

Abstract
The spindle assembly checkpoint (SAC) is a conserved mitotic regulator that preserves genome stability by monitoring kinetochore–microtubule attachments and blocking anaphase onset until chromosome biorientation is achieved. Despite its central role in maintaining mitotic fidelity, the ability of the SAC to delay mitotic exit in the presence of kinetochore–microtubule attachment defects (SAC “strength”) appears to vary widely. How different cellular aspects drive this variation remains largely unknown. Here we show that SAC strength is correlated with cell fate during development of Caenorhabditis elegans embryos, with germline-fated cells experiencing longer mitotic delays upon spindle perturbation than somatic cells. These differences are entirely dependent on an intact checkpoint and only partially attributable to differences in cell size. In two-cell embryos, cell size accounts for half of the difference in SAC strength between the larger somatic AB and the smaller germline P1 blastomeres. The remaining difference requires asymmetric cytoplasmic partitioning downstream of PAR polarity proteins, suggesting that checkpoint-regulating factors are distributed asymmetrically during early germ cell divisions. Our results indicate that SAC activity is linked to cell fate and reveal a hitherto unknown interaction between asymmetric cell division and the SAC.
INTRODUCTION
The fidelity of mitosis depends upon equal partitioning of the replicated genome between daughter cells. During mitosis, sister chromatid pairs connect to the mitotic spindle via kinetochore–microtubule attachments. Stable attachment of sister chromatids to opposite spindle poles (biorientation) ensures that, upon chromatid separation, one copy segregates to each daughter cell. Attachment of sister chromatids to the mitotic spindle is an inherently stochastic process of variable duration (Musacchio and Salmon, 2007). Thus, to safeguard against chromosome segregation errors, the spindle assembly checkpoint (SAC) monitors kinetochore–microtubule attachments and prevents anaphase onset until stable biorientation has been achieved. Weakening of the SAC can lead to aneuploidy and has been associated with tumor development in both model systems and human cancers (Cahill et al., 1998; Michel et al., 2001). Conversely, a major class of anti-mitotic chemotherapeutics (spindle poisons) depends on robust SAC activity to disrupt mitosis in cancer cells (Weaver and Cleveland, 2005).
Interfering with the formation of stable kinetochore–microtubule attachments prevents cells from satisfying the SAC and leads to prolonged mitotic delays. However, the SAC cannot block anaphase indefinitely, and most cells, even in the complete absence of spindle microtubules, will eventually exit mitosis (Rieder and Maiato, 2004). The duration of mitotic arrest under conditions that preclude satisfaction of the SAC is often used as an indication of the “strength” of the SAC. Variation in SAC strength has been observed between cancer cell lines from different tissues (Gascoigne and Taylor, 2008; Shi et al., 2008), cells from different organisms (Rieder and Maiato, 2004), and cells at different developmental stages, with early embryonic cells generally displaying a weak checkpoint (Sluder, 1979; Hara et al., 1980; Clute and Masui, 1995, 1997; Zhang et al., 2015). Although variation in the strength of the SAC is seemingly widespread, how different cellular attributes influence SAC activity remains largely unknown.
The core SAC module consists of the proteins Bub1, Bub3, BubR1, Mad1, and Mad2, which are recruited to unattached kinetochores in a stepwise manner to promote assembly of the mitotic checkpoint complex (MCC), which acts as a diffusible “wait-anaphase” signal (Lara-Gonzalez et al., 2012; London and Biggins, 2014). The MCC consists of Mad2, BubR1, and Bub3 bound to the anaphase-promoting complex/cyclosome (APC/C) cofactor Cdc20 (Sudakin et al., 2001). The MCC binds to the APC/C (Chao et al., 2012; Primorac and Musacchio, 2013), blocking degradation of APC/C substrates, namely, cyclin B (Clute and Pines, 1999) and securin (Nasmyth et al., 2000), and maintaining a mitotic state. Once all kinetochores are stably bound by microtubules, disassembly of the MCC (Mansfeld et al., 2011; Westhorpe et al., 2011), dephosphorylation of SAC proteins (Lesage et al., 2011), and stripping of Mad1 and Mad2 from all kinetochores (Howell et al., 2001) effectively silences the SAC and permits anaphase onset and mitotic exit.
Mitotic exit, despite failure to satisfy the SAC, occurs when activity of cyclin B/Cdk1 falls below the threshold necessary to maintain a mitotic state (Rieder and Maiato, 2004). In mammalian cells, this mitotic “slippage” occurs because the active cytoplasmic pool of MCC is not sufficient to completely inhibit the APC/C, and the progressive degradation of cyclin B eventually enables mitotic exit (Brito and Rieder, 2006). The steady-state concentration of the MCC pool is influenced by the rate of new, kinetochore-catalyzed MCC generation and disassembly of existing cytoplasmic MCC (Musacchio and Ciliberto, 2012; Joglekar, 2016), the former being intimately related to the severity of the spindle defect (Brito et al., 2008; Collin et al., 2013; Dick and Gerlich, 2013). Thus, variation in the strength of the SAC, as assayed by the duration of mitotic delay following spindle perturbation, may be linked to differences in MCC production, activity, or stability.
One factor that contributes to SAC strength in certain circumstances is cell size. In vitro experiments using Xenopus laevis egg extracts suggested that an increased nuclear to cytoplasmic ratio, as would be found in smaller cells, could increase SAC activity (Minshull et al., 1994). Recent work in Caenorhabditis elegans embryos and mouse oocytes has shown that the strength of the SAC indeed scales with cell size, with smaller cells exhibiting a stronger SAC (Galli and Morgan, 2016; Kyogoku and Kitajima, 2017; Lane and Jones, 2017). However, in other organisms, the SAC remains inactive until the midblastula transition and acquisition of SAC activity is neither accelerated by decreasing cell volume (X. laevis; Clute and Masui, 1995, 1997) nor delayed by increasing cell volume (Danio rerio; Zhang et al., 2015), indicating that SAC activity can also be developmentally regulated independently of changes in cell volume. How this developmental regulation of the SAC is achieved is unknown.
We have reported that the adult germline stem cells (GSCs) of C. elegans exhibit a stronger SAC relative to early embryonic cells (Gerhold et al., 2015) providing a tractable model in which to examine variability in SAC strength. Here we use an inducible monopolar spindle assay to investigate the developmental origins of enhanced germline SAC activity. In agreement with Galli and Morgan (2016), we find that the duration of SAC-dependent mitotic delays increases as cell size decreases during embryogenesis. However, the relationship between cell size and SAC activity is strongly influenced by cell fate, with cells in the germline lineage displaying a stronger SAC relative to their cell size than cells with a somatic fate. At the two-cell stage, we find that differential SAC activity in the somatic AB vs. germline P1 blastomere requires asymmetric cytoplasmic partitioning during division of the zygote, P0, downstream of the highly conserved PAR protein polarity regulators. Our results are consistent with a model in which a determinant or determinants of checkpoint strength are asymmetrically distributed between somatic and germline cells.
RESULTS
Germline blastomeres have longer mitotic delays upon spindle perturbation
C. elegans GSCs are derived from a single founder cell (P4), which is specified during embryogenesis by a series of asymmetric cell divisions (Deppe et al., 1978). As the C. elegans embryonic lineage is invariant and fully mapped (Figure 1A; Sulston et al., 1983; Bao et al., 2008), we asked whether the embryonic precursors to GSCs, cells in the germline P lineage, also exhibited an enhanced SAC, by comparing the duration of mitosis following spindle perturbation between each of the founding cell lineages. As C. elegans embryos are largely refractory to treatment with small molecule spindle poisons without physical or genetic manipulations to permeabilize the egg shell (Strome and Wood, 1983; Carvalho et al., 2011), we opted for a genetic method to induce spindle perturbations with temporal control. We used a recessive temperature-sensitive allele of the polo-related kinase zyg-1, which prevents centriole duplication and produces cells with monopolar spindles (zyg-1[or297], hereafter zyg-1[ts]; O’Connell et al., 2001; O’Rourke et al., 2011), combined with fluorescent markers to visualize the mitotic spindle (β-tubulin fused to green fluorescent protein [GFP], hereafter β-tubulin::GFP) and/or the nucleus/chromatin (histone H2B fused to mCherry, hereafter H2B::mCH). This allowed us to disrupt spindle formation with a simple temperature shift and to follow mitotic progression by monitoring changes in nuclear and chromosome morphology (Figure 1, B and C). zyg-1(ts) is fast-acting (O’Rourke et al., 2011) and readily permitted the induction of monopolar spindles in all embryonic cells up to the 16-cell stage. Following nuclear envelope breakdown (NEBD), condensed chromosomes spread radially around the single spindle pole and remained dispersed until the start of decondensation (Figure 1C and Supplemental Video 1).
FIGURE 1:

Germline blastomeres have longer mitotic delays than somatic cells when spindle formation is perturbed. (A) Schematic depiction of the first four rounds of division in C. elegans embryogenesis, with cells color-coded as in D, E, H, and I. The germline (P) lineage is in red. (B, C) Representative cropped time-lapse images showing a bipolar (B) and monopolar (C) mitosis in two P1 blastomeres expressing H2B::mCH (cyan) and β-tubulin::GFP (red). (D, E) The duration of bipolar (D) and monopolar (E) mitoses (NEBD to DECOND) in cells from 2- to 16-cell stage embryos, grouped by lineage and stage. (F, G) Representative cropped time-lapse images showing the duration of mitosis in two P2 cells from perm-1(RNAi) permeabilized embryos expressing H2B::mCH (cyan) and β-tubulin::GFP (red) and treated with DMSO (F) or 33 μM nocodazole (G). (H, I) The duration of mitosis (NEBD to DECOND) in cells from 1- to 16-cell stage embryos, grouped by lineage and stage, and treated with DMSO (H) or 33 μM nocodazole (I). For B, C, F and G, frames corresponding to NEBD and DECOND are boxed in magenta, time is expressed in minutes relative to NEBD, and scale bars = 5 μm. For D, E, H, and I, cells are color-coded as in A. Black bars show the mean, with error bars representing the 95% confidence interval for the mean. ** = 0.001 < p < 0.01; *** = p < 0.001 by an Anova1 with Tukey-Kramer post hoc test. See Supplemental Table S2 for summary statistics.
Movie S1.
zyg-1(ts)-induced monopolar spindles in 2- and 4-cell stage embryos.
As reportedly previously, the duration of mitosis in cells without spindle perturbations was invariant across embryonic stages and cell lineages (Figure 1D; Arata et al., 2014; Galli and Morgan, 2016). In cells with monopolar spindles, however, cells in the germline P lineage remained in mitosis for significantly longer than both their immediate somatic siblings and somatic cells at later embryonic stages (Figure 1E). A similar result was obtained using a temperature-sensitive allele of the microtubule subunit β-tubulin (tbb-2[or362]; Ellis et al., 2004), which disrupts microtubule dynamics and/or stability (Supplemental Figure S1, A and B, and Supplemental Video 2).
Movie S2.
tbb-2(ts)-induced spindle defects in 2- and 4-cell stage embryos.
Cells with monopolar spindles fail to satisfy the SAC due to the persistence of monotelic and syntelic kinetochore–microtubule attachments (Kapoor et al., 2000). The presence of these attachments, albeit erroneous, may reduce the proportion of checkpoint-signaling kinetochores, thereby weakening the SAC response (Collin et al., 2013). Variation in the attachment error correction pathway could further modulate the SAC under these conditions (Krenn and Musacchio, 2015). In addition, a microtubule-dependent checkpoint silencing mechanism functions in C. elegans embryos (Espeut et al., 2012). Thus, longer monopolar mitoses in germline cells could be related to differences in kinetochore–microtubule attachment or in microtubule-dependent checkpoint silencing, rather than to increases in the strength of the SAC per se. To test this possibility, we measured the length of mitotic delays in 1- to 16-cell stage, permeabilized embryos (Carvalho et al., 2011), in which spindle microtubules were largely eliminated by treatment with the microtubule destabilizing agent nocodazole (Figure 1, F and G, and Supplemental Videos 3 and 4). In control, dimethyl sulfoxide (DMSO)–treated embryos the duration of mitosis was constant across embryonic stages and cell lineages (Figure 1H), suggesting that, despite permeabilization, the majority of cell divisions occur normally. In nocodazole-treated embryos, we observed 3- to 10-fold mitotic delays, with cells in the germline lineage delaying for significantly longer than somatic cells at the same and at later embryonic stages (Figure 1I). As kinetochore–microtubule attachments are largely absent under these conditions and therefore unlikely to contribute to the duration of mitotic delays, these results suggest that SAC strength itself is enhanced in germline-fated cells.
Movie S3.
A P2 cell from a permeabilized embryo treated with DMSO.
Movie S4.
A P2 cell from a permeabilized embryo treated with 33μM Nocodazole.
The difference in mitotic delay between germline and somatic cells is not solely due to cell size
Specification of the germline is achieved via a series of asymmetric cell divisions, such that the germline blastomere is always smaller than its immediate somatic sibling (Deppe et al., 1978). Thus, the increased duration of monopolar mitoses in germline blastomeres could be related to their relatively small size. To address this possibility we considered cell size in two ways. We estimated the average volume for each cell in the different lineages relative to the volume of the founding P0 blastomere by measuring the position of the spindle midpoint along the division axis in all cells from one- to eight-cell stage embryos (Figure 2, A and B). In both lineages, mitotic delay increased as cell volume decreased (Figure 2C; n = 44, r = −0.62, p = 5.85 × 10−6 for the AB lineage; n = 22, r = −0.72, p = 1.36 × 10−4 for the P lineage). However, the relationship between cell volume and the duration of monopolar mitoses differed significantly between the two lineages (AB vs. P regression slope: p = 0.028; y-intercept: p = 0), with germline cells displaying longer mitotic delays relative to their cell volume than somatic AB cells (see also Supplemental Figure S3A). To approximate the size of monopolar cells themselves, we used the presence of H2B::mCH in our zyg-1(ts) strain to measure nuclear area just before NEBD (Figure 2, D and E). Nuclear area scales with cell size in many organisms including C. elegans (Figure 2F; Hara and Kimura, 2009; Edens et al., 2013) and can be used as a proxy for cell volume. Although the duration of monopolar mitoses negatively correlated with nuclear area in both lineages, germline cells exhibited longer mitotic delays than somatic AB lineage cells with similar nuclear areas (Figure 2G; n = 22, r = −0.73, p = 9.89 × 10−5 and n = 40, r = −0.61, p = 2.82 × 10−5, respectively), suggesting that, between comparably sized cells, the SAC is stronger in germline cells.
FIGURE 2:
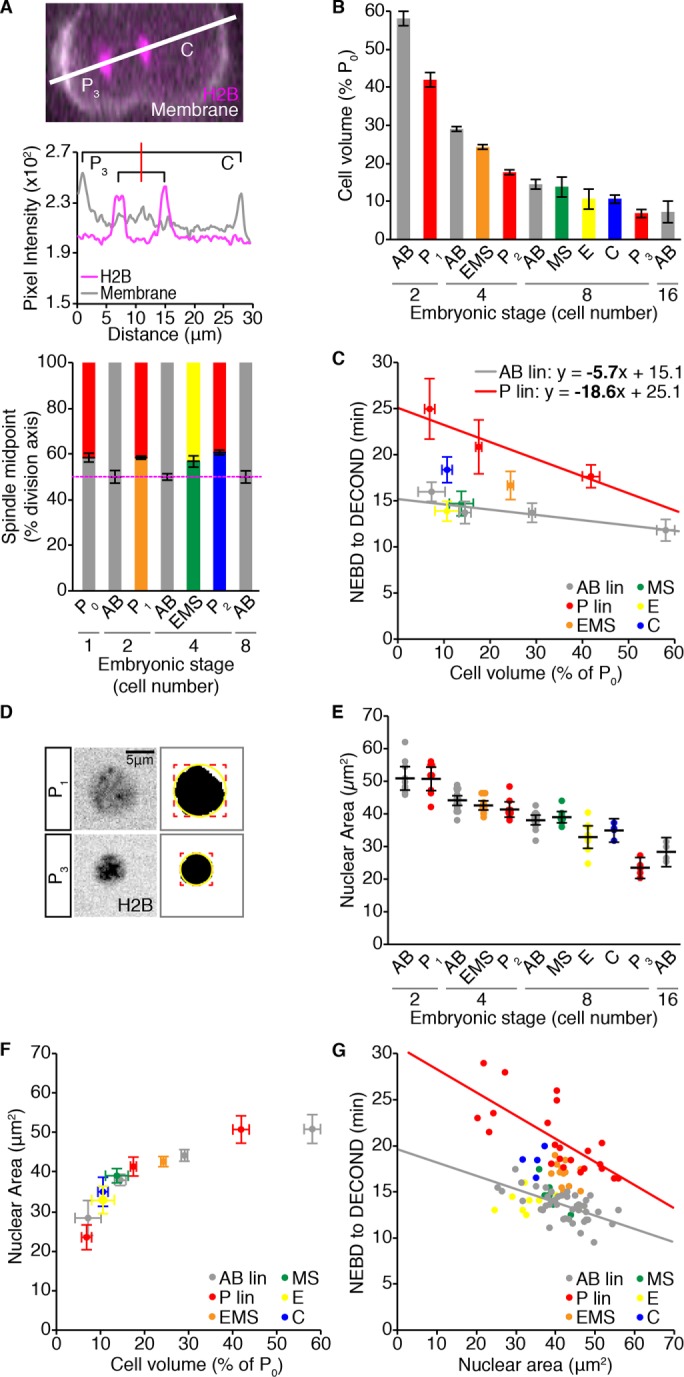
Differences in cell size do not account for the difference in duration of monopolar mitoses between germline and somatic cells during embryonic development. (A) Representative image of a dividing P2 cell expressing the cell membrane marker mNeonGreen::PH (mNG, white) and H2B::mCH (magenta). White line shows the axis of cell division. P3 is to the left and C is to the right. Pixel intensity values for each channel along this line are shown below. The red line indicates the spindle midpoint. Displacement of the spindle midpoint along the axis of division was used to calculate the average symmetry or asymmetry of division for the listed cells, which is graphed below. Error bars show the SD of the mean. (B) Volume estimates for the listed cells, represented as a percentage of the starting embryonic volume (P0) and calculated from the spindle displacement measurements shown in A. Error bars show the SD of the mean. (C) Mean cell volume vs. mean duration of monopolar mitoses for cells from 2- to 16-cell stage embryos. Monopolar mitosis duration measurements are reproduced from Figure 1E. Horizontal error bars show SD. Vertical error bars show the 95% confidence interval for the mean. Linear least -squares regression models for germline P (red; n = 22, r = −0.72, p = 1.36 × 10−4) and somatic AB (gray; n = 44, r = −0.62, p = 5.85 × 10−6) lineages are shown. Statistically different regression coefficients are written in bold and were compared using a nonparametric bootstrap (p = 0.028 for slope). (D) Sum projections through the center of an H2B::mCH-marked P1 (top) and P3 (bottom) nucleus 1 min before NEBD, with the corresponding segmented image to the right. A bounding box (red) was fit to the segmented nucleus and used to calculate the radius of a circle (yellow) that approximates nuclear area. Scale bar = 5 μm. (E) Pre-NEBD nuclear area measurements for cells from 2- to 16-cell stage embryos, for which the duration of monopolar mitoses was measured in Figure 1E. Black bars show the mean. Error bars represent the 95% confidence interval for the mean. (F) The scaling relationship between mean nuclear area and mean cell volume. Horizontal error bars show SD. Vertical error bars show the 95% confidence interval for the mean. (G) Nuclear area vs. the duration of monopolar mitoses. Lines show the linear least-squares regression fit for germline P lineage cells (red; n = 22, r = −0.73, p = 9.89 × 10−5) vs. somatic AB lineage cells (gray; n = 40, r = −0.61, p = 2.82 × 10−5). (C) For B and D, p values for Pearson’s coefficient (r) were determined using Student’s t distribution. For all panels, cells are color-coded as in Figure 1A. See Supplemental Table S2 for summary statistics.
Both cell size and lineage-specific differences in the duration of monopolar mitoses are checkpoint dependent
The increased duration of monopolar mitoses in germline blastomeres could be due to a stronger SAC; alternatively, factors downstream of checkpoint regulation could contribute to a delayed mitotic exit in these cells. To discriminate between these possibilities, we examined the duration of monopolar mitoses in the germline P and somatic AB lineages when checkpoint activity was eliminated using RNA-mediated interference (RNAi) depletion of the C. elegans orthologue of Mad1, mdf-1(RNAi) (Kitagawa and Rose, 1999), or a null allele of the C. elegans orthologue of Mad2, mdf-2(tm2190) (Tarailo-Graovac et al., 2010; Consortium, 2012). Mad1 and Mad2 are essential components of the core SAC module and absolutely required for checkpoint activity (Lara-Gonzalez et al., 2012; London and Biggins, 2014). In mdf-1(RNAi) and mdf-2(tm2190) embryos, cells with monopolar spindles rapidly exited mitosis, with chromosome decondensation evident within 4 min of NEBD (Figure 3A and Supplemental Video 5). Plotting the duration of monopolar mitoses in the germline P and somatic AB lineages relative to cell size (as approximated by nuclear area) revealed that all cells, irrespective of their size or lineage, exited mitosis with the same timing (Figure 3, B and C). The average duration of checkpoint-deficient monopolar mitoses was comparable to that of normal bipolar divisions (3.3 ± 0.5 vs. 3.4 ± 0.3 min [mean ± SD], respectively). Thus, monopolar spindle-induced mitotic delays are entirely checkpoint dependent and differences in the duration of these delays are likely to reflect differences in the strength of the SAC rather than downstream factors.
FIGURE 3:
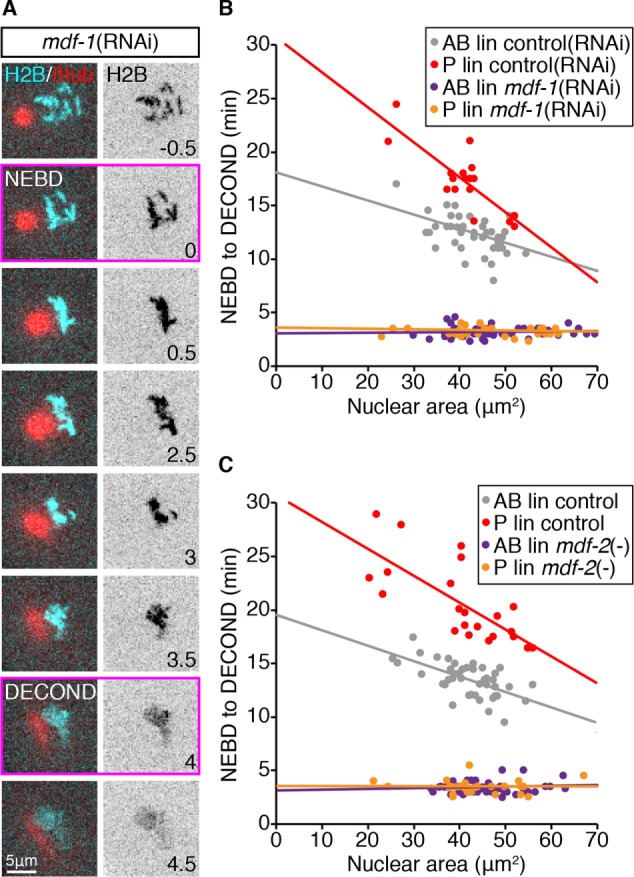
Cell size and cell fate–related differences in the duration of monopolar mitoses require the SAC. (A) Representative cropped time-lapse images showing NEBD and DECOND, as assessed by changes in H2B::mCH fluorescence, in a monopolar germline P1 cell from a mdf-1(RNAi) embryo. Time is expressed in minutes relative to NEBD. Scale bar = 5 μm. (B, C) The relationship between nuclear area and the length of monopolar mitoses in somatic AB and germline P lineage cells in control embryos (red = P/germline, gray = AB/somatic) compared with embryos in which SAC activity has been impaired (orange = P/germline, purple = AB/somatic). (B) mdf-1(RNAi) vs. Control(RNAi). (C) mdf-2(null) vs. mdf-2(+). Control data in C are reproduced from Figure 2G. See Supplemental Table S2 for summary statistics.
Movie S5.
zyg-1(ts)-induced monopolar spindles in 2- and 4-cell stage embryos following RNAi depletion of MDF-1.
Cell size accounts for approximately half of the difference in SAC strength between the somatic AB and germline P1 cell of two-cell embryos
Our results suggest that both cell size and cell fate contribute to checkpoint activity during C. elegans embryogenesis. To evaluate the relative contribution of each, we genetically modified embryo size and measured the duration of monopolar mitoses at a single developmental stage, in the somatic AB and germline P1 blastomeres of two-cell stage embryos. We used RNAi depletion of ANI-2 and PTC-1 to generate small embryos, and C27D9.1 to generate large embryos (Figure 4A and unpublished data; Maddox et al., 2005; Sonnichsen et al., 2005; Hara and Kimura, 2009; Green et al., 2011). Altering embryo size by these means gave a fivefold range of embryo volumes, from slightly smaller than a control P1 cell, to about twice the size of an average control embryo (see Supplemental Table S2), without disrupting basal mitotic timing and other defining features of AB and P1, such as cell cycle asynchrony and cell size asymmetry (Supplemental Figure S2, A–C). Hereafter, we will express cell volume as the radius of the equivalent sphere (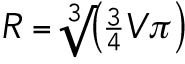 ), which we will call “cell size.”
), which we will call “cell size.”
FIGURE 4:
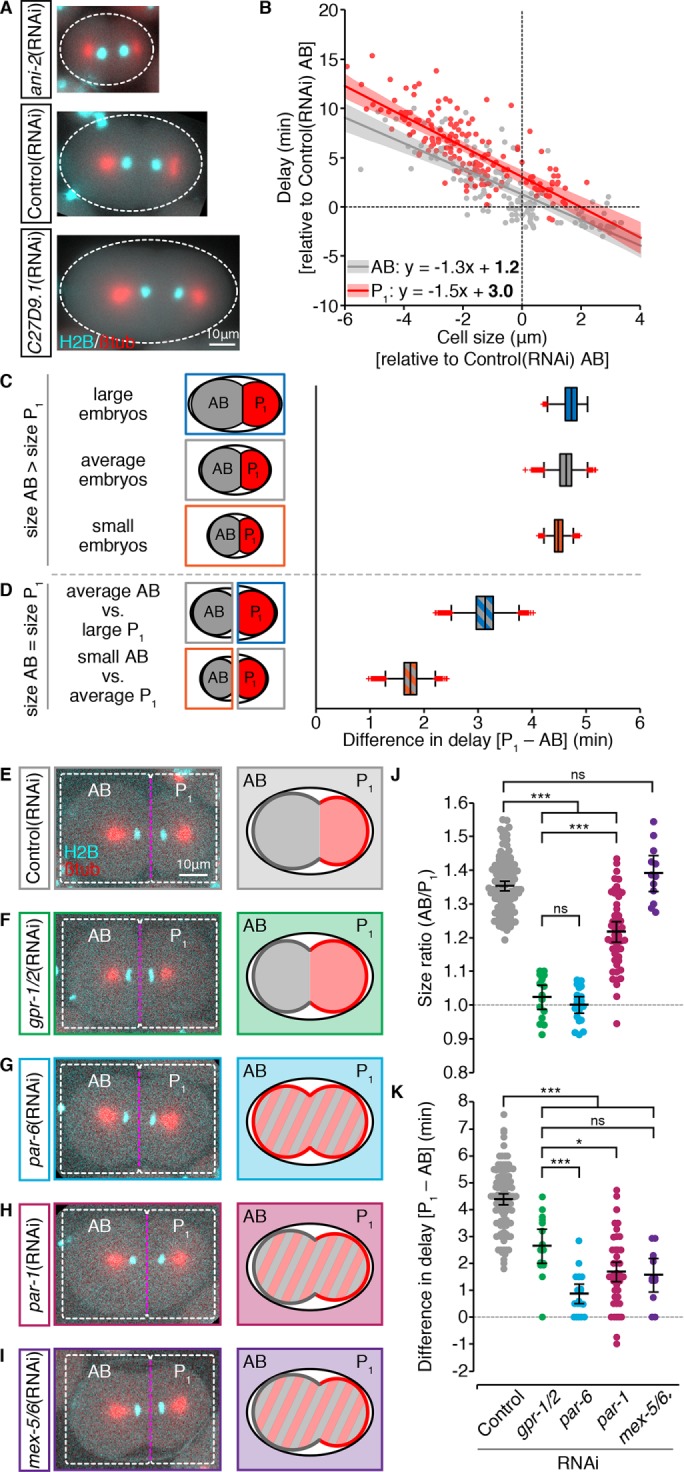
In two-cell stage embryos cell size and asymmetric cytoplasmic partitioning contribute equally to differential checkpoint strength between the somatic AB and germline P1 cell. (A) Representative images of zyg-1(or297ts), H2B::mCH (cyan), β-tubulin::GFP (red) embryos following RNAi depletion of ANI-2 or C27D9.1, as compared with control. Scale bar = 10 μm. Anterior is to the left. (B) The duration of monopolar mitoses, NEBD to DECOND, relative to cell size, in somatic AB (gray; n = 155) and germline P1 (red; n = 167) cells following RNAi-induced changes in embryo volume. Cell volume measurements were converted into the radius of the corresponding sphere (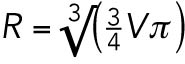 ), which we call “cell size.” All measurements are expressed relative to (±) the mean value for corresponding Control(RNAi) AB cells. Lines represent the linear least-squares regression fit with 95% confidence interval (shaded regions). Linear regression models are shown with statistically different (p < 0.05) coefficients in bold. Regression coefficients were compared using a nonparametric bootstrap (p = 0.055 for slope; p = 0 for y-intercept). (C, D) Schematic and resulting boxplot showing the nonparametric bootstrap analysis of data from B to estimate the difference in the duration of monopolar mitoses between somatic AB and germline P1 cells when AB is larger than P1 (C) vs. when AB and P1 are the same size (D). See Materials and Methods and Supplemental Table S2 for details. Box edges are the 25th and 75th percentiles; whiskers extend to the most extreme data points. Outliers are plotted individually in red. (E–I) Representative images of zyg-1(or297ts), H2B::mCH (cyan), β-tubulin::GFP (red) embryos following RNAi depletion of GPR-1/2 (green), PAR-6 (blue), PAR-1 (maroon), and MEX-5/6 (purple) compared with Control(RNAi) (gray), showing the position of the spindle midpoint (magenta dashed line) along the anterior-to-posterior axis of P0. Scale bar is 10 μm. Anterior is to the left. A schematic representing the expected cell sizes, cytoplasmic asymmetry, and cortical polarity resulting from each RNAi condition is shown on the right. Anterior cortex and cytoplasm are in gray. Posterior cortex and cytoplasm are in red. (J, K) Cell size ratio [AB/P1] (J) and the difference in the duration of monopolar mitoses [P1-AB] (K) for AB/P1 sibling pairs from embryos treated as in E–I. Black bars represent the mean. Error bars show the 95% confidence interval for the mean. ns = p ≥ 0.05; * = 0.01 < p < 0.05; *** = p < 0.001 by an Anova1 with Tukey-Kramer post hoc test. See Supplemental Table S2 for summary statistics.
), which we call “cell size.” All measurements are expressed relative to (±) the mean value for corresponding Control(RNAi) AB cells. Lines represent the linear least-squares regression fit with 95% confidence interval (shaded regions). Linear regression models are shown with statistically different (p < 0.05) coefficients in bold. Regression coefficients were compared using a nonparametric bootstrap (p = 0.055 for slope; p = 0 for y-intercept). (C, D) Schematic and resulting boxplot showing the nonparametric bootstrap analysis of data from B to estimate the difference in the duration of monopolar mitoses between somatic AB and germline P1 cells when AB is larger than P1 (C) vs. when AB and P1 are the same size (D). See Materials and Methods and Supplemental Table S2 for details. Box edges are the 25th and 75th percentiles; whiskers extend to the most extreme data points. Outliers are plotted individually in red. (E–I) Representative images of zyg-1(or297ts), H2B::mCH (cyan), β-tubulin::GFP (red) embryos following RNAi depletion of GPR-1/2 (green), PAR-6 (blue), PAR-1 (maroon), and MEX-5/6 (purple) compared with Control(RNAi) (gray), showing the position of the spindle midpoint (magenta dashed line) along the anterior-to-posterior axis of P0. Scale bar is 10 μm. Anterior is to the left. A schematic representing the expected cell sizes, cytoplasmic asymmetry, and cortical polarity resulting from each RNAi condition is shown on the right. Anterior cortex and cytoplasm are in gray. Posterior cortex and cytoplasm are in red. (J, K) Cell size ratio [AB/P1] (J) and the difference in the duration of monopolar mitoses [P1-AB] (K) for AB/P1 sibling pairs from embryos treated as in E–I. Black bars represent the mean. Error bars show the 95% confidence interval for the mean. ns = p ≥ 0.05; * = 0.01 < p < 0.05; *** = p < 0.001 by an Anova1 with Tukey-Kramer post hoc test. See Supplemental Table S2 for summary statistics.
Cell size and the duration of monopolar mitoses were negatively correlated in both AB and P1 blastomeres (Figure 4B; n = 155, r = −0.79, p = 8.96 × 10−34 and n = 167, r = −0.78, p = 1.69 × 10−35, respectively); however, the average duration of monopolar mitoses was always higher in P1 cells, indicating that P1 cells have a stronger SAC regardless of cell size. To determine how much of the difference in SAC strength between AB and P1 was due to cell size, we used a bootstrap analysis to determine the average difference in mitotic delay between AB and P1 cells that were the same size versus AB and P1 cells where the normal cell size ratio was preserved (i.e., AB is larger than P1; Supplemental Figure S2, D and E). Monopolar mitoses were 4.5 min longer, on average, in P1 cells when AB was larger than P1, irrespective of embryo volume (Figure 4C). When AB and P1 were the same size, the difference in mitotic delay between AB and P1 was approximately halved (Figure 4D). In both cases, if lineage labels were scrambled, the average difference in delay approached 0 (see Supplemental Table S2). Thus, cell size and cell identity make roughly equal contributions to the difference in SAC strength between AB and P1.
The linear regression models for the duration of monopolar mitoses relative to cell size for AB and P1 were nonoverlapping and roughly parallel over the range of cell sizes measured (Figure 4B; AB vs. P1 regression slope: p = 0.055), suggesting that cell identity sets a baseline of SAC activity, which is then scaled according to cell size similarly in AB and P1. In contrast, SAC strength increases more rapidly with decreasing cell size in germline P versus somatic AB lineage cells during early embryonic development (Figure 2C and Supplemental Figure S2F; AB vs. P1 regression slope: p = 0.028 and p = 0.013, respectively). Distinct relationships between cell size and SAC strength could occur if cells within the AB and P lineages interpret their cell size differently (i.e., lineage-specific size scaling) or if SAC strength were progressively modified during lineage development (i.e., enhanced or suppressed in germline or somatic cells, respectively). As somatic AB and germline P1 cells in two-cell stage embryos showed roughly equivalent rates of increase in SAC strength with decreasing cell size, we favor the latter hypothesis. We note, however, that genetically reducing embryo volume tended to produce longer mitotic delays than comparable developmental reductions in cell size, a trend which was most pronounced when comparing AB cells from small embryos to similarly sized cells later in development of the AB lineage (Supplemental Figure S3, B and C). Thus, reducing cell size by a reduction in embryo volume may not be equivalent to reducing cell size via cleavage during early embryogenesis. Consequently, we cannot exclude the possibility that lineage-specific size scaling of SAC strength may contribute to differential SAC activity between germline and somatic cells.
PAR protein–mediated cytoplasmic asymmetries are required for differential SAC activity in AB versus P1
The pattern of checkpoint activity in 2- to 16-cell stage embryos mirrors the asymmetric inheritance of certain germline factors, raising the possibility that a determinant of SAC activity could be similarly regulated. The asymmetric inheritance of germline determinants is regulated by the highly conserved PAR proteins, which lead us to ask whether asymmetries in SAC activity also required PAR proteins.
The polarized, cortical distribution of PAR proteins regulates both cell size and the cytoplasmic distribution of cell fate determinants during the division of the zygote P0. PAR-6, PAR-3, and the atypical protein kinase C (PKC) PKC-3 localize to the anterior cortex, while PAR-2 localizes to the posterior cortex (Etemad-Moghadam et al., 1995; Boyd et al., 1996; Tabuse et al., 1998; Hung and Kemphues, 1999). The asymmetric cortical distribution of PAR proteins depends on mutual antagonism between the anterior and posterior PARs. Consequently, in the absence of anterior PARs, posterior PARs move into the anterior, and vice versa (Goldstein and Macara, 2007). The PAR-1 kinase is also enriched in the posterior and is both cytoplasmic and cortical (Guo and Kemphues, 1995). PAR protein–mediated cytoplasmic partitioning is regulated by PAR-1, which drives the anterior enrichment of two closely related proteins MEX-5 and MEX-6 (hereafter, MEX-5/6; Griffin et al., 2011). MEX-5/6 promote the posterior enrichment and anterior degradation of certain germline determinants (Schubert et al., 2000; Cuenca et al., 2003; DeRenzo et al., 2003; Wu et al., 2015; Han et al., 2018) and anchor and enrich other factors in the anterior (Nishi et al., 2008; Han et al., 2018). By limiting the accumulation and activity of MEX-5/6 in the posterior (Schubert et al., 2000; Cuenca et al., 2003; Griffin et al., 2011) and through other MEX-5/6–independent mechanisms (e.g., Cheeks et al., 2004; Gallo et al., 2010; Benkemoun et al., 2014), PAR-1 promotes germline traits in the posterior P1 cell.
To determine whether PAR-mediated asymmetries during the division of P0 contribute to differential SAC activity between AB and P1, we compared sibling AB/P1 pairs from embryos in which PAR proteins or MEX-5/6 were depleted by RNAi. Although disrupting PAR proteins in P0 will change the identity of the resulting cells, for simplicity we will continue to refer to the anterior blastomere as AB and the posterior blastomere as P1. In control embryos, the position of the mitotic spindle in P0 is shifted toward the posterior, resulting in an average size ratio (AB/P1) of 1.35 (Figure 4, E and J), and P1 delays for 4.4 min longer, on average, than its sibling AB cell (Figure 4K). In gpr-1/2(RNAi) embryos, in which the two blastomeres have the same size (Figure 4, F and J) but maintain somatic versus germline fate (Colombo et al., 2003; Gotta et al., 2003; Srinivasan et al., 2003), the difference in the duration of monopolar mitoses between AB/P1 sibling pairs was halved to an average of 2.7 min (Figure 4K), supporting our earlier bootstrap analysis. In par-6(RNAi) embryos, in which both cortical and cytoplasmic asymmetries are lost (Watts et al., 1996; Nishi et al., 2008), AB and P1 were the same size (Figure 4, G and J), while the difference in duration of monopolar mitoses was reduced to an average of 0.87 min (Figure 4K). RNAi depletion of PAR-1 had an intermediate impact on spindle positioning in P0 (Figure 4H), and par-1(RNAi) embryos showed an incomplete loss of cell size asymmetry (Figure 4J; AB/P1= 1.16). Despite this, the average difference in the duration of monopolar mitoses between AB/P1 sibling pairs in par-1(RNAi) embryos was only 1.7 min (Figure 4K). Similarly, in mex-5/6(RNAi) embryos, in which the difference in size between AB and P1 was indistinguishable from controls (Figure 4, I and J; AB/ P1 = 1.39 for mex-5/6(RNAi), compared with 1.35 for controls), the average difference in the duration of delay between sibling AB and P1 cells was only 1.57 min (Figure 4K). Thus, at the two-cell stage, PAR protein–mediated asymmetries in both cell size and cytoplasmic composition contribute to differential checkpoint strength between the somatic AB and germline P1 cell.
Loss of either anterior or posterior PAR proteins increases SAC activity in AB
The mutually antagonistic behavior of the anterior and posterior PARs supports a simplified model wherein removing anterior PARs permits PAR-1 activity in the anterior, resulting in two cells with germline P1 traits. Conversely, eliminating posterior PARs results in two cells with somatic AB traits (Goldstein and Macara, 2007). If this “mutual antagonism” model were applicable to SAC strength, depletion of anterior PARs should yield two cells with a stronger P1-like SAC, whereas depletion of posterior PARs should lead to two cells with a weaker AB-like SAC. To ask whether SAC activity was subject to this mode of regulation, we measured the absolute duration of monopolar mitoses (hereafter, “delay”) in PAR protein–depleted AB and P1 cells. We found that although depletion of the anterior PAR-6 and PKC-3 appeared to increase delay in AB, depletion of the posterior PAR-2 and PAR-1 did not decrease delay in P1 (Figure 5, A–E). Instead, depletion of PAR-1 led to a moderate increase in delay in AB (Figure 5D), while depletion of PAR-2 had an intermediate effect (Figure 5, E). All par(RNAi) embryos displayed a loss of cell cycle asynchrony, with both cells in par-1(RNAi) and par-2(RNAi) embryos exhibiting an AB-like cell cycle duration (Supplemental Figure S4A; Budirahardja and Gonczy, 2008; Rivers et al., 2008) indicating that our depletions were efficient.
FIGURE 5:

Depleting anterior PARs or PAR-1 increases the duration of monopolar mitoses in the somatic AB cell. (A–E) The average duration of monopolar mitoses relative to average cell size for AB and P1 cells from GPR-1/2 and PAR-depleted embryos compared with controls. All measurements are expressed relative to (±) the mean value for corresponding Control(RNAi) AB cells. (F) Boxplot showing the duration of monopolar mitoses in AB and P1 cells from GPR-1/2 and PAR-depleted embryos expressed relative to (±) the predicted mean value for comparably sized control AB (left) or P1 (right) cells. Values that fall within the darker or lighter gray shaded regions are shorter or longer, respectively, than the predicted mean for comparably sized control cells. Box edges are the 25th and 75th percentiles; whiskers extend to the most extreme data points. Outliers are plotted individually. Statistical significance was evaluated using a Student’s t test with the null hypothesis that the mean of the queried set is 0. (G–J) The average duration of monopolar mitoses vs. cell size for AB and P1 cells from embryos of the given genotypes, relative to the mean value for corresponding control AB cells. (G) par-3(it71). (H) par-2(lw32)±par-1(RNAi). (I) par-1(it51). (J) par-1(it51)±C27D9.1(RNAi). For A–E and G–J, circles = P1, squares = AB, open shapes = control cells, and closed shapes = par(-) and/or RNAi treated; error bars show the 95% confidence interval for the mean, cell size was derived from cell volume as in Figure 4B, and statistical significance was determined using an Anova1 with Tukey-Kramer post hoc test. For all panels, ns = p ≥ 0.05; * = 0.01 < p < 0.05; *** = p < 0.001. See Supplemental Table S2 for summary statistics.
Loss of PAR proteins disrupts spindle positioning during the division of P0 and has varying effects on embryo size (unpublished data), both of which will impact cell size in AB and P1. Thus, to distinguish between the effect of PAR protein depletion on cell size versus SAC strength, we utilized data from Figure 4B (hereafter referred to as “control” cells) to compare delay duration in par(RNAi) AB and P1 cells with comparably sized, but normally polarized and fated, control AB and P1 cells (see Materials and Methods). This analysis indicated that delays in par-6(RNAi) and pkc-3(RNAi) AB cells were more similar in duration to those found in comparably sized P1 than AB control cells (Figure 5F), supporting the idea that depletion of anterior PARs generates two cells with a stronger P1-like SAC. However, we did not observe the converse phenotype in par-1(RNAi) and par-2(RNAi) embryos. Instead, the duration of delay was more or less unchanged in par-2(RNAi) AB and P1 and par-1(RNAi) P1 cells, but was increased in par-1(RNAi) AB cells, relative to their respective, comparably sized control cells (Figure 5F). Overall, depletion of PAR-6, PKC-3, or PAR-1 all appeared to increase SAC strength, relative to cell size, in AB, while depletion of PAR-2, similar to depletion of GPR-1/2, did not appear to affect SAC strength in either AB or P1, apart from its effect on cell size.
We confirmed these results using a kinase-dead allele of par-1 (it51; Guo and Kemphues, 1995) and strong loss of function alleles of par-2 (lw32; Levitan et al., 1994; Boyd et al., 1996) and par-3 (it71; Etemad-Moghadam et al., 1995), with their respective genetic controls (see Supplemental Table S1 for complete genotypes). par-3(it71) embryos resembled par-6(RNAi) and pkc-3(RNAi) embryos with longer delays in AB cells (Figure 5G). Similarly, par-1(it51) AB cells displayed delays that were comparable to control P1 cells and significantly longer than control AB cells (Figure 5H). In par-2(lw32) embryos, differential delay between AB and P1 was largely maintained (Figure 5I). RNAi depletion of PAR-1 in par-2(lw32) embryos eliminated this difference almost entirely (the difference in delay between sibling AB/P1 pairs, as in Figure 4K, was 0.06 ± 0.9 min, mean ± SD) largely via an increase in delay duration in AB cells (Figure 5I). par-3(it71), par-1(it51), par-2(lw32) and par-2(lw32); par-1(RNAi) embryos all exhibited changes in cell cycle duration and asynchrony consistent with their genotype (Supplemental Figure S4, B–D).
Under all conditions, par-1(-) AB cells were significantly smaller than control AB cells and comparable in size to control P1 cells (Figure 5, D, H, and I); however, in light of our AB/P1 size scaling and gpr-1/2(RNAi) results (Figure 4), small AB cells should have a weaker SAC than comparably sized control P1 cells. As delays in par-1(-) AB cells are largely indistinguishable in duration from control P1 cells, it is unlikely that the impact of losing PAR-1 is due exclusively to its effect on cell size. However, to fully exclude this possibility, we used RNAi depletion of C27D9.1 to increase the size of par-1(it51) embryos and to measure the duration of monopolar mitoses in par-1(it51) AB cells that were similar in size to control AB cells (Figure 5J). Here again, par-1(it51) AB cells displayed longer delays than control AB cells. Overall, our genetic and RNAi data are closely aligned and strongly suggest that the regulation of SAC activity downstream from PAR proteins does not conform to the mutual antagonism model.
DISCUSSION
Although variation in checkpoint strength has been noted in a variety of contexts, the driving factors that lead to this variability are poorly understood. Here we show that, during C. elegans embryogenesis, the strength of the SAC correlates with both cell size and cell fate. Spindle perturbations produce longer SAC-dependent mitotic delays in smaller cells, supporting the long-standing hypothesis that the strength of the SAC can be influenced by cell size (Minshull et al., 1994), whereas cells in the germline P lineage delay for longer than comparably sized somatic cells, indicating that SAC strength is further subject to cell fate–specific regulation.
Variation in the strength of the SAC could arise at many points in the checkpoint-signaling mechanism. Conceptually, we envision two primary possibilities: variation at the level of signal generation or variation in the rate of signal degradation. Changes in the efficacy of MCC generation at the kinetochore or the number of MCC-generating kinetochores would impact signal generation, whereas changes that occur away from the kinetochore, including differences in the rate of cytoplasmic MCC disassembly or the efficiency of APC/C inhibition would influence signal degradation. A third possibility could arise downstream from the SAC if, for example, cell size or cell fate changed the threshold of APC/C substrates at which cells exit mitosis. We think this unlikely, as the timing of mitotic exit is invariant across cells of different sizes and fates when SAC regulation is removed and APC/C activity is unimpeded.
Changes in cell size have been proposed to influence SAC strength via two different mechanisms. The first occurs via increased signal generation. Smaller cells have a higher ratio of DNA to cytoplasm (Minshull et al., 1994) and, effectively, a higher concentration of kinetochores per unit cytoplasm, thereby favoring MCC production (Galli and Morgan, 2016). The second incorporates the contribution of premitotic, Mad1/nuclear pore–derived anaphase inhibitor (Rodriguez-Bravo et al., 2014) and predicts that the ratio of pre-NEBD inhibitor-containing nucleoplasm to cytoplasm determines final SAC strength (Kyogoku and Kitajima, 2017). The first mechanism is likely to apply to size scaling of SAC strength during normal embryonic cleavage divisions, where the DNA-to-cytoplasm ratio increases, but nucleoplasm and cytoplasm are partitioned proportionally. The second is relevant in situations where the nucleoplasm-to-cytoplasm ratio is perturbed (e.g., by the removal of cytoplasm before NEBD; Kyogoku and Kitajima, 2017).
Our results raise the possibility that both mechanisms may play a role in C. elegans embryos. In agreement with Galli and Morgan (2016), we find that SAC strength increases as cell size decreases during cleavage of the early embryo. However, we also find that reducing cell size via a genetic reduction in embryo volume has a more profound effect on SAC strength than comparable cleavage-driven reductions in cell size. The genetic manipulations that we used to reduce embryo volume do so by reducing oocyte volume via premature oocyte cellularization (Maddox et al., 2005; Green et al., 2011). At the time of cellularization, oocyte nuclei are intact and, at least in the case of the nucleolus, fully formed (Weber and Brangwynne, 2015), such that both the nucleoplasm-to-cytoplasm and the kinetochore-to-cytoplasm ratio may be increased in small embryos. Although the relationship between the nucleoplasm-to-cytoplasm ratio and SAC strength has not been investigated in C. elegans, the C. elegans orthologue of Mad1, MDF-1, is enriched at the nuclear periphery in interphase cells (Moyle et al., 2014) and depletion of MDF-1 accelerates basal mitotic timing in GSCs (Gerhold et al., 2015), consistent with the presence of premitotic anaphase inhibitor (Rodriguez-Bravo et al., 2014). Thus, we hypothesize that the kinetochore-to-cytoplasm and nucleoplasm-to-cytoplasm ratios may be additive, such that cells from small two-cell stage embryos have a stronger SAC than comparably sized cells from later stage, normally sized embryos. Further unraveling of the mechanism(s) of SAC size scaling is necessary to explore this possibility.
Although our results support the idea that SAC activity scales with cytoplasmic volume, they also indicate that the ratio of DNA or kinetochore to cytoplasm is not necessarily the primary determinant of checkpoint strength during cleavage in C. elegans embryos. Instead, we find that lineage-specific factors are equally important in the final determination of SAC strength. This is perhaps most evident when comparing nocodazole-treated P3 germline blastomeres, which have an average mitotic duration of 40.2 min, to comparably sized somatic AB cells, which have an average mitotic duration of 19.4 min. How germline-fated cells produce a stronger SAC from the same DNA-to-cytoplasm ratio remains an open question. More MCC could be produced per an unattached kinetochore in germline cells or MCC activity in the cytoplasm could be enhanced. Alternatively, differential production of Mad1/nuclear pore–derived anaphase inhibitor during interphase could lead to increased SAC strength in germline cells. As patterning of the early embryo is largely controlled by the distribution of maternally encoded mRNAs and proteins, germline fate at these early developmental stages essentially equates to the distinct cytoplasmic composition of germline cells. Germline precursors are transcriptionally repressed until roughly the 100-cell embryonic stage (Seydoux and Dunn, 1997; Tenenhaus et al., 1998) and defects in germ cell identity in the absence of germline determinants largely manifest during or after the division of the P3 blastomere (e.g., in pos-1 mutants; Tabara et al., 1999). As such, it is highly likely that the factor or factors that enhance SAC activity in germline blastomeres are inherited as part of the germ plasm. Similar factors may then be expressed during the “active” stages of germline specification to augment SAC activity in adult GSCs. Identification of the relevant factor(s) in germline blastomeres will permit testing of this hypothesis.
Consistent with the presence of a SAC enhancing factor (or factors) in the germ plasm, enhanced checkpoint strength in the germline P1 cell, as compared with its somatic AB sibling, requires PAR protein–mediated asymmetric cell division. We found that depletion of PAR-1 and MEX-5/6 both diminished the difference in mitotic delay between AB and P1 sibling pairs without overly impacting cell size. As asymmetries in the cortical localization of PAR proteins are largely maintained in these embryos (Etemad-Moghadam et al., 1995; Boyd et al., 1996; Cuenca et al., 2003), while cytoplasmic asymmetries are lost entirely (Kemphues et al., 1988; Guo and Kemphues, 1995; Schubert et al., 2000; Griffin et al., 2011), our results suggest that PAR-dependent cytoplasmic asymmetries, but not cortical polarity per se, drive differential checkpoint activity in AB and P1.
In addition, our results suggest that PAR-mediated asymmetries in checkpoint strength between AB and P1 are not regulated according to the mutual antagonism model, by which depletion of the anterior or posterior PARs give rise to opposing phenotypes. Instead, we find that removing anterior PARs or the posterior PAR-1 all increase checkpoint activity in AB, whereas removing the posterior PAR-2 has a minimal effect. In the absence of PAR-2, anterior PAR proteins expand into the posterior, but are present in a graded manner, and the posterior enrichment of PAR-1 and some germline factors (e.g., P granules) is often maintained (Boyd et al., 1996; Griffin et al., 2011; Beatty et al., 2013). In addition, although PAR-1 is no longer cortical in par-2(-) embryos, its activity is not lost (Boyd et al., 1996; Watts et al., 1996; Labbé et al., 2006; Griffin et al., 2011). As differential mitotic delays between AB and P1 cells in par-2(lw32) embryos were entirely dependent on PAR-1, we conclude that cytoplasmic asymmetries downstream of PAR-1, and partially independent of PAR-2, are largely responsible for differential SAC activity between AB and P1.
Overall, our results are consistent with a model in which a checkpoint-promoting factor is sequestered in P1 via a PAR-1–dependent mechanism. In the absence of PAR-6, PKC-3, or PAR-1, this factor is equally inherited by AB and P1, thereby increasing checkpoint activity in AB while maintaining it in P1 (Figure 4E). This model requires that levels of this factor are not limiting for checkpoint activity and that it is not degraded in the absence of PAR-1, at least at the two-cell stage. As degradation of germline factors is delayed in par-1 mutants, such that germ plasm proteins persist in all cells until the four-cell stage (Reese et al., 2000), the latter condition is not without precedent. We note that although AB/P1-like reversals upon anterior versus posterior PAR protein depletions have been observed for certain asymmetries, notably cell cycle duration (Budirahardja and Gonczy, 2008; Rivers et al., 2008) and spindle orientation (Kemphues et al., 1988), this may be linked to the particularities of how these specific behaviors are regulated and does not necessarily equate to a whole-scale transformation of cell fate. Intermediate phenotypes may be observed (e.g., PIE-1 diffusivity in par-3(it71) mutant embryos [Wu et al., 2015]) and it has been shown that no single par gene can account for all AB/P1 asymmetries (Bowerman et al., 1997). Our results suggest that although asymmetries in SAC strength between AB and P1 require PAR-mediated asymmetric cytoplasmic partitioning, the mechanism by which this asymmetry is achieved is different from that of cell cycle asynchrony.
Altogether, our results suggest that a checkpoint-regulating factor (or factors) is partitioned during the asymmetric division of germline blastomeres, downstream of PAR-mediated cell polarity, such that germline cells possess a stronger SAC relative to their DNA/kinetochore-to-cytoplasmic ratio than their somatic siblings. SAC activity may be further tuned within each lineage as development progresses. Future work to identify the relevant molecular asymmetries between somatic and germline cells driving differential SAC activity will permit further investigation of this model and identification of where PAR-mediated cell polarity and checkpoint regulation intersect. Finally, that checkpoint strength is seemingly enhanced in germline cells raises the interesting possibility that variation in checkpoint strength may be adaptive. As germline cells give rise to all future generations, checkpoint strength could be increased in these cells to ensure reproductive and thus evolutionary success.
MATERIALS AND METHODS
C. elegans strains and culture
All strains were maintained at 15°C (excepting those used for perm-1[RNAi], DMSO/nocadozole treatments, which were maintained at 20°C) on nematode growth media (NGM) plates, seeded with Escherichia coli bacteria (OP50) following standard procedures (Brenner, 1974). All genotypes are listed in Supplemental Table S1. For all zyg-1(ts) experiments not involving RNAi depletion, L4 stage larvae were transferred to fresh OP50 plates at 15°C for 2 d, after which time the embryos were harvested from gravid adults for imaging. Experiments using the temperature-sensitive allele tbb-2(or362) (Supplemental Figure S1) followed the same procedure. zyg-1(ts) RNAi depletions were performed at 15°C by feeding (Kamath et al., 2001) as follows. Synchronized L1 larvae were obtained by sodium hypochlorite treatment (1.2% NaOCl, 250 mM KOH) and were plated onto 35 mm NGM plates containing 1–1.5 mM isopropyl β-d-thiogalactoside (IPTG) and 25 μg/ml carbenicillin (Carb), seeded with HT115 bacteria containing the empty feeding vector L4440. After 3 d, early L4 larvae were transferred to fresh RNAi feeding plates seeded with bacteria expressing double-stranded RNA (dsRNA) for gene inactivation or the empty vector L4440 for controls. RNAi feeding plates were prepared 1 d before L4 larvae transfer, by adding 100 μl of a bacterial “preculture” (a 1:200 dilution of a fresh overnight culture in Lysogeny/Luria Broth (LB) with 100 μg/ml ampicillin, grown for ∼3 h at 37°C to reach log phase) per 35 mm NGM/ITPG/Carb plate and leaving plates overnight at room temperature to induce dsRNA expression. After 3 d on an RNAi feeding plate, embryos were isolated from gravid adults. The following clones from the Arhinger RNAi library were used: sjj_C50F4.11 (mdf-1/Mad1), sjj_K10B2.5 (ani-2), sjj_ZK675.1 (ptc-1), sjj_C38C10.4 (gpr-1/2), sjj_T26E3.3 (par-6), sjj_F09E5.1 (pkc-3), sjj_H39E23.1 (par-1), sjj_F58B6.3 (par-2), and sjj_W02A2.7 (mex-5/6). The clone targeting C27D9.1 was kindly provided by B. Lacroix, Institute Jacques-Monod. For permeabilization of embryos using perm-1(RNAi) (Carvalho et al., 2011), L4 larvae were picked from OP50-seeded NGM plates at 20°C, washed in M9 buffer, and transferred onto NGM plates containing 2 mM IPTG and 25 μg/ml carbenicillin, seeded with HT115 bacteria containing the plasmid targeting perm-1 (Ahringer clone sjj_TH01H3.4) prepared as described above. After 20–24 h at 20°C, embryos were harvested from gravid adults. All clones were verified by sequencing.
Embryo permeabilization, mounting, drug treatment, and live imaging
Embryos were permeabilized by RNAi depletion of PERM-1 by feeding, as described above. We initially titrated the perm-1(RNAi) dose by serial dilution of the perm-1(RNAi) bacteria with bacteria carrying the L4440 empty vector and examined eggshell permeability by staining with the lipophilic dye FM4-64 (Molecular Probes). Dilution of perm-1(RNAi) bacteria led to increased embryo-to-embryo variability in permeability (unpublished data), and we elected to proceed with nondiluted cultures. Permeabilized embryos were collected as follows. Gravid adults were washed once in the blastomere culturing media, Shelton growth media (SGM; Shelton and Bowerman, 1998), and transferred to a glass-bottom dish (MatTek; 35 mm dish, No. 1.5 coverslip, 14 mm glass diameter) containing 270 μl of SGM, the central region of which had been coated with polylysine (Sigma), washed with ultrapure water (Milli-Q), and dried. Gravid adults were cut using two 25-gauge needles. Embryos of the desired stage were floated over the central, polylysine-coated region of the glass-bottom dish using an eyelash, and allowed to settle. Embryos were imaged at room temperature (∼20°C), on a Cell Observer SD spinning disk confocal (Zeiss; Yokogawa), with an AxioCam 506 Mono camera (Zeiss) and a 40×/1.4 NA Plan Apochromat DIC (UV) VIS-IR oil immersion objective (Zeiss), in Zen software (Zeiss), using 488 nm, 30 mW and 561 nm, 50 mW solid-state lasers, 466/523/600/677 multiple band pass emission filter, 100 ms exposure, and 3 × 3 binning. Either 1.5 or 2.0 μm z-sectioning was used to acquire a ∼25–30-μm-thick z-stack at each time point. Time sampling varied between movies—30–40 s for DMSO-treated embryos and 30–80 s for nocodazole-treated embryos—with a total acquisition time of 1–1.5 h. After the fourth or fifth time point, image acquisition was paused and 30 μl of 10× nocodazole or DMSO in SGM was added for a final concentration of 33 μM nocodazole or 1:1000 DMSO. Under these conditions, we saw highly penetrant eggshell permeabilization, and defects consistent with a weakened eggshell in ∼25% of both DMSO and nocodazole-treated embryos, the most common of which was polar body reabsorption at the one- to two-cell stage, into the embryo anterior or AB cell. Any cells exhibiting abnormal nuclear morphology, cell cycle timing, or extra—presumably polar body–derived—DNA were excluded from analysis. All experiments were carried out in embryos carrying GFP-tagged β-tubulin (β-tubulin::GFP) and mCherry-tagged histone H2B (H2B::mCH; strain UM399; see Supplemental Table S1). The efficiency of nocodazole treatment was assessed visually by loss of β-tubulin::GFP-positive spindle formation.
zyg-1(ts) and tbb-2(or362) embryo mounting and live imaging
Embryos were harvested by cutting open gravid hermaphrodites in M9 buffer using two 25-gauge needles. Embryos were transferred by mouth pipette to a 2% agarose pad, positioned using an eyelash, and covered with a coverslip. The chamber was backfilled with M9 and sealed using VaLaP (1:1:1 Vaseline, lanolin, and paraffin). Embryos were mounted at room temperature in solutions that had been cooled to 15°C. Slides were transferred to a temperature-controlled imaging chamber set to 26°C and time-lapse images were acquired using either a Cell Observer SD spinning disk confocal (Zeiss; Yokogawa) equipped with a stage-top incubator (Pecon) or a DeltaVision inverted microscope (GE Healthcare Life Sciences) equipped with a WeatherStation environmental chamber (PrecisionControl). Spinning disk images were captured using an AxioCam 506 Mono camera (Zeiss) and a 63×/1.4 NA Plan Apochromat DIC oil immersion objective (Zeiss) in Zen software (Zeiss), with 488 nm, 30 mW and 561 nm, 50 mW solid-state lasers, a 466/523/600/677 multiple band pass emission filter, 200 ms exposure time, and 4 × 4 binning. DeltaVision images were captured using a Coolsnap HQ2 charge-coupled device camera (Photometrics) and a 60×/1.42 NA Plan Apo N oil immersion objective (Olympus) in softWoRx software (GE Healthcare Life Sciences), a xenon arc lamp with GFP (470/40 excitation, 525/50 emission, 10% neutral density) and mCherry (572/35 excitation, 632/60 emission, 32% neutral density) filters, 100 ms exposure, and 2 × 2 binning. Time-lapse acquisitions were 45–60 min in duration, with ∼30 s time sampling. At each time point a roughly 20-μm-thick z-stack was acquired, using either 1 or 1.5 μm sectioning. To exclude potentially confounding effects of accumulated mitotic errors due to any partial conditionality of our temperature-sensitive alleles, only cells in which the preceding parental division was observed to be normal were analyzed.
Image processing and measurements
Image processing and analysis was carried out in ImageJ (National Institutes of Health [NIH]). For nocodazole-treated embryos, we defined the duration of mitosis as the time from nuclear envelope breakdown (NEBD), when nonchromatin-associated H2B::mCH is lost from and cytoplasmic β-tubulin::GFP appears within the nuclear space, to the start of chromosome decondensation (DECOND), when the intensity of H2B::mCH begins to decrease and the size of the nuclear mass begins to increase (Figure 1, F and G). Similarly, the duration of bipolar, monopolar, and tbb-2(ts) mitoses were determined visually by monitoring H2B::mCH fluorescence. NEBD was defined as the first frame in which nonincorporated H2B::mCH was lost from the nuclear area. DECOND was defined as the first frame in which the distribution of H2B::mCH shifted from bright and compact to fainter and diffuse (Figure 1C and Supplemental Figure S1A). For all zyg-1(ts) experiments, excepting par-1(it51) experiments and related controls, the timing of NEBD was confirmed by the appearance of microtubules in the nuclear space and the start of DECOND was concomitant with the start of spindle disassembly and shrinking of the spindle pole (see Supplemental Video 1). The par-1(it51) strain and its associated controls lack the β-tubulin::GFP marker, and timing was scored by H2B::mCH fluorescence exclusively. For all experiments, maximal z-stack projections of unprocessed image files were scored. Single z-slices were examined if projected images were ambiguous.
To measure nuclear area, a sum projection of the central three z-slices of the nucleus of interest was processed and segmented. The average of the dimensions of a bounding box fit to the segmented nucleus was taken as representative of nuclear diameter, and nuclear area was then calculated as the area of the corresponding circle (Figure 2D). Reported values reflect the average of measurements made at three time points, 1–2 min before NEBD.
To determine the relative volume of cells in each lineage from the 2–16-cell stage, the position of the mitotic spindle midpoint along the division axis, relative to the midpoint of the dividing cell was used to determine what proportion of each dividing cell was allocated to each daughter (Figure 2A). The Plot Profile tool in ImageJ was used to generate signal intensity profiles along a line drawn parallel to the division axis, through the segregating sister nuclei, in cells in which the cell membrane was labeled with mNeonGreen::PH (mNG::PH) and nuclei were marked by H2B::mCH. Spindle displacement was defined as the offset of the center point between the two membrane mNG::PH peaks versus the center point between the two H2B::mCH peaks. When segregating sister nuclei were not in the same xy plane (i.e., the cell division axis was tilted relative to the plane of imaging), the Stack Reslice tool in ImageJ was used to construct an image of the long axis of the dividing cell from the encompassing z-slices. Measurements were made at three time points during anaphase, after the start of membrane ingression. Nearly identical volume relationships were obtained when cell volume was calculated from cross-sectional area and cell height measurements using the mNG::PH membrane signal to manually outline cells (unpublished data).
The cell volume for AB and P1 in Figures 4 and 5 was estimated by combining measurements of spindle displacement in P0, with measurements of embryo volume. Embryo volume was measured by manually outlining each embryo using the Polygon Selection Tool in ImageJ and fitting an ellipsoid to the resulting area. Spindle displacement was measured using the Plot Profile tool in ImageJ to generate signal intensity profiles along the anterior-to-posterior axis of the embryo. The spindle midpoint was defined as the center point between the two peaks of β-tubulin::GFP fluorescence, corresponding to the spindle poles, and/or the two peaks of H2B::mCH fluorescence, corresponding to the segregating sister nuclei. The position of the spindle midpoint relative to the embryo midpoint was used as an indication of what proportion of the zygote would be inherited by each cell. Final values reflect the average of three measurements made at three different time points during anaphase in P0. For gpr-1/2(RNAi), par-6(RNAi), pkc-3(RNAi), and par-2(RNAi) experiments only embryos in which the position of the mitotic spindle in P0 was roughly centered (50 ± 2% of embryo length) were considered. AB and P1 cell volume measurements (μm3) were converted to the length of the radius (μm) of the corresponding sphere (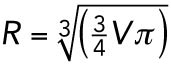 ), which we term “cell size.” To control for variation in experimental conditions and/or scoring bias, measurements for both the duration of monopolar mitoses (NEBD to DECOND) and cell size in AB and P1 cells were normalized by subtracting the mean value for Control(RNAi) AB cells within the same experimental cohort. Normalized NEBD to DECOND values are referred to as “delay.”
), which we term “cell size.” To control for variation in experimental conditions and/or scoring bias, measurements for both the duration of monopolar mitoses (NEBD to DECOND) and cell size in AB and P1 cells were normalized by subtracting the mean value for Control(RNAi) AB cells within the same experimental cohort. Normalized NEBD to DECOND values are referred to as “delay.”
Graphing and statistical analysis
Graphing and statistical analysis were carried out in MATLAB (MathWorks). A comparison of multiple means was performed using an Anova1 with a Tukey-Kramer post hoc test, using the anova1 and multcompare (with α = 0.05, 0.01, or 0.001) functions in the Statistics Toolbox. In Supplemental Figure S2, D and E, sample population means were compared with a two-sample, two-tailed t test, using the ttest2 function in the Statistics Toolbox. In Figure 5F, each sample was evaluated by a t test of the null hypothesis that the sample mean equals 0 using the ttest function in the Statistics Toolbox. Linear least-squares regression models were calculated using the fit function in the Curve Fitting Toolbox, and Pearson’s linear correlation coefficient (r) was reported, with statistical significance (p) assessed using Student’s t distribution, using the corr function in the Statistics Toolbox. Regression coefficients were compared by a nonparametric bootstrap, using a custom MATLAB script, in which the difference between the regression coefficients when true lineage labels were ascribed to the data was compared with the difference in regression coefficients when lineage labels were scrambled over 105 iterations. For Figure 2C, linear regression models were calculated by plotting the duration of mitosis for each cell by the average volume for its respective lineage/cell stage. For all figures ns = p ≥ 0.05, * = 0.01 < p < 0.05, ** = 0.001 < p < 0.01, and *** = p < 0.001. Unless otherwise specified, “n” refers to the number of cells for a given condition. For AB, P1, EMS, P2, MS, E, C, and P3, “n” also reflects the number of individual embryos from which cells were analyzed. For AB lineage cells at the 4-, 8-, and 16-cell stage, between one and three cells of a given class may have been analyzed per a single embryo. For all experiments, embryos were collected and imaged on at least three separate days. Summary statistics for each figure panel are listed in Supplemental Table S2.
For Figure 4, C and D, the data presented in Figure 4A, in which the durations of monopolar mitoses were measured in AB and P1 cells across a wide range of cell volumes, were used to generate five paired data sets of AB and P1 cells (Supplemental Figure S2, D and E). 1) P1 cells within ±2 SDs of the mean cell size for Control(RNAi) AB cells, with their sibling AB cells = “large” embryos; 2) AB cells within ±2 SDs of the mean cell size for Control(RNAi) P1 cells with their sibling P1 cells = “small” embryos; 3) embryos where both AB and P1 were within ±2 SDs of the mean cell size for Control(RNAi) AB and P1 cells, respectively = “average” embryos; 4) P1 and AB cells both within ±2 SDs of the mean cell size for Control(RNAi) AB cells = “large” P1 versus “average” AB; and 5) P1 and AB cells within ±2 SDs of the mean cell size for Control(RNAi) P1 cells = “small” AB versus “average” P1. We approximated the distribution for the difference in delay for each class using a custom MATLAB script to randomize the pairing of AB and P1 cells and calculate the average difference in delay over 105 iterations, the outcome of which is presented as a boxplot in Figure 4, C and D. For each class, the mean difference in delay was compared with the distribution of values for “average” size embryos. This process was repeated for each class with lineage labels scrambled (see Supplemental Table S2), and the mean difference in delay when lineage was ignored was compared with the distribution of values obtained when lineage was correctly ascribed. For both comparisons, p values (see Supplemental Table S2) represent the frequency at which the queried mean is greater than values within the distribution to which it is being compared.
For Figure 5F, data from Figure 4A were combined with all control AB and P1 measurements from Figure 5, A–E, to derive a set of values for the duration of monopolar mitoses and cell size for normally polarized and fated AB (n = 282) and P1 (n = 293) cells. Using a custom MATLAB script, we calculated the distribution of cell size values for each experimental condition (e.g., AB cells from par-6[RNAi] embryos) and used the resulting bin edges and counts to create an analogous cell size distribution using randomly sampled control cells over 104 iterations. For each iteration, the average duration of monopolar mitoses was determined and the resulting collection of means was used to estimate the population mean (μ). Each set (AB or P1) of depleted cells was then normalized to the predicted population mean for comparably sized control AB or P1 cells (measured value, μ), under the assumption that, if the effect of given RNAi treatment on the duration of monopolar mitoses was due exclusively to its impact on cell size, the mean of the resulting, normalized data set should approach 0.
Supplementary Material
Acknowledgments
We thank A. Golden, B. Goldstein, D. Dickinson, and the Caenorhabditis Genetics Center for strains, C. Charbonneau of IRIC’s Bio-imaging facility for technical assistance, and E. D. Salmon and members of the Labbé and Maddox labs for helpful discussions. This work was funded by grants from the Canadian Institutes of Health Research (Grants no. MOP-115171 and no. PJT-153283) to J-C.L. and P.S.M. J-C.L. held the Canada Research Chair in Cell Division and Differentiation. P.S.M. is supported as a William Burwell Harrison Fellow of Biology and by a National Science Foundation Career Award (1652512). IRIC is supported in part by the Canadian Center for Excellence in Commercialization and Research, the Canada Foundation for Innovation, and the Fonds de Recherche du Québec–Santé.
Abbreviations used:
- APC/C
anaphase-promoting complex/cyclosome
- DECOND
chromosome decondensation
- GSC
germline stem cell
- MCC
mitotic checkpoint complex
- NEBD
nuclear envelope breakdown
- PAR
partitioning-defective
- RNAi
RNA-mediated interference
- SAC
spindle assembly checkpoint
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-04-0215) on April 24, 2018.
REFERENCES
- Arata Y, Takagi H, Sako Y, Sawa H. (2014). Power law relationship between cell cycle duration and cell volume in the early embryonic development of Caenorhabditis elegans. Front Physiol , 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z, Zhao Z, Boyle TJ, Murray JI, Waterston RH. (2008). Control of cell cycle timing during C. elegans embryogenesis. Dev Biol , 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty A, Morton DG, Kemphues K. (2013). PAR-2, LGL-1 and the CDC-42 GAP CHIN-1 act in distinct pathways to maintain polarity in the C. elegans embryo. Development , 2005–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkemoun L, Descoteaux C, Chartier NT, Pintard L, Labbé JC. (2014). PAR-4/LKB1 regulates DNA replication during asynchronous division of the early C. elegans embryo. J Cell Biol , 447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman B, Ingram MK, Hunter CP. (1997). The maternal par genes and the segregation of cell fate specification activities in early Caenorhabditis elegans embryos. Development , 3815–3826. [DOI] [PubMed] [Google Scholar]
- Boyd L, Guo S, Levitan D, Stinchcomb DT, Kemphues KJ. (1996). PAR-2 is asymmetrically distributed and promotes association of P granules and PAR-1 with the cortex in C.elegans embryo. Development , 3075–3084. [DOI] [PubMed] [Google Scholar]
- Brenner S. (1974). The genetics of Caenorhabditis elegans. Genetics , 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito DA, Rieder CL. (2006). Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol , 1194–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito DA, Yang Z, Rieder CL. (2008). Microtubules do not promote mitotic slippage when the spindle assembly checkpoint cannot be satisfied. J Cell Biol , 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budirahardja Y, Gönczy P. (2008). PLK-1 asymmetry contributes to asynchronous cell division of C. elegans embryos. Development , 1303–1313. [DOI] [PubMed] [Google Scholar]
- Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. (1998). Mutations of mitotic checkpoint genes in human cancers. Nature , 300–303. [DOI] [PubMed] [Google Scholar]
- Carvalho A, Olson SK, Gutierrez E, Zhang K, Noble LB, Zanin E, Desai A, Groisman A, Oegema K. (2011). Acute drug treatment in the early C. elegans embryo. PLoS One , e24656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao WC, Kulkarni K, Zhang Z, Kong EH, Barford D. (2012). Structure of the mitotic checkpoint complex. Nature , 208–213. [DOI] [PubMed] [Google Scholar]
- Cheeks RJ, Canman JC, Gabriel WN, Meyer N, Strome S, Goldstein B. (2004). C. elegans PAR proteins function by mobilizing and stabilizing asymmetrically localized protein complexes. Curr Biol , 851–862. [DOI] [PubMed] [Google Scholar]
- Clute P, Masui Y. (1995). Regulation of the appearance of division asynchrony and microtubule-dependent chromosome cycles in Xenopus laevis embryos. Dev Biol , 273–285. [DOI] [PubMed] [Google Scholar]
- Clute P, Masui Y. (1997). Microtubule dependence of chromosome cycles in Xenopus laevis blastomeres under the influence of a DNA synthesis inhibitor, aphidicolin. Dev Biol , 1–13. [DOI] [PubMed] [Google Scholar]
- Clute P, Pines J. (1999). Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol , 82–87. [DOI] [PubMed] [Google Scholar]
- Collin P, Nashchekina O, Walker R, Pines J. (2013). The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat Cell Biol , 1378–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo K, Grill SW, Kimple RJ, Willard FS, Siderovski DP, Gönczy P. (2003). Translation of polarity cues into asymmetric spindle positioning in Caenorhabditis elegans embryos. Science , 1957–1961. [DOI] [PubMed] [Google Scholar]
- Consortium CEDM. (2012). Large-scale screening for targeted knockouts in the Caenorhabditis elegans genome. G3 , 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenca AA, Schetter A, Aceto D, Kemphues K, Seydoux G. (2003). Polarization of the C. elegans zygote proceeds via distinct establishment and maintenance phases. Development , 1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deppe U, Schierenberg E, Cole T, Krieg C, Schmitt D, Yoder B, von Ehrenstein G. (1978). Cell lineages of the embryo of the nematode Caenorhabditis elegans. Proc Natl Acad Sci USA , 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRenzo C, Reese KJ, Seydoux G. (2003). Exclusion of germ plasm proteins from somatic lineages by cullin-dependent degradation. Nature , 685–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick AE, Gerlich DW. (2013). Kinetic framework of spindle assembly checkpoint signalling. Nat Cell Biol , 1370–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edens LJ, White KH, Jevtic P, Li X, Levy DL. (2013). Nuclear size regulation: from single cells to development and disease. Trends Cell Biol , 151–159. [DOI] [PubMed] [Google Scholar]
- Ellis GC, Phillips JB, O’Rourke S, Lyczak R, Bowerman B. (2004). Maternally expressed and partially redundant β-tubulins in Caenorhabditis elegans are autoregulated. J Cell Sci , 457–464. [DOI] [PubMed] [Google Scholar]
- Espeut J, Cheerambathur DK, Krenning L, Oegema K, Desai A. (2012). Microtubule binding by KNL-1 contributes to spindle checkpoint silencing at the kinetochore. J Cell Biol , 469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etemad-Moghadam B, Guo S, Kemphues KJ. (1995). Asymmetrically distributed PAR-3 protein contributes to cell polarity and spindle alignment in early C. elegans embryos. Cell , 743–752. [DOI] [PubMed] [Google Scholar]
- Galli M, Morgan DO. (2016). Cell size determines the strength of the spindle assembly checkpoint during embryonic development. Dev Cell , 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo CM, Wang JT, Motegi F, Seydoux G. (2010). Cytoplasmic partitioning of P granule components is not required to specify the germline in C. elegans. Science , 1685–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, Taylor SS. (2008). Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell , 111–122. [DOI] [PubMed] [Google Scholar]
- Gerhold AR, Ryan J, Vallee-Trudeau JN, Dorn JF, Labbe JC, Maddox PS. (2015). Investigating the regulation of stem and progenitor cell mitotic progression by in situ imaging. Curr Biol , 1123–1134. [DOI] [PubMed] [Google Scholar]
- Goldstein B, Macara IG. (2007). The PAR proteins: fundamental players in animal cell polarization. Dev Cell , 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotta M, Dong Y, Peterson YK, Lanier SM, Ahringer J. (2003). Asymmetrically distributed C. elegans homologs of AGS3/PINS control spindle position in the early embryo. Curr Biol , 1029–1037. [DOI] [PubMed] [Google Scholar]
- Green RA, Kao HL, Audhya A, Arur S, Mayers JR, Fridolfsson HN, Schulman M, Schloissnig S, Niessen S, Laband K, et al (2011). A high-resolution C. elegans essential gene network based on phenotypic profiling of a complex tissue. Cell , 470–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin EE, Odde DJ, Seydoux G. (2011). Regulation of the MEX-5 gradient by a spatially segregated kinase/phosphatase cycle. Cell , 955–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Kemphues KJ. (1995). par-1, a gene required for establishing polarity in C. elegans embryos, encodes a putative Ser/Thr kinase that is asymmetrically distributed. Cell , 611–620. [DOI] [PubMed] [Google Scholar]
- Han B, Antkowiak KR, Fan X, Rutigliano M, Ryder SP, Griffin EE. (2018). Polo-like kinase couples cytoplasmic protein gradients in the C. elegans zygote. Curr Biol , 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Kimura A. (2009). Cell-size-dependent spindle elongation in the Caenorhabditis elegans early embryo. Curr Biol , 1549–1554. [DOI] [PubMed] [Google Scholar]
- Hara K, Tydeman P, Kirschner M. (1980). A cytoplasmic clock with the same period as the division cycle in Xenopus eggs. Proc Natl Acad Sci USA , 462–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BJ, McEwen BF, Canman JC, Hoffman DB, Farrar EM, Rieder CL, Salmon ED. (2001). Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol , 1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung TJ, Kemphues KJ. (1999). PAR-6 is a conserved PDZ domain-containing protein that colocalizes with PAR-3 in Caenorhabditis elegans embryos. Development , 127–135. [DOI] [PubMed] [Google Scholar]
- Joglekar AP. (2016). A cell biological perspective on past, present and future investigations of the spindle assembly checkpoint. Biology , 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. (2001). Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol , research0002.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor TM, Mayer TU, Coughlin ML, Mitchison TJ. (2000). Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol , 975–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemphues KJ, Priess JR, Morton DG, Cheng NS. (1988). Identification of genes required for cytoplasmic localization in early C. elegans embryos. Cell , 311–320. [DOI] [PubMed] [Google Scholar]
- Kitagawa R, Rose AM. (1999). Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans. Nat Cell Biol , 514–521. [DOI] [PubMed] [Google Scholar]
- Krenn V, Musacchio A. (2015). The Aurora B kinase in chromosome bi-orientation and spindle checkpoint signaling. Front Oncol , 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyogoku H, Kitajima TS. (2017). Large cytoplasm is linked to the error-prone nature of oocytes. Dev Cell , 287–298. [DOI] [PubMed] [Google Scholar]
- Labbé JC, Pacquelet A, Marty T, Gotta M. (2006). A genomewide screen for suppressors of par-2 uncovers potential regulators of PAR protein-dependent cell polarity in Caenorhabditis elegans. Genetics , 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane SIR, Jones KT. (2017). Chromosome biorientation and APC activity remain uncoupled in oocytes with reduced volume. J Cell Biol , 3949–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Gonzalez P, Westhorpe FG, Taylor SS. (2012). The spindle assembly checkpoint. Curr Biol , R966–R980. [DOI] [PubMed] [Google Scholar]
- Lesage B, Qian J, Bollen M. (2011). Spindle checkpoint silencing: PP1 tips the balance. Curr Biol , R898–R903. [DOI] [PubMed] [Google Scholar]
- Levitan DJ, Boyd L, Mello CC, Kemphues KJ, Stinchcomb DT. (1994). par-2, a gene required for blastomere asymmetry in Caenorhabditis elegans, encodes zinc-finger and ATP-binding motifs. Proc Natl Acad Sci USA , 6108–6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N, Biggins S. (2014). Signalling dynamics in the spindle checkpoint response. Nat Rev Mol Cell Biol , 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddox AS, Habermann B, Desai A, Oegema K. (2005). Distinct roles for two C. elegans anillins in the gonad and early embryo. Development , 2837–2848. [DOI] [PubMed] [Google Scholar]
- Mansfeld J, Collin P, Collins MO, Choudhary JS, Pines J. (2011). APC15 drives the turnover of MCC-CDC20 to make the spindle assembly checkpoint responsive to kinetochore attachment. Nat Cell Biol , 1234–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R. (2001). MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature , 355–359. [DOI] [PubMed] [Google Scholar]
- Minshull J, Sun H, Tonks NK, Murray AW. (1994). A MAP kinase-dependent spindle assembly checkpoint in Xenopus egg extracts. Cell , 475–486. [DOI] [PubMed] [Google Scholar]
- Moyle MW, Kim T, Hattersley N, Espeut J, Cheerambathur DK, Oegema K, Desai A. (2014). A Bub1-Mad1 interaction targets the Mad1-Mad2 complex to unattached kinetochores to initiate the spindle checkpoint. J Cell Biol , 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, Ciliberto A. (2012). The spindle-assembly checkpoint and the beauty of self-destruction. Nat Struct Mol Biol , 1059–1061. [DOI] [PubMed] [Google Scholar]
- Musacchio A, Salmon ED. (2007). The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol , 379–393. [DOI] [PubMed] [Google Scholar]
- Nasmyth K, Peters LM, Uhlmann F. (2000). Splitting the chromosome: Cutting the ties that bind sister chromatids. Science , 1379–1384. [DOI] [PubMed] [Google Scholar]
- Nishi Y, Rogers E, Robertson SM, Lin R. (2008). Polo kinases regulate C. elegans embryonic polarity via binding to DYRK2-primed MEX-5 and MEX-6. Development , 687–697. [DOI] [PubMed] [Google Scholar]
- O’Connell KF, Caron C, Kopish KR, Hurd DD, Kemphues KJ, Li Y, White JG. (2001). The C. elegans zyg-1 gene encodes a regulator of centrosome duplication with distinct maternal and paternal roles in the embryo. Cell , 547–558. [DOI] [PubMed] [Google Scholar]
- O’Rourke SM, Carter C, Carter L, Christensen SN, Jones MP, Nash B, Price MH, Turnbull DW, Garner AR, Hamill DR, et al (2011). A survey of new temperature-sensitive, embryonic-lethal mutations in C. elegans: 24 alleles of thirteen genes. PLoS One , e16644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primorac I, Musacchio A. (2013). Panta rhei: the APC/C at steady state. J Cell Biol , 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese KJ, Dunn MA, Waddle JA, Seydoux G. (2000). Asymmetric segregation of PIE-1 in C.elegans is mediated by two complementary mechanisms that act through separate PIE-1 protein domains. Mol Cell , 445–455. [DOI] [PubMed] [Google Scholar]
- Rieder CL, Maiato H. (2004). Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell , 637–651. [DOI] [PubMed] [Google Scholar]
- Rivers DM, Moreno S, Abraham M, Ahringer J. (2008). PAR proteins direct asymmetry of the cell cycle regulators Polo-like kinase and Cdc25. J Cell Biol , 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Bravo V, Maciejowski J, Corona J, Buch HK, Collin P, Kanemaki MT, Shah JV, Jallepalli PV. (2014). Nuclear pores protect genome integrity by assembling a premitotic and Mad1-dependent anaphase inhibitor. Cell , 1017–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert CM, Lin R, de Vries CJ, Plasterk RH, Priess JR. (2000). MEX-5 and MEX-6 function to establish soma/germline asymmetry in early C. elegans embryos. Mol Cell , 671–682. [DOI] [PubMed] [Google Scholar]
- Seydoux G, Dunn MA. (1997). Transcriptionally repressed germ cells lack a subpopulation of phosphorylated RNA polymerase II in early embryos of Caenorhabditis elegans and Drosophila melanogaster. Development , 2191–2201. [DOI] [PubMed] [Google Scholar]
- Shelton CA, Bowerman B. (1998). Simplified embryonic growth medium for Caenorhabditis elegans. Technical Tips Online , 46–47. [Google Scholar]
- Shi J, Orth JD, Mitchison T. (2008). Cell type variation in responses to antimitotic drugs that target microtubules and kinesin-5. Cancer Res , 3269–3276. [DOI] [PubMed] [Google Scholar]
- Sluder G. (1979). Role of spindle microtubules in the control of cell cycle timing. J Cell Biol , 674–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnichsen B, Koski LB, Walsh A, Marschall P, Neumann B, Brehm M, Alleaume AM, Artelt J, Bettencourt P, Cassin E, et al (2005). Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature , 462–469. [DOI] [PubMed] [Google Scholar]
- Srinivasan DG, Fisk RM, Xu H, van den Heuvel S. (2003). A complex of LIN-5 and GPR proteins regulates G protein signaling and spindle function in C.elegans. Genes Dev , 1225–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strome S, Wood WB. (1983). Generation of asymmetry and segregation of germ-line granules in early C. elegans embryos. Cell , 15–25. [DOI] [PubMed] [Google Scholar]
- Sudakin V, Chan GK, Yen TJ. (2001). Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol , 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE, Schierenberg E, White JG, Thomson JN. (1983). The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol , 64–119. [DOI] [PubMed] [Google Scholar]
- Tabara H, Hill RJ, Mello CC, Priess JR, Kohara Y. (1999). pos-1 encodes a cytoplasmic zinc-finger protein essential for germline specification in C. elegans. Development , 1–11. [DOI] [PubMed] [Google Scholar]
- Tabuse Y, Izumi Y, Piano F, Kemphues KJ, Miwa J, Ohno S. (1998). Atypical protein kinase C cooperates with PAR-3 to establish embryonic polarity in Caenorhabditis elegans. Development , 3607–3614. [DOI] [PubMed] [Google Scholar]
- Tarailo-Graovac M, Wang J, Chu JS, Tu D, Baillie DL, Chen N. (2010). Spindle assembly checkpoint genes reveal distinct as well as overlapping expression that implicates MDF-2/Mad2 in postembryonic seam cell proliferation in Caenorhabditis elegans. BMC Cell Biol , 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenhaus C, Schubert C, Seydoux G. (1998). Genetic requirements for PIE-1 localization and inhibition of gene expression in the embryonic germ lineage of Caenorhabditis elegans. Dev Biol , 212–224. [DOI] [PubMed] [Google Scholar]
- Watts JL, Etemad-Moghadam B, Guo S, Boyd L, Draper BW, Mello CC, Priess JR, Kemphues KJ. (1996). par-6, a gene involved in the establishment of asymmetry in early C. elegans embryos, mediates the asymmetric localization of PAR-3. Development , 3133–3140. [DOI] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW. (2005). Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell , 7–12. [DOI] [PubMed] [Google Scholar]
- Weber SC, Brangwynne CP. (2015). Inverse size scaling of the nucleolus by a concentration-dependent phase transition. Curr Biol , 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westhorpe FG, Tighe A, Lara-Gonzalez P, Taylor SS. (2011). p31comet-mediated extraction of Mad2 from the MCC promotes efficient mitotic exit. J Cell Sci , 3905–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Zhang H, Griffin EE. (2015). Coupling between cytoplasmic concentration gradients through local control of protein mobility in the Caenorhabditis elegans zygote. Mol Biol Cell , 2963–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Kothari P, Lampson MA. (2015). Spindle assembly checkpoint acquisition at the mid-blastula transition. PLoS One , e0119285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.