, Shaked A, Fouser LA, Weiner DB, Tessier PA, Friedman JR, Kiyono H, Bushman FD, Chang KM, Artis D. 2012. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 336:1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Basu R, O'Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, Hatton RD, Weaver CT. 2012. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 37:1061–1075. doi: 10.1016/j.immuni.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lindemans CA, Calafiore M, Mertelsmann AM, O'Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G, Ivanov JA, Fu YY, Takashima S, Hua G, Martin ML, O'Rourke KP, Lo YH, Mokry M, Romara-Hernandez M, Cupedo T, Low L, Nieuwenhuis EE, Shroyer NF, Liu C, Kolesnick R, van den Brink MRM, Hanash AM. 2015. Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528:560–564. doi: 10.1038/nature16460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hasegawa M, Yada S, Liu MZ, Kamada N, Muñoz-Planillo R, Do N, Núñez G, Inohara N. 2014. Interleukin-22 regulates the complement system to promote resistance against pathobionts after pathogen-induced intestinal damage. Immunity 41:620–632. doi: 10.1016/j.immuni.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Simonian PL, Wehrmann F, Roark CL, Born WK, O'Brien RL, Fontenot AP. 2010. γδ T cells protect against lung fibrosis via IL-22. J Exp Med 207:2239–2253. doi: 10.1084/jem.20100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoegl S, Bachmann M, Scheiermann P, Goren I, Hofstetter C, Pfeilschifter J, Zwissler B, Muhl H. 2011. Protective properties of inhaled IL-22 in a model of ventilator-induced lung injury. Am J Respir Cell Mol Biol 44:369–376. doi: 10.1165/rcmb.2009-0440OC. [DOI] [PubMed] [Google Scholar]
- 28.Liang M, Wang J, Chu H, Zhu X, He H, Liu Q, Qiu J, Zhou X, Guan M, Xue Y, Chen X, Zhou X. 2013. Interleukin-22 inhibits bleomycin-induced pulmonary fibrosis. Mediators Inflamm 2013:209179. doi: 10.1155/2013/209179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Besnard AG, Sabat R, Dumoutier L, Renauld JC, Willart M, Lambrecht B, Teixeira MM, Charron S, Fick L, Erard F, Warszawska K, Wolk K, Quesniaux V, Ryffel B, Togbe D. 2011. Dual role of IL-22 in allergic airway inflammation and its cross-talk with IL-17A. Am J Resp Crit Care Med 183:1153–1163. doi: 10.1164/rccm.201008-1383OC. [DOI] [PubMed] [Google Scholar]
- 30.Nakagome K, Imamura M, Kawahata K, Harada H, Okunishi K, Matsumoto T, Sasaki O, Tanaka R, Kano MR, Chang H, Hanawa H, Miyazaki J, Yamamoto K, Dohi M. 2011. High expression of IL-22 suppresses antigen-induced immune responses and eosinophilic airway inflammation via an IL-10-associated mechanism. J Immunol 187:5077–5089. doi: 10.4049/jimmunol.1001560. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi K, Hirose K, Kawashima S, Niwa Y, Wakashin H, Iwata A, Tokoyoda K, Renauld JC, Iwamoto I, Nakayama T, Nakajima H. 2011. IL-22 attenuates IL-25 production by lung epithelial cells and inhibits antigen-induced eosinophilic airway inflammation. J Allergy Clin Immunol 128:1067–1076. doi: 10.1016/j.jaci.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 32.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK. 2008. IL-22 mediates mucosal host defense against gram-negative bacterial pneumonia. Nat Med 14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, Khader SA, Dubin PJ, Enelow RI, Kolls JK, Alcorn JF. 2011. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol 186:1666–1674. doi: 10.4049/jimmunol.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu X, Weiss ID, Zhang HH, Singh SP, Wynn TA, Wilson MS, Farber JM. 2014. Conventional NK cells can produce IL-22 and promote host defense in Klebsiella pneumoniae pneumonia. J Immunol 192:1778–1786. doi: 10.4049/jimmunol.1300039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng Y, Gao X, Yang J, Shekhar S, Wang S, Fan Y, Zhao W, Yang X. 2014. Interleukin-22 promotes T helper 1 (Th1)/Th17 immunity in chlamydial lung infection. Mol Med 20:109–119. doi: 10.2119/molmed.2013.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Van Maele L, Carnoy C, Cayet D, Ivanov S, Porte R, Deruy E, Chabalgoity JA, Renauld JC, Eberl G, Benecke AG, Trottein FF, Faveeuw C, Sirard JC. 2014. Activation of type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J Infect Dis 210:493–503. doi: 10.1093/infdis/jiu106. [DOI] [PubMed] [Google Scholar]
- 37.Gauguet S, D'Ortona S, Ahnger-Pier K, Duan B, Surana NK, Lu R, Cywes-Bentley C, Gadjeva M, Shan Q, Priebe GP, Pier GB. 2015. Intestinal microbiota of mice influences resistance to Staphylococcus aureus pneumonia. Infect Immun 83:4003–4014. doi: 10.1128/IAI.00037-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pichavant M, Sharan R, Le Rouzic O, Olivier C, Hennegrave F, Rémy G, Pérez-Cruz M, Koné B, Gosset P, Just N, Gosset P. 2015. IL-22 Defect during Streptococcus pneumoniae infection triggers exacerbation of chronic obstructive pulmonary disease. EBioMedicine 2:1686–1696. doi: 10.1016/j.ebiom.2015.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sharan R, Perez-Cruz M, Kervoaze G, Gosset P, Weynants V, Godfroid F, Hermand P, Trottein F, Pichavant M, Gosset P. 2017. Interleukin-22 protects against non-typeable Haemophilus Influenzae Infection: Alteration during chronic obstructive pulmonary disease. Mucosal Immunol 10:139–149. doi: 10.1038/mi.2016.40. [DOI] [PubMed] [Google Scholar]
- 40.Mulcahy ME, Leech JM, Renauld J-C, Hg Mills K, McLoughlin RM. 2016. Interleukin-22 regulates antimicrobial peptide expression and keratinocyte differentiation to control Staphylococcus aureus colonization of the nasal Mucosa. Mucosal Immunol 9:1429–1441. doi: 10.1038/mi.2016.24. [DOI] [PubMed] [Google Scholar]
- 41.Trevejo-Nunez G, Elsegeiny W, Conboy P, Chen K, Kolls JK. 2016. Critical role of IL-22/IL22-RA1 signaling in pneumococcal pneumonia. J Immunol 197:1877–1883. doi: 10.4049/jimmunol.1600528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Treerat P, Prince O, Cruz-Lagunas A, Muñoz-Torrico M, Salazar-Lezama MA, Selman M, Fallert-Junecko B, Reinhardt TA, Alcorn JF, Kaushal D, Zuñiga J, Rangel-Moreno J, Kolls JK, Khader SA. 2017. Novel role for IL-22 in protection during chronic Mycobacterium tuberculosis HN878 infection. Mucosal Immunol 10:1069–1081. doi: 10.1038/mi.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broquet A, Jacqueline C, Davieau M, Besbes A, Roquilly A, Martin J, Caillon J, Dumoutier L, Renauld JC, Heslan M, Josien R, Asehnoune K. 2017. Interleukin-22 level is negatively correlated with neutrophil recruitment in the lungs in a Pseudomonas aeruginosa pneumonia model. Sci Rep 7:11010. doi: 10.1038/s41598-017-11518-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guillon A, Brea D, Morello E, Tang A, Jouan Y, Ramphal R, Korkmaz B, Perez-Cruz M, Trottein F, O'Callaghan RJ, Gosset P, Si-Tahar M. 2017. Pseudomonas aeruginosa proteolytically alters the interleukin 22-dependent lung mucosal defense. Virulence 8:810–820. doi: 10.1080/21505594.2016.1253658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gessner MA, Werner JL, Lilly LM, Nelson MP, Metz AE, Dunaway CW, Chan YR, Ouyang W, Brown GD, Weaver CT, Steele C. 2012. Dectin-1-dependent interleukin-22 contributes to early innate lung defense against Aspergillus fumigatus. Infect Immun 80:410–417. doi: 10.1128/IAI.05939-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wozniak KL, Hole CR, Yano J, Fidel PL, Wormley FL. 2014. Characterization of IL-22 and antimicrobial peptide production in mice protected against pulmonary Cryptococcus neoformans infection. Microbiology 160:1440–1452. doi: 10.1099/mic.0.073445-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gimeno Brias S, Stack G, Stacey MA, Redwood AJ, Humphreys IR. 2016. The role of IL-22 in viral infections: paradigms and paradoxes. Front Immunol 7:211–219. doi: 10.3389/fimmu.2016.00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo H, Topham DJ. 2010. Interleukin-22 (IL-22) production by pulmonary natural killer cells and the potential role of IL-22 during primary influenza virus infection. J Virol 84:7750–7759. doi: 10.1128/JVI.00187-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paget C, Ivanov S, Fontaine J, Renneson J, Blanc F, Pichavant M, Dumoutier L, Ryffel B, Renauld JC, Gosset P, Gosset P, Si-Tahar M, Faveeuw C, Trottein F. 2012. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: potential role in protection against lung epithelial damages. J Biol Chem 287:8816–8829. doi: 10.1074/jbc.M111.304758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar P, Thakar MS, Ouyang W, Malarkannan S. 2013. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol 6:69–82. doi: 10.1038/mi.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ivanov S, Renneson J, Fontaine J, Barthelemy A, Paget C, Macho-Fernandez E, Blanc F, De Trez C, Van Maele L, Dumoutier L, Huerre MR, Eberl G, Si-Tahar M, Gosset P., Renauld JC, Sirard JC, Faveeuw C, Trottein F. 2013. Interleukin-22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. J Virol 87:6911–6924. doi: 10.1128/JVI.02943-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pociask DA, Scheller EV, Mandalapu S, McHugh KJ, Enelow RI, Fattman CL, Kolls JK, Alcorn JF. 2013. IL-22 is essential for lung epithelial repair following influenza infection. Am J Pathol 182:1286–1296. doi: 10.1016/j.ajpath.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio L, Eidenschenk C, Zhang J, Lesch J, Lee WP, Ross J, Diehl L, van Bruggen N, Koluman G, Ouyang W. 2014. Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 514:237–241. doi: 10.1038/nature13564. [DOI] [PubMed] [Google Scholar]
- 54.Herold S, Steinmueller M, von Wulffen W, Cakarova L, Pinto R, Pleschka S, Mack M, Kuziel WA, Corazza N, Brunner T, Seeger W, Lohmeyer J. 2008. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med 205:3065–3077. doi: 10.1084/jem.20080201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin KL, Sweeney S, Kang BD, Ramsburg E, Gunn MD. 2011. CCR2-antagonist prophylaxis reduces pulmonary immune pathology and markedly improves survival during influenza infection. J Immunol 186:508–515. doi: 10.4049/jimmunol.1001002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ellis GT, Davidson D, Crotta S, Branzk N, Papayannopoulos V, Wack A. 2015. TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection. EMBO Rep 16:1203–1218. doi: 10.15252/embr.201540473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barthelemy A, Ivanov S, Fontaine J, Soulard D, Bouabe H, Paget C, Faveeuw C, Trottein F. 2017. Influenza A virus-induced release of interleukin-10 inhibits the anti-microbial activities of invariant natural killer T cells during invasive pneumococcal superinfection. Mucosal Immunol 10:460–469. doi: 10.1038/mi.2016.49. [DOI] [PubMed] [Google Scholar]
- 58.Ghoneim HE, Thomas PG, McCullers JA. 2013. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J Immunol 191:1250–1259. doi: 10.4049/jimmunol.1300014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koluman G, Wu X, Lee WP, Hackney JA, Solorio-Zavala J, Gangham V, Danilenko DM, Arora P, Wang X, Ouyang W. 2017. IL-22R ligands IL-20, IL-22, and IL-24 promote wound healing in diabetic db/db mice. PLoS One 12:e0170639. doi: 10.1371/journal.pone.0170639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He X, Li H, Chen Y, Chen A, Shan K, Chen J, Zhao H, Zhang X, Cai T. 2016. The effects of IL-22 on the inflammatory mediator production, proliferation, and barrier function of HUVECs. Inflammation 39:1099–1107. doi: 10.1007/s10753-016-0341-3. [DOI] [PubMed] [Google Scholar]
- 61.Wu Z, Hu Z, Cai X, Ren W, Dai F, Liu H, Chang J, Li B. 2017. Interleukin 22 attenuated angiotensin II induced acute lung injury through inhibiting the apoptosis of pulmonary microvascular endothelial cells. Sci Rep 7:2210–2221. doi: 10.1038/s41598-017-02056-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guillon A, Jouan Y, Brea D, Gueugnon F, Dalloneau E, Baranek T, Henry C, Morello E, Renauld JC, Pichavant M, Gosset P, Courty Y, Diot P, Si-Tahar M. 2015. Neutrophil proteases alter the interleukin-22-receptor-dependent lung antimicrobial defence. Eur Respir J 46:771–782. doi: 10.1183/09031936.00215114. [DOI] [PubMed] [Google Scholar]
- 63.Sun K, Metzger DW. 2014. Influenza infection suppresses NADPH oxidase-dependent phagocytic bacterial clearance and enhances susceptibility to secondary methicillin-resistant Staphylococcus aureus infection. J Immunol 192:3301–3307. doi: 10.4049/jimmunol.1303049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sun K, Metzger DW. 2008. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med 14:558–564. doi: 10.1038/nm1765. [DOI] [PubMed] [Google Scholar]
- 65.McNamee LA, Harmsen AG. 2006. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun 74:6707–6721. doi: 10.1128/IAI.00789-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, Belperio JA, Cheng G, Deng JC. 2009. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest 119:1910–1920. doi: 10.1172/JCI35412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Robollo A, Buch T, Heppner FL, Renauld JC, Becher B. 2007. IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol 179:8098–8104. doi: 10.4049/jimmunol.179.12.8098. [DOI] [PubMed] [Google Scholar]
- 68.Paget C, Ivanov S, Fontaine J, Blanc F, Pichavant M, Renneson J, Bialecki E, Pothlichet J, Vendeville C, Barba-Spaeth G, Huerre MR, Faveeuw C, Si-Tahar M, Trottein FF. 2011. Potential role of invariant NKT cells in the control of pulmonary inflammation and CD8+ T cell response during acute influenza A virus H3N2 pneumonia. J Immunol 186:5590–5602. doi: 10.4049/jimmunol.1002348. [DOI] [PubMed] [Google Scholar]
- 69.Leymarie O, Jouvion G, Hervé PL, Chevalier C, Lorin V, Lecardonnel J, Da Costa B, Delmas B, Escriou N, Le Goffic R. 2013. Kinetic characterization of PB1-F2-mediated immunopathology during highly pathogenic avian H5N1 influenza virus infection. PLoS One 8:e57894. doi: 10.1371/journal.pone.0057894. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.