

Abstract
Optic atrophy 1 (OPA1) is the A-kinase anchoring protein targeting the pool of protein kinase A (PKA) responsible for perilipin 1 phosphorylation, a gatekeeper for lipolysis. However, the involvement of OPA1-bound PKA in the downstream regulation of lipolysis is unknown. Here we show up-regulation and relocation of OPA1 from mitochondria to lipid droplets during adipocytic differentiation of human adipose stem cells. We employed various biochemical and immunological approaches to demonstrate that OPA1-bound PKA phosphorylates perilipin 1 at S522 and S497 on lipolytic stimulation. We show that the first 30 amino acids of OPA1 are essential for its lipid droplet localization as is OMA1-dependent processing. Finally, our results indicate that presence of OPA1 is necessary for lipolytic phosphorylation of downstream targets. Our results show for the first time, to our knowledge, how OPA1 mediates adrenergic control of lipolysis in human adipocytes by regulating phosphorylation of perilipin 1.
INTRODUCTION
White adipose tissue (WAT) is the major energy storage organ in humans where triacylglycerol (TAG) accumulates in lipid droplets (LDs) through the process of lipogenesis. On energy demand, lipolytic pathways are activated by sympathetic stimuli to hydrolyze triacylglycerol into glycerol and free fatty acids (FFA), which in turn fuels energy production (Frayn, 2002). Adrenergic stimuli acting through intracellular cAMP signaling pathways are essential for lipolysis and involve protein kinase A (PKA) phosphorylation of key targets such as perilipin 1 (PLIN1), comparative gene identification-58 (CGI-58), adipose triglyceride lipase (ATGL), and hormone-sensitive lipase (HSL). On the other hand, PKA also phosphorylates phosphodiesterase 3B (PDE3B) to counteract lipolysis (Kawamura et al., 1981; Rahn et al., 1996; Brasaemle, 2007).
Spatiotemporal regulation of PKA-mediated phosphorylation is organized by A-kinase anchoring proteins (AKAPs), which direct the PKA holoenzyme to specific subcellular microdomains close to its substrates to enable their efficient phosphorylation (Tasken and Aandahl, 2004; Scott et al., 2013). AKAPs target pools of PKA type I and type II, distinguished by the type I or type II regulatory (R) subunit, RI or RII, respectively. The PKA isoforms display different subcellular localization due to their affinity for specific AKAPs (Tasken and Aandahl, 2004; Ilouz et al., 2017). AKAPs also scaffold supramolecular signaling complexes with multiple signaling enzymes. All AKAPs contain an A-kinase-binding domain and a unique targeting domain directing the PKA–AKAP complex to defined subcellular structures, membranes, or organelles (Carr et al., 1992; Wong and Scott, 2004; Gold et al., 2006; Kinderman, Kim, et al., 2006; Pidoux and Tasken, 2010).
LDs can occupy up to 90–95% of the cell volume in mature adipocytes and consist of a neutral lipid core consisting mostly of TAG coated by a phospholipid and cholesterol monolayer with numerous LD-associated proteins embedded. Proteomics and cellular biological approaches have identified several hundred proteins that may be involved in the regulation of LD formation and breakdown, although at present only a fraction of these proteins have confirmed LD functions (Guo, Walther, et al., 2008; Meex et al., 2009; Thiam et al., 2013; Meyers et al., 2017). Perilipin 1, the most abundant protein on the surface of LDs and a major substrate for PKA in adipose cells, is known to function as a gatekeeper in catecholamine-stimulated lipolysis by controlling the access of lipases to LDs in a phosphorylation-dependent manner (Greenberg et al., 1991; Blanchette-Mackie et al., 1995; Subramanian et al., 2004; Miyoshi et al., 2006; Yamaguchi et al., 2007; Kuo et al., 2017; Sztalryd and Brasaemle, 2017). Human perilipin 1 has five established consensus PKA phosphorylation sites, although recent phosphoproteomic analysis on 3T3-L1 and primary murine adipocytes found as many as 27 phosphorylated S and T residues in perilipin 1, some of these being potential new PKA targets (Sztalryd et al., 2003; Sztalryd and Brasaemle, 2017; Itabe et al., 2017). However, at present it remains unclear whether all of the five established sites are phosphorylated in vivo by PKA on lipolytic activation in humans. In mice, mutagenesis studies provide indirect evidence for phosphorylation of at least three of the six PKA phosphorylation sites in perilipin 1 on stimulation of lipolysis. Moreover, studies of monkey fibroblasts ectopically expressing mutated forms of the perilipin 1 gene indicate that phosphorylation of the two C-terminal PKA phosphorylation sites S492 and S517 (corresponding to S497 and S522 in the human orthologue) is essential for PKA-stimulated lipolysis, as it is necessary for the release of CGI-58 from perilipin-1 and subsequent activation of ATGL (Granneman et al., 2009). At the same time, simultaneous mutation of the three N-terminal PKA phosphorylation sites in perilipin 1 also reduces its ability to induce lipolysis (Brasaemle et al., 2009; McDonough et al., 2013).
PKA phosphorylation alters perilipin 1 conformation, which allows lipases to access LDs and degrade neutral fat. A consequence of perilipin 1 phosphorylation by PKA is that comparative gene identification-58 (CGI-58) binding to perilipin 1 is disrupted with subsequent CGI-58-mediated activation and LD-translocation of ATGL (Granneman et al., 2007, 2009; Yamaguchi et al., 2007), an enzyme essential for the initial step of lipolysis involving triacylglycerol hydrolysis to diacylglycerol (Zechner et al., 2005; Huijsman, van de Par, et al., 2009; Zimmermann et al., 2009). Phosphorylation of two residues in ATGL (S406 and S430) has been shown, and S406 has been indicated to be regulated both by AMP-activated kinase (AMPK) and PKA (Ahmadian et al., 2011; Pagnon, Matzaris, et al., 2012). Phosphorylation of ATGL on S406 is functionally important in vivo as mutation of S406 abrogates adrenergic stimulation of lipolysis (Pagnon, Matzaris, et al., 2012).
Translocation of HSL from the cytosol to the surface of LDs and its activation is essential for the second step of TAG breakdown from diacylglycerol to monoacylglycerols and FFAs (Clifford et al., 2000; Su et al., 2003; Sztalryd et al., 2003). Perilipin 1 has to be fully phosphorylated (in part by PKA) to enable HSL translocation to LDs and its lipolytic activation (Sztalryd et al., 2003; Miyoshi et al., 2006). Several reports indicate five PKA-phosphorylation sites in rat HSL, including S659 and S660 (S649 and S650 in human HSL, respectively), considered to be the major PKA-phosphorylation sites required for HSL localization and activation at LDs (Anthonsen et al., 1998; Krintel et al., 2008). In contrast, the S563 (S552 in humans) PKA-phosphorylation site appears to be dispensable for LD localization, while mutations in S655 (S554 in humans), a site not phosphorylated by PKA but possibly by glycogen synthase kinase (GSK), AMPK, and Ca2+/calmodulin-dependent protein kinase II (CaMK II) (Carmen and Victor, 2006), also prevents HSL translocation to LDs. Although little is known about HSL phosphorylation in humans, there is significant sequence conservation in the regions where PKA phosphorylation takes place, and available evidence indicates that S649 and S650 are essential for lipolytic activation of human HSL (Contreras et al., 1998; Watt et al., 2003; Talanian et al., 2006).
Genetic alterations in OPA1 are responsible for autosomal dominant optic atrophy (ADOA), a neurological disorder causing visual defects and/or blindness and with a fraction of patients showing more severe neurodegeneration leading to deafness, paralysis, and/or myopathy (Delettre et al., 2001; Lenaers et al., 2012). OPA1 encodes a mitochondrial GTPase essential for proper organization of mitochondrial fission–fusion events and regulation of apoptosis (Olichon et al., 2002; Kamei et al., 2005; Gottlieb, 2006). OPA1 is ubiquitously expressed with highest levels in neurons, heart, and adipose cells, where we showed that OPA1 is targeted to LDs in addition to its mitochondrial targeting (Pidoux, Witczak, et al., 2011). Moreover, in murine classical brown adipocytes, the OPA1 cleavage (conversion of long to short form of OPA1) and energy expenditure, induced by a combined norepinephrine and palmitate stimulation, go together with Drp1-mediated fission and upstream of depolarization in mitochondria (Wikstrom, Mahdaviani, et al., 2014).
The essential role of PKA in activation of lipolysis in adipocytes has been known for decades; however, AKAPs responsible for targeting PKA remains largely unknown. Prevention of isoproterenol-stimulated PKA activation of lipolysis in adipose cells with the PKA anchoring disruptor peptide Ht31 (Zhang et al., 2005; Pidoux, Witczak, et al., 2011) strongly argues for an important role for AKAPs in these cells. AKAP1 is up-regulated during adipogenesis although the substrate for AKAP1-bound PKA remains unknown (Chaudhry et al., 2002; Rodriguez-Cuenca et al., 2012). Recently, we identified OPA1 as the adipocyte AKAP responsible for organizing a complex consisting of OPA1, perilipin 1, and PKA for anchoring PKA in close proximity to perilipin 1 to enable its phosphorylation on lipolytic stimulation in murine cells (Pidoux, Witczak, et al., 2011).
Here we describe how PKA control of lipolysis depends on OPA1 anchoring in human adipose stem cells (hASC) that have been differentiated to adipocytes. We demonstrate, by the use of complementary immunological and biochemical approaches, that perilipin 1 S522 and S497 are phosphorylated by OPA1-anchored PKA on adrenergic stimulation and the cascade effects on ATGL and HSL.
RESULTS
OPA1 is up-regulated during adipocyte differentiation and forms a complex with perilipin 1, RIα, and RIIβ
We first characterized the involvement of OPA1 as an AKAP in the process of lipolysis in human adipose cells derived from hASCs. Localization of RIα and RIIβ was examined in primary as well as cultured hASCs (Figure 1, A and B). RIα is expressed at low levels and localizes in the cytoplasm, and RIIα is robustly expressed and localizes to Golgi membranes as indicated by costaining with the Golgi marker Giantin (Figure 1B). RIIβ is expressed only in a minor fraction of undifferentiated hASCs, where it localizes to Golgi membranes (Figure 1B). The Golgi localization of the RII regulatory subunits is consistent with previous reports demonstrating that PKA is stored in Golgi membranes in interphase at basal state (Muniz et al., 1997; Keryer et al., 1999; Cabrera et al., 2003; Nigg and Raff, 2009).
FIGURE 1:
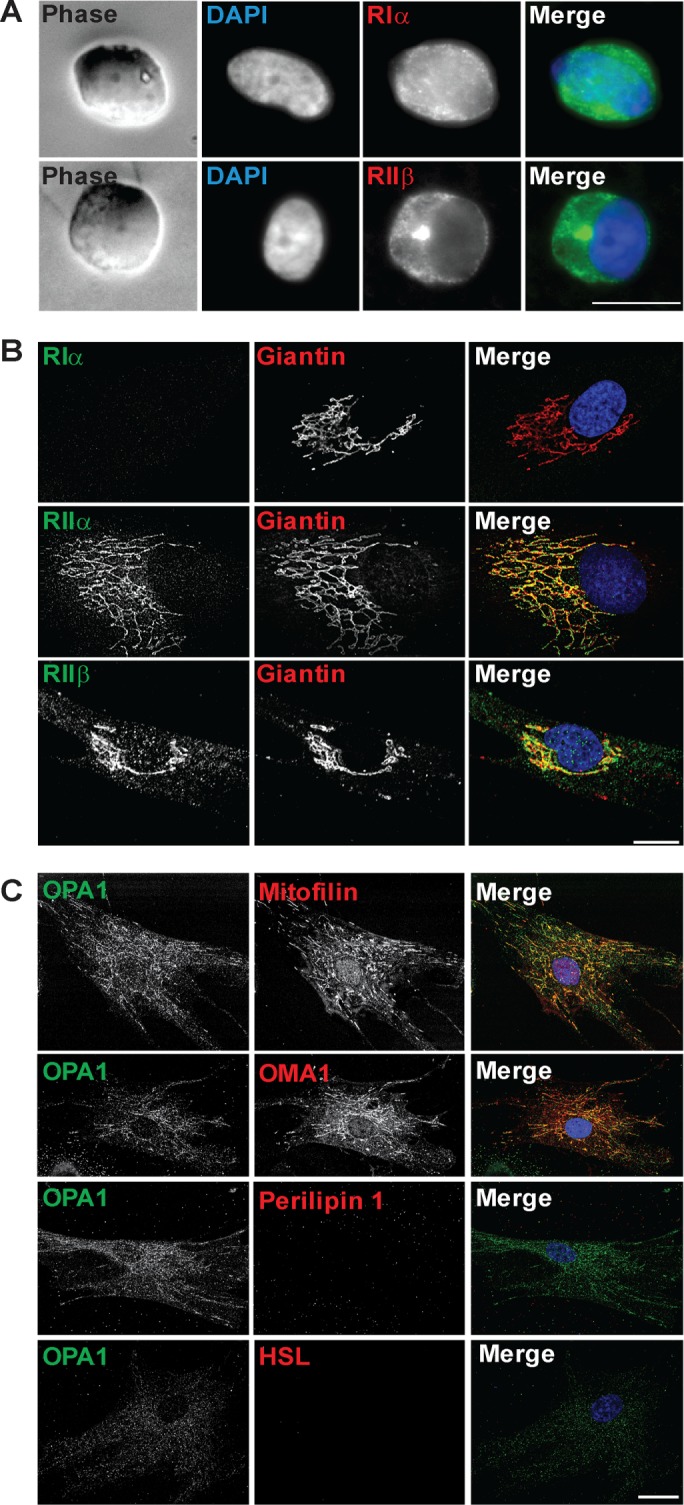
Subcellular localization of PKA and OPA1 in hASCs. Representative images show (A) uncultured isolated hASC stained with RIα or RIIβ (green) and DAPI (blue); (B) cultured hASCs (P7-8) stained for RIα, RIIα, or RIIβ (green) and Giantin (red); and (C) hASCs (P7-8) stained for OPA1 (green) and Mitofilin, OMA1, perilipin, or HSL (red). DAPI (blue) included in merged pictures (n = 3 donors). Scale bar: 10 µm.
Undifferentiated hASCs express low levels of OPA1 (Figure 1C). OPA1 displays a mitochondrial localization as indicated by costaining with the mitochondrial markers (Mitofilin and OMA1). HSL and perilipin 1 as markers for mature adipocytes are not expressed in undifferentiated hASCs (Figure 1C). Thus, while mitochondrial proteins are expressed at robust levels in undifferentiated hASCs, OPA1 and the adipocyte proteins HSL and perilipin 1 are absent or expressed at very low levels.
The majority of experiments aimed to unravel the role of OPA1 in lipogenesis and lipolysis have been conducted in mouse cells or in vivo in murine models. Therefore, we first examined the expression levels of these proteins during human adipocytic cell differentiation. LDs, visualized by Nile Red, appear after ∼1 wk of adipocytic differentiation, and mature adipose cells with large (>5–10 µm) LDs are obtained after 3 wk of differentiation (Figure 2A). Cells were harvested after 0, 1, 2, and 3 wk of differentiation (Figure 2, B and C, and Supplemental Figure 1) for Western blot analysis. We observed an eight- to ninefold up-regulation of OPA1 during adipogenesis from the low levels found in undifferentiated hASCs (Figure 2, B and C, and Supplemental Figure 1). Markers of mature adipocytes such as perilipin 1, HSL, and RIIβ are absent in undifferentiated hASCs but become expressed at high levels during adipogenesis (Kawamura et al., 1981; Greenberg et al., 1991; Planas, Cummings, et al., 1999). We observed a more than threefold up-regulation of RIα while RIIα levels remain constant throughout differentiation. As expected, the early adipogenic marker CEBPα shows a strong up-regulation after 1 wk of differentiation with expression decreasing to lower levels during later stages of adipocyte maturation. ATGL initially shows decreased levels on hormonal stimulation of adipogenesis, in agreement with a previous observation (Chakrabarti et al., 2013), but expression levels are restored during adipocyte maturation. In contrast, CGI-58 is not expressed in undifferentiated ASCs but becomes up-regulated 1 wk into adipogenic stimulation. In addition, we analyzed mitochondrial markers Mitofusin 2 (Mfn2), Mitofilin, and OMA1 (Wilson-Fritch et al., 2003; Gandotra et al., 2011), which display increased protein levels in week 1 and are maintained throughout adipogenesis (Figure 2, B and C, and Supplemental Figure 1). Thus, OPA1 levels increase during adipogenic differentiation concomitantly with perilipin 1 and before other adipocytic markers such as HSL and RIIβ.
FIGURE 2:
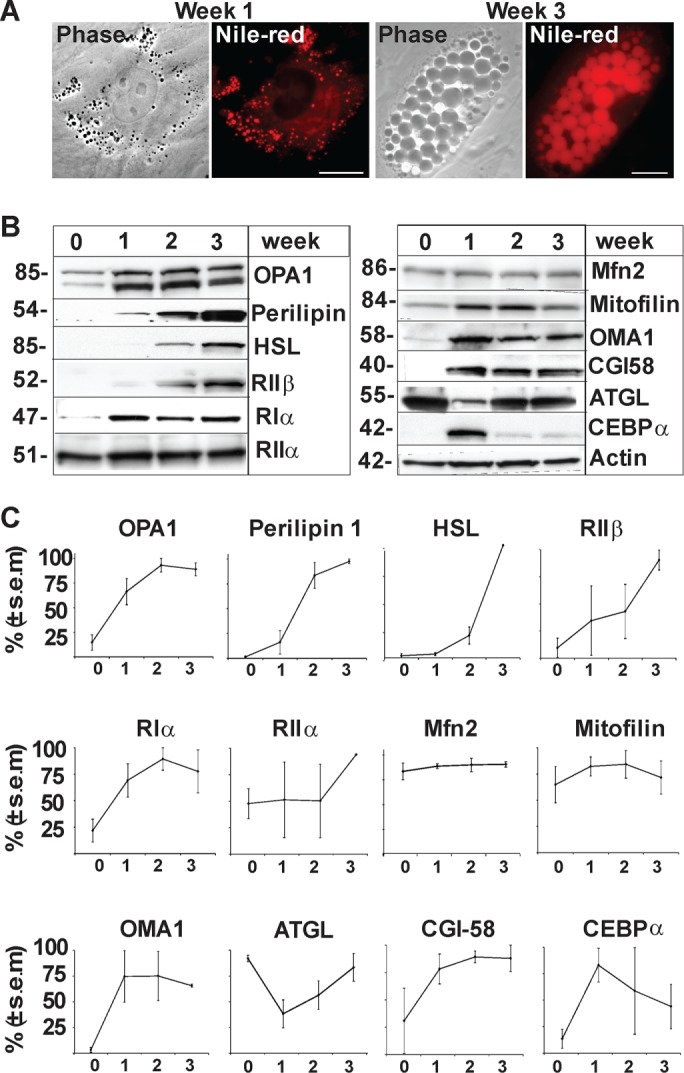
Regulation of OPA1 and adipocytic markers during adipocyte differentiation of hASCs. (A) LDs in hACSs were visualized with Nile Red at weeks 1 and 3 of adipocyte differentiation. (B) Levels of endogenous OPA1, perilipin 1, HSL, RIIβ, RIα, RIIα, Mfn2, Mitofilin, OMA1, CGI-58, ATGL, CEBPα and Actin detected by Western blot analysis in one representative donor (Donor B) during adipocyte differentiation (weeks 0, 1, 2, and 3). (C) Amalgamated data from densitometric analysis of three donors (Donor B in panel B and Donors A and C in Supplemental Figure 1) (mean ± SEM, n = 3 donors).
To examine whether OPA1 forms a complex with perilipin 1 in human differentiated adipocytes, we subjected adipose cell lysates to immunoprecipitation with endogenous OPA1. Immunoblots for presence of perilipin 1, RIIβ, and RIα revealed their presence in the precipitated protein complex (Figure 3A). Conversely, the other macromolecular complex components are also coimmunoprecipitated with RIIβ (Rb) (Figure 3B) and perilipin 1 (GP) (Figure 3C). Thus, OPA1, perilipin 1, and RIIβ form a physical complex in adipocyte-differentiated hASCs.
FIGURE 3:

Association of OPA1 with perilipin and PKA in adipocyte-differentiated hASCs. Lysates from hASC-derived adipocytes were immunoprecipitated with antibodies to OPA1 (A), RIIβ (B), perilipin 1 (C), RIα (D), and CGI-58 (E) and the appropriate igg control (Mo/Rb/GP igg). Precipitates were analyzed for the presence of the indicated protein interaction partners. Lysates from adipocytes incubated in the absence or presence of 10 nM isoproterenol were subjected to immunoprecipitation for OPA1 (F) and ATGL (G) and blotted for presence of the indicated protein interaction partners. Dotted lines indicate images merged from the same gel (different exposure time for input lysate) (n = 3 donors). In situ proximity studies using PLA for the OPA1-perilipin 1, OPA1-CGI-58, and OPA1-ATGL associations (red, top and bottom rows), lipid droplet visualization (green, top row), and DAPI (blue, top row) in hASC-derived adipocytes (H). Scale bars: 10 µm. Statistical analysis of in situ proximity experiments as in H of hASC incubated in the absence or presence of 100 nM of isoproterenol for 20 min and stained for OPA1-perilipin 1 (I), OPA1-CGI-58 proximity signal (J), and OPA1-ATGL proximity signal (K); *p < 0.05, **p < 0.005, ***p < 0.0005. Fifty cells were counted from each condition and for every donor (mean ± SEM, n = 3 donors).
As perilipin 1 also forms a complex with HSL, we examined whether HSL is part of the OPA1-perilipin 1-PKA supramolecular complex but did not observe its binding to OPA1 (Figure 3, A and F). In contrast, OPA1, perilipin 1, and HSL are present in RIα immunoprecipitates which indicates that RIα in addition to its binding to OPA1 also could partition into a macromolecular complex that phosphorylates HSL (Figure 3D). Moreover, ATGL does not coimmunoprecipitate with OPA1 despite its phosphorylation by PKA (Figure 3, F and G). However, we observe the presence of perilipin 1 in ATGL immunoprecipitates after adrenergic stimulation (Figure 3G), suggesting partition into the same complex. We also observe presence of CGI-58 in OPA1 and RIIβ immunoprecipitates (Figure 3, A and B) and vice versa (Figure 3E). This indicates that CGI-58 is a part of the OPA1 (Pidoux, Witczak, et al., 2011)/perilipin 1/PKA complex prior to adrenergic stimulation, as expected since CGI-58 dissociates from perilipin 1 after S497 and S522 phosphorylation (Granneman et al., 2009).
Given the role of OPA1 in lipolysis and the above described supramolecular complex of OPA1 and perilipin 1 in human differentiated adipocytes, we used proximity ligation assay (PLA) (Soderberg et al., 2008) to assess the spatial proximity of OPA1 with perilipin 1/CGI-58/ATGL at LDs (Figure 3, H–K). We found robust OPA1-perilipin 1 PLA signal, demonstrating that OPA1 and perilipin 1 are in close proximity to each other at the LDs in adipocytes differentiated from hASCs (Figure 3, H and I). Moreover, we found significant OPA1-CGI-58 PLA signals under basal conditions supporting presence of CGI-58 in the OPA11-perilipin 1 supramolecular complex under these conditions. Interestingly, OPA1-CGI-58 PLA signals are absent after adrenergic stimulation (Figure 3J). We do not observe OPA1-ATGL PLA signals over background levels neither under basal nor stimulated conditions, strengthening our observation that OPA1 does not associate with ATGL at LDs.
OPA1 relocalizes from mitochondria to perilipin 1–coated LDs during adipocyte differentiation
Since OPA1 localizes to mitochondria in undifferentiated hASCs (Figure 1) and to LDs in mature mouse adipose cells (Satoh et al., 2003; Pidoux, Witczak, et al., 2011), we examined the subcellular distribution of OPA1 during adipocytic differentiation. After 1 wk of differentiation OPA1 displays a mitochondrial localization (Figure 4A, first row) with little or no colocalization with the small perilipin 1–coated LDs (≈1 µm) that now occur. Mitochondrial localization of OPA1 was confirmed by costaining with Mitofilin and OMA1 (Supplemental Figure 2, A and B). As the differentiating adipocytes become filled with LDs in week 2, OPA1 localizes in the vicinity of LDs and partly colocalizes with perilipin 1 (Figure 4A, second row). Immunofluorescence also shows that the localization of OPA1 and Mitofilin/OMA1 diverge (Supplemental Figure 2, A and B). After 3 wk of differentiation LDs with a diameter of more than 5–10 µm fill the cytoplasm. At this stage OPA1 mainly colocalizes with perilipin 1 around LDs as also evident from the z-line scans and correlation (R = 0.91) (Figure 4A, third row).
FIGURE 4:

Subcellular localization of OPA1 during adipocyte differentiation. Representative images show hASC-derived adipocytes after 1, 2, and 3 wk and immunostained for OPA1 (green) and perilipin 1 (red) (A) or for OPA1 (green) and RIIβ, HSL or Mitofilin (red) (B). On the right-hand side, enlarged pictures with details from the area indicated by dotted squares in the original image is shown. Z-lines for overlay diagrams are indicated as lines in the representative image. DAPI is included in merged pictures (n = 3 donors). Scale bars: 10 µm.
OPA1 and RIIβ (Rb) show a good overlap in their subcellular distribution around LDs in mature adipose cells (Figure 4B, first line). Since the existence of fragmented mitochondria (defined as <0.3 µm) has been reported between mature LDs (Zhang et al., 2009; Goldman et al., 2011), we costained for OPA1 and Mitofilin in mature adipose cells to distinguish OPA1 pools in LDs and mitochondria. We observe that while OPA1 mainly localizes to LDs, the majority of Mitofilin-stained mitochondria localizes at the edges of the adipocytes. We detect some fragmented mitochondria in between LDs where they partly colocalizes with OPA1 (Figure 4B, second line). Thus, the majority of OPA1 colocalizes with perilipin 1 and RIIβ to LDs in mature adipocytes.
OPA1 phosphorylates perilipin 1 at pS522 and pS497 to induce lipolysis
Human perilipin 1 has five established PKA phosphorylation sites, of which S497 and S522 are shown to be essential for lipolytic induction (Greenberg et al., 1993; Tansey et al., 2001; Souza et al., 2002; Sztalryd et al., 2003; Tansey et al., 2003; McDonough et al., 2013). We therefore examined whether phosphorylation at these two sites requires an OPA1-bound pool of PKA by transient OPA1 small interfering RNA (siRNA) knockdown in human differentiated adipose cells followed by stimulation with 0–100 nM of isoproterenol. In cells transfected with scrambled control, a concentration-dependent increase in the phosphorylation levels of perilipin 1 S522 and S497 is observed (Figure 5, A and B). Consistent with a previous study in mouse adipocytes (McDonough et al., 2013), maximum was reached after 3 min stimulation with 10 nM isoproterenol (Figure 5, A, right, and B). In OPA1 knocked-down cells, basal phosphorylation of perilipin 1 S522 is absent and isoproterenol-stimulated phosphorylation of both S522 and S497 are abrogated or strongly inhibited (Figure 5, A, left, and B).
FIGURE 5:
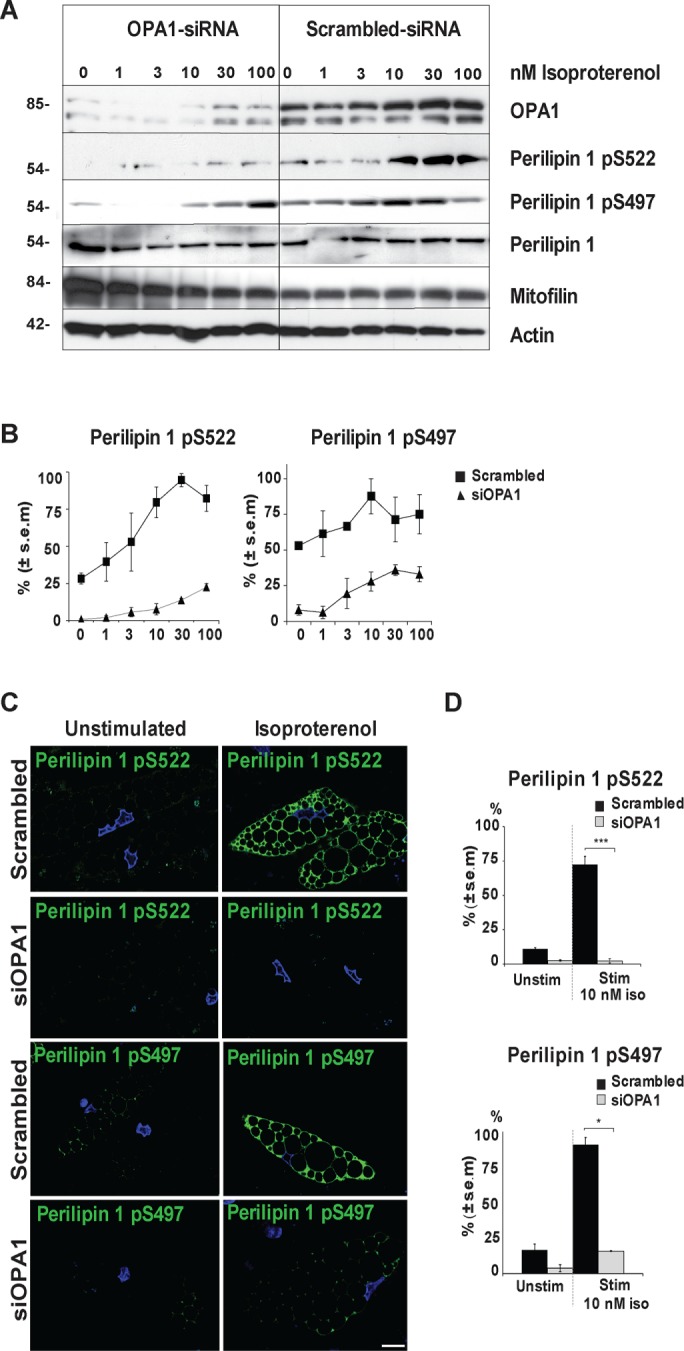
Perilipin S497 and S522 phosphorylation upon isoproterenol stimulation in OPA1 knockdown and control conditions. hASC-derived adipocytes were transfected with OPA1 siRNA or scrambled control 3 wk after initiation of differentiation and stimulated with 0–100 nM isoproterenol for 3 min before immunoblotting for the presence of OPA1, perilipin 1 pS522 and pS497, perilipin 1, mitofilin, and actin (A). Densitometric analysis of perilipin 1 phosphorylation levels at S522 and S497 as in A normalized against actin (mean ± SEM from 3 donors) under scrambled control (▪) or siOPA1 (▴) conditions (B). Immunofluorescence staining for perilipin 1 pS522 or pS497 (green) and DAPI (blue) in hASC-derived adipocytes transfected with OPA1 siRNA or scrambled control, ± 10 nM of isoproterenol for 3 min (C). Scale bars: 10 µm. Statistical analysis of experiments as in C; ***p < 0.0005, *p < 0.05 (D). One hundred cells were scored from each condition and for every donor (mean ± SEM, n = 3 donors).
Similarly, examination of phosphorylation levels of perilipin 1 S522 and S497 by immunofluorescence revealed an effective phosphorylation-response in control-transfected cells on adrenergic stimulation with perilipin 1 in 75–80 and 90% of cells being phosphorylated at S522 and S497, respectively. This in sharp contrast to OPA1 knockdown conditions where only 3–5 and 15% of the cells display full phosphorylation of perilipin 1 S522 and S497, respectively (Figure 5, C and D). Collectively, these results demonstrate that OPA1 is essential for perilipin 1 S522 and S497 phosphorylation upon adrenergic stimulation of lipolysis.
Amino acids 1–30 of OPA1 are essential for LD localization
To map the region of OPA1 responsible for the targeting to LDs, various truncation variants of OPA1 were generated. We transfected either full-length OPA1-Flag, OPA1Δ1-30-Flag lacking the first 30 amino acids or OPA1ΔMTS-Flag lacking the mitochondrial targeting domain (1–87) (Misaka et al., 2002) into differentiated adipocytes (Figure 6, A and B). To distinguish LD and mitochondrial localization of the Flag-tagged OPA1 variants, dual-immunofluorescence staining of the OPA1 truncations with perilipin 1 as LD marker and Mitofilin as mitochondrial marker was performed 24 h posttransfection. While full-length OPA1-Flag localizes to both LDs and mitochondria (Figure 6, A and B, left panels), the OPA1Δ1-30-Flag truncation variant loses LD localization while mitochondrial targeting is retained (Figure 6, A and B, middle panels). As anticipated from previous reports OPA1ΔMTS-Flag is neither targeted to mitochondria nor LDs (Figure 6, A and B, right panels). Transfection of differentiated adipocytes with either of the OPA1-Flag, OPA1Δ1-30-Flag or OPA1ΔMTS-Flag constructs together with a GFP-tagged truncated mitochondrial deadbox helicase (ΔMDDX28-GFP) as another mitochondrial marker confirm the localization pattern observed with Mitofilin (Supplemental Figure 3). Collectively, these observations show that OPA1 localization to LDs depends on its first 30 amino acids.
FIGURE 6:

Effect of N-terminal truncations on localization of OPA1. hASC-derived adipocytes were transfected with OPA1-Flag, OPA1D1-30-Flag, or OPA1DMTS-Flag and immunostained for Flag (green in A, red in B) together with endogenous perilipin 1 (red) (A) or mitofilin (green) (B). Zoomed images with details from split channels (A and B, bottom). DAPI is included in merged pictures (representative of n = 3 donors). Scale bars: 10 µm.
OMA1 expression is required for OPA1 LD-localization and perilipin 1 S522 phosphorylation
A recent report indicated that knockdown of OMA1, a mitochondrial zinc metallopeptidase that cleaves OPA1, is required for OPA1 localization to mitochondrial-ER contact points (Anand, Wai, et al., 2014). To investigate whether OMA1 cleavage of OPA1 could affect its LD localization, we transiently knocked down OMA1 72 h prior to immunolocalization analysis of OPA1 (green) and OMA1 (red) (Figure 7A). OMA1 silencing leads to a shift in subcellular localization of OPA1 from LDs in scrambled control cells (95% LD staining) to a mitochondrial localization pattern for OPA1 in cells with OMA1 knockdown (75% mitochondrial staining, Figure 7B). Furthermore, OMA1 knockdown leads to a change in the OPA1 long to short isoform ratio with the longer OPA1 isoform being most prominent in OMA1 knocked down cells, whereas the opposite is the case in cells transfected with scrambled siRNA (Figure 7, C and D) as observed previously (Anand, Wai, et al., 2014). Thus, OMA1 cleavage of OPA1 is necessary for LD association of OPA1.
FIGURE 7:

Consequences of OMA1 knockdown for LD localization of OPA1 and perilipin 1 S522 phosphorylation. hASC-derived adipocytes were transfected with OMA1 siRNA or scrambled control and subjected to immunostaining for OPA1 (green) and OMA1 (red). Bottom: zoom from the area indicated by dotted squares in the original image. DAPI is included in merged pictures (A). Scale bars: 10 µm. Statistical analysis of OPA1 localization in cells immunostained as representative images in A. OPA1 staining was categorized as 1) in defined mitochondrial structures only or 2) localized to LDs in addition to mitochondria (100 cells counted for each donor and condition; shown mean ± SEM of n = 3 donors, **p < 0.005) (B). hASC-derived adipocytes transfected with OMA1 siRNA or scrambled control were stimulated with 0–100 nM isoproterenol as indicated to assess the presence of OMA1, perilipin 1 pS522, OPA1, and actin (C). Statistical analysis of the presence of the long vs. the short OPA1 isoforms in scrambled control vs. OMA1 knockdown as in C (shown mean ± SEM of n = 18, ***p < 0.0005 of scrambled control vs. OMA1 knockdown) (D). Densitometric analysis of perilipin 1 S522 phosphorylation as in C normalized to actin expression under scrambled control (▪) or siOMA1 (▴) conditions (mean ± SEM of n = 3) (E). Immunofluorescence of perilipin 1 pS522 (green) in hASC-derived adipocytes transfected with OMA1 siRNA or scrambled control incubated with or without 10 nM of isoproterenol counterstained with DAPI (blue) (F). Scale bars: 10 µm. Statistical analysis of immunostained cells as in F (100 cells counted for each donor and condition; shown mean ± SEM of n = 3 donors) (G).
To explore whether the presence of OMA1 affected perilipin 1 S522 phosphorylation, we knocked down OMA1 before stimulation with 0–100 nM of isoproterenol for 3 min (Figure 7C). While cells transfected with scrambled siRNA have strong perilipin 1 S522 phosphorylation in response to isoproterenol, perilipin 1 S522 phosphorylation is ablated in cells with OMA1 knockdown as evident from immunoblot analysis (Figure 7, C and E) and immunofluorescence staining (Figure 7, F and G). Collectively, these data suggest that OPA1 maturation by OMA1 in mitochondria is a prerequisite for the LD association of OPA1 and perilipin 1 S522 phosphorylation.
OPA1 impacts on HSL recruitment to LDs and its activation
It has been shown that phosphorylated perilipin 1 is required for HSL to translocate from the cytoplasm to LDs and become fully activated for lipolysis by PKA at the lipid droplet. Therefore, we investigated the localization of HSL in differentiated human adipocytes in the presence or absence of OPA1. We stimulated lipolysis with 10 nM isoproterenol for 3 min before fixation and staining of OPA1 and HSL. While HSL translocates to LDs on lipolytic induction in more than 90% of the control cells transfected with scrambled siRNA, only 15% of OPA1 siRNA-transfected cells (siOPA1 cells) display LD localization of HSL (Figure 8, A and B). Thus, expression of OPA1 appears to be necessary to get HSL translocation to LDs on induction of lipolysis.
FIGURE 8:

Effect of OPA1 knockdown on HSL translocation to LDs upon adrenergic stimulation. hASC-derived adipocytes transfected with OPA1 siRNA or scrambled control were stimulated with or without 10 nM of isoproterenol followed by immunostaining for OPA1 (green) and HSL (red). Right: Zoomed images with details are shown from the areas indicated by dotted squares in the original image. DAPI is included in merged pictures (A). Scale bars: 10 µm. Statistical analysis of cells immunostained for HSL as in A. HSL localization in cells was categorized as 1) cytoplasmic (stained strongest in the cytoplasm) or 2) LD associated (strongest staining around LDs). One hundred cells were counted for each donor and condition (shown mean ± SEM of n = 3 donors); ***p < 0.0005 (B).
Perilipin 1 and HSL must both be phosphorylated prior to lipolytic translocation and activation of HSL and it has been shown that double mutations of the two C-terminal phosphorylation sites in mouse HSL (S659 and S660 [human S650]) prevent HSL translocation and lipolytic activity (Su et al., 2003). For this reason, we investigated HSL S650 phosphorylation in the presence and absence of OPA1 after stimulation with 0–100 nM of isoproterenol for 5 min to induce lipolysis (Figure 9A, right panel). Cells transfected with scrambled siRNA shows a concentration-dependent increase in HSL S650 phosphorylation upon stimulation, while both basal and stimulated HSL S650 phosphorylation are absent in OPA silenced cells (Figure 9A). We next evaluated HSL phosphorylation at S650, S552 as well as the PKA-independent phosphorylation at S554 by immunofluorescence in OPA1 knockdown cells and scrambled control in combination with adrenergic stimulation. While the majority of scrambled siRNA-transfected adipocytes are phosphorylated at HSL S650 and S552 upon stimulation (80% or more), only 10–15% of OPA1 knockdown cells display phosphorylation at HSL S650 and S552 (Figure 9, B and C, and Supplemental Figure 4, A and B). As expected, the PKA-independent phosphorylation at HSL S554 is not affected by OPA1 knockdown (Supplemental Figure 4C). Collectively, these results indicate that OPA1 is indispensable for HSL translocation to LDs and its phosphorylation on S650 and S552. Similarly, phosphorylation of ATGL on S404 in response to adrenergic stimulation requires presence OPA1 (Figure 9A).
FIGURE 9:

Requirement of OPA1 for HSL phosphorylation on adrenergic stimulation. Adipocyte-differentiated hASCs were transfected with OPA1 siRNA or scrambled control, stimulated with 0–100 nM of isoproterenol, and subjected to immunoblot analysis for the presence of OPA1, HSL S650 phosphorylation and actin (A). Cells as in A with or without 10 nM of isoproterenol were analyzed by immunofluorescence for endogenous HSL pS650 (red) and OPA1 (green) (B). DAPI is included in merged pictures. Scale bars: 10 µm. (C) Statistical analysis of experiments as in B (100 cells counted for each condition and donor, mean ± SEM, n = 3 donors); ***p < 0.0005. (D) Schematic illustration of our proposed model for induction of lipolysis. When lipolysis is induced through the β-adrenergic receptor, OPA1-anchored PKA phosphorylates perilipin 1 leading to recruitment and activation of HSL at the lipid droplets and start the breakdown of triglycerides. When OPA1 is knocked down, perilipin 1 cannot be fully phosphorylated, and HSL is trapped in the cytoplasm, unable to become translocated and activated for lipolysis.
DISCUSSION
Here we demonstrate that OPA1 in its function as an AKAP is necessary for perilipin 1 phosphorylation on S522 and S497 and indispensable for phosphorylation of ATGL and HSL, which catalyzes the breakdown of TAGs.
The human perilipin 1 protein has five established consensus PKA phosphorylation sites. Although only a few studies have been performed in human cells, the two C-terminal phosphorylation sites S497 and S522 (S492 and S517 in mice) have been demonstrated to be essential for PKA-stimulated lipolysis (Greenberg et al., 1993; Tansey et al., 2003; Brasaemle et al., 2009; Granneman et al., 2009). Studies in mouse fibroblasts ectopically expressing perilipin 1 mutants indicated that coordinated mutation of the three N-terminal PKA- phosphorylation sites also may reduce the ability of perilipin 1 to induce lipolysis but does not affect basal lipolysis (Souza et al., 2002). Here we show that OPA1 knockdown abolishes both basal and isoproterenol-induced perilipin 1 phosphorylation at S522 and S497. Lack of specific antibodies to the three N-terminal sites precluded us from studying the phosphorylation status of these sites separately. However, this does not exclude their potential role in the lipolytic activation of HSL (Zhang et al., 2003; Aboulaich et al., 2006). Given the dominant role of OPA1 knockdown, we speculate that OPA1 may also play a role in phosphorylation of one or more of the N-terminal phosphorylation sites.
By immunoblotting we observe that while OPA1 knockdown abolishes perilipin 1 phosphorylation at S522 under all isoproterenol-stimulated conditions, we still observe some phosphorylation of S497 after stimulation with the higher concentrations of isoproterenol. This finding could be explained by the easier accessibility of perilipin 1 S497 for phosphorylation. Alternatively, residual OPA1 protein at low levels may be sufficient to facilitate some degree of pS497 phosphorylation on stimulation. However, it is tempting to speculate that the binding of CGI-58 at the S522 phosphorylation site could hinder unspecific phosphorylation at this site and, consequently, a complete absence of phosphorylation is observed. As it recently has been shown that CGI-58 also becomes phosphorylated by PKA on lipolytic stimulation (Sahu-Osen et al., 2015), it would be interesting to examine whether OPA1 could function as the AKAP that facilitates this PKA phosphorylation.
We observed that HSL is retained in the cytoplasm and remains unphosphorylated at S650 and S552 on adrenergic stimulation in cells with transient OPA1 knockdown. Consistent with this observation, both perilipin 1 and HSL have to be phosphorylated to enable HSL translocation from the cytoplasm to LDs and lipolytic activation (Su et al., 2003; Sztalryd et al., 2003). It has been shown that mouse HSL S660 becomes phosphorylated rapidly in the cytosol, while HSL S563 occurs at the lipid droplet probably after HSL relocalizes to the LDs (Martin et al., 2009). However, a variant of perilipin 1 that cannot be phosphorylated on any of the known phosphorylation sites was also reported to relocalize HSL to LDs, suggesting that part of the LD targeting could be independent of perilipin 1 phosphorylation under some conditions (Miyoshi et al., 2006). Moreover, a study in rat showed direct interaction between HSL and perilipin 1 through amino acids 463–517 (S517 corresponding to the S522 phosphorylation site in the human protein), which is particularly important for LD localization of HSL on lipolytic stimulation (Shen et al., 2009). Whether HSL is retained in the cytoplasm of OPA1-silenced cells due to their inability to phosphorylate perilipin 1 on S522 (or potentially some of the N-terminal phosphorylation sited) or because sustained binding of CGI-58 to perilipin 1 prevents LD localization of HSL remains to be elucidated.
In OPA1 knockdown cells we observe that lack of phosphorylation of perilipin 1 at S522 and S497 is associated with abolished phosphorylation of HSL S650 and S552. However, OPA1 and HSL do not coimmunoprecipitate (Figure 3). Thus, the inability of HSL to be phosphorylated by PKA in OPA1 knockdown cells is probably not due to lack of substrate-binding to the AKAP but could be an indirect consequence of loss of perilipin 1 phosphorylation. These results are in line with the observation that phosphorylation of both perilipin 1 and HSL is required to facilitate phosphorylation of HSL at S660 and S563 (S650 and S552 in the human protein) (Miyoshi et al., 2006; Shen et al., 2009). Another observation in mice shows rapid phosphorylation of HSL in the cytoplasm before its translocation to LDs on lipolytic stimulation, while S563 phosphorylation is suggested to regulate sustained lipolytic activation of HSL at the LDs (Martin et al., 2009). As our results indicate that OPA1 does not directly affect HSL phosphorylation and is not functioning as the AKAP facilitating the lipolytic phosphorylation of HSL, the downstream effect on HLS phosphorylation our observations make it tempting to speculate that OPA1’s role in the lipolytic initiation is upstream of HSL S650 and S552 phosphorylation.
Perilipin 1 S522 phosphorylation is a prerequisite for CGI-58 translocation to the cytoplasm, which is essential for activation of ATGL (Zechner et al., 2005; Granneman et al., 2007; Pagnon, Matzaris, et al., 2012). We show that CGI-58 is in close proximity to the OPA1-perilipin 1 complex under basal conditions but is absent after lipolytic activation, supporting the established mechanism with release of CGI-58 from the perilipin 1 complex on lipolytic activation. Interestingly, CGI-58 is also a substrate for PKA, but further analysis would be required to reveal potential effects of OPA1 knockdown on CGI-58 subcellular distribution and ATGL activation. ATGL was recently suggested to be phosphorylated by PKA or AMPK upon stimulation of lipolysis. In agreement with this, we observed ablated ATGL S404 phosphorylation in response to adrenergic stimulation on OPA1 knockdown. As with HSL, ATGL coprecipitation with OPA1 was neither observed under basal or stimulated conditions; in situ proximity experiments further strengthened this observation as no proximity between OPA1 and ATGL was seen under basal or stimulated conditions. These results indicate that OPA1 does not form a complex with ATGL as a substrate for PKA.
OPA1 was previously shown to be a dual-specificity AKAP with the ability to complex with both type I and type II PKA (Pidoux, Witczak, et al., 2011). Here we observed up-regulation of both RIα and RIIβ during adipogenesis, and both isozymes coimmunoprecipitated with OPA1 in mature adipocytes. Since OPA1 has affinity for both isoenzymes, availability and/or ratio between the two isoenzymes could determine the relative level of RIα and RIIβ bound to OPA1 (Pidoux, Witczak, et al., 2011). In our experiments, OPA1 displayed clear colocalization with RIIβ in differentiated hASC adipocytes (Supplemental Figure 2C). The adipocytic differentiation used in the present study is considered to yield cells that resemble white adipocytes, and RIIβ has been shown to control lipolysis in WAT, making it tempting to speculate that OPA1-bound RIIβ could initiate the lipolysis in our system (Beebe et al., 1984; Robinson-Steiner et al., 1984; Park et al., 2014). Whether lipolysis is controlled by type I or type II PKA appears to be important as the activation constant (Kact) for the two isozymes differ (50–100 and 200–400 nM for type I and type II, respectively). In turn, this affects the sensitivity to catecholamine-stimulated lipolysis as exemplified in RIIβ knockout mice where rescue by up-regulation of RIα providing type I PKA to control lipolysis results in lean mice (Cummings et al., 1996). Furthermore, regulation of RIα levels may be a way to control adipocyte sensitivity to cAMP-regulated lipolysis. This was demonstrated in mice with high expression levels of transcription factor FoxC2, which controls RIα expression, resulting in mice protected against diet-induced obesity and insulin resistance (Cederberg et al., 2001).
The mitochondrial localization of OPA1 and its essential role in mitochondrial dynamics is well characterized (Hall et al., 2014). We have previously demonstrated that OPA1 is also localized to LDs in mice adipocytes (Pidoux, Witczak, et al., 2011). By immunolocalization studies of endogenous OPA1 in human adipose stem cells we show that OPA1 expression increases and its localization shifts from mitochondria to LDs during adipocyte maturation as LDs increase in size. Previous reports have indicated presence of fragmented mitochondria (mitochondria <0,3 µm) localized in close proximity to LDs in adipocytes (Kita et al., 2009), a phenomenon we also observe here. This phenotype is characterized by small mitochondria with clearly defined organelle structure distinct from the more uniform staining of LDs observed for OPA1. In addition, we observe a major part of the mitochondrial network in the peripherally localized cytoplasm of the adipocytes. A recent finding shows that OPA1 was redistributed from membranes to extending membrane tubulations depending on the lipid binding properties of OPA1 (Ban et al., 2010) or lost during ER stress (Zhang et al., 2008), indicating some mobility of OPA1 after mitochondrial maturation. It is tempting to speculate that OPA1 localization on both mitochondria and LDs could coordinate the positioning of LDs in close proximity to mitochondria to facilitate the supply of FFA from lipolysis to mitochondria for beta-oxidation to support ATP synthesis while avoiding toxic effects of liberated FFAs. Interestingly, OPA1 knockdown in mouse models show accumulation of LDs in muscle biopsies (Sarzi et al., 2012). Moreover, inducible reduction of OPA1 expression in murine skeletal muscle has been shown to not only cause progressive mitochondrial dysfunction and loss of muscle mass but also resistance towards age- and diet-induced weight gain and insulin resistance in the mice. This is by mechanisms that involved the activation of ER stress and secretion of fibroblast growth factor 21 (FGF21) from the skeletal muscle (Pereira et al., 2017). Knockdown of Mfn2 (another mitochondrial fusion protein) in adipocytes also leads to accumulation of LDs, whereas silencing of fission proteins such as Drp1 is responsible for a decrease in TAG content (Kita et al., 2009), indicating an important interplay between mitochondrial dynamics and lipid storage in adipocytes.
Our observations indicate that the first 30 amino acids of OPA1 are essential for OPA1 LD localization as OPA1∆1-30-Flag lacking amino acids 1-30 colocalized completely with Mitofilin-stained mitochondria and not with LDs. As anticipated from previous reports (Misaka et al., 2002; Olichon et al., 2002; Satoh et al., 2003; Kita et al., 2009; Pidoux, Witczak, et al., 2011), OPA1ΔMTS-Flag lacking the entire mitochondrial localization signal (1–87) was neither targeted to mitochondria nor LDs in human adipose cells. The differences in subcellular distribution between wild type and N-terminally truncated OPA1 could reflect the fact that the lipophilic nature of the OPA1 membrane targeting domain favors its association with LDs. Furthermore, the observation that deletion of the first 87 amino acids of OPA1 led to loss of both mitochondrial and LD targeting suggests a mitochondrial transit route. Another observation that OPA1 is retained in mitochondria on OMA1 knockdown is compatible with this notion (Pidoux, Witczak, et al., 2011). A recent report indicated that OPA1 localization to mitochondrial-ER contact points also was lost on OMA1 knockdown (Anand, Wai, et al., 2014). In addition, we observed attenuated perilipin 1 S522 phosphorylation on adrenergic stimulation in OMA1 knockdown cells. Collectively, these results suggest that OPA1 matures in the mitochondria before targeting to LDs.
At the cellular level, obesity leads to increased numbers of adipocytes with enlarged size and a pathological accumulation of TAG. Accordingly, understanding the dysregulation of lipolysis in obesity and obesity-related diseases has great interest from a public health perspective. Several of the genes encoding key proteins implicated in lipolysis, like perilipin 1, CGI-58, HSL, and ATGL, harbor known mutations that cause lipodystrophies in humans for which there, at present, is no treatment that targets the pathogenic mechanisms (Lefevre et al., 2001; Gandotra et al., 2011; Wu et al., 2015). Thus, more in-depth knowledge in the area of control of fat storage and breakdown is needed. We report for the first time, to our knowledge, on how OPA1-anchored PKA controls lipolysis in human adipocytes by regulating phosphorylation of perilipin 1 S522 and S497 on adrenergic stimulation and the cascade effects on the translocalization and activation of the lipolytic lipases ATGL and HSL.
MATERIALS AND METHODS
Isolation of adipose stem cells from human adipose tissue
ASCs (CD45-CD31-CD34+CD105+) were isolated from the stromal vascular fraction of human adipose tissue as described previously (Boquest et al., 2005). In short, lipoaspirates obtained from the hip or thigh regions of female donors less than 40 yr of age were digested with collagenase and DNase I, and adipocytes were separated from stromal vascular cells by centrifugation. Stromal cells were strained through filters and CD45- CD31- cells were retrieved by negative selection for both cellular markers. Isolated ASCs are CD34+CD105+ so no negative selection was performed for these markers (Boquest et al., 2005).
The collection and preparation of hASC for research from liposuction material was approved by the regional ethics committee (REK S-06387) and samples were collected after informed consent.
Cell culture and adipocyte differentiation
Cells were cultured at 37°C in an atmosphere of 5% CO2 in humidified air. Cells were passaged approximately every 7th day (1:5) by trypsination and cultured in DMEM/F12 Glutamax (Life Technologies, Thermo Fisher Scientific, MA) containing 10% fetal calf serum (FCS) and 1% Pen/Strep. Undifferentiated hASCs were passaged at around 60% confluence. For adipogenic differentiation, cells were grown to confluence before addition of adipose differentiation medium (DMEM/F12 Glutamax containing 10% FCS, 0.5 mM 1-methyl-3 isobutylxanthine (IBMX), 1 μM dexamethasone (DEX), 10 μg/ml insulin and 200 μM indomethacin (Sigma-Aldrich, St. Louis, MO) for 3 wk as described (Boquest et al., 2005). For isoproterenol induction experiments and immunofluorescence staining, IBMX, DEX and Indomethacin were removed from the differentiation medium the last 5 d before the start of experiments.
Extract preparation
To prepare total cell lysates cells were lysed by addition of RIPA buffer (50 mM Tris-HCl, pH 8.0, 1% NP-40, 0.5% Na-deoxycholate, 0.5% Triton X-100, 100 mM NaCl, 2 mM EDTA [Sigma], and Protease Inhibitors [Roche, Basel, Switzerland]) directly into the well, cells were left 5 min to swell before harvesting by cell scraping. Cell suspension was homogenized for 30 s while still at room temperature with a Pellet Pestle homogenizer (Sigma). Protein concentration was measured using BCA protein assay kit (Pierce, MA) and 10 µg protein extract was run on SDS–PAGE and immunoblotted against indicated antibodies.
Cloning and expression of OPA1 proteins
Human OPA1 clones (full length and truncations) were created using the gateway cloning system. OPA1 fragments were amplified by PCR and inserted into a pENTR vector with ATG start site intact N terminally and without stop codon C terminally. Subsequently, the OPA1 fragments from the pENTR clones were digested with appropriate restriction enzymes and cloned into a pDEST-CMV5-Flag vector. For mutagenesis, we used the Quick change Site-Directed Mutagenesis Kit (Stratagene).
Antibodies
Antibodies for immunoblotting (WB) or immunofluorescence (IF) generated in the mouse (Mo), guinea pig (GP), or rabbit (Rb) to indicated proteins were obtained and used at dilutions as follows: OPA1 (Mo) (1:1000 WB, 1:100 IF; BD Biosciences, CA), Perilipin 1 (GP) (1:1000 WB; Fitzgerald Industries, MA), Perilipin 1 (Rb) (1:300 IF; Fitzgerald Industries), HSL (Rb) (1:1000 WB, 1:100 IF; Cell Signaling, MA), pHSL (p660) (Rb) (1:1000 WB, 1:100 IF; Cell Signaling cat. no. 4126), pHSL (p563) (Rb) (1:100 IF; Cell Signaling cat. no. 4139), pHSL (p565) (1:100 IF; Cell Signaling cat. no. 1437), Mitofilin (Rb) (1:2000 WB, 1:500 IF; Abcam, Cambridge, UK), pPerilipin 1 (Mo) (Ser522) (1:5000 WB, 1:1000 IF; Vala Sciences, CA), pPerilipin 1 (Ser497) (Mo) (1:1000, 1:100 IF; Vala Sciences), Mfn2 (Mo) (1:1000 WB, 1:100 IF; Abcam), RIα (Mo) (1:1000 WB, 1:100 IF; BD Biosciences), RIIα (Mo) (1:1000 WB, 1:100 IF; BD Biosciences), RIIβ (Mo) (1:1000 WB, 1:100 IF; BD Biosciences), CEBPα (Rb) (1:2000; LifeSpan BioSciences, WA), ATGL (Rb) (1:1000; Novus Biologicals, CO, cat. no. 2138), CGI-58 (Rb) (1:2000; Novusbio cat. no NB110-41576), pATGL S406 (Rb) (1:1000; Abcam) and Flag (Rb) (1:300 IF, Cell Signaling cat. no. 2368S).
Immunoprecipitation
Protein G magnetic beads (Invitrogen, CA) were washed two times in lysis buffer before preincubation with 5 µg antibodies against OPA1 (Mo) (BD Biosciences), RIα (Mo) (BD Biosciences), RIIβ (Mo) (BD Biosciences), Perilipin 1 (GP) (Fitzgerald Industries), or IGG (Mo)/(GP) (Jackson laboratories, ME, USA) for 1 h (± cross-linking before proceeding with immunoprecipitation (Pierce)). Cell lysates were prepared as above in RIPA buffer or IPB (10 mM HEPES, pH 7.5, 100 mM NaCl, 1% NP40, 2 mM EDTA and protease inhibitors) (±250 mM sucrose) or lysis/wash buffer (from IP Cross-Linking Kit, Pierce) depending on the protein immunoprecipitated. Lysates (500–1000 µg) were added to the antibody/beads complexes and incubated at 4°C overnight or following the instructions from the manufacturer of the IP Cross-Linking Kit (Pierce) on a rotating wheel. Immune complexes were washed three times in lysis buffer before SDS–PAGE and immunoblotting with indicated antibodies.
Immunoblot analysis
After sample preparation proteins were resolved by SDS–PAGE gel (BioRad Criterion Gels) and transferred to polyvinylidene difluoride membranes (1 h, 100 W). Membranes were blocked in 5% fatty acid free milk or 3% bovine serum albumin (BSA) for 30 min before probing with indicated antibodies. After incubation with appropriate horseradish peroxidase–conjugated secondary antibody (1:10,000; Jackson laboratories), blots were washed five times and then developed using Super Signal West Pico/Dura substrate (Pierce).
Immunolocalization studies
Immunofluorescence stainings were performed on undifferentiated and adipocyte-differentiated ASC at 1, 2, and 3 wk under adipogenic differentiation. Cells were fixed for 30 min in 3% paraformaldehyde, permabilized for 15 min with 0.1% saponin (Sigma), blocked for 15 min in phosphate-bufferend saline (PBS) with 0.01% Tween-20 (PBST) with 3% (fatty acid free) BSA and 0.1% saponin (PBST-BSA-SAP) (±3 min incubation with 6 M guanidine HCl (Sigma) in 50 mM Tris, pH = 7.5), and washed four times in PBS before reblocking in PBST-BSA-SAP for 30 min. Primary antibody solution containing the indicated antibody or Nile Red (as described in Fink et al., 2004) were prepared in PBST-BSA-SAP incubated over night at room temperature. Coverslips were washed three times in PBST-BSA-SAP before incubation in secondary Alexa fluor 488 donkey anti-mouse (1:500) (Invitrogen), Alexa fluor 546 donkey anti-rabbit (1:500) (Invitrogen), and/or Alexa fluor 488 anti-rabbit (1:500) (Invitrogen) antibodies for 5 h. Coverslips were washed three times for 5 min in PBS with 0.1% Saponin, incubated 5 min with 4’,6-diamidino-2-phenylindole (DAPI) (Sigma), rinsed in dH2O, and mounted with Fluromount G. Images were acquired with an LSM510 META confocal microscope (Zeiss, Oberkochen, Germany) fitted with a 100× NA 1.45 oil plan Apochromat objective and using Zen 2009 software.
In situ proximity ligation studies
Cells were fixed and immunolabeled as for immunolocalization studies described above until incubated with primary antibodies (OPA1 1:100), perilipin1 (Rb) (1:100), CGI-58 (Rb) (1:100), and ATGL (Rb) (1:100) before being exposed to PLA linkers (Duolink In Situ PLA Probe Anti-Goat MINUS Affinity purified Donkey and Duolink In Situ PLA Probe Anti-Rabbit PLUS Affinity purified Donkey; Sigma) and Duolink In Situ Detection Reagents Orange (Sigma, Darmstadt, Germany) according to the manufacturer's instruction. Imaging was performed using a 60×/1.4 DIC oil immersion objective on a LSM800 airyscan confocal microscope (Zeiss). Pictures of cells included in PLA statistics were subjected to maximum intensity projections of z-stack pictures (maximal resolution, 0.2 µm slicing) manual integration of cellular regions of interest were established and in situ proximity signals registered.
siRNA, overexpression, and transfection
siRNA transfection was performed using Effectene Transfection Reagent (Qiagen, Hilden, Germany) according to the manufacturer's protocol. We transfected 1.2 µg siOPA1 or siOMA at 0, 24, and 48 h before harvesting the cells at 72 h. Knockdown efficiency for OPA1 and OMA1 was 80–90% and 50–60%, respectively. For overexpression experiments, hASCs were trypsinized and detached before transfected with 2 µg OPA1-Flag, OPA1Δ1-30-Flag, or OPA1ΔMTS-Flag and/or 1 µg ΔMDDX28-GFP with the Human MSC Nucleofector Kit (Lonza, Basel, Switzerland) according to the manufactured protocol before the cells were replated and incubated 24 h before immunofluorescence staining.
Statistics
Quantitative data are presented as average ± SEM. Differences were identified by analysis of variance and considered significant when p < 0.05.
Supplementary Material
Acknowledgments
This work was supported by grants from The Research Council of Norway (grant numbers 187615 and 191744 [K.T.] and 239854, 247668, and 249734 [P.C.]), the Novo Nordic Foundation, and the K.G. Jebsen Foundation. D.T.C. is a postdoc under the SCIENTIA FELLOWS program cofunded by Faculty of Medicine, University of Oslo, and the EU Seventh Framework Program (FP7) Marie S. Curie scheme—People: Cofunding of Regional, National and International Programs (COFUND), grant number 609020. We are grateful to Jorun Solheim and Gladys Marie Tjørhom for technical assistance and to Ruth Valgardsdottir for the gift of the ΔMDDX28-GFP plasmid.
Abbreviations used:
- ADOA
autosomal dominant optical atrophy
- AKAP
A-kinase anchoring protein
- ATGL
adipose triglyceride lipase
- CamKII
CA 2+/calmodulin-dependent protein kinase II
- CGI-58
comparative gene identification 58
- DAPI
4’,6-diamidino-2-phenylindole
- DEX
dexamethasone
- FCS
fetal calf serum
- FFA
free fatty acid
- GP
guinea pig
- GSK
glycogen synthase kinase
- hASC
human adipose stem cells
- HSL
hormone-sensitive lipase
- IF
immunofluorescence
- IMBX
1-methyl-3-isobutylxanthine
- IP
immunoprecipitation
- LD
lipid droplet
- Mfn2
mitofusin 2
- Mo
mouse
- OPA1
optic atrophy 1
- PDE3B
phosphodiesterase 3B
- PKA
protein kinase A
- PLA
proximity ligation assay
- PLIN1
perlipin 1
- R
regulatory subunit of PKA
- Rb
rabbit
- siRNA
small interfering RNA
- TAG
triacylglycerol
- WAT
white adipose tissue
- WB
Western blot
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-09-0538) on April 24, 2018.
REFERENCES
Boldface names denote co–first authors.
- Aboulaich N, Vener AV, Stralfors P. (2006). Hormonal control of reversible translocation of perilipin B to the plasma membrane in primary human adipocytes. J Biol Chem , 11446–11449. [DOI] [PubMed] [Google Scholar]
- Ahmadian M, Abbott MJ, Tang T, Hudak CS, Kim Y, Bruss M, Hellerstein MK, Lee HY, Samuel VT, Shulman GI, et al. (2011). Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab , 739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T. (2014). The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol , 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthonsen MW, Ronnstrand L, Wernstedt C, Degerman E, Holm C. (1998). Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J Biol Chem , 215–221. [DOI] [PubMed] [Google Scholar]
- Ban T, Heymann JA, Song Z, Hinshaw JE, Chan DC. (2010). OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation. Hum Mol Genet , 2113–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe SJ, Holloway R, Rannels SR, Corbin JD. (1984). Two classes of cAMP analogs which are selective for the two different cAMP-binding sites of type II protein kinase demonstrate synergism when added together to intact adipocytes. J Biol Chem , 3539–3547. [PubMed] [Google Scholar]
- Blanchette-Mackie EJ, Dwyer NK, Barber T, Coxey RA, Takeda T, Rondinone CM, Theodorakis JL, Greenberg AS, Londos C. (1995). Perilipin is located on the surface layer of intracellular lipid droplets in adipocytes. J Lipid Res , 1211–1226. [PubMed] [Google Scholar]
- Boquest AC, Shahdadfar A, Fronsdal K, Sigurjonsson O, Tunheim SH, Collas P, Brinchmann JE. (2005). Isolation and transcription profiling of purified uncultured human stromal stem cells: alteration of gene expression after in vitro cell culture. Mol Biol Cell , 1131–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasaemle DL. (2007). Thematic review series: adipocyte biology. The perilipin family of structural lipid droplet proteins: stabilization of lipid droplets and control of lipolysis. J Lipid Res , 2547–2559. [DOI] [PubMed] [Google Scholar]
- Brasaemle DL, Subramanian V, Garcia A, Marcinkiewicz A, Rothenberg A. (2009). Perilipin A and the control of triacylglycerol metabolism. Mol Cell Biochem , 15–21. [DOI] [PubMed] [Google Scholar]
- Cabrera M, Muniz M, Hidalgo J, Vega L, Martin ME, Velasco A. (2003). The retrieval function of the KDEL receptor requires PKA phosphorylation of its C-terminus. Mol Biol Cell , 4114–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmen GY, Victor SM. (2006). Signalling mechanisms regulating lipolysis. Cell Signal , 401–408. [DOI] [PubMed] [Google Scholar]
- Carr DW, Hausken ZE, Fraser ID, Stofko-Hahn RE, Scott JD. (1992). Association of the type II cAMP-dependent protein kinase with a human thyroid RII-anchoring protein. Cloning and characterization of the RII-binding domain. J Biol Chem , 13376–13382. [PubMed] [Google Scholar]
- Cederberg A, Gronning LM, Ahren B, Tasken K, Carlsson P, Enerback S. (2001). FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell , 563–573. [DOI] [PubMed] [Google Scholar]
- Chakrabarti P, Kim JY, Singh M, Shin YK, Kim J, Kumbrink J, Wu Y, Lee MJ, Kirsch KH, Fried SK, et al (2013). Insulin inhibits lipolysis in adipocytes via the evolutionarily conserved mTORC1-Egr1-ATGL-mediated pathway. Mol Cell Biol , 3659–3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry A, Zhang C, Granneman JG. (2002). Characterization of RII(beta) and D-AKAP1 in differentiated adipocytes. Am J Physiol Cell Physiol , C205–C212. [DOI] [PubMed] [Google Scholar]
- Clifford GM, Londos C, Kraemer FB, Vernon RG, Yeaman SJ. (2000). Translocation of hormone-sensitive lipase and perilipin upon lipolytic stimulation of rat adipocytes. J Biol Chem , 5011–5015. [DOI] [PubMed] [Google Scholar]
- Contreras JA, Danielsson B, Johansson C, Osterlund T, Langin D, Holm C. (1998). Human hormone-sensitive lipase: expression and large-scale purification from a baculovirus/insect cell system. Protein Expr Purif , 93–99. [DOI] [PubMed] [Google Scholar]
- Cummings DE, Brandon EP, Planas JV, Motamed K, Idzerda RL, McKnight GS. (1996). Genetically lean mice result from targeted disruption of the RII beta subunit of protein kinase A. Nature , 622–626. [DOI] [PubMed] [Google Scholar]
- Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B, Faivre L, Lenaers G, Belenguer P, Hamel CP. (2001). Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet , 584–591. [DOI] [PubMed] [Google Scholar]
- Fink T, Abildtrup L, Fogd K, Abdallah BM, Kassem M, Ebbesen P, Zachar V. (2004). Induction of adipocyte-like phenotype in human mesenchymal stem cells by hypoxia. Stem Cells , 1346–1355. [DOI] [PubMed] [Google Scholar]
- Frayn KN. (2002). Adipose tissue as a buffer for daily lipid flux. Diabetologia , 1201–1210. [DOI] [PubMed] [Google Scholar]
- Gandotra S, Lim K, Girousse A, Saudek V, O'Rahilly S, Savage DB. (2011). Human frame shift mutations affecting the carboxyl terminus of perilipin increase lipolysis by failing to sequester the adipose triglyceride lipase (ATGL) coactivator AB-hydrolase-containing 5 (ABHD5). J Biol Chem , 34998–35006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MG, Lygren B, Dokurno P, Hoshi N, McConnachie G, Tasken K, Carlson CR, Scott JD, Barford D. (2006). Molecular basis of AKAP specificity for PKA regulatory subunits. Mol Cell , 383–395. [DOI] [PubMed] [Google Scholar]
- Goldman SJ, Zhang Y, Jin S. (2011). Autophagic degradation of mitochondria in white adipose tissue differentiation. Antioxid Redox Signal , 1971–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb E. (2006). OPA1 and PARL keep a lid on apoptosis. Cell , 27–29. [DOI] [PubMed] [Google Scholar]
- Granneman JG, Moore HP, Granneman RL, Greenberg AS, Obin MS, Zhu Z. (2007). Analysis of lipolytic protein trafficking and interactions in adipocytes. J Biol Chem , 5726–5735. [DOI] [PubMed] [Google Scholar]
- Granneman JG, Moore HP, Krishnamoorthy R, Rathod M. (2009). Perilipin controls lipolysis by regulating the interactions of AB-hydrolase containing 5 (Abhd5) and adipose triglyceride lipase (Atgl). J Biol Chem , 34538–34544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C. (1991). Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem , 11341–11346. [PubMed] [Google Scholar]
- Greenberg AS, Egan JJ, Wek SA, Moos MC, Jr, Londos C, Kimmel AR. (1993). Isolation of cDNAs for perilipins A and B: sequence and expression of lipid droplet-associated proteins of adipocytes. Proc Natl Acad Sci USA , 12035–12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Walther TC, Rao M, Stuurman N, Goshima G, Terayama K, Wong JS, Vale RD, Walter P, Farese RV. (2008). Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature , 657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AR, Burke N, Dongworth RK, Hausenloy DJ. (2014). Mitochondrial fusion and fission proteins: novel therapeutic targets for combating cardiovascular disease. Br J Pharmacol , 1890–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijsman E, van de Par C, Economou C, van der Poel C, Lynch GS, Schoiswohl G, Haemmerle G, Zechner R, Watt MJ. (2009). Adipose triacylglycerol lipase deletion alters whole body energy metabolism and impairs exercise performance in mice. Am J Physiol Endocrinol Metab , E505–E513. [DOI] [PubMed] [Google Scholar]
- Ilouz R, Lev-Ram V, Bushong EA, Stiles TL, Friedmann-Morvinski D, Douglas C, Goldberg JL, Ellisman MH, Taylor SS. (2017). Isoform-specific subcellular localization and function of protein kinase A identified by mosaic imaging of mouse brain. Elife , e17681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itabe H, Yamaguchi T, Nimura S, Sasabe N. (2017). Perilipins: a diversity of intracellular lipid droplet proteins. Lipids Health Dis , 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei S, Chen-Kuo-Chang M, Cazevieille C, Lenaers G, Olichon A, Belenguer P, Roussignol G, Renard N, Eybalin M, Michelin A, et al. (2005). Expression of the Opa1 mitochondrial protein in retinal ganglion cells: its downregulation causes aggregation of the mitochondrial network. Invest Ophthalmol Vis Sci , 4288–4294. [DOI] [PubMed] [Google Scholar]
- Kawamura M, Jensen DF, Wancewicz EV, Joy LL, Khoo JC, Steinberg D. (1981). Hormone-sensitive lipase in differentiated 3T3-L1 cells and its activation by cyclic AMP-dependent protein kinase. Proc Natl Acad Sci USA , 732–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keryer G, Skalhegg BS, Landmark BF, Hansson V, Jahnsen T, Tasken K. (1999). Differential localization of protein kinase A type II isozymes in the Golgi-centrosomal area. Exp Cell Res , 131–146. [DOI] [PubMed] [Google Scholar]
- Kinderman FS, Kim C, von Daake S, Ma Y, Pham BQ, Spraggon G, Xuong NH, Jennings PA, Taylor SS. (2006). A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol Cell , 397–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita T, Nishida H, Shibata H, Niimi S, Higuti T, Arakaki N. (2009). Possible role of mitochondrial remodelling on cellular triacylglycerol accumulation. J Biochem , 787–796. [DOI] [PubMed] [Google Scholar]
- Krintel C, Osmark P, Larsen MR, Resjo S, Logan DT, Holm C. (2008). Ser649 and Ser650 are the major determinants of protein kinase A-mediated activation of human hormone-sensitive lipase against lipid substrates. PLoS One , e3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo T, Chen TC, Lee RA, Nguyen NHT, Broughton AE, Zhang D, Wang JC. (2017). Pik3r1 is required for glucocorticoid-induced perilipin 1 phosphorylation in lipid droplet for adipocyte lipolysis. Diabetes , 1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefevre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, Lakhdar H, Wollenberg A, Verret JL, Weissenbach J, et al. (2001). Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet , 1002–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaers G, Hamel C, Delettre C, Amati-Bonneau P, Procaccio V, Bonneau D, Reynier P, Milea D. (2012). Dominant optic atrophy. Orphanet J Rare Dis , 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, Okano S, Kistler C, Fernandez-Rojo MA, Hill MM, Parton RG. (2009). Spatiotemporal regulation of early lipolytic signaling in adipocytes. J Biol Chem , 32097–32107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough PM, Maciejewski-Lenoir D, Hartig SM, Hanna RA, Whittaker R, Heisel A, Nicoll JB, Buehrer BM, Christensen K, Mancini MG, et al. (2013). Differential phosphorylation of perilipin 1A at the initiation of lipolysis revealed by novel monoclonal antibodies and high content analysis. PLoS One , e55511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meex RC, Schrauwen P, Hesselink MK. (2009). Modulation of myocellular fat stores: lipid droplet dynamics in health and disease. Am J Physiol Regul Integr Comp Physiol , R913–R924. [DOI] [PubMed] [Google Scholar]
- Meyers A, Weiskittel TM, Dalhaimer P. (2017). Lipid droplets: formation to breakdown. Lipids , 465–475. [DOI] [PubMed] [Google Scholar]
- Misaka T, Miyashita T, Kubo Y. (2002). Primary structure of a dynamin-related mouse mitochondrial GTPase and its distribution in brain, subcellular localization, and effect on mitochondrial morphology. J Biol Chem , 15834–15842. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Souza SC, Zhang HH, Strissel KJ, Christoffolete MA, Kovsan J, Rudich A, Kraemer FB, Bianco AC, Obin MS, et al. (2006). Perilipin promotes hormone-sensitive lipase-mediated adipocyte lipolysis via phosphorylation-dependent and -independent mechanisms. J Biol Chem , 15837–15844. [DOI] [PubMed] [Google Scholar]
- Muniz M, Martin ME, Hidalgo J, Velasco A. (1997). Protein kinase A activity is required for the budding of constitutive transport vesicles from the trans-Golgi network. Proc Natl Acad Sci USA , 14461–14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Raff JW. (2009). Centrioles, centrosomes, and cilia in health and disease. Cell , 663–678. [DOI] [PubMed] [Google Scholar]
- Olichon A, Emorine LJ, Descoins E, Pelloquin L, Brichese L, Gas N, Guillou E, Delettre C, Valette A, Hamel CP, et al. (2002). The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett , 171–176. [DOI] [PubMed] [Google Scholar]
- Pagnon J, Matzaris M, Stark R, Meex RC, Macaulay SL, Brown W, O’Brien PE, Tiganis T, Watt MJ. (2012). Identification and functional characterization of protein kinase A phosphorylation sites in the major lipolytic protein, adipose triglyceride lipase. Endocrinology , 4278–4289. [DOI] [PubMed] [Google Scholar]
- Park A, Kim WK, Bae KH. (2014). Distinction of white, beige and brown adipocytes derived from mesenchymal stem cells. World J Stem Cells , 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira RO, Tadinada SM, Zasadny FM, Oliveira KJ, Pires KMP, Olvera A, Jeffers J, Souvenir R, McGlauflin R, Seei A, et al. (2017). OPA1 deficiency promotes secretion of FGF21 from muscle that prevents obesity and insulin resistance. EMBO J , 2126–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pidoux G, Tasken K. (2010). Specificity and spatial dynamics of protein kinase A signaling organized by A-kinase-anchoring proteins. J Mol Endocrinol , 271–284. [DOI] [PubMed] [Google Scholar]
- Pidoux G, Witczak O, Jarnaess E, Myrvold L, Urlaub H, Stokka AJ, Kuntziger T, Tasken K. (2011). Optic atrophy 1 is an A-kinase anchoring protein on lipid droplets that mediates adrenergic control of lipolysis. EMBO J , 4371–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas JV, Cummings DE, Idzerda RL, McKnight GS. (1999). Mutation of the RIIbeta subunit of protein kinase A differentially affects lipolysis but not gene induction in white adipose tissue. J Biol Chem , 36281–36287. [DOI] [PubMed] [Google Scholar]
- Rahn T, Ronnstrand L, Leroy MJ, Wernstedt C, Tornqvist H, Manganiello VC, Belfrage P, Degerman E. (1996). Identification of the site in the cGMP-inhibited phosphodiesterase phosphorylated in adipocytes in response to insulin and isoproterenol. J Biol Chem , 11575–11580. [DOI] [PubMed] [Google Scholar]
- Robinson-Steiner AM, Beebe SJ, Rannels SR, Corbin JD. (1984). Microheterogeneity of type II cAMP-dependent protein kinase in various mammalian species and tissues. J Biol Chem , 10596–10605. [PubMed] [Google Scholar]
- Rodriguez-Cuenca S, Carobbio S, Velagapudi VR, Barbarroja N, Moreno-Navarrete JM, Tinahones FJ, Fernandez-Real JM, Oresic M, Vidal-Puig A. (2012). Peroxisome proliferator-activated receptor gamma-dependent regulation of lipolytic nodes and metabolic flexibility. Mol Cell Biol , 1555–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu-Osen A, Montero-Moran G, Schittmayer M, Fritz K, Dinh A, Chang YF, McMahon D, Boeszoermenyi A, Cornaciu I, Russell D, et al. (2015). CGI-58/ABHD5 is phosphorylated on Ser239 by protein kinase A: control of subcellular localization. J Lipid Res , 109–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarzi E, Angebault C, Seveno M, Gueguen N, Chaix B, Bielicki G, Boddaert N, Mausset-Bonnefont AL, Cazevieille C, Rigau V, et al. (2012). The human OPA1delTTAG mutation induces premature age-related systemic neurodegeneration in mouse. Brain , 3599–3613. [DOI] [PubMed] [Google Scholar]
- Satoh M, Hamamoto T, Seo N, Kagawa Y, Endo H. (2003). Differential sublocalization of the dynamin-related protein OPA1 isoforms in mitochondria. Biochem Biophys Res Commun , 482–493. [DOI] [PubMed] [Google Scholar]
- Scott JD, Dessauer CW, Tasken K. (2013). Creating order from chaos: cellular regulation by kinase anchoring. Annu Rev Pharmacol Toxicol , 187–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen WJ, Patel S, Miyoshi H, Greenberg AS, Kraemer FB. (2009). Functional interaction of hormone-sensitive lipase and perilipin in lipolysis. J Lipid Res , 2306–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, Larsson LG, Landegren U. (2008). Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods , 227–232. [DOI] [PubMed] [Google Scholar]
- Souza SC, Muliro KV, Liscum L, Lien P, Yamamoto MT, Schaffer JE, Dallal GE, Wang X, Kraemer FB, Obin M, et al. (2002). Modulation of hormone-sensitive lipase and protein kinase A-mediated lipolysis by perilipin A in an adenoviral reconstituted system. J Biol Chem , 8267–8272. [DOI] [PubMed] [Google Scholar]
- Su CL, Sztalryd C, Contreras JA, Holm C, Kimmel AR, Londos C. (2003). Mutational analysis of the hormone-sensitive lipase translocation reaction in adipocytes. J Biol Chem , 43615–43619. [DOI] [PubMed] [Google Scholar]
- Subramanian V, Rothenberg A, Gomez C, Cohen AW, Garcia A, Bhattacharyya S, Shapiro L, Dolios G, Wang R, Lisanti MP, Brasaemle DL. (2004). Perilipin A mediates the reversible binding of CGI-58 to lipid droplets in 3T3-L1 adipocytes. J Biol Chem , 42062–42071. [DOI] [PubMed] [Google Scholar]
- Sztalryd C, Brasaemle DL. (2017). The perilipin family of lipid droplet proteins: gatekeepers of intracellular lipolysis. Biochim Biophys Acta , 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztalryd C, Xu G, Dorward H, Tansey JT, Contreras JA, Kimmel AR, Londos C. (2003). Perilipin A is essential for the translocation of hormone-sensitive lipase during lipolytic activation. J Cell Biol , 1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talanian JL, Tunstall RJ, Watt MJ, Duong M, Perry CG, Steinberg GR, Kemp BE, Heigenhauser GJ, Spriet LL. (2006). Adrenergic regulation of HSL serine phosphorylation and activity in human skeletal muscle during the onset of exercise. Am J Physiol Regul Integr Comp Physiol , R1094–R1099. [DOI] [PubMed] [Google Scholar]
- Tansey JT, Huml AM, Vogt R, Davis KE, Jones JM, Fraser KA, Brasaemle DL, Kimmel AR, Londos C. (2003). Functional studies on native and mutated forms of perilipins. A role in protein kinase A-mediated lipolysis of triacylglycerols. J Biol Chem , 8401–8406. [DOI] [PubMed] [Google Scholar]
- Tansey JT, Sztalryd C, Gruia-Gray J, Roush DL, Zee JV, Gavrilova O, Reitman ML, Deng CX, Li C, Kimmel AR, Londos C. (2001). Perilipin ablation results in a lean mouse with aberrant adipocyte lipolysis, enhanced leptin production, and resistance to diet-induced obesity. Proc Natl Acad Sci USA , 6494–6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasken K, Aandahl EM. (2004). Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev , 137–167. [DOI] [PubMed] [Google Scholar]
- Thiam AR, Farese RV, Jr, Walther TC. (2013). The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol , 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt MJ, Stellingwerff T, Heigenhauser GJ, Spriet LL. (2003). Effects of plasma adrenaline on hormone-sensitive lipase at rest and during moderate exercise in human skeletal muscle. J Physiol , 325–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikstrom JD, Mahdaviani K, Liesa M, Sereda SB, Si Y, Las G, Twig G, Petrovic N, Zingaretti C, Graham A, et al. (2014). Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J , 418–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson-Fritch L, Burkart A, Bell G, Mendelson K, Leszyk J, Nicoloro S, Czech M, Corvera S. (2003). Mitochondrial biogenesis and remodeling during adipogenesis and in response to the insulin sensitizer rosiglitazone. Mol Cell Biol , 1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, Scott JD. (2004). AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol , 959–970. [DOI] [PubMed] [Google Scholar]
- Wu JW, Yang H, Wang SP, Soni KG, Brunel-Guitton C, Mitchell GA. (2015). Inborn errors of cytoplasmic triglyceride metabolism. J Inherit Metab Dis , 85–98. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Omatsu N, Morimoto E, Nakashima H, Ueno K, Tanaka T, Satouchi K, Hirose F, Osumi T. (2007). CGI-58 facilitates lipolysis on lipid droplets but is not involved in the vesiculation of lipid droplets caused by hormonal stimulation. J Lipid Res , 1078–1089. [DOI] [PubMed] [Google Scholar]
- Zechner R, Strauss JG, Haemmerle G, Lass A, Zimmermann R. (2005). Lipolysis: pathway under construction. Curr Opin Lipidol , 333–340. [DOI] [PubMed] [Google Scholar]
- Zhang HH, Souza SC, Muliro KV, Kraemer FB, Obin MS, Greenberg AS. (2003). Lipase-selective functional domains of perilipin A differentially regulate constitutive and protein kinase A-stimulated lipolysis. J Biol Chem , 51535–51542. [DOI] [PubMed] [Google Scholar]
- Zhang J, Hupfeld CJ, Taylor SS, Olefsky JM, Tsien RY. (2005). Insulin disrupts beta-adrenergic signalling to protein kinase A in adipocytes. Nature , 569–573. [DOI] [PubMed] [Google Scholar]
- Zhang M, Huang Q, Huang Y, Wood O, Yuan W, Chancey C, Daniel S, Rios M, Hewlett I, Clouse KA, Dayton AI. (2008). beta-Estradiol attenuates the anti-HIV-1 efficacy of Stavudine (D4T) in primary PBL. Retrovirology , 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. (2009). Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci USA , 19860–19865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann R, Lass A, Haemmerle G, Zechner R. (2009). Fate of fat: the role of adipose triglyceride lipase in lipolysis. Biochim Biophys Acta , 494–500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.