

Peroxisomal dysfunction is often associated with mitochondrial abnormalities for unknown reasons. We found that peroxisomal loss upon Pex19 mutation in Drosophila results in Hnf4 hyperactivation with free fatty acid accumulation and mitochondrial damage as a consequence. Genetic reduction of Hnf4 in Pex19 mutants improves all phenotypes, including lethality.
Abstract
Inherited peroxisomal biogenesis disorders (PBDs) are characterized by the absence of functional peroxisomes. They are caused by mutations of peroxisomal biogenesis factors encoded by Pex genes, and result in childhood lethality. Owing to the many metabolic functions fulfilled by peroxisomes, PBD pathology is complex and incompletely understood. Besides accumulation of peroxisomal educts (like very-long-chain fatty acids [VLCFAs] or branched-chain fatty acids) and lack of products (like bile acids or plasmalogens), many peroxisomal defects lead to detrimental mitochondrial abnormalities for unknown reasons. We generated Pex19 Drosophila mutants, which recapitulate the hallmarks of PBDs, like absence of peroxisomes, reduced viability, neurodegeneration, mitochondrial abnormalities, and accumulation of VLCFAs. We present a model of hepatocyte nuclear factor 4 (Hnf4)-induced lipotoxicity and accumulation of free fatty acids as the cause for mitochondrial damage in consequence of peroxisome loss in Pex19 mutants. Hyperactive Hnf4 signaling leads to up-regulation of lipase 3 and enzymes for mitochondrial β-oxidation. This results in enhanced lipolysis, elevated concentrations of free fatty acids, maximal β-oxidation, and mitochondrial abnormalities. Increased acid lipase expression and accumulation of free fatty acids are also present in a Pex19-deficient patient skin fibroblast line, suggesting the conservation of key aspects of our findings.
INTRODUCTION
Peroxisomes, while rather simply structured organelles delimited by a single membrane, harbor complex metabolic functions, which are still incompletely understood. In mammalian cells, they are involved in the β-oxidation of very-long-chain fatty acids (VLCFAs), the formation of ether phospholipids (like plasmalogens), the catabolism of branched-chain fatty acids, the production of bile acids, polyamine oxidation, and amino acid catabolism (Lodhi and Semenkovich, 2014). Consequently, inherited peroxisomal defects lead to complex metabolic alterations. For example, peroxisomal biogenesis disorders (PBDs) of the Zellweger syndrome spectrum, which are caused by the complete loss of functional peroxisomes due to mutation of one of the ∼15 Pex genes, are characterized by distinctive facial features, hypotonia, feeding problems, neurodegenerative symptoms, and defects in the liver, heart, and kidneys. Zellweger syndrome, the most severe form of PBDs, typically leads to lethality within the first year of life (Steinberg et al., 2006). PBDs can be distinguished from single enzyme deficiencies of the peroxisomal β-oxidation machinery, which also lead to VLCFA accumulation, although other peroxisomal functions are preserved. However, more severe forms, like bifunctional protein deficiency, closely resemble Zellweger syndrome, suggesting that VLCFA accumulation is an important factor in the pathology of PBDs (Ferdinandusse et al., 2006).
Although fatty acids up to a chain length of C20 can be used by the mitochondrial β-oxidation machinery for energy gain, VLCFAs of C22 and more are not substrates for the mitochondrial transporter carnitine palmitoyltransferase (CPT-I), and thus cannot enter mitochondria for β-oxidation. Instead, VLCFAs are exclusively catabolized in peroxisomes, where they are shortened by the peroxisomal β-oxidation machinery. The resulting short- or medium-chain fatty acids (typically C8) are transported out of the peroxisome via specialized carnitine shuttles and then can enter the mitochondria, where they are further oxidized to acetyl-CoA while feeding the tricarboxylic acid (TCA) cycle and the electron transport chain (Wanders, 2014). It is unclear whether and to what extent the shortened peroxisomal products contribute to mitochondrial energy production, but overall the energy gain from VLCFAs is minor in comparison to shorter fatty acids due to their low abundance and the fact that they fuel mitochondria only after substantial shortening in peroxisomes (Wanders et al., 2001).
Peroxisomes exhibit a functional interplay with mitochondria by employing shared metabolic pathways and coordination of activity (Thoms et al., 2009; Wanders et al., 2009; Mohanty and McBride, 2013). They interact via mitochondria-derived vesicles (MDVs), which allow for the exchange of lipid as well as protein content between the two compartments (Neuspiel et al., 2008), and they share regulators controlling organelle biogenesis (Bagattin et al., 2010) and fission (Pan and Hu, 2001; Koch et al., 2005). Recently, it has been described that mitochondria are important for peroxisome de novo biogenesis by forming preperoxisomes (Sugiura et al., 2017). The interplay between peroxisomes and mitochondria is illustrated by the fact that loss of peroxisomes leads to defects in mitochondrial metabolism and structure (Baes et al., 1997; Peeters et al., 2011, 2015; Salpietro et al., 2015), highlighting a role of mitochondrial dysfunction in PBD pathology. The mechanism behind these defects, however, remains elusive in spite of some major efforts in the field.
The machinery involved in the inheritance, assembly, division, and maintenance of peroxisomes is encoded by peroxin (Pex) genes, which are highly conserved in evolution from yeast to mammals (Pieuchot and Jedd, 2012). Pex19 is a predominantly cytoplasmic peroxisomal core factor and essential for both the import of peroxisomal membrane proteins (PMPs) and the de novo formation of peroxisomes (Léon et al., 2006; Brown and Baker, 2008). Together with Pex3 and Pex16, it is responsible for the translocation of membrane proteins and membrane vesicle assembly (Fujiki et al., 2014). Mutations in Pex genes lead to the loss of peroxisomes and cause PBDs, and Pex19 loss of function specifically leads to Zellweger syndrome, the severest form of PBDs (Braverman et al., 2016).
The peroxisomal biogenesis and assembly machinery as well as metabolic functions are well conserved in Drosophila melanogaster (Mast et al., 2011; Faust et al., 2012, 2014; Sivachenko et al., 2016). We therefore generated and characterized a Drosophila Pex19 mutant, which recapitulates all major hallmarks of Zellweger syndrome, such as absence of peroxisomes, VLCFA accumulation, mitochondrial defects, neurodegeneration, and early lethality, therefore serving as a model for PBDs. This efficient and genetically tractable model system was used in order to identify metabolic abnormalities downstream from peroxisomal loss. We were able to identify severely increased lipolysis in Pex19 mutants driven by hyperactive Hnf4 signaling, which promotes mitochondrial damage by increasing mitotoxic free fatty acid levels. Our data thereby contribute to the current efforts in the field aiming at unraveling pathological cascades causing mitochondrial damage in PBDs.
RESULTS
Generation of a Drosophila Peroxin19 mutant as a model for the human disease class of PBDs
The single Pex19 orthologue in D. melanogaster is well conserved on the sequence and structural level. It contains the typical Pex19 core domain (Figure 1A; Sato et al., 2010) and a highly conserved Pex3 binding domain, both required in vertebrate Pex19 for its activity. Deletion of one of the ∼15 peroxins leads to the disruption of peroxisome assembly and prevents the formation of functional peroxisomes, resulting in humans in PBDs. When Pex19 is affected, the consequence is the severest form of PBDs, Zellweger syndrome, consistent with a dual function of Pex19 in de novo formation of peroxisomal vesicles together with Pex3, and its role in the import of PMPs. We generated a deletion of the coding region of the gene by imprecise excision (Figure 1B), and molecularly characterized and identified the resulting Pex19ΔF7 flies as transcript null mutants (Supplemental Figure S1; subsequently referred to as Pex19 mutants). Because Drosophila deposits maternal-derived mRNAs and proteins in the zygote, we also generated maternal-zygotic Pex19 mutants using FRT40A recombinase-driven germline mutation to ensure the absence of maternal Pex19. The resulting Pex19−/− (m–/z–) mutants, lacking both maternal and zygotic components of Pex19, die at the end of embryogenesis, and ultrastructural analysis revealed the absence of peroxisomes (Figure 1, C and D). In contrast, the zygotic mutants survive longer and lose peroxisomes during larval development (Figure 1, E and F). Most larvae develop to pupae (∼70%), but only 20% adults develop and most of them die during the first 24 h (Figure 1G).
FIGURE 1:

(A) Protein structure and sequence of Pex19 from human (HsPex19) and Drosophila melanogaster (DmPex19) and their conservation (ClustalOmega alignment; * indicates identical amino acids; : indicates conservation between groups of strongly similar properties—scoring >0.5 in the Gonnet PAM 250 matrix; . indicates conservation between groups of weakly similar properties—scoring ≤0.5 in the Gonnet PAM 250 matrix). (B) Schematic representation of the Pex19 gene locus and the deletion in the Pex19ΔF7 mutant. (C, D) Ultrastructural analysis of diaminobenzidine (DAB) stained peroxisomes of w- and maternal-zygotic Pex19−/− (m–/z–) mutant embryos reveals electron dense DAB-positive peroxisomes (white arrows in C) only in w- but not in Pex19−/− (m–/z–) embryos. m, mitochondria; n, nucleus; ld, lipid droplets; ER, endoplasmatic reticulum. (E, F) Zygotic Pex19−/− larvae do not have peroxisomes, as shown by the absence of a punctate pattern of SKL-GFP–positive peroxisomes in mutants (F) as compared with w- larvae (E). (G) Lethality profile of Pex19−/− mutants, indicating the number of pupae, adults including pharates, and viable adults which survived for more than 24 h. n = 20 in groups of 25 individuals. (H, I) Pex19 mutant adult escapers show high numbers of apoptotic cells in the optic lobe of the brain. (J) Climbing speed of Pex19−/− shows low performance in the negative geotaxis assay as a readout for neurodegeneration. Scale bars as indicated. Error bars represent SD. *, p < 0.05; ***, p < 0.001.
A common feature of PBDs is neurodegeneration. We analyzed apoptosis in the CNS of adult Pex19 mutant escapers by AnnexinV-FITC staining, and found that the number of apoptotic cells is highly increased compared with age-matched wild types (Figure 1, H and I). A reliable readout for neurodegeneration in Drosophila is the negative geotaxis assay or climbing assay. We found that adult Pex19 mutants are weak, do not inflate their wings properly, are unable to fly, and show low performance in the climbing assay (Figure 1J).
Pex19 mutants show aberrant lipid metabolism
A genetic rescue with a Pex19 expression construct could be achieved with the ubiquitous tubulin-Gal4 driver and with pumpless-Gal4, which drives expression in the fatbody and gut. Expression with neuronal (elav-Gal4) or glia (repo-Gal4) drivers failed to rescue the lethality of Pex19 mutants, which highlights an important role for Pex19 in metabolic organs rather than the CNS (Table 1).
TABLE 1:
Driver lines used to express UAS-Pex19: expression in metabolic organs, but not in the CNS, rescues Pex19 mutants.
| Driver line | Expression | Rescue | Phenotype |
|---|---|---|---|
| Tubulin-Gal4 | Ubiquitous | Yes | Male sterile |
| cg-Gal4 | Fatbody, hemocytes | No | – |
|---|---|---|---|
| repo-Gal4 | Glia | No | – |
| elav-Gal4 | Neurons | No | – |
|---|---|---|---|
| pumpless-Gal4 | Metabolic organs: Fatbody, gut, salivary glands, proventriculus, Malpighian tubes | Yes | Fertile |
To assess the consequences of peroxisome loss in metabolic organs, we stained neutral lipids (such as triacylglycerols) in larval tissue, and found that storage fat in the gut is depleted (Figure 2, A and C) and oenocytes (hepatocyte-like cells; Gutierrez et al., 2007) are lipid-filled, which indicates up-regulated lipolysis and lipid mobilization in Pex19 mutants (Figure 2, B and D). Peroxisome loss is often associated with lipid imbalance, like accumulation of peroxisomal educts such as VLCFAs (>C20). To analyze the lipid profile of Pex19 mutants, we determined the fatty acid chain lengths of fatty acid methylesters (FAMEs) by gas chromatography/mass spectrometry (GC/MS). FAMEs represent the sum of all fatty acids from all lipid classes, such as, for example, triglycerides, phospholipids, acyl-CoAs, and free fatty acids. Consistent with a loss of peroxisomal function, GC/MS analysis of FAMEs prepared from larvae shows that lipids containing VLCFAs are present at increased levels in flies lacking peroxisomes, similar to the situation in PBD patients. In addition, we found that lipids containing MCFAs (≤C12) and LCFAs (C14–C20) are reduced (Figure 2E). Also, the total amount of FAMEs per milligram of tissue is lowered (Figure 1F), indicating reduced amounts of lipids. VLCFAs are present at ∼0.01–0.2 nmol/mg tissue and their potential contribution to energy gain is therefore negligible, whereas M- and LCFAs are present at ∼1−40 nmol/mg tissue (depending on length; Supplemental Figure S1, B and C) and can provide fuel for energy gain by the mitochondria.
FIGURE 2:

(A, C) OilRed O staining of neutral lipids in anterior midguts of third instar larvae shows depleted fat stores in Pex19 mutants. (B, D) OilRed O staining of neutral lipids in larval oenocytes shows increased lipid content in Pex19 mutants’ oenocytes. (E) Fatty acid methyl esters (FAMEs) from Pex19−/− larvae, normalized to w- control. n = 6 in groups of 20 individuals. VLCFAs are enriched, whereas shorter fatty acids (C12–C20) are depleted in Pex19 mutants. (F) Sum of FAMEs measured in Pex19 mutants and w-, corresponding to overall lipid content, shows lipid depletion in Pex19 mutants (for a detailed FAME profile, see Supplemental Figure S1, B and C). Error bars represent SD. *, p < 0.05. (G, H) Staining of third instar larval Malpighian tubes with MitoTracker Red CM-H2XRos to detect ROS. (I, J) Staining of third instar larval Malpighian tubes with MitoSOX to detect production of superoxide anions by mitochondria. Scale bars as indicated.
Formation of ROS/RNS in Pex19 mutants
Peroxisomes have an important role in the cellular redox system, and peroxisome deficiency is characterized by increased levels of reactive oxygen species (ROS). We visualized ROS levels by staining Malpighian tubules of third instar larvae with MitoTracker Red CM-H2XRos, a dye which emits fluorescence only upon oxidation in the cell before entering the mitochondrion. It thus detects cellular oxidative capacity. We found that cells of Pex19 mutants show increased staining with MitoTracker Red CM-H2XRos compared with wild types (Figure 2, G and H). The dye MitoSOX enters the mitochondrion and emits fluorescence in the presence of superoxide anions. It thus detects mitochondrial production of this ROS species. We could not detect MitoSOX staining in wild-type mitochondria, while the large, swollen mitochondria in Pex19 mutants are positive for MitoSOX, indicating increased mitochondrial superoxide production (Figure 2, I and J).
Reactive nitrogen species (RNS) arise from a reaction of nitric oxide (NO) with ROS to form peroxynitrite (O2− + NO). We therefore measured nitrite concentrations in larval lysates, as nitrite is an indirect measure for NO (oxidation product of NO). NO is produced by nitric oxide synthases (NOS) and serves as a signaling molecule and in immune defense (Thomas, 2015). We measured reduced amounts of nitrite in Pex19 larvae (Supplemental Figure S1D), which is consistent with increased oxidation of NO to peroxynitrite in the presence of increased amounts of ROS, but could also hint at reduced production of NO, fitting to the fact of peroxisomal NOS being an important source for NO production (Fransen et al., 2012). Taking the results together, while ROS production is clearly increased in Pex19 mutants (Figure 2, E–H), we cannot draw clear conclusions with regard to RNS production.
Gene expression of metabolic enzymes is altered in Pex19 mutants
To further characterize the lipid imbalance and metabolic state of Pex19 mutants, we analyzed the expression of several genes encoding for metabolic enzymes reportedly regulated on the transcript level (Chambers et al., 2012), as well as other enzymes of interest (summarized in Figure 3 and Supplemental Table S1). We suspected that Pex19 mutants are in a state of starvation (as indicated by the lack of M/LCFAs and lipid mobilization in gut/oenocytes) and therefore low insulin signaling levels, but instead found that insulin signaling is elevated, as indicated by reduced expression of 4E-BP and the insulin receptor (InR; Supplemental Table S1). We next analyzed the expression of lipases, which could be involved in the observed lipid mobilization. Surprisingly, the majority of lipases analyzed, including lipase 4 (Vihervaara and Puig, 2008) and the ATGL homologue brummer (Grönke et al., 2005), are reduced in Pex19 mutants (Supplemental Table S1). By contrast, expression levels of lipase 3 were strongly elevated (∼250-fold compared with wt control; Figure 3 and Supplemental Table S1). Of note, lipase 4 and brummer are negative targets of insulin signaling and therefore up-regulated under starvation to allow lipolysis for energy gain. lipase 3, on the other hand, is also up-regulated in starved animals, but not as a direct target of insulin signaling (Becker et al., 2010). It is instead regulated by the hepatic nuclear factor Hnf4 (Palanker et al., 2009). Increased lipase3 expression (indicative of starvation) while insulin signaling is high is therefore a surprising contradiction and raises the question whether lipid sensing is impaired in Pex19 mutants, because lipolysis is up-regulated in spite of high insulin signaling. Among the glycolytic and gluconeogenic enzymes, we found hexokinase c, which catalyzes the initial step of glycolysis (conversion of glucose to glucose-6-phosphate), ∼30-fold up-regulated. We analyzed three genes involved in mitochondrial acyl-CoA import, carnitine-O-octanoyltransferase (CROT) and the carnitine-palmitoyltransferases 1 (CPT1) and 2 (colt), and found all of them up-regulated in Pex19 mutants (Figure 3 and Supplemental Table S1). Other genes for enzymes involved in lipid catabolism (mitochondrial β-oxidation, fatty acid activation, TCA cycle, etc.), most prominently the acetyl-CoA acyltransferase yip2 (yippee interacting protein 2, ∼20-fold) and the long-chain fatty acid CoA ligase (Acsl, ∼15-fold), were also up-regulated. Strikingly, most of the up-regulated genes in Pex19 mutants, including lip3, are targets of the hepatocyte nuclear factor 4 (Hnf4), a transcription factor that regulates metabolic processes such as lipolysis and mitochondrial fatty acid β-oxidation in response to starvation (summarized in Figure 3; Palanker et al., 2009), and stimulates insulin secretion (Barry and Thummel, 2016). This prompted us to investigate Hnf4 in the context of peroxisome deficiency.
FIGURE 3:
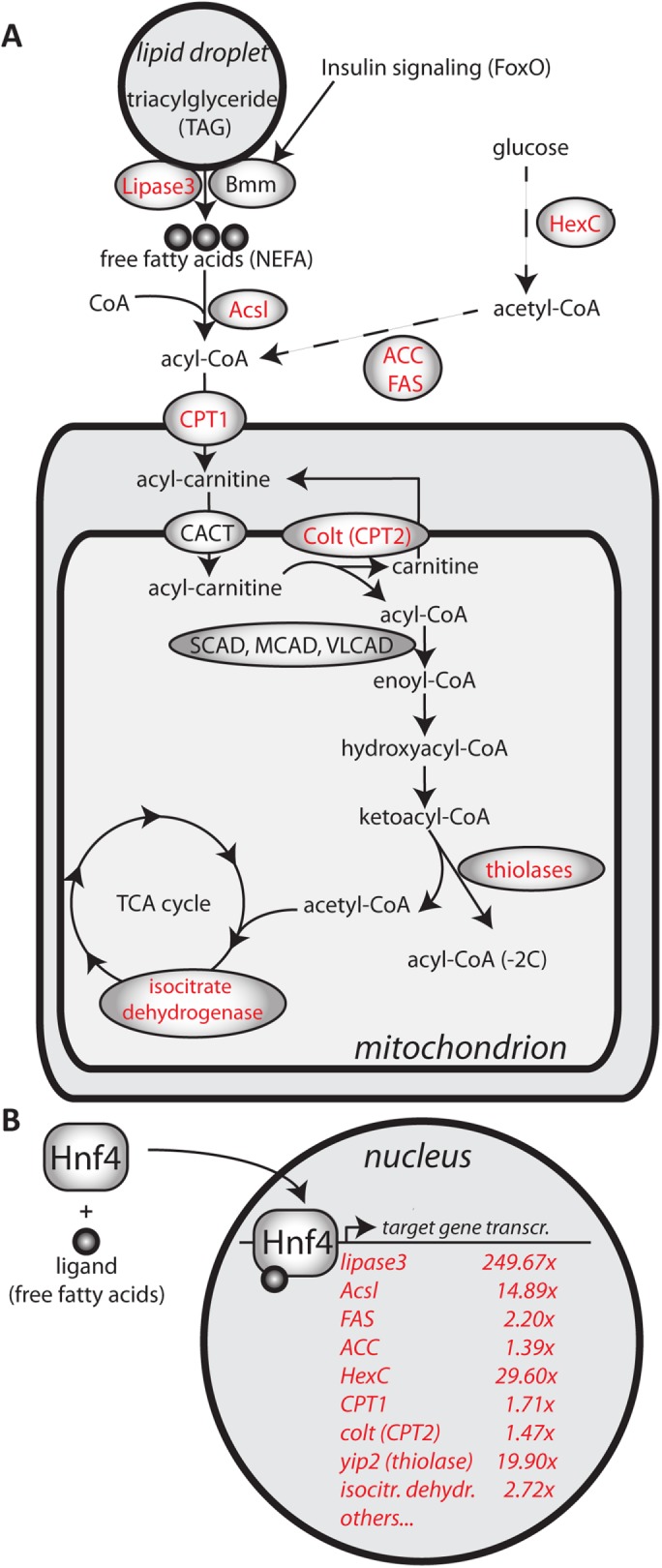
(A) Schematic representation of metabolic enzymes analyzed in this study. Enzymes depicted in red are transcriptionally regulated by Hnf4. (B) Transcriptional regulation of Hnf4 target genes in Pex19 mutants upon ligand activation of Hnf4. Values represent fold change, normalized to w-, determined by real-time reverse transcriptase (RT)-PCR analysis. n ≥ 5 in groups of five individuals.
Hnf4 is hyperactive in Pex19 mutants
Hnf4 is an important regulator of mitochondrial β-oxidation and lipolysis. Upon starvation, insulin-responsive lipases (like the ATGL homologue brummer) release free fatty acids from storage fat, which bind Hnf4 as activating ligands. Hnf4 then translocates to the nucleus and induces a transcriptional program to up-regulate β-oxidation for energy gain (compare Figure 3 for an overview of Hnf4 targets). The striking transcriptional up-regulation of several Hnf4 target genes prompted us to determine the ligand-dependent activation of Hnf4 in Pex19 mutants. To this end, we used a LacZ reporter under the control of a heat shock–inducible Hnf4:Gal4 fusion protein that expresses LacZ in the presence of Hnf4-specific ligands (Palanker et al., 2009). We did not observe Hnf4 activity in fatbodies of third instar wild-type larvae, but high levels of Hnf4 reporter induction in Pex19 mutants, confirming that Hnf4 is hyperactive in Pex19 mutants (Figure 4A).
FIGURE 4:

(A, B) An Hnf4 reporter, inducible by heat shock, which expresses LacZ under the control of the Hnf4 ligand-binding domain in the presence of ligands (Palanker et al., 2009), was crossed into the Pex19 mutant background. The reporter indicates Hnf4 induction in fatbodies of third instar larvae in Pex19 mutants. (C) Amount of nonesterified fatty acids (NEFAs) in whole third instar larvae. Dots represent single experiments in groups of three larvae; black bars represent median. (D) Determination of de novo lipid synthesis as the amount of incorporated 1-14C-acetate in lipid extracts of w- and Pex19 third instar larvae. (E) Percentage of de novo synthesized lipids contributing to the free fatty acid pool. (F) Lethality profile of Hnf4+/−, Pex19−/− mutants, indicating the number of pupae, adults including pharates, and viable adults that survived for more than 24 h. n ≥ 10 in groups of 25 individuals. (G) Representation of putative Hnf4 target gene expression levels: all of the measured Hnf4 target genes are up-regulated in Pex19 mutants (fold change normalized to w- in black), but less so in Hnf4+/−, Pex19−/− mutants (fold change normalized to w- in red). n ≥ 5 in groups of five individuals. (H–J) OilRed O staining of neutral lipids in anterior midguts of third instar larvae indicates restored lipid storage in Hnf4+/−, Pex19−/− guts (compare H to G). (K–M) Apoptotic cells in the optic lobes of adult brains stained with AnnexinV-FITC indicates reduction of neurodegeneration in Hnf4+/−, Pex19−/− (compare K to J). Scale bars as indicated. Error bars indicate SD. *, p < 0.05; ***, p < 0.001.
Nonesterified fatty acids accumulate in Pex19 mutants
Fatty acids occur in lipids mainly esterified to glycerol (in TAGs or phospholipids), or to coenzyme A (CoA) in their activated form as acyl-CoAs. In contrast, free fatty acids are not esterified and therefore often referred to as nonesterified fatty acids (NEFAs), and occur upon lipolysis of storage fat (triacylglycerols) under starvation. In order for them to be used by metabolic pathways/in enzymatic reactions, they must be activated subsequently by acyl-CoA synthetases (for summary, see Figure 3). Free fatty acids/NEFAs act as ligands for Hnf4, which as a reaction starts a lipid catabolic transcriptional program of increased mitochondrial β-oxidation and further lipolysis (Palanker et al., 2009). Because increased Hnf4 activity from the Hnf4 ligand-binding reporter line (Figure 4, A and B) indicates increased free fatty acid levels, we wanted to analyze the free fatty acid content of tissue of Pex19 mutant larvae. To this end, we developed an adapted protocol of the copper-triethanolamine method (Tinnikov and Boonstra, 1999) suitable for D. melanogaster tissue. We found that the free fatty acid content is indeed significantly elevated in Pex19 samples compared with control (Figure 4C). We considered if elevated VLCFA levels associated with the loss of peroxisomes might contribute to the observed free fatty acid levels. However, we ruled this explanation out, because, as FAMEs, they account for only 0.34 nmol/mg tissue (C20:0–C26:0) in control larvae, and 0.43 nmol/mg tissue in Pex19 mutants (C24:0–C26:0: 0.028 and 0.046 nmol/mg tissue in control and Pex19 mutant larvae, respectively), whereas the free fatty acid content is ∼4 nmol/mg tissue on average in control and ∼6 nmol/mg tissue in Pex19 mutant larvae (see Supplemental Figure S1E for comparison and summary). These numbers mean that, even if one would assume that all VLCFAs measured as FAMEs in the mutant were actually free fatty acids, they could only account for a small fraction of the observed increase of ∼2 nmol/mg tissue. Thus, lipolysis of medium- and long-chain fatty acid–containing lipids contributes to the elevated free fatty acid levels, consistent with highly increased lipase3 levels as a lipolytic enzyme.
Increased fatty acid synthesis in Pex19 mutants
Although Pex19 mutants are clearly in a state of high lipolysis, indicated by increased lip3 expression and empty lipid stores, we wanted to determine fatty acid synthesis, because this process is also regulated by Hnf4 via the target genes Fas (fatty acid synthase) and ACC (acetyl-CoA carboxylase), which are up-regulated in Pex19 mutants. We measured lipogenesis as the incorporation of 1-14C-acetate and found that it is increased in lipid extracts of Pex19 mutants compared with wild types (Figure 4D). The percentage of radioactivity incorporated into newly synthesized free fatty acids is the same in wild types and Pex19 mutants (Figure 4E). The total free fatty acid pool is thus supplied by both lipolysis and lipogenesis. These results indicate that lipolysis rates surpass increased lipogenesis, consistent with extremely high lip3 expression levels. The result is also consistent with highly increased expression levels of HexC, which could funnel glucose into the lipogenesis pathway.
Genetic reduction of Hnf4 rescues Pex19 mutants to adulthood
To test the impact of Hnf4 hyperactivity on the lethality of Pex19 mutants, we generated double knockouts of Hnf4 and Pex19 by genetic recombination of Pex19ΔF7 with the Hnf4Δ33 allele (Palanker et al., 2009). We found that mutation of one copy of Hnf4 is sufficient to rescue Pex19 mutants to adulthood (Figure 4F). The resulting Hnf4+/−, Pex19−/− (from now on referred to as Hnf4, Pex19) mutants showed a rate of hatched adults of >60%. Adult Hnf4, Pex19 mutants were viable and fertile, but died 2–3 wk after hatching from the pupa. This result indicates that elevated Hnf4 signaling indeed contributes substantially to the lethality of Pex19 mutants.
Hnf4 mutation rescues gut lipid storage and neurodegeneration
To analyze the effect of reduced Hnf4 activity on the Pex19 phenotype, we stained neutral lipids in Hnf4, Pex19 mutant larval midguts with OilRed O and found that gut lipid storage is restored in Hnf4, Pex19 mutants, suggesting reduced lipolysis (Figure 4, H–J). In addition, we found that expression of lip3, as well as most other target genes of Hnf4, is reduced, suggesting normalized lipid metabolic processes (Figure 4G). When we analyzed neurodegeneration in the CNS of adult Hnf4, Pex19 mutants by staining apoptotic cells with AnnexinV-FITC, we found that neurodegeneration is also reduced (Figure 4, K–M).
Elevated levels of mitochondrial β-oxidation in Pex19 mutants
Hyperactive Hnf4 not only provokes further lipolysis, but also increases mitochondrial β-oxidation. Therefore, we first measured citrate synthase activity and mitochondrial DNA to analyze mitochondrial abundance, and found that both were unchanged in Pex19 mutants (Figure 5, A and B). Next, we measured mitochondrial β-oxidation in control and Pex19 mutant larvae by direct quantification of permeabilized tissue with a Clark electrode (Oxygraph). β-oxidation levels were measured as the amount of oxygen consumption (in the presence of palmitoyl-CoA substrate, the necessary TCA cycle intermediates, and ADP, i.e., state 3 respiration) that is sensitive to etomoxir, a standard carnitine palmitoyl transferase (CPT-I) inhibitor (Kuznetsov et al., 2008; Xu et al., 2012). We found that Pex19 mutants have elevated levels of etomoxir-sensitive oxygen consumption and therefore increased mitochondrial β-oxidation rates compared with control animals (Figure 5C and Supplemental Figure S2A). These findings suggest that Pex19 mutants display a shortage in M- and LCFAs as a result of their degradation at high rates in mitochondrial β-oxidation, consistent with hyperactive Hnf4 signaling and increased lipolysis.
FIGURE 5:

(A) Determination of citrate synthase activity. (B) Mitochondrial DNA relative to genomic DNA in w- and Pex19−/−. (C) Etomoxir-sensitive mitochondrial β-oxidation rate. n = 4 in groups of 10 individuals. (D–G) TMRE staining to visualize active mitochondria in Malpighian tubules of third instar larvae. Scale bars represent 20 µm. (H) Quantification of mitochondrial particle size. (I) Concentration of nonesterified fatty acids (NEFAs) in whole third instar larvae. Dots represent single experiments; black bars represent median. (J, K) Ultrastructural analysis of maternal-zygotic Pex19−/− mutant embryos (K) shows mitochondrial swelling as compared with normal mitochondrial morphology in w- embryos (J). ld, lipid droplet; n, nucleus. Black arrows point to normal mitochondria with intact cristae, arrowheads indicate mitochondria with altered cristae morphology, asterisks indicate swollen mitochondria with lost cristae structure. White arrows indicate DAB-positive peroxisomes. (L) Percentage of adult survivors upon lipase 3 overexpression compared to controls. Dots represent single experiments (group of 25 individuals); black bars represent median. Error bars represent SD. *, p < 0.05; **, p < 0.01.
Hnf4 hyperactivity induces mitochondrial alterations and promotes excess free fatty acids
In most peroxisomal diseases, mitochondrial abnormalities are reported. We therefore analyzed mitochondrial morphology using mitotracker green (Supplemental Figure S2B; stains all mitochondria) and tetramethylrhodamine ethylester (TMRE; incorporated into active mitochondria as a measure for membrane potential) to stain mitochondria in live tissue samples. We found that mitochondria are small and more tubular in wild types (Figure 5D), whereas they are increased in size and balloon-shaped in Pex19 mutants (Figure 5, E and H). To test whether this phenotype is dependent on Hnf4 hyperactivity, we analyzed tissue of Hnf4, Pex19 double mutants and found that here, mitochondria are smaller and resemble more the wild-typic mitochondria (Figure 5F). Restored mitochondrial shape in Hnf4, Pex19 mutants is concomitant with lowered levels of free fatty acids (Figure 5I). These results show that hyperactive Hnf4 is responsible for accumulating free fatty acids and mitochondrial alterations in Pex19 mutants.
To further characterize the observed mitochondrial alterations, we analyzed embryos of Pex19 mutants lacking both maternal and zygotic Pex19 (termed Pex19(m-z-), as described above) by transmission electron microscopy (TEM) to observe mitochondrial ultrastructure. Although the majority of mitochondria in wild-typic embryos show intact cristae and tubular shapes (Figure 5J, black arrows), with only a few looking slightly swollen with distorted cristae morphology, most mitochondria in Pex19 (m-z-) embryos display round shapes and cristae abnormalities (Figure 5K, arrowheads), with some swollen mitochondria completely devoid of recognizable cristae structures (Figure 5K, asterisks).
Lipase 3 overexpression phenocopies some aspects of Pex19 deletion
Because we argue that increased levels of free fatty acids are a consequence of high transcript levels of lip3, we induced lip3 expression in a wild-typic background using the Gal4/UAS system. We first tested our overexpression system and found that with the fatbody drivers pumpless (ppl-) and combgap (cg-) Gal4 as well as the ubiquitous, steroid-inducible driver Tubulin-GeneSwitch (TubGS), we were able to increase the transcript levels of lip3 ~100- to 150-fold compared with wild type (Supplemental Figure S3, A and B). We decided to use the fatbody driver cg-Gal4 to overexpress lip3 and analyzed mitochondrial morphology. We found that in a wild-typic background, overexpression of lip3 induces mitochondrial alterations: mitochondria are fragmented and balloon-shaped (Figure 5, G and H, and Supplemental Figure 3C), resembling the situation in Pex19 mutants. Furthermore, we found that free fatty acids accumulate (Figure 5I) and that overexpression of lip3 leads to reduced survival rates both with the fatbody driver cg-Gal4 (Figure 5L) and the ubiquitous driver TubGS upon expression induction with mifepristone/RU486 (Supplemental Figure 3D). Therefore, inducing lipolysis by overexpressing lip3 alone can phenocopy key aspects of Pex19 mutants, which highlights the role of increased lipolysis by lip3 in their pathology.
Accumulating free fatty acids and mitochondrial swelling are also present in human fibroblasts lacking PEX19
To elucidate the relevance of our findings in a mammalian system, we analyzed mitochondrial morphology in a human skin fibroblast line (Δ19T) derived from a patient with a PEX19 mutation (Muntau et al., 2003) and compared it to a fibroblast line from a healthy person. We performed immunostaining using an antibody against the mitochondrial marker TOMM-20 (translocase of outer membrane). Although the mitochondria are organized in filamentous, network-like structures in control cells, they resemble the mitochondria from our Pex19 fly mutant and appear swollen in Δ19T cells (Figure 6, A and B). Because we argue that increased lipolysis and high free fatty acid levels as a consequence of peroxisome loss have an impact on mitochondrial morphology and induce swelling, we also analyzed free fatty acid concentration in Δ19T. We found that free fatty acid levels are indeed elevated in Δ19T cells to a comparable extent as in Drosophila mutant larvae (Figure 6C). We also found that the mammalian lip3 homologue acid lipase LIPA is slightly up-regulated (Figure 6D). Interestingly, the insulin signaling regulated lipase ATGL, as well as other acid lipases, are down-regulated in Δ19T cells, which is a surprising similarity to our Drosophila model, in which all other lipases (ATGL homologue brummer, gastric and acid lipases) except for lip3 are down-regulated. Taking the results together, we conclude that increased lipolysis and accumulation of free fatty acids and their damage to mitochondria upon peroxisome loss is conserved between flies and mammals.
FIGURE 6:

(A) Immunostaining with an α-TOMM-20 antibody to visualize mitochondria in human skin fibroblasts from a healthy person (control) and (B) a patient with a Pex19 mutation (Δ19T). (C) Concentration of nonesterified fatty acids (NEFAs) in human skin fibroblasts. (D) Transcript levels of human lipases in Δ19T fibroblasts, normalized to control cells. Scale bars represent 20 µm. ***, p < 0.001.
DISCUSSION
The tight interconnection of peroxisomes and mitochondria has long been established, and an impact of peroxisome loss on mitochondria has been proposed before. However, the reason for mitochondrial damage as a result of peroxisome deficiency has remained elusive. Here we propose a mechanism by which peroxisome loss due to Pex19 mutation leads to mitochondrial swelling through dysregulated lipolysis. We show that peroxisome loss leads not only to VLCFA accumulation, but also depletion of shorter fatty acids, probably provoked by enhanced lipolysis by massively increased expression of lipase3, and their subsequent degradation in mitochondria. Pex19 mutants therefore are in a constant state of lipid catabolism, with highly increased fatty acid release from fat stores and their subsequent β-oxidation in mitochondria. However, in spite of increased mitochondrial flux, free fatty acids accumulate in Pex19 mutants, suggesting that their availability surpasses the turnover rate by the subsequent lipid catabolic steps (from activation with CoA by acyl-CoA synthetases to their shortening in β-oxidation). Free fatty acids are activating ligands for the lipid sensor Hnf4, which accounts for further up-regulation of lipase3 and induction of enzymes for mitochondrial acyl-CoA import and β-oxidation. Subsequently, a vicious cycle of Hnf4-driven increased lipolysis/high free fatty acid amounts ensues, which in turn can further activate Hnf4 as its ligands. This spiral results in extreme up-regulation of lip3, depletion of the lipid stores, and free fatty acid accumulation, whereas mitochondrial β-oxidation runs at maximal capacity. At the same time, Hnf4 hyperactivity also drives increased lipogenesis, further contributing to the free fatty acid load. Ultimately, the described effects cause mitochondrial swelling and damage, leading to energy deficiency and lethality (Figure 7). High β-oxidation rates and increased mitochondrial flux, as well as accumulating free fatty acids, cause stress for mitochondria for a number of reasons. First, β-oxidation produces superoxide anions, thereby increasing oxidative damage of mitochondria. Second, increased mitochondrial flux/β-oxidation rates induce mitochondrial fragmentation, which is, for example, observed in metabolic syndrome when constant overfeeding leads to a metabolic shift in mitochondria, resulting in preferential oxidation of fatty acids as energy substrates at all times. As a result, mitochondrial quality control by mitophagy is impaired, because it depends on mitochondrial dynamics of fusion and fission cycles (Liesa and Shirihai, 2013). Third, increased free fatty acid levels can harm mitochondria for a number of reasons, among them their effect on membrane permeability with mitochondrial swelling as a possible result (Sultan and Sokolove, 2001; Schönfeld and Reiser, 2013).
FIGURE 7:

Summary of the role of Hnf4 in the pathology of Pex19 mutants. Our data indicate that massively increased expression of lipase3 (∼250-fold up-regulation) leads to the release of nonesterified fatty acids (NEFAs), which are toxic for mitochondria and can cause mitochondrial swelling. Additionally, NEFAs act as ligands for Hnf4, which as a result enters the nucleus to start a lipid catabolic program (compare Figure 3 for an overview of relevant Hnf4 target genes). Of note, lipase3 itself is a target gene of Hnf4, resulting in even higher amounts of lipase3 activity and further increase of NEFA levels, starting a detrimental cycle of Hnf4 hyperactivity. Hnf4 also regulates enzymes involved in the degradation of NEFAs, among them Acsl and mitochondrial enzymes (compare Figure 3 for details), leading to increased mitochondrial flux and increased ROS production with potentially harmful effects on mitochondria. However, increased NEFA levels indicate that their production surpasses the degradation capacity of mitochondria in Pex19 mutants. Besides lipolysis, increased amounts of acetyl-CoA from glycolysis (HexC up-regulation as a result of Hnf4 hyperactivation) feed into NEFA production via lipogenesis (Hnf4-dependent up-regulation of ACC and FAS). Green indicates pathways contributing to free fatty acid production. Red indicates pathways that potentially reduce free fatty acids by esterification to CoA by Acsl and subsequent degradation in mitochondria (red arrows).
Although our study provides an explanation for mitochondrial damage upon peroxisome loss and characterizes increased lipolysis and free fatty acid accumulation as a novel feature of the pathophysiology of PBDs, it remains unclear how this aberrant metabolic program is induced, because Hnf4-driven lipid catabolism seems to be a consequence of free fatty acid accumulation (via their activity as Hnf4-ligands), although lip3 is also a transcriptional target of Hnf4. However, lip3 expression is not reduced to wild-type levels in Hnf4, Pex19 double mutants; thus, other factors might be involved in transcriptional lip3 (dys-)regulation. One candidate is the ceramide synthase Schlank, which has been shown very recently to function as an unusual transcription factor, with lip3 as a negative target gene (Sociale et al., 2018). Furthermore, Schlank exerts its function as a transcription factor in response to fatty acids. Future studies will exploit possible links between Pex19, Hnf4, and Schlank.
Although lipase 3 overexpression alone leads to accumulation of toxic free fatty acids and mitochondrial swelling, which we consider a major contribution to the lethality of the mutant, it does not phenocopy the lethality of Pex19 mutants fully (although it does reduce viability to some extent). One explanation could be that peroxisomes, which are still present in lipase 3 overexpressing animals, but not in Pex19 mutants, play a role in free fatty acid clearance or mitochondrial turnover rate, thereby counteracting the detrimental effects of dysregulated lipolysis. This hypothesis fits to the observation that peroxisomes and mitochondria share fission and fusion factors and thereby might influence each other’s quality control. Additionally, other Hnf4 target genes might contribute to lipotoxicity; for example, Acsl and yip2. Our preliminary data (not shown) indicate that Acsl overexpression reduces the free fatty acid concentration in wild-typic flies and rescues Pex19 mutants to adulthood.
Taking the results together, we show that loss of peroxisomes in flies as well as a human cell line leads to increased lipolysis and accumulated mitotoxic free fatty acids, which results in mitochondrial swelling and dysfunction. We discover a pathological mechanism driven by the lipid sensor Hnf4 in Pex19 deficient flies and thereby unravel a novel cascade from peroxisome deficiency to mitochondrial damage, which can be ameliorated by genetic intervention targeting Hnf4. Our results thereby shed light on mitochondrial phenotype development in the absence of peroxisomes and promote our understanding of metabolic defects in peroxisome deficiency.
MATERIALS AND METHODS
Flywork
The Pex19 mutant was generated by transposase-induced P-element mobilization and imprecise excision. The line Pex19ΔF7 was chosen from a jump-out screen and tested as a transcript null. To detect homozygous animals, they were crossed with a CyO-twi-GFP marker. To generate mutants that lack both the zygotic and the maternal component of Pex19, we recombined the Pex19ΔF7 locus on an FRT chromosome and crossed it to a line carrying ovoD, FRT. Progeny of ovoD FRT40A/Pex19∆F7 FRT40A flies lack the maternal component of Pex19. As control flies we used the strain w1118 (Bloomington stock #3605). Wild-type and heterozygous Pex19ΔF7 flies were reared on standard fly food. Hnf4Δ33 and hs-Gal4: Hnf4, UAS LacZ flies were kindly provided by Carl Thummel, University of Utah, Salt Lake City. Hnf4Δ33 Pex19ΔF7 double mutants were generated by genomic recombination, and Hnf4Δ33/+ Pex19ΔF7/ΔF7 mutants by crossing the double mutant with Pex19ΔF7. The hs-Gal4;;Act5C>CD2>Gal4, UAS GFP line for clones was described previously (Bülow et al., 2014). For survival assays, larvae were collected as first instars and transferred to fresh apple juice agar plates (2 g Agar Kobe I [Roth], 2.5 g sucrose, 25 ml apple juice, 75 ml water) with yeast paste (1 cube [42 g] fresh yeast with 10 ml water). 25 larvae were collected for each condition, and at least five independent experiments were conducted. The number of surviving pupae, adults including pharates and viable adults (survivors), which were able to move and lived at least 24 h, was counted.
Cell culture
Human fibroblast control and Δ19T cells were kept in DMEM (Life Technologies) with 10% fetal bovine serum, 10,000 U of penicillin, and 10 mg streptomycin per ml. For NEFA measurement, 1 × 105 cells were seeded in six well plates and harvested after 48 h. Cells were pelleted and cell pellets were treated like larval tissue (see Free fatty acids section). For stainings, cells were seeded into eight-well slides for microscopy and immunostained after 48 h with α-TOMM20 (Sigma-Aldrich).
Imaging
Antibodies used were α-GFP (Sigma), α-spectrin (Developmental Studies Hybridoma Bank), and α-Tomm20 (Sigma-Aldrich). Secondary antibodies coupled to Alexa dyes were from Molecular Probes, and 4’,6-diamidin-2-phenylindol (DAPI) from Sigma-Aldrich. Stainings were analyzed using a Zeiss LSM 710 confocal microscope. For immunohistochemistry, we dissected tissue of interest from third instar larvae. Tissue was fixed for 30 min in 3.7% formaldehyde and washed with phosphate-buffered saline (PBS) with 0.1% Tween-20 (PBT) before and after incubation with primary antibody and Alexa dye-coupled secondary antibody. Tissue was mounted in Fluoromont G and analyzed using a Zeiss LSM 710 confocal microscope. For stainings of neutral lipids with OilRed O, larval tissue was dissected in PBS by inverting the cuticle to provide access to the oenocytes. Tissue was fixed for 20 min in 3.7% formaldehyde and washed with PBS. Before and after staining with an OilRed O solution containing 60% isopropanol for 30 min, tissue was incubated for 5 min with 60% isopropanol. Tissue was washed with PBS, mounted in glycerol, and immediately analyzed using an Olympus AX70 microscope. For visualization of ROS production with MitoTracker CM-H2XRos and MitoSOX (ThermoFisher), 96-h-old larvae were dissected in PBS and their Malpighian tubules were stained at room temperature for 30 and 10 min, respectively, according to the manufacturer’s protocol. For staining of mitochondria, 96-h-old L3 larvae were dissected in ice-cold PBS, and their Malpighian tubules were stained for 20 min at room temperature (RT) with 50 nM TMRE (Sigma-Aldrich) or MitoTracker Green (ThermoFisher) in PBS according to the manufacturer’s protocol. The Malpighian tubules were then directly mounted in Fluoromount G and analyzed with a Zeiss LSM 710 confocal microscope. Picture analysis and quantification was done using ImageJ. Each staining was done at least five times.
TEM analysis
For ultrastructural analysis by TEM, embryos were dechorionated and fixed in 50% Karnovsky fixative. Embryos were then devitellinized and postfixed with 5% glutaraldehyde for 2 h at RT. After being washed with 0.01 M Teorell Stenhagen buffer, embryos were stained with diaminobenzidine (DAB) to reveal peroxisomes. Embryos were incubated in 1% osmium tetroxide as contrasting agent and postfixed in 1% uranylacetate. Then they were dehydrated with an ethanol series and embedded in Durcupan. Ultrathin sections were prepared and analyzed using a Zeiss Libra 120 electron microscope.
Lipid profile
For quantification of FAMEs, 15 third instar larvae were homogenized in 1N MeHCl in a Precellys 24 homogenizer (Peqlab). A minimum of n = 7 were analyzed for each condition. C15:0 and C27:0 standards were added and samples were incubated for 45 min at 80°C. Methylesters were collected by addition of hexane and a 0.9% NaCl solution. The hexane phase was collected in a new glass vial and concentrated by vaporization. Samples were analyzed by gas chromatography/mass spectrometry using an Agilent HP 6890 with a HP-5MS column.
Free fatty acids
NEFAs were measured by an adaptation of the copper-soap method (Tinnikov and Boonstra, 1999). In brief, three third instar larvae were weighed and homogenized in 20 µl of 1 M phosphate buffer per mg tissue. Supernatant (25 µl) was transferred to 500 µl of chloroform/heptane 4:3, and lipids were extracted by shaking the vial for 5 min. Unspecific background provoked by phospholipids was circumvented by the addition of 23 mg of activated silicic acid. Chloroform phase (300 µl) was transferred to 250 µl of Cu-TEA (copper-triethanolamine). After shaking and centrifuging, 150 µl of the organic phase was transferred to fresh cups. Liquid was evaporated in a 60°C heat block, and lipids were dissolved in 100 µl of 100% ethanol. Copper was detected by complexation with a mixture of dicarbazone-dicarbazide, and the color intensity was measured in a 96-well plate at 550 nm in a TECAN plate reader.
β-Oxidation measurements
Six larvae per genotype were washed with PBS and their weight was recorded for normalization purposes. The larvae were inverted in ice-cold PBS and permeabilized in ice-cold BIOPS buffer (2.77 mM CaK2EGTA, 7.23 mM K2EGTA, 5.77 mM Na2ATP, 6.56 mM MgCl2∙6H2O, 20 mM taurine, 15 mM Na2∙phosphocreatine, 20 mM imidazole, 0.5 mM dithiothreitol [DTT], and 50 mM 2-(N-morpholino)ethanesulfonic acid [MES]) containing 100 μg/ml saponin (fresh) at 4°C with gentle rocking for 10 mins. Then the larvae were equilibrated in respiration medium (MiR05, 0.5 mM EGTA, 3 mM MgCl2∙6H2O, 60 mM K-lactobionate [lactobionic acid is dissolved in H2O and pH is adjusted to pH 7.4 with KOH], 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, and 1 g/l fatty acid–free BSA) supplemented with 0.5 mM carnitine. The larvae were added into the oxygraph chambers and oxygen concentration was brought to around 500 μM by using catalase and H2O2. After basal respiration was recorded, 5 µM palmitoyl CoA was added to the chamber. Fatty acid β-oxidation was induced by adding complex I substrates, ETF (electron transfer flavoprotein) substrates, and ADP (10 mM proline, 10 mM pyruvate, 5 mM malate, 5 mM glutamate, 2 mM ADP, and 15 mM glycerol-3-phosphate). After that, etomoxir was added at the indicated concentrations to block fatty acid transfer into mitochondria via CPT1, thereby blocking β-oxidation and leaving complex I–dependent respiration. Finally, residual oxygen consumption (ROX) was measured by inhibiting complex III with antimycin A. All values were corrected for ROX. β-Oxidation was calculated by subtracting etomoxir-resistant respiration from respiration in the presence of all substrates.
Fatty acid synthesis measurement
For metabolic labeling, third instar w- and Pex19−/− larvae were fed with [1–14C] sodium acetate (61 Ci/mol; Amersham, Braunschweig, Germany) in heat-inactivated yeast (10 µCi/agar dish containing 150 larvae) for 6 h. Afterward, larvae were homogenized and extracted as described for adult Drosophila in Sellin et al. (2017). The incorporated radioactivity was determined in the resulting lipid extract.
The same amounts of radioactivity were applied to each lane on TLC plates. The chromatograms were developed in hexane/diethyl ether/glacial acetic acid (70:30:1) for fatty acids. Radioactive bands were visualized with a Typhoon FLA 7000 (GE Healthcare), and quantification was performed with ImageQuant TL software (GE Healthcare). Lipids were identified using commercially available standards.
Real-time qPCR
Whole RNA of five third instar larvae was isolated using TriFast reagent (Peqlab). Tissue was homogenized using a Precellys 24 homogenizer (Peqlab). Transcription to cDNA was performed using the Quantitect Reverse Transcription Kit (Qiagen). Quantitative PCR was performed with a CFX Connect cycler (Bio-Rad). Each experiment was repeated at least five times.
Statistics
Bar graphs represent average and SD. Scatter plots represent single data points and median. We used the statistics software GraphPad InStat for our statistical analyses. The Kolmogorov–Smirnov test was applied to test normality. Two-sided Student’s t test was applied for normally distributed data in single comparisons. Asterisk representations are as follows: * = p < 0.05, ** = p < 0.01, and *** = p < 0.001. The number of replicates indicated by n represents biological replicates.
Supplementary Material
Acknowledgments
We thank the Bloomington Drosophila Stock Center for fly strains, Carl Thummel for providing Hnf4 fly lines and α-Hnf4 antibody, and Ingo Zinke and Michael Pankratz for the ppl-Gal4 and UAS lip3 line. We thank Christoph Thiele for input and help with the NEFA measurements. We thank Gabriele Dodt and Ronald Wanders for human fibroblast lines, Mélisande Richard for help with the ultrastructural analysis, Fatmire Bujupi for help with the characterization of the Pex19 mutant, the group of Peter Dörmann for help with mass spectrometry analysis of FAMEs, and members of the Hoch group for discussion. The work was funded by grants from the Deutsche Forschungsgemeinschaft (DFG) to M.H. (SFB 645, TP B1, SFB 704, TP A9, and TR 83, TP A7) and by the Helmholtz Portfolio grant “metabolic dysfunction” to M.H. and A.A.T. M.H. is a member of the Bonn Excellence Cluster ImmunoSensation.
Abbreviations used:
- FAMEs
fatty acid methylesters
- Hnf4
hepatocyte nuclear factor 4
- LCFAs
long-chain fatty acids
- lip3
lipase 3
- MCFAs
medium-chain fatty acids
- NEFAs
nonesterified fatty acids
- PBDs
peroxisomal biogenesis disorders
- Pex
peroxin
- VLCFAs
very-long-chain fatty acids.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-08-0535) on December 27, 2017.
REFERENCES
- Baes M, Gressens P, Baumgart E, Carmeliet P, Casteels M, Fransen M, Evrard P, Fahimi D, Declercq PE, Collen D, et al. A mouse model for Zellweger syndrome Nat Genet 1997. 49 57 [DOI] [PubMed] [Google Scholar]
- Bagattin A, Hugendubler L, Mueller E. Transcriptional coactivator PGC-1α promotes peroxisomal remodeling and biogenesis. Proc Natl Acad Sci USA. 2010:20376–20381. doi: 10.1073/pnas.1009176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry WE, Thummel CS. The Drosophila HNF4 nuclear receptor promotes glucose-stimulated insulin secretion and mitochondrial function in adults. eLife. 2016:e11183. doi: 10.7554/eLife.11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T, Loch G, Beyer M, Zinke I, Aschenbrenner AC, Carrera P, Inhester T, Schultze JL, Hoch M. FOXO-dependent regulation of innate immune homeostasis. Nature. 2010:369–373. doi: 10.1038/nature08698. [DOI] [PubMed] [Google Scholar]
- Braverman NE, Raymond GV, Rizzo WB, Moser AB, Wilkinson ME, Stone EM, Steinberg SJ, Wangler MF, Rush ET, Hacia JG, Bose M. Peroxisome biogenesis disorders in the Zellweger spectrum: an overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab. 2016:313–321. doi: 10.1016/j.ymgme.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LA, Baker A. Shuttles and cycles: transport of proteins into the peroxisome matrix. Mol Membr Biol. 2008:363–375. doi: 10.1080/09687680802130583. [DOI] [PubMed] [Google Scholar]
- Bülow MH, Bülow TR, Hoch M, Pankratz MJ, Jünger MA. Src tyrosine kinase signaling antagonizes nuclear localization of FOXO and inhibits its transcription factor activity. Sci Rep. 2014:4048. doi: 10.1038/srep04048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MC, Song KH, Schneider DS. Listeria monocytogenes infection causes metabolic shifts in Drosophila melanogaster. PLoS One. 2012:e50679. doi: 10.1371/journal.pone.0050679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust JE, Manisundaram A, Ivanova PT, Milne SB, Summerville JB, Brown HA, Wangler M, Stern M, McNew JA. Peroxisomes are required for lipid metabolism and muscle function in Drosophila melanogaster. PLoS One. 2014:e100213. doi: 10.1371/journal.pone.0100213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust JE, Verma A, Peng C, McNew JA. An inventory of peroxisomal proteins and pathways in Drosophila melanogaster. Traffic. 2012:1378–1392. doi: 10.1111/j.1600-0854.2012.01393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdinandusse S, Ylianttila MS, Gloerich J, Koski MK, Oostheim W, Waterham HR, Hiltunen JK, Wanders RJ, Glumoff T. Mutational spectrum of D-bifunctional protein deficiency and structure-based genotype-phenotype analysis. Am J Hum Genet. 2006:112–124. doi: 10.1086/498880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen M, Nordgren M, Wang B, Apanasets O. Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim Biophys Acta. 2012:1363–1373. doi: 10.1016/j.bbadis.2011.12.001. [DOI] [PubMed] [Google Scholar]
- Fujiki Y, Okumoto K, Mukai S, Honsho M, Tamura S. Peroxisome biogenesis in mammalian cells. Front Physiol. 2014:307. doi: 10.3389/fphys.2014.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grönke S, Mildner A, Fellert S, Tennagels N, Petry S, Müller G, Jäckle H, Kühnlein RP. Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metab. 2005:323–330. doi: 10.1016/j.cmet.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Gutierrez E, Wiggins D, Fielding B, Gould AP. Specialized hepatocyte-like cells regulate Drosophila lipid metabolism. Nature. 2007:275–280. doi: 10.1038/nature05382. [DOI] [PubMed] [Google Scholar]
- Koch A, Yoon Y, Bonekamp NA, McNiven MA, Schrader M. A role for Fis1 in both mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2005:5077–5086. doi: 10.1091/mbc.E05-02-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov AV, Veksler V, Gellerich FN, Saks V, Margreiter R, Kunz WS. Analysis of mitochondrial function in situ in permeabilized muscle fibers, tissues and cells. Nat Protoc. 2008:965–976. doi: 10.1038/nprot.2008.61. [DOI] [PubMed] [Google Scholar]
- Léon S, Goodman JM, Subramani S. Uniqueness of the mechanism of protein import into the peroxisome matrix: transport of folded, co-factor-bound and oligomeric proteins by shuttling receptors. Biochim Biophys Acta. 2006:1552–1564. doi: 10.1016/j.bbamcr.2006.08.037. [DOI] [PubMed] [Google Scholar]
- Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013:491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodhi IJ, Semenkovich CF. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab. 2014:380–392. doi: 10.1016/j.cmet.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast FD, Li J, Virk MK, Hughes SC, Simmonds AJ, Rachubinski RA. A Drosophila model for the Zellweger spectrum of peroxisome biogenesis disorders. Dis Model Mech. 2011:659–672. doi: 10.1242/dmm.007419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty A, McBride HM. Emerging roles of mitochondria in the evolution, biogenesis, and function of peroxisomes. Front Physiol. 2013:268. doi: 10.3389/fphys.2013.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntau AC, Roscher AA, Kunau WH, Dodt G. The interaction between human PEX3 and PEX19 characterized by fluorescence resonance energy transfer (FRET) analysis. Eur J Cell Biol. 2003:333–342. doi: 10.1078/0171-9335-00325. [DOI] [PubMed] [Google Scholar]
- Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol. 2008:102–108. doi: 10.1016/j.cub.2007.12.038. [DOI] [PubMed] [Google Scholar]
- Palanker L, Tennessen JM, Lam G, Thummel CS. Drosophila HNF4 regulates lipid mobilization and β-oxidation. Cell Metab. 2009:228–239. doi: 10.1016/j.cmet.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan R, Hu J. The conserved fission complex on peroxisomes and mitochondria. Plant Signal Behav. 2001:870–872. doi: 10.4161/psb.6.6.15241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peeters A, Shinde AB, Dirkx R, Smet J, De Bock K, Espeel M, Vanhorebeek I, Vanlander A, Van Coster R, Carmeliet P, et al. Mitochondria in peroxisome-deficient hepatocytes exhibit impaired respiration, depleted DNA, and PGC-1α independent proliferation Biochim Biophys Acta 2015. 285 298 [DOI] [PubMed] [Google Scholar]
- Peeters A, Swinnen JV, Van Veldhoven PP, Baes M. Hepatosteatosis in peroxisome deficient liver despite increased β-oxidation capacity and impaired lipogenesis. Biochimie. 2011:1828–1838. doi: 10.1016/j.biochi.2011.06.034. [DOI] [PubMed] [Google Scholar]
- Pieuchot L, Jedd G. Peroxisome assembly and functional diversity in eukaryotic microorganisms. Annu Rev Microbiol. 2012:237–263. doi: 10.1146/annurev-micro-092611-150126. [DOI] [PubMed] [Google Scholar]
- Salpietro V, Phadke R, Saggar A, Hargreaves IP, Yates R, Fokoloros C, Mankad K, Hertecant J, Ruggieri M, McCormick D, Kinali M. Zellweger syndrome and secondary mitochondrial myopathy. Eur J Pediatr. 2015:557–563. doi: 10.1007/s00431-014-2431-2. [DOI] [PubMed] [Google Scholar]
- Sato Y, Shibata H, Nakatsu T, Nakano H, Kashiwayama Y, Imanaka T, Kato H. Structural basis for docking of peroxisomal membrane protein carrier Pex19p onto its receptor Pex3p. EMBO J. 2010:4083–4093. doi: 10.1038/emboj.2010.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönfeld P, Reiser G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J Cereb Blood Flow Metab. 2013:1493–1499. doi: 10.1038/jcbfm.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellin J, Schulze H, Paradis M, Gosejacob D, Papan C, Shevchenko A, Psathaki OE, Paululat A, Thielisch M, Sandhoff K, Hoch M. Characterization of Drosophila Saposin-related mutants as a model for lysosomal sphingolipid storage diseases. Dis Model Mech. 2017:737–750. doi: 10.1242/dmm.027953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivachenko A, Gordon HB, Kimball SS, Gavin EJ, Bonkowsky JL, Letsou A. Neurodegeneration in a Drosophila model of adrenoleukodystrophy: the roles of the Bubblegum and Double bubble acyl-CoA synthetases. Dis Model Mech. 2016:377–387. doi: 10.1242/dmm.022244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sociale M, Wulf AL, Breiden B, Klee K, Thielisch M, Eckardt F, Sellin J, Bülow MH, Löbbert S, Weinstock N, et al. Ceramide synthase Schlank is a transcriptional regulator adapting gene expression to energy requirements Cell Rep 2018. 1 12 [DOI] [PubMed] [Google Scholar]
- Steinberg SJ, Dodt G, Raymond GV, Braverman NE, Moser AB, Moser HW. Peroxisome biogenesis disorders. Biochim Biophys Acta. 2006:1733–1748. doi: 10.1016/j.bbamcr.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Sugiura A, Mattie S, Prudent J, McBride HM. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature. 2017:251–254. doi: 10.1038/nature21375. [DOI] [PubMed] [Google Scholar]
- Sultan A, Sokolove PM. Free fatty acid effects on mitochondrial permeability: an overview. Arch Biochem Biophys. 2001:52–61. doi: 10.1006/abbi.2000.2195. [DOI] [PubMed] [Google Scholar]
- Thomas DD. Breathing new life into nitric oxide signaling: a brief overview of the interplay between oxygen and nitric oxide. Redox Biol. 2015:225–233. doi: 10.1016/j.redox.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoms S, Grønborg S, Gärtner J. Organelle interplay in peroxisomal disorders. Trends Mol Med. 2009:293–302. doi: 10.1016/j.molmed.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Tinnikov AA, Boonstra R. Colorimetric micro-determination of free fatty acids in plasma using microplate readers. Clin Chim Acta. 1999:159–162. doi: 10.1016/s0009-8981(98)00216-2. [DOI] [PubMed] [Google Scholar]
- Vihervaara T, Puig O. dFOXO regulates transcription of a Drosophila acid lipase. J Mol Biol. 2008:1215–1223. doi: 10.1016/j.jmb.2007.12.042. [DOI] [PubMed] [Google Scholar]
- Wanders RJ. Metabolic functions of peroxisomes in health and disease. Biochimie. 2014:36–44. doi: 10.1016/j.biochi.2013.08.022. [DOI] [PubMed] [Google Scholar]
- Wanders RJ, Vreken P, Ferdinandusse S, Jansen GA, Waterham HR, van Roermund CW, Van Grunsven EG. Peroxisomal fatty acid α- and β-oxidation in humans: enzymology, peroxisomal metabolite transporters and peroxisomal diseases. Biochem Soc Trans. 2001;(Pt 2):250–267. doi: 10.1042/0300-5127:0290250. [DOI] [PubMed] [Google Scholar]
- Wanders RJ, Waterham HR, Ferdinandusse S. Metabolic interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Front Cell Dev Biol. 2009:83. doi: 10.3389/fcell.2015.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Gopalacharyulu P, Seppänen-Laakso T, Ruskeepää AL, Aye CC, Carson BP, Mora S, Orešicˇ M, Teleman AA. Insulin signaling regulates fatty acid catabolism at the level of CoA activation. PLoS Genet. 2012:e1002478. doi: 10.1371/journal.pgen.1002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.