

The force spectra for various LFA-1/Mac-1–ligand bonds were compared and their functions tested in mediating PMN recruitment under flow. Multiple endothelial ligands present distinct bond rupture forces and lifetimes, which correlate well with their biological phenotypes. In addition to ICAM, JAM or RAGE is also crucial in cell adhesion and crawling.
Abstract
Lymphocyte function–associated antigen-1 (LFA-1) and macrophage-1 antigen (Mac-1) and their counterreceptors such as intercellular cell adhesion molecules (ICAM-1 and ICAM-2), junctional adhesion molecules (JAM-A, JAM-C), and receptors for advanced glycation end products (RAGE) are crucial for promoting polymorphonuclear leukocyte (neutrophil, PMN) recruitment. The underlying mechanisms of ligand-specific bindings in this cascade remain incompletely known. We compared the dynamic force spectra for various LFA-1/Mac-1–ligand bonds using single-molecule atomic force microscopy (AFM) and tested their functions in mediating PMN recruitment under in vitro shear flow. Distinct features of bond rupture forces and lifetimes were uncovered for these ligands, implying their diverse roles in regulating PMN adhesion on endothelium. LFA-1 dominates PMN adhesion on ICAM-1 and ICAM-2, while Mac-1 mediates PMN adhesion on RAGE, JAM-A, and JAM-C, which is consistent with their bond strength. All ligands can trigger PMN spreading and polarization, in which Mac-1 seems to induce outside-in signaling more effectively. LFA-1–ICAM-1 and LFA-1/Mac-1–JAM-C bonds can accelerate PMN crawling under high shear stress, presumably due to their high mechanical strength. This work provides new insight into basic molecular mechanisms of physiological ligands of β2 integrins in PMN recruitment.
INTRODUCTION
Polymorphonuclear leukocyte (neutrophil, PMN) recruitment from blood circulation to the sites of infection or sterile injury, where PMNs exhibit numerous effector functions such as phagocytosis, degranulation, and release of neutrophil extracellular traps (NETs), is the first line of host defense against invading pathogens and tissue injury (Zarbock and Ley, 2009; Robert et al., 2013; Voisin and Nourshargh, 2013; Lyck and Enzmann, 2015). The conventional PMN recruitment cascade is a multistep process including capture (or tethering), rolling, slow rolling, arrest (firm adhesion), adhesion strengthening and spreading, intraluminal crawling, and paracellular or transcellular transmigration (Ley et al., 2007; Zarbock and Ley, 2009). In addition to selectin-mediated capture and rolling, two β2 integrin members, lymphocyte function–associated antigen-1 (LFA-1, αLβ2, CD11a/CD18) and macrophage-1 antigen (Mac-1, αMβ2, CD11b/CD18), and their counterreceptors are crucial in promoting the following steps of the PMN recruitment cascade (Ley et al., 2007; Zarbock and Ley, 2009; Lyck and Enzmann, 2015). LFA-1 and Mac-1 are αβ heterodimeric transmembrane glycoproteins expressed on PMNs. Besides their common endothelial ligands of intercellular cell adhesion molecules (ICAM-1 and ICAM-2), additional counterreceptors of junctional adhesion molecules (JAM-A and JAM)-C and receptors for advanced glycation end products (RAGE) are also identified as LFA-1 or Mac-1 ligands on endothelial cell surfaces (Ostermann et al., 2002; Chavakis et al., 2003, 2004; Gorina et al., 2014; Lyck and Enzmann, 2015). JAM-A has a direct interaction with LFA-1 but not with Mac-1 (Ostermann et al., 2002), while JAM-C and RAGE bind to Mac-1 but not LFA-1 (Chavakis et al., 2003, 2004). Thus, it is physiologically relevant to elucidate the ligand-specific functions of β2 integrins in PMN recruitment.
A body of evidence suggests that PMN recruitment is tissue- and stimulus-specific, mainly depending on expression and function of β2 integrins and their diverse ligands (also seen in the summaries in Supplemental Table 1; Ostermann et al., 2002; Chavakis et al., 2003, 2004; Shaw et al., 2004; Aurrand-Lions et al., 2005; Pullerits et al., 2006; Orlova et al., 2007; Sircar et al., 2007; Menezes et al., 2009; Frommhold et al., 2010; McDonald et al., 2010; Woodfin et al., 2010; Li et al., 2012; Jenne et al., 2013; Robert et al., 2013; Gorina et al., 2014; Halai et al., 2014). On one hand, LFA-1 and Mac-1 present distinct functions in different tissues with their respective ligands. For example, time-lapse cinematography within postcapillary venules in cremaster muscle reveals that LFA-1 and Mac-1 play distinct and sequential roles in MIP-2-triggered PMN recruitment cascade, in which LFA-1 mediates initial adhesion whereas Mac-1 is required for intraluminal crawling (Robert et al., 2013). However, LFA-1–ICAM-1 and Mac-1–RAGE interactions can also work cooperatively in mediating trauma-induced leukocyte adhesion, crawling, and transmigration in cremaster muscle venules (Frommhold et al., 2010). In locally fMLF-stimulated hepatic sinusoids, Mac-1 is dominant in PMN adhesion and crawling, while LFA-1 only affects crawling velocity slightly. In contrast, Mac-1 is selectively down-regulated via IL-10 in systemically LPS-induced inflammation, leading to integrin-independent, CD44-dependent hepatic PMN recruitment (Menezes et al., 2009). On the other hand, the same ligand of β2 integrins works differentially under various physiological stimuli. For instance, ICAM-2 is required for IL-1β–, but not for MIP-2–induced PMN crawling in cremaster muscle microcirculation (Robert et al., 2013; Halai et al., 2014). JAM-A contributes to LFA-1–mediated transmigration of PMNs triggered by CXCR2 on human umbilical vein endothelial cells (HUVECs) stimulated with TNF-α and IFN-γ under shear flow in vitro (Ostermann et al., 2002), but does not participate in emigration of PMA-stimulated PMNs on TNF-α-activated HUVECs (Shaw et al., 2004). Mac-1–JAM-C binding is involved in PMN transendothelial migration toward MCP-1 but not in PMA-stimulated PMN-endothelial adhesion in a static assay in vitro (Chavakis et al., 2004). While JAM-C has no effect on PMN adhesion and transmigration on HUVECs under flow in vitro (Sircar et al., 2007), JAM-C overexpression raises PMN adhesion and transmigration through IL-1β-stimulated cremasteric venules in vivo (Aurrand-Lions et al., 2005). These seemingly controversial observations of PMN recruitment may stem from tissue- and stimulus-specific interactions between LFA-1 or Mac-1 and their multiple ligands, in which mechanical strength (rupture force) and binding kinetics (on- or off-rate and affinity) of these receptor-ligand interactions, together with molecular expression and distribution, govern how strongly the interacting PMNs remain adhered on and how fast the cells crawl over and transmigrate across the endothelium under blood flow.
LFA-1 and Mac-1 can adjust their conformations with different mechanical strengths and binding affinities to their ligands: low-affinity bent conformation with closed headpiece, intermediate-affinity extended conformation with closed headpiece, and high-affinity extended conformation with open headpiece (Luo and Springer, 2006; Lyck and Enzmann, 2015). Single-molecule force spectroscopy of ligand-specific binding of LFA-1 and Mac-1 with different activation states is important in unraveling the underlying mechanisms of PMN recruitment, since bond formation and dissociation of β2 integrin/ligand binding are prerequisite for PMN adhesion, crawling, and transmigration under external forces. Although bond lifetimes and forced activation of LFA-1–ICAM-1 bindings (Chen et al., 2010, 2012; Evans et al., 2010; Kinoshita et al., 2010), dynamic strength of LFA-1–ICAM-1, LFA-1–ICAM-2, or Mac-1–ICAM-1 interactions (Zhang et al., 2002; Wojcikiewicz et al., 2006; Yang et al., 2007), and binding kinetics of LFA-1–ICAM-1 and Mac-1–ICAM-1 complexes (Zhang et al., 2005; Fu, Tong, et al., 2011; Li et al., 2013) have been extensively investigated, much less is known for mechanical strength or kinetic rates of other LFA-1/Mac-1–ligand interactions. Moreover, PMN adhesion, crawling, and transmigration are mechanically sensitive at cellular level since blood flow varies from one to another tissue. For example, shear stress regulates leukocyte adhesion (Simon and Green, 2005), induces activation and cleavage of β2 integrins during PMN migration (Makino et al., 2007), triggers guanine nucleotide exchange factor (GEF)-H1–dependent spreading and crawling of PMNs (Fine et al., 2016), and promotes lymphocyte transmigration (Cinamon et al., 2001). Thus, it is necessary to coordinate molecular mechanical forces and binding kinetics with cellular functions for resting or activated PMN recruitment.
In the current study, we elucidated the force spectroscopy of various β2 integrin-ligand bonds in different LFA-1 and Mac-1 activation states using single-molecule atomic force microscopy (AFM). Force dependence of bond lifetimes was also determined to compare the abilities of these multiple types of bonds to resist external forces. We further tested whether the respective ligand-specific binding of LFA-1 and Mac-1 could mediate adhesion or initiate PMN spreading, polarization, and crawling under shear flow. Ligand-specific dependence of forced PMN recruitment and its underlying molecular mechanisms are also discussed.
RESULTS
Dynamic force spectrum for LFA-1 and Mac-1 binding to respective ligands
Direct measurements of molecule adhesion and bond rupture forces between human LFA-1 or Mac-1 and their respective ligands were conducted at given parameter setting (Figure 1, A–C). Here the adhesion probability, Pa, was measured with allosterically inhibitory monoclonal antibody (mAb) TS1/18 (Figure 1D) or Mn2+ (Figure 1E) to mimic their respective conformation and function of low- or high-affinity β2 integrins (Lu et al., 2001; Kong et al., 2009; Xie, Zhu, et al., 2010). Since Mn2+ can enhance LFA-1 or Mac-1 binding activity (Lu et al., 2001), 10-fold lower ligand-coating concentration was used for the tests in Mn2+ in order to maintain a similar level of Pa. The adhesion frequencies of LFA-1-RAGE and Mac-1-RAGE are extremely high compared with other ligands, and therefore the coating concentration of RAGE was further reduced 10-fold. A 16.3–36.7% Pa is observed when LFA-1- or Mac-1–coated tips contact their ligands but the adhesion is significantly abrogated to 1.2–12.9% in the presence of LFA-1 or Mac-1 blocking mAbs (Figure 1, D and E). These data demonstrate that the observed adhesion is specifically mediated by the bindings between LFA-1/Mac-1 and their ligands. Moreover, all ligands are found to present direct interactions with both LFA-1 and Mac-1.
FIGURE 1:

AFM tests for β2 integrin-ligand bonds. (A) Schematic of AFM setup. A PZT was used to drive the movement of an AFM cantilever. Adhesion events and forced bond rupture signals were collected from a QPD that measures the deflection of a laser beam reflected on the cantilever. (B) AFM functionalization. Recombinant human LFA-1s or Mac-1s were adsorbed onto the AFM tip and treated with TS1/18 mAbs or Mn2+ to obtain low- or high-affinity conformation integrins, respectively. Soluble ligand (ICAM-1, ICAM-2, RAGE, JAM-A, or JAM-C)–IgG Fc chimeras were coated via anti-IgG Fc secondary antibodies precoated on a Petri dish. (C) Typical force–displacement curves. An LFA-1– or Mac-1–captured AFM tip was driven to approach to (from left to right, blue lines), contact, and retract from (from right to left, red lines) a ligand-coated Petri dish. Adhesion was visualized from cantilever deflection and rupture force (fr) was measured from the force–displacement curve (middle and lower trajectories). ks is the system spring constant derived from the slope of the adhesive event. (D, E) Binding specificity. The LFA-1– or Mac-1–captured tip was pretreated with isotype control (closed bars) or LFA-1/Mac-1 blocking (open bars) mAbs. Adhesion probabilities were measured between the tip and various ligand-coated Petri dishes with TS1/18 (D) or Mn2+ (E). All measurements were acquired at a cantilever approach and retraction velocity of 1 μm/s, a contact duration of 50 ms, and a compression force of 200 pN. An anti-IgG Fc secondary antibody–coated substrate was used as a negative control. Data are presented as the mean ± SEM of three or four tips in each case. Significant differences are indicated by *, p < 0.05; **, p < 0.01; ***, p < 0.001 between each ligand and negative control, by #, p < 0.05; ##, p < 0.01; ###, p < 0.001 between each blocking group and isotype control, or by $, p < 0.05; $$, p < 0.01; $$$, p < 0.001 among various ligands. (F) Typical rupture force distributions for the interactions between LFA-1 and ICAM-1 with TS1/18 at the indicated loading rates. Total 251–394 single-bond rupture force data at each loading rate were collected and analyzed using a force bin of 50 pN.
We next measured the dependence of bond rupture forces on varied retraction velocities (corresponding loading rate ranging from 3171–114,852 pN/s). In all the measurements, an adhesion frequency of <36.7% ensures that there is a >78.9% probability that the adhesion is achieved by single-bond events with Poisson distribution statistics (Tees et al., 2001). Experimentally, 65 ± 1% of adhesive events are mediated by a single LFA-1/Mac-1–ligand bond in all cases observed from the force–displacement curves (Figure 1C), which is similar to the theoretical prediction. Force histograms of those single bonds exhibit a single peak at each loading rate, fit well with Eq. 2 (see AFM assay; Evans and Ritchie, 1997), and shift toward the higher values with increased loading rates (Figures 1F and 2). Mean rupture forces of individual LFA-1/Mac-1–ligand bonds display a linear increase with the logarithm of loading rates (Figure 3A), which is consistent with dynamic force spectroscopy (DFS) theory (Evans and Ritchie, 1997; Lü et al., 2006; Zhang et al., 2008). The significant differences in force data in Figure 3A were analyzed with two-way ANOVA (followed by a Holm–Sidak test) and summarized in Supplemental Table 2. Since Mn2+-activation can up-regulate bond mechanical strength (Yang et al., 2007; Chen et al., 2012) of LFA-1/Mac-1–ligand interactions, the expected augment of rupture forces (Figure 3A, Supplemental Table 2) are observed in most cases. The difference in mean rupture forces at each loading rate between low- and high-affinity β2 integrins is 56.1 ± 4.1, 37.7 ± 2.0, 17.7 ± 3.1, 37.4 ± 4.3, and 34.0 ± 5.9 pN for LFA-1–ICAM-1, –ICAM-2, –RAGE, –JAM-A, and –JAM-C bonds, or yields 18.0 ± 2.6, 20.0 ± 0.8, 25.2 ± 4.2, 11.5 ± 1.4, and 32.0 ± 2.8 pN for Mac-1–ICAM-1, –ICAM-2, –RAGE, –JAM-A, and –JAM-C complexes, suggesting a more effective impact of Mn2+-activation on LFA-1–ICAM-1, LFA-1–ICAM-2, LFA-1–JAM-A, LFA-1–JAM-C, and Mac-1–JAM-C bindings (force difference > 30 pN) than on the others (force difference < 30 pN). No matter whether β2 integrin is activated or not, the mechanical strength of LFA-1–ICAM-1/ICAM-2 complexes is significantly stronger than that of Mac-1–ICAM-1/ICAM-2 complexes, while Mac-1–RAGE/JAM-C bonds undergo greater external forces than LFA-1–RAGE/JAM-C bonds. Specifically, the bond strength of the LFA-1–JAM-A pair is less at low-affinity conformation but greater at high-affinity conformation than that for the Mac-1–JAM-A pair, since Mn2+-activation seems more impactful for the LFA-1–JAM-A pair. To evaluate the ligand specificity of LFA-1/Mac-1–ligand interactions, the rupture forces in Figure 3A were normalized and compared among various ligands (Supplemental Figure 1). The bond strength of LFA-1–ICAM-1 and LFA-1/Mac–1-JAM-C pairs is significantly higher than that of the others, especially after Mn2+-activation.
FIGURE 2:

Typical rupture force distributions (histograms) of LFA-1–ICAM-1 (A, C) or Mac-1–ICAM-1 (B, D) bonds prebound with TS1/18 mAbs (A, B) or activated with Mn2+ (C, D) at the indicated loading rates. Total 109–474 single-bond rupture force data at each loading rate were collected and analyzed using a force bin of 20 pN. The fitted curves (lines) were obtained using Eq. 2 (see AFM assay).
FIGURE 3:

Rupture force and lifetime of LFA-1/Mac-1–ligand bonds. (A) Dependence of rupture force on loading rate. Data are presented as the mean ± SEM of 109–474 force data at each loading rate and fitted by Eq. 3 (lines; see AFM assay). (B) Bond lifetime as a function of applied force (lines), obtained using a Bell model (Eq. 1) and the parameters koff0 and a obtained from Figure 2A (Supplemental Table 3). (C) Lifetime as a function of applied force (points), obtained from the variance of rupture force distribution using Eq. 4. The fitted curves (lines) were obtained using Eq. 1. Measurements were acquired between LFA-1 (red squares and lines) or Mac-1 (blue circles and lines) and the indicated ligand with TS1/18 (closed points, solid lines) or Mn2+ (open points, dashed lines).
All the data (Figure 3A) were further fitted with DFS theory (Eq. 3; see AFM assay) and the parameters so predicted are summarized in Supplemental Table 3. The equilibrium off-rate koff0 and reactive compliance a that characterize the spontaneous force-free dissociation and the width of the energy well of the LFA-1/Mac-1–ligand bond were substituted into Eq. 1 and then used to obtain linear dependence of the logarithm of lifetimes (1/koff) on applied external forces (Figure 3B). Alternatively, those force-dependent bond lifetimes were transformed directly from the variance of rupture force distribution measured at different loading rates by Eq. 4 (Figure 3C) (Dudko et al., 2008), which fits the Bell model well, as expected (lines in Figure 3C). The lifetime values calculated by the two methods are quite similar, and, more importantly, the force-dependent lifetimes (Figure 3, B and C) of various LFA-1/Mac-1–ligand bonds follow an order consistent with rupture forces (Figure 3A). For example, the lifetimes of LFA-1–ICAM-1 complexes are longer than those of Mac-1–ICAM-1 complexes, while LFA-1–JAM-C bonds are more transient than Mac-1-JAM-C bonds. Therefore, high rupture forces of LFA-1/Mac-1-ligand bonds can give rise to long lifetimes.
PMN adhesion mediated by LFA-1/Mac-1–ligand interactions
To better understand the physiological roles of ligand-specific β2 integrin binding in PMN recruitment, we next compared the abilities of different ligands to mediate PMN adhesion under shear flow (Figure 4). Here we used mouse bone marrow (BM)–derived PMNs because of their greater availability and lesser individual difference than for human subjects. The interactions of mouse LFA-1–ICAM-1 bonds are also stronger than that of Mac-1–ICAM-1 bonds, whether the β2 integrins are activated or not, suggesting similar patterns of LFA-1/Mac-1–ligand bond strengths between mouse and human species (Supplemental Figure 2; Figure 3A).
FIGURE 4:

Adhesion of PMNs to respective ligands in a parallel flow chamber test. Adherent resting (A) or fMLF-activated (B) PMNs pretreated with isotype control (black bars) or LFA-1 (red bars)/Mac-1 (blue bars) blocking mAbs on respective ligand-coated substrates after being perfused into the flow chamber and incubated for 5 min without flow. Data are presented as the mean ± SEM of five to nine independent experiments and analyzed with two-way ANOVA (followed by a Holm–Sidak test). Significant differences are indicated by *, p < 0.05; **, p < 0.01; ***, p < 0.001 among various ligands, by #, p < 0.05; ##, p < 0.01; ###, p < 0.001 between each blocking group and isotype control, or by $, p < 0.05; $$, p < 0.01; $$$, p < 0.001 between LFA-1– and Mac-1–blocking groups.
Resting or fMLF-activated PMNs were allowed to adhere to various ligands for 5 min without flow, washed at a wall shear stress of τw = 1 dyn/cm2 for 1 min, and counted for those cells undergoing firm adhesion. All ligands are able to induce firm adhesion for resting or activated PMNs (Figure 4). Specifically, PMN adhesion on ICAM-1 and ICAM-2 is mainly LFA-1–dependent, while Mac-1 dominates PMN adhesion on RAGE, JAM-A, and JAM-C, which is consistent with their rupture forces in most cases (Figure 3A). Most LFA-1 or Mac-1 molecules remain a low-affinity conformation on resting PMNs (Luo and Springer, 2006). While the expression of Mac-1 (but not of LFA-1) is up-regulated after fMLF stimulation via translocation of Mac-1-containing granules up to cell membranes (Supplemental Figure 3), most of LFA-1s and 10% of Mac-1s are switched to a high-affinity conformation (Diamond and Springer, 1999; Lum et al., 2002).
ICAM-1 is the most effective ligand for mediating PMN adhesion, mainly due to the high bond strength of LFA-1–ICAM-1 pairs (Figure 3A; Supplemental Figure 1). Even resting PMNs can nearly all adhere to ICAM-1–coated substrates in flow chamber tests (Supplemental Figure 4), and thereby fMLF activation cannot further raise the number of adherent PMNs (from 62 ± 3 to 64 ± 3, p = 0.301). LFA-1–ICAM-1 and Mac-1–ICAM-1 interactions mediate specific adhesion of resting PMN cooperatively (Figure 4A), but, after fMLF activation, the bond strength for high-affinity LFA-1 is sufficient to mediate PMN adhesion alone, and hence Mac-1 blocking cannot reduce adhesion (Figure 4B). LFA-1, but not Mac-1, can mediate resting PMN adhesion on ICAM-2 (Figure 4A). After fMLF activation, the adherent PMNs on ICAM-2 are significantly increased (from 48 ± 2 to 60 ± 2, p < 0.001) (Figure 4B), but the impact of Mac-1–ICAM-2 bonds still cannot compare with that of LFA-1–ICAM-2 bonds because of their lower mechanical strength (Figure 3A). It should be noticed that Mac-1 blocking increases moderately fMLF-activated PMN adhesion on ICAM-1 but reduces the adhesion on ICAM-2 (Figure 4B), possibly due to the greater difference of mechanical strength between LFA-1– and Mac-1–ICAM-1 bonds than between LFA-1– and Mac-1–ICAM-2 bonds. The increased LFA-1–ICAM-1 bonds due to Mac-1 blocking can enhance the magnitude of PMN adhesion, while the augmented LFA-1–ICAM-2 bonds may not be sufficient to compensate for the loss of Mac-1–ICAM-2 interactions. When both LFA-1 and Mac-1 were blocked, the fMLF-activated PMN adhesion was significantly abrogated to 16 ± 1 and 7 ± 0 (adherent PMNs/FOV) on ICAM-1 and ICAM-2, indicating that Mac-1 also plays a role in PMN adhesion. Although no significant differences are found for the adhesion of resting and fMLF-activated PMNs on RAGE (Figure 4), quite a few resting PMNs present unstable adhesion on RAGE that is easily washed away by shear flow and quickly rebounds from flow (Supplemental Figure 4), which is consistent with the low bond strength and high binding activity of LFA-1/Mac-1–RAGE interactions (Supplemental Figure 1). Mac-1–RAGE binding is effective to mediate most and all PMN adhesion in resting and activated states, respectively, because of its higher rupture forces than the LFA-1–RAGE pair (Figure 3A). The capability of JAM-A to mediate PMN adhesion is lowest among all ligands (Figure 4) mainly due to the low bond strength of LFA-1/Mac-1–JAM-A interactions (Supplemental Figure 1). Here Mac-1 plays a major role in JAM-A–dependent adhesion of resting or activated PMNs (Figure 4). For resting PMNs, the bond strength for low-affinity LFA-1 is weaker than that for low-affinity Mac-1 (Figure 3A), and therefore LFA-1 blocking can increase adhesion probably by raising more Mac-1–JAM-A bonds instead (Figure 4A). For activated PMNs, however, the increased Mac-1–JAM-A interactions may equalize the reduced mechanical strength by LFA-1 blocking, so that no significant difference is seen between control and LFA-1 blocking group (Figure 4B). The capability of JAM-C to mediate adhesion of resting PMNs is quite low and similar to JAM-A (Figure 4A) despite of the high rupture forces observed (Figure 3A), while this capability can be significantly enhanced after fMLF stimulation (from 39 ± 2 to 55 ± 2, p < 0.001) (Figure 4B). Collectively, these data indicate that the capability of LFA-1/Mac-1–ligand to mediate PMN adhesion is directly related to their bond strength.
PMN spreading, polarization, and intraluminal crawling on various ligands
In addition to their adhesive functions, ligand–specific binding of LFA-1 or Mac-1 also triggers outside-in signaling in PMNs, leading to morphological change and cytoskeletal reorganization that are crucial for postadhesion functions such as cell spreading, polarization, and intraluminal crawling (Zhan et al., 2012; Robert et al., 2013). To analyze the differential contributions of endothelial ligands to PMN spreading and polarization, we compared shear-induced morphological alteration of PMNs on distinct ligands (Figures 5 and 6). All ligands are able to enhance the spreading area of resting or fMLF-activated PMNs significantly at low (1 dyn/cm2) or high shear stress (10 dyn/cm2) (Figure 5). Meanwhile, most of the ligands are capable of enhancing cell elongation at the two typical shear stresses (Figure 6), indicating a polarized change of cell morphology. Specifically, Mac-1 plays a more important role in cell spreading and polarization than LFA-1 in most cases, especially for RAGE, JAM-A, and JAM-C (Figures 6 and 7). This is not surprising for RAGE and JAM-C since they form stronger bonds with Mac-1 than LFA-1 (Figure 3A). Mac-1–JAM-A bonds are more effective in mediating resting PMN spreading and polarization (Figures 5, A and B, and 6, A and B) possibly due to their higher bond strength than LFA-1–JAM-A bonds in a low affinity state (Figure 3A). For fMLF-activated PMNs (Figures 5, C and D, and 6, C and D), however, LFA-1 blocking enhances (but does not reduce) PMN spreading and polarization by creating more Mac-1–JAM-A bonds instead, although high-affinity LFA-1–JAM-A bonds are stronger than high-affinity Mac-1–JAM-A bonds (Figure 3A). For ICAM-1 and ICAM-2, the importance of LFA-1 is only increased after fMLF activation (Figures 5, C and D, and 6, C and D) or under high shear stress (Figures 5, B and D, and 6, B and D), which may be attributed to the higher bond strength and more valid activation than for Mac-1 (Figure 3A). These data indicate that, although stronger bonds may enhance outside-in signaling slightly, Mac-1-ligand bonds are more effective in inducing PMN spreading and polarization.
FIGURE 5:

Shear-induced spreading of adherent PMNs on distinct ligands. Spreading area of resting (A, B) or fMLF-activated (C, D) PMNs pretreated with isotype control (black bars) or LFA-1 (red bars)/Mac-1 (blue bars) blocking mAbs on various ligands after being exposed to a shear flow of 1 dyn/cm2 (A, C) or 10 dyn/cm2 (B, D) for 10 min. Data are presented as the mean ± SEM from a total of 178–2003 cells in 50–90 FOVs of five to nine independent experiments and analyzed with two-way ANOVA (followed by a Holm–Sidak test). Significant differences are indicated by *, p < 0.05; **, p < 0.01; ***, p < 0.001 among various ligands, by #, p < 0.05; ##, p < 0.01; ###, p < 0.001 between each blocking group and isotype control, or by $, p < 0.05; $$, p < 0.01; $$$, p < 0.001 between LFA-1– and Mac-1–blocking groups.
FIGURE 6:

Shear-induced polarization of adherent PMNs on distinct ligands. Elongation of resting (A,B) or fMLF-activated (C, D) PMNs pretreated with isotype control (black bars) or LFA-1 (red bars)/Mac-1 (blue bars) blocking mAbs on various ligands after being exposed to a shear flow of 1 dyn/cm2 (A, C) or 10 dyn/cm2 (B, D) for 10 min. Data are presented as the mean ± SEM from a total of 178–2003 cells in 50–90 FOVs of five to nine independent experiments and analyzed with two-way ANOVA (followed by a Holm–Sidak test). Significant differences are indicated by *, p < 0.05; **, p < 0.01; ***, p < 0.001 among various ligands, by #, p < 0.05; ##, p < 0.01; ###, p < 0.001 between each blocking group and isotype control, or by $, p < 0.05; $$, p < 0.01; $$$, p < 0.001 between LFA-1– and Mac-1–blocking groups.
FIGURE 7:

Comparison of PMN crawling speed on various ligands. Crawling speed of resting (A, B) or fMLF-activated (C, D) PMNs pretreated with isotype control (black bars) or LFA-1 (red bars)/Mac-1 (blue bars) blocking mAbs on various ligands at 1 dyn/cm2 (A, C) or 10 dyn/cm2 (B, D). Data are presented as the mean ± SEM from a total of 18–139 cells of five to nine independent experiments and analyzed with two-way ANOVA (followed by a Holm–Sidak test). Significant differences were indicated by *, p < 0.05; **, p < 0.01; ***, p < 0.001 among various ligands, by #, p < 0.05; ##, p < 0.01; ###, p < 0.001 between each blocking group and isotype control, or by $, p < 0.05; $$, p < 0.01; $$$, p < 0.001 between LFA-1– and Mac-1–blocking groups.
To investigate the roles of different endothelial ligands in cell crawling, we next evaluated PMN crawling dynamics on various ligands (Supplemental Figure 5; Figure 7). Here crawling trajectories of resting or fMLF-activated PMNs are random at low shear stress (Supplemental Figure 5, A, C, E, and G) and preferentially oriented along the direction of flow at high shear stress (Supplemental Figure 5, B, D, F, and H). No significant difference in the x forward migration index (xFMI) is found after LFA-1 or Mac-1–blocking (Supplemental Figure 5, E–H), suggesting that the impact of LFA-1/Mac-1–ligand bonds on the directionality of PMN crawling along shear flow is not significant when the ligands are uniformly placed on a planar substrate.
Another key feature for cell crawling dynamics is crawling speed (Figure 7). It is seen that, at low shear stress, ICAM-1 plays a more important role than other ligands in the crawling of resting PMNs by Mac-1–ICAM-1 bonds (Figure 7A). At high shear stress, however, LFA-1/Mac-1–JAM-C and LFA-1/Mac-1–ICAM-1 bonds speed up the crawling of resting (Figure 7B) and fMLF-activated (Figure 7D) PMNs, respectively. Mac-1–JAM-A bonds are also found to accelerate PMN crawling in some cases (Figure 7, B and C). On one hand, these data confirm strong outside-in signaling through Mac-1–ligand bonds. On the other hand, they also imply the important role of bond strength in PMN crawling under high shear stress, since the mechanical strength of LFA-1–ICAM-1 and LFA-1/Mac-1–JAM-C bonds is highest among all LFA-1/Mac-1–ligand bonds.
Taken together, all ligands of LFA-1 or Mac-1 can trigger PMN spreading, polarization, and crawling in different degrees, and Mac-1 seems to induce outside-in signaling more effectively. Moreover, LFA-1–ICAM-1 and LFA-1/Mac-1–JAM-C bonds can accelerate PMN crawling under high shear stress due to their high mechanical strength.
DISCUSSION
Diversity of ligand-specific binding to β2 integrin complicates PMN recruitment on endothelium in distinct tissues. Little is known about how these multiple ligands regulate β2 integrin–mediated PMN adhesion and crawling under shear flow. In this study, we attempted to correlate the forces binding LFA-1 or Mac-1 to respective ligands with their biological phenotypes. High rupture forces of LFA-1/Mac-1–ligand bonds indicate long lifetimes, and high strength to support PMN adhesion under physiological-like flow. Moreover, LFA-1/Mac-1–ligand bonds with high rupture forces might bring up intense outside-in signaling and play a dominant role in PMN spreading, polarization, and crawling under high shear stress.
Biophysical tests of ligand-specific binding of β2 integrins provide the bases for elucidating their biological functions. As summarized in Supplemental Table 4, single-molecule measurements of LFA-1/ICAM-1, LFA-1/ICAM-2, or Mac-1/ICAM-1 interactions have been conducted extensively by AFM studies (Zhang et al., 2002; Wojcikiewicz et al., 2006; Yang et al., 2007). A fast (7000–60,000 pN/s) and a slow (50–7000 pN/s) linear loading regime are separately observed in the dynamic force spectra of LFA-1/ICAM-1 and LFA-1/ICAM-2 bonds, which supports the existence of two energy barriers (Zhang et al., 2002; Wojcikiewicz et al., 2006). Here we performed the measurements at loading rates ranging from 3171 to 114,852 pN/s and uncovered a single linear regime in this range, which is similar to results of previous tests of Mac-1/ICAM-1 interactions in the loading rate range of 100–10,000 pN/s (Yang et al., 2007). Our data also support the previous observations of LFA-1 binding, since the known facts of high rupture forces for LFA-1/ICAM-1 bonds but low forces for LFA-1/ICAM-2 bonds remain unchanged in these two loading regimes (Wojcikiewicz et al., 2006). Thus, the loading rates used and rupture forces obtained in the current study are comparable to those for the fast loading regime in the literature (Supplemental Table 4). The reason we did not perform measurements at loading rates <3171 pN/s is mainly concern with physiological flow and stress. PMN recruitment usually occurs at postcapillary venules, where the shear stress is measured to be 1–10 dyn/cm2 (Heisig, 1968), and therefore the force (= 32τwr2) applied to PMNs by physiological shear flow is 120–1200 pN (radius r is calculated as 6 μm) (McEver and Zhu, 2010). The rupture forces obtained in the slow loading regime are usually less than 100 pN, which seems to be less physiologically relevant, especially when we are focusing on linking bond mechanical strength with PMN adhesion and migration. More importantly, our measurements presented the first sets of forced dissociation data for JAM and RAGE ligands, which provide a global picture for endothelial ligands in the distinct ligand-specific binding of β2 integrins.
Bond lifetime is another regulating factor for ligand-specific β2 integrin bindings. The lifetimes of LFA-1/ICAM-1 bonds measured by biomembrane force probe (BFP) first increase (catch bonds) and then decrease (slip bonds) with increasing forces (Chen et al., 2010, 2012). Those catch bond features only exist at low forces (<15 pN) and are not observed at rupture forces >15 pN in previous BFP studies (Evans et al., 2010; Kinoshita et al., 2010). In this study, we transformed the rupture forces measured in AFM experiments at a constant pulling speed into force-dependent lifetimes by two methods (Figure 3, B and C). Since the collected rupture forces are all above 100 pN, which are considered to be more physiologically relevant tor PMNs, as aforementioned, the lifetimes exhibit slip bond features in all cases (Figure 3, B and C). In contrast to those in the literature (Chen et al., 2010, 2012), our data specified the biomechanical features of distinct β2 integrin-ligand bonds and implied different phenotypes of various endothelial ligands at given high loading rates with the majority of single bonds. Evidently, examining the effects of distinct ligands on bond rupture forces in the catch-bond regime is biophysically meaningful in future work.
Bond rupture force and lifetime are determined by the conformations and valencies of molecular pairs of interest. As a typical allosterically inhibitory mAb to β2 integrins, TS1/18 is reported to bind to βI residues on the α1- and α7-helices and stable low affinity conformations of LFA-1 and Mac-1 (Xie, Zhu, et al., 2010). Metal ions such as Mn2+ and Mg2+ are known to increase integrin affinity by replacing Ca2+ at the LIMBS (ligand-induced metal ion binding site) and ADMIDAS (adjacent to metal ion dependent adhesion site) of the β I-like domain (Chen et al., 2003; Mould et al., 2003). Mn2+ activation–induced augment of rupture forces and prolongation of lifetimes are found in all β2 integrin-ligand bonds in comparison with TS1/18 binding groups (Figure 3; Supplemental Table 2), which are similarly observed in previous AFM and BFP studies (Zhang et al., 2002; Wojcikiewicz et al., 2006; Yang et al., 2007; Evans et al., 2010; Kinoshita et al., 2010; Chen et al., 2012). Meanwhile, the ligands used in this work are all dimeric proteins with immunoglobulin G (IgG)-Fc chimeras. ICAM-1 dimerization appears to augment its binding to LFA-1 (Miller et al., 1995; Reilly et al., 1995) and facilitate the presentation of the binding site in the D1 domain (Yang et al, 2004). Since the majority of adhesive events are dominated by single bonds and only the rupture forces and lifetimes for those single bonds were analyzed, the dimerization of ligands is not likely to overturn the conclusions drawn from the current tests.
Different β2 integrin ligands present distinct functions in PMN recruitment. ICAM-1 and ICAM-2, common for both LFA-1 and Mac-1, are the most important and widely tested ligands. Our previous work found that the binding affinity for LFA-1–ICAM-1 complexes is much higher than that for Mac-1–ICAM-1 complexes, mainly due to the highly enhanced on-rate (Li et al., 2013). Here we further demonstrate that the mechanical strength of LFA-1–ICAM-1/ICAM-2 bonds is also much higher than that of Mac-1–ICAM-1/ICAM-2 bonds (Figure 3A), so that LFA-1–ICAM-1/ICAM-2 (but not Mac-1–ICAM-1/ICAM-2) binding mainly contributes to maintaining PMN adhesion, especially after fMLF-activation (Figure 4). LFA-1–ICAM-2 bonds are less resistant to applied forces than LFA-1–ICAM-1 bonds (Supplemental Figure 1), which is consonant with previous work measured by AFM (Wojcikiewicz et al., 2006), but they are still effective in mediating adhesion (Figure 4). Meanwhile, ICAM-1s and ICAM-2s can promote or restrict PMN crawling. ICAM-1- or ICAM-2–knockout (KO) reduces MIP-2– or IL-1β–induced PMN crawling speed in vivo, respectively (Robert et al., 2013; Halai et al., 2014). In contrast, PMN movement is accelerated along the flow direction on LPS-stimulated ICAM-1-KO brain microvascular endothelial cells under flow in vitro (Gorina et al., 2014). Under static in vitro conditions, PMNs exhibit randomized crawling on ICAM-1- or ICAM-2–coated surfaces and depress crawling speed and distance by Mac-1 blocking (Halai et al., 2014). Evidently, diverse functionality of these ligands in PMN crawling is closely related to physiological shear stress. In this work, the role of ICAM-1 but not ICAM-2 in accelerating PMN crawling is observed under physiological-like flow (Figure 7). Crawling trajectories are random at low shear stress and directed along the flow direction at high shear stress (Supplemental Figure 5). In addition to those Mac-1–ICAM-1 bonds found to dominate PMN crawling in previous works (Menezes et al., 2009; Robert et al., 2013), LFA-1–ICAM-1 interactions are also found to speed up the crawling of fMLF-activated PMNs under high shear flow (Figure 7D), which may be related to the strong bond strength of high-affinity LFA-1–ICAM-1 (Figure 3; Supplemental Figure 1). ICAM-1s and ICAM-2s can also enhance PMN spreading and polarization (Figures 5 and 6), which is consistent with previous work (Gorina et al., 2014).
RAGE is identified as a counterligand specific for Mac-1 and engaged in leukocyte recruitment to peritoneum in a thioglycollate-induced acute inflammation model (Chavakis et al., 2003). It regulates leukocyte adhesion with effectiveness equal to that of ICAM-1 in preterm and term infants (Buschmann et al., 2014). Mac-1–RAGE and LFA-1–ICAM-1 interactions also cooperate in mediating trauma-induced leukocyte adhesion and crawling in cremaster muscle venules (Frommhold et al., 2010). In this study, RAGE is found to be a common ligand for LFA-1 and Mac-1 from molecular mechanics tests (Figure 1, D and E), and the bond strength of Mac-1–RAGE interactions is much higher than for LFA-1–RAGE, especially after Mn2+ activation (Figure 3A). In shear-induced cell adhesion tests, Mac-1s appear to dominate PMN adhesion on immobilized RAGEs, supporting in vivo observations qualitatively (Chavakis et al., 2003; Frommhold et al., 2010; Buschmann et al., 2014). Mac-1–RAGE bonds also induce effective PMN spreading and polarization under shear flow, particularly after fMLF activation, suggesting strong outside-in signaling (Figures 5 – 7). LFA-1–RAGE bonds affect adhesion (Figure 4A) and spreading (Figure 5B) of resting PMNs only slightly, which is consistent with their low rupture forces (Figure 3A). These findings are specifically meaningful for Mac-1s due to their complicated functionality of binding to quite diverse ligands.
JAM-A and JAM-C serve as another ligand family of β2 integrins specific for LFA-1 and Mac-1, respectively. They are mainly localized at endothelial junctions and have been shown to play a role in PMN transmigration (Ostermann et al., 2002; Chavakis et al., 2004; Aurrand-Lions et al., 2005; Sircar et al., 2007). A combination of the inflammatory cytokines TNF-α and IFN-γ can induce the redistribution of JAM-As to the endothelial apical surface (Ozaki et al., 1999), which strengthens the adhesion mediated by LFA-1–JAM-A binding (Ostermann et al., 2002). Although JAM-C is independent in PMN adhesion on HUVECs under shear flow in vitro (Sircar et al., 2007), it is involved in PMN adhesion on IL-1β-stimulated cremasteric venules in vivo (Aurrand-Lions et al., 2005). Here we find that both LFA-1 and Mac-1 can bind to JAM-A and JAM-C specifically (Figure 1, D and E). On one hand, the Mac-1–JAM-A bonds are stronger than LFA-1–JAM-A bonds in the low-affinity state and weaker in the high-affinity state (Figure 3). Moreover, Mac-1s dominate PMN adhesion, spreading, polarization, and crawling on JAM-A in current work (Figures 4 – 7), which seems inconsistent with previous observations (Ostermann et al., 2002) displaying LFA-1- but not Mac-1-transfected J-β2.7 cell adhesion on CHO cells. Noting that the adhesion frequency was quite low in the literature (2% for Mac-1 and 4% for LFA-1), it is hard to make a direct comparison for drawing reliable conclusion. LFA-1–JAM-A bonds are not effective in mediating PMN adhesion in current work, which is also consistent with previous work (Ostermann et al., 2002). On the other hand, the mechanical strength of Mac-1–JAM-C bonds is especially high for mediating PMN adhesion, spreading, polarization, and crawling (Figures 3 – 7), while the LFA-1–JAM-C bonds are not effective in maintaining PMN adhesion (Figure 4) due to their lower bond strength (Figure 3A), confirming the independence of LFA-1s on JAM-C–mediated adhesion in previous works (Sircar et al., 2007). Thus, our data on PMN adhesion mediated by JAM-As and JAM-Cs are at least partially consistent with those previous observations.
Collectively, our results quantified the respective contributions of diverse ligands of β2 integrins in bond mechanical strength and PMN recruitment. All ligands are able to induce PMN adhesion, spreading, polarization, and crawling in different degrees. LFA-1s and Mac-1s play dominant roles for specified ligands. Moreover, these LFA-1- and Mac-1–specific functions are related to high rupture forces of receptor–ligand interactions. This work links mechanical strength of LFA-1/Mac-1–ligand bonds to their biological functions, which provides insight into the ligand-specific mechanisms of PMN recruitment.
MATERIALS AND METHODS
Antibodies and reagents
Recombinant human LFA-1 (CD11a/CD18) or Mac-1 (CD11b/CD18), soluble human ICAM-1, ICAM-2, RAGE, JAM-A, and JAM-C with IgG Fc chimeras, and mouse LFA-1, Mac-1, and ICAM-1 were purchased from R&D Systems. Fluorescein isothiocyanate (FITC)-conjugated rat anti-mouse CD11a (M17/4) and CD11b (M1/70) for flow cytometry staining, blocking mAbs against human or mouse LFA-1 (HI111 or M17/4) and Mac-1 (ICRF44 or M1/70), allosterically inhibitory mAbs TS1/18, and isotype control (MOPC-21, RTK2758, RTK4530) mAbs were all from Biolegend. Goat anti-human IgG Fc polyclonal antibodies, LPS, fMLF, MnCl2, and BSA were from Sigma-Aldrich.
PMN isolation from mouse BM
Mouse BM-derived PMNs were isolated from 8–12-wk male C57BL/6 mice obtained from Vital River Laboratories (Beijing, China). All experiments with mice were approved by the Institutional Animal and Medicine Ethical Committee (IAMEC) at the Institute of Mechanics, Chinese Academy of Sciences. Briefly, BM cells were isolated by gently flushing femurs and tibias with Dulbecco’s phosphate-buffered saline (DPBS) supplemented with 0.5% bovine serum albumin (BSA) and 2 mM EDTA. After filtration with a cell strainer of mesh size 70 µm (Corning), the cell suspension was centrifuged at 300 × g for 10 min. Packed cells were resuspended and layered on top of a Histopaque-1077/1119 (Sigma-Aldrich) two-layer density gradient. After centrifugation at 700 × g for 30 min, the PMNs were harvested from the Histopaque-1077/1119 interface. After being washed twice with DPBS at 300 × g for 10 min, the cells were suspended in ice-cold HBSS containing Ca2+, Mg2+, and 0.1% BSA and kept on ice until use. In some cases, the PMNs were incubated with 10 μg/ml isotype control or LFA-1/Mac-1 blocking mAbs at 4°C for 30 min or stimulated with 1 μM fMLF at 37°C for 20 min immediately before use. To detect the expression of LFA-1 or Mac-1, resting and fMLF-stimulated PMNs were incubated with 10 μg/ml FITC-labeled isotype control or respective specific mAbs at 4°C for 30 min. Washed PMNs were analyzed by FACSCanto II flow cytometer (BD Biosciences).
AFM functionalization
Soluble LFA-1s or Mac-1s and their respective ligands were coupled onto AFM tips and Petri dish surfaces as described (Zhang et al., 2002; Lü et al., 2006). Briefly, soluble LFA-1s or Mac-1s were adsorbed onto a silicon nitride cantilever tip (MLCT; Bruker AFM Probes) by incubating the cantilever for 2 h at 37°C in a 100 μg/ml protein solution. After being washed and blocked by 1% BSA in DPBS for 1 h at 37°C, the cantilever was incubated in 20 μg/ml mAbs TS1/18 or 1 mM Mn2+ for 1 h at 37°C to obtain low- or high-conformation integrins, respectively. In some cases, the cantilever was washed three times and then incubated with 20 μg/ml isotype control or LFA-1/Mac-1 blocking mAbs for 1 h at 37°C. Separately, goat anti-human IgG Fc polyclonal antibodies were adsorbed onto a small spot on a 60-mm Petri dish for 2 h at 37°C in a 2 mg/ml or 200 μg/ml protein solution for AFM tests in either the TS1/18 or Mn2+ group, except for the RAGE-Fc group in 20 μg/ml protein solution. The washed dish was blocked with 1% BSA in DPBS for 1 h at 37°C and rinsed three times. The dish was then incubated with 100 or 10 μg/ml ligand–Fc chimera for 1 h at 37°C. The cantilever and the dish were washed three times under the desired cationic condition, namely Ca2+/Mg2+ (HBSS containing 0.1% BSA and 1 mM Ca2+ plus 1 mM Mg2+) for the TS1/18 group or Mn2+ (HBSS containing 0.1% BSA and 1 mM Mn2+) used for the Mn2+ group and used for AFM tests immediately.
AFM assay
Bond rupture force measurements were carried out at room temperature on a BioScope Catalyst AFM (Bruker Corporation) (Figure 1, A and B; Lü et al., 2006). The ligand-coated dish was placed on the AFM stage, while an LFA-1- or Mac-1–captured cantilever was repeatedly driven by a piezoelectric translator (PZT) to approach the substrate of the dish, to make contact to allow reversible bond formation and dissociation, and to retract to detect the presence of adhesive event(s) from the force–displacement curves (Figure 1C). The rupture force of the β2 integrin–ligand bond was obtained from the deflection of the cantilever reflecting a laser beam focused on the largest triangular cantilever (“C” cantilever 320 μm long and 22 μm wide) into the position of a sensitive quad photodetector (QPD) (Figure 1A). The cantilever deflection was converted to a bond force using the cantilever spring constant, which was experimentally determined using thermal fluctuation analysis and proved to be consistent with the nominal value of 0.01 N/m specified by the manufacturer. AFM measurements were acquired at a cantilever approach velocity of 1 μm/s, a contact duration of 50 ms, a compression force of 200 pN, and a retraction velocity ranging from 0.5 to 8 μm/s for varying loading rates. Under each cationic condition (Ca2+/Mg2+ or Mn2+), three or four independent experiments with different AFM tips were conducted at 5–10 different locations on each dish. Fifty to one hundred cycles were tested at each location to collect a set of adhesion events and rupture forces. The rupture force, fr, was determined from the peak force for single rupture events (Figure 1C). The system spring constant, ks, was derived from the slope of the force–displacement trajectory just before each rupture event (Figure 1C). The loading rate, rf, was estimated by multiplying the system spring constant by the retraction velocity. According to the Bell model, the off-rate, koff, is assumed to follow an exponential function of applied force, F (Bell, 1978):
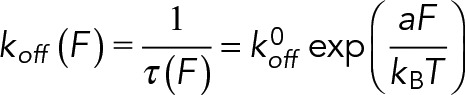 (1) (1)
|
Here τ is the bond lifetime, which is the reciprocal of the instantaneous off-rate koff, kB is the Boltzmann constant, T is the absolute temperature, koff0 is the equilibrium off-rate, and a is the reactive compliance, which measures the width of the energy well that traps the interacting molecules in a bound state. Under conditions of constant loading rate, the probability density for the dissociation of a complex at force f is given by (Bell, 1978; Evans and Ritchie, 1997; Zhang et al., 2008):
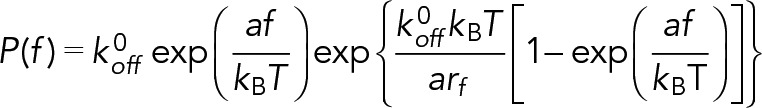 (2) (2)
|
Dynamic force spectroscopy theory was used to predict the dependence of rupture forces on loading rates (Evans and Ritchie, 1997; Lü et al., 2006; Zhang et al., 2008):
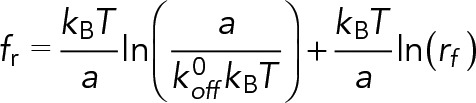 (3) (3)
|
The rupture-force histograms measured at different loading rates were directly transformed into the force dependence of bond lifetime measurable in constant-force experiments as described by Dudko et al. (2008). The lifetime is assumed to be a function of applied force equal to mean rupture force, obtained from the variance of rupture force distribution:
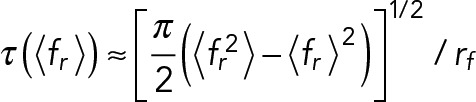 (4) (4)
|
Here <∙> denotes the mean value.
PMN adhesion and crawling assay
PMN adhesion and crawling were measured at 37°C in a parallel plate flow chamber (GlycoTech) connected to a PHD22/2000 syringe pump (Harvard Apparatus) and mounted on an inverted microscope (CKX41; Olympus) equipped with a charge-coupled device (CCD) camera. Resting or fMLF-activated PMNs (2 × 106 or 1 × 106/ml) pretreated with isotype control or LFA-1-/Mac-1–blocking mAbs were perfused with Ca2+/Mg2+ solution into the chamber and incubated for 5 min without flow. After nonadherent or weakly adherent PMNs were washed out at 1 dyn/cm2 for 1 min, the number of adherent PMNs was recorded from 10 fields of view (FOVs) through a 10 ×/NA 0.25 objective using a CCD camera and counted using ImageJ software (National Institutes of Health). Aggregated adherent cells (<15%) were counted as one cell. PMN movement was then recorded sequentially every 15 s via a 20 ×/NA 0.4 objective for a total of 10 min at low (1 dyn/cm2) and high shear stress (10 dyn/cm2). At the endpoint of either phase, the images of adherent PMNs were acquired from 10 FOVs and the shear-induced morphological alteration was analyzed using NIS-ELEMENT software (Nikon, Tokyo, Japan) to obtain their spreading area and elongation (= length/width). Migration trajectories of individual PMNs presented within the FOV for ≥2.5 min were manually tracked and also analyzed using NIS-ELEMENT. The parameters crawling speed (in μm/min) and xFMI (= Dx/Dacc, where Dx is the vectorial distance along the x-axis and Dacc is the accumulated scalar distance of cell movement) were then quantified systematically.
Statistical analysis
Data are presented as the mean ± SEM. Significant differences among multiple groups were identified by one-way or two-way analysis of variance (ANOVA), followed by a Holm–Sidak test. Differences between any two groups were analyzed by the unpaired two-tailed Student’s t test or the Mann–Whitney rank sum test depending on the readouts of the normality test (Kolmogorov–Smirnov test). P values less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by National Natural Science Foundation of China Grants 31230027, 31110103918, and 31300776, Strategic Priority Research Program and Frontier Science Key Project of Chinese Academy of Sciences Grants XDA01030102, XDB22040101, and QYZDJ-SSW-JSC018, National Key Research and Development Program of China Grant 2016YFA0501601, and the Visiting Scholar Foundation of the Key Laboratory of Biorheological Science and Technology (Chongqing University), Ministry of Education (CQKLBST-2015-002).
Abbreviations used:
- AFM
atomic force microscopy
- BM
bone marrow
- DFS
dynamic force spectroscopy
- DPBS
Dulbecco’s phosphate-buffered saline
- FOV
field of view
- ICAM-1
intercellular adhesive molecule 1
- ICAM-2
intercellular adhesive molecule 2
- JAM-A
junctional adhesion molecule-A
- JAM-C
junctional adhesion molecule-C
- LFA-1
lymphocyte function-associated antigen-1
- mAb
monoclonal antibody
- Mac-1
macrophage-1 antigen
- PMNs
neutrophils, polymorphonuclear leukocytes
- PZT
piezoelectric translator
- QPD
quad photodetector
- RAGE
receptor for advanced glycation end products
- xFMI
x forward migration index.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-12-0827) on December 27, 2017.
REFERENCES
- Aurrand-Lions M, Lamagna C, Dangerfield JP, Wang SJ, Herrera P, Nourshargh S, Imhof BA. Junctional adhesion molecule-C regulates the early influx of leukocytes into tissues during inflammation. J Immunol. 2005:6406–6415. doi: 10.4049/jimmunol.174.10.6406. [DOI] [PubMed] [Google Scholar]
- Bell GI. Models for specific adhesion of cells to cells. Science. 1978:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- Buschmann K, Tschada R, Metzger MS, Braach N, Kuss N, Hudalla H, Poeschl J, Frommhold D. RAGE controls leukocyte adhesion in preterm and term infants. BMC Immunol. 2014:53. doi: 10.1186/s12865-014-0053-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavakis T, Bierhaus A, AI-Fakhri N, Schneider D, Witte S, Linn T, Nagashima M, Morser J, Arnold B, Preissner KT, et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment J Exp Med 2003. 1507 1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavakis T, Keiper T, Matz-Westphal R, Hersemeyer K, Sachs UJ, Nawroth PP, Preissner KT, Santoso S. The junctional adhesion molecule-C promotes neutrophil transendothelial migration in vitro and in vivo. J Biol Chem. 2004:55602–55608. doi: 10.1074/jbc.M404676200. [DOI] [PubMed] [Google Scholar]
- Chen JF, Salas A, Springer TA. Bistable regulation of integrin adhesiveness by a bipolar metal ion cluster. Nat Struct Biol. 2003:995–1001. doi: 10.1038/nsb1011. [DOI] [PubMed] [Google Scholar]
- Chen W, Lou JZ, Evans EA, Zhu C. Observing force-regulated conformational changes and ligand dissociation from a single integrin on cells. J Cell Biol. 2012:497–512. doi: 10.1083/jcb.201201091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Lou JZ, Zhu C. Forcing switch from short- to intermediate- and long-lived states of the alpha A domain generates LFA-1/ICAM-1 catch bonds. J Biol Chem. 2010:35967–35978. doi: 10.1074/jbc.M110.155770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinamon G, Grabovsky V, Winter E, Franitza S, Feigelson S, Shamri R, Dwir O, Alon R. Novel chemokine functions in lymphocyte migration through vascular endothelium under shear flow. J Leukocyte Biol. 2001:860–866. [PubMed] [Google Scholar]
- Diamond MS, Springer TA. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J Cell Biol. 1999:545–866. doi: 10.1083/jcb.120.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudko OK, Hummer G, Szabo A. Theory, analysis, and interpretation of single-molecule force spectroscopy experiments. Proc Natl Acad Sci USA. 2008:15755–15760. doi: 10.1073/pnas.0806085105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E, Kinoshita K, Simon S, Leung A. Long-lived, high-strength states of ICAM-1 bonds to beta(2) integrin: I. Lifetimes of bonds to recombinant alpha(L) beta(2) under force. Biophys J. 2010:1458–1466. doi: 10.1016/j.bpj.2009.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E, Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys J. 1997:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fine N, Dimitriou ID, Rullo J, Sandi MJ, Petri B, Haitsma J, Ibrahim H, La Rose J, Glogauer M, Kubes P, et al. GEF-H1 is necessary for neutrophil shear stress-induced migration during inflammation J Cell Biol 2016. 107 119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommhold D, Kamphues A, Hepper I, Pruenster M, Lukic IK, Socher I, Zablotskaya V, Buschmann K, Lange-Sperandio B, Schymeinsky J, et al. RAGE and ICAM-1 cooperate in mediating leukocyte recruitment during acute inflammation in vivo Blood 2010. 841 849 [DOI] [PubMed] [Google Scholar]
- Fu CL, Tong CF, Wang ML, Gao YX, Zhang Y, Lü SQ, Liang S, Dong C, Long M. Determining β2-integrin and intercellular adhesion molecule 1 binding kinetics in tumor cell adhesion to leukocytes and endothelial cells by a gas-driven micropipette assay. J Biol Chem. 2011:34777–34787. doi: 10.1074/jbc.M111.281642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorina R, Lyck R, Vestweber D, Engelhardt B. Beta(2) Integrin-mediated crawling on endothelial ICAM-1 and ICAM-2 is a prerequisite for transcellular neutrophil diapedesis across the inflamed blood–brain barrier. J Immunol. 2014:324–337. doi: 10.4049/jimmunol.1300858. [DOI] [PubMed] [Google Scholar]
- Halai K, Whiteford J, Ma B, Nourshargh S, Woodfin A. ICAM-2 facilitates luminal interactions between neutrophils and endothelial cells in vivo. J Cell Sci. 2014:620–629. doi: 10.1242/jcs.137463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heisig N. Functional analysis of microcirculation in the exocrine pancreas. Adv Microcirc. 1968:89–94. [Google Scholar]
- Jenne CN, Wong CHY, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, McFadden G, Kubes P. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe. 2013:169–180. doi: 10.1016/j.chom.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Kinoshita K, Leung A, Simon S, Evans E. Long-lived, high-strength states of ICAM-1 bonds to beta(2) integrin, II: lifetimes of LFA-1 bonds under force in leukocyte signaling. Biophys J. 2010:1467–1475. doi: 10.1016/j.bpj.2009.12.4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F, Garcia AJ, Mould AP, Humphries MJ, Zhu C. Demonstration of catch bonds between an integrin and its ligand. J Cell Biol. 2009:1275–1284. doi: 10.1083/jcb.200810002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- Li N, Mao DB, Lü SQ, Tong CF, Zhang Y, Long M. Distinct binding affinities of Mac-1 and LFA-1 in neutrophil activation. J Immunol. 2013:4371–4381. doi: 10.4049/jimmunol.1201374. [DOI] [PubMed] [Google Scholar]
- Li WJ, Nava RG, Bribriesco AC, Zinselmeyer BH, Spahn JH, Gelman AE, Krupnick AS, Miller MJ, Kreisel D. Intravital 2-photon imaging of leukocyte trafficking in beating heart. J Clin Invest. 2012:2499–2508. doi: 10.1172/JCI62970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C, Shimaoka M, Ferzly M, Oxvig C, Takagi J, Springer TA. An isolated, surface-expressed I domain of the integrin alpha L beta 2 is sufficient for strong adhesive function when locked in the open conformation with a disulfide bond. Proc Natl Acad Sci USA. 2001:2387–2392. doi: 10.1073/pnas.041606398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lü SQ, Ye ZY, Zhu C, Long M. Quantifying the effects of contact duration, loading rate, and approach velocity on P-selectin-PSGL-1 interactions using AFM. Polymer. 2006:2539–2547. [Google Scholar]
- Lum AFH, Green CE, Lee GR, Staunton DE, Simon SI. Dynamic regulation of LFA-1 activation and neutrophil arrest on intercellular adhesion molecule 1 (ICAM-1) in shear flow. J Biol Chem. 2002:20660–20670. doi: 10.1074/jbc.M202223200. [DOI] [PubMed] [Google Scholar]
- Luo BH, Springer TA. Integrin structures and conformational signaling. Curr Opin Cell Biol. 2006:579–586. doi: 10.1016/j.ceb.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyck R, Enzmann G. The physiological roles of ICAM-1 and ICAM-2 in neutrophil migration into tissues. Curr Opin Hematol. 2015:53–59. doi: 10.1097/MOH.0000000000000103. [DOI] [PubMed] [Google Scholar]
- Makino A, Shinb HY, Komai Y, Fukuda S, Coughlin M, Sugihara-Seki M, Schmid-Schönbein GW. Mechanotransduction in leukocyte activation: a review. Biorheology. 2007:221–249. [PubMed] [Google Scholar]
- McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CCM, Beck PL, Muruve DA, Kube P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010:362–366. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- McEver RP, Zhu C. Rolling cell adhesion. Annu Rev Cell Dev Biol. 2010:363–396. doi: 10.1146/annurev.cellbio.042308.113238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menezes GB, Lee WY, Zhou H, Waterhouse CCM, Cara DC, Kubes P. Selective down-regulation of neutrophil Mac-1 in endotoxemic hepatic microcirculation via IL-10. J Immunol. 2009:7557–7568. doi: 10.4049/jimmunol.0901786. [DOI] [PubMed] [Google Scholar]
- Miller J, Knorr R, Ferrone M, Houdei R, Carron CP, Dustin ML. Intercellular adhesion molecule-1 dimerization and its consequences for adhesion mediated by lymphocyte function addociated-1. J Exp Med. 1995:1231–1241. doi: 10.1084/jem.182.5.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mould AP, Barton SJ, Askari JA, Craig SE, Humphries MJ. Role of ADMIDAS cation-binding site in ligand recognition by integrin α5β1. J Biol Chem. 2003:51622–51629. doi: 10.1074/jbc.M306655200. [DOI] [PubMed] [Google Scholar]
- Orlova VV, Choi EY, Xie CP, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin EMBO J 2007. 1129 1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermann G, Weber KSC, Zernecke A, Schroder A, Weber C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol. 2002:151–158. doi: 10.1038/ni755. [DOI] [PubMed] [Google Scholar]
- Ozaki H, Ishii K, Horiuchi H, Arai H, Kawamoto T, Okawa K, Iwamatsu A, Kita T. Cutting edge: combined treatment of TNF-α and IFN-γ causes redistribution of junctional adhesion molecule in human endothelial cells. J Immunol. 1999:553–557. [PubMed] [Google Scholar]
- Pullerits R, Brisslert M, Jonsson IM, Tarkowski A. Soluble receptor for advanced glycation end products triggers a proinflammatory cytokine cascade via β2 integrin Mac-1. Arthritis Rheum. 2006:3898–3907. doi: 10.1002/art.22217. [DOI] [PubMed] [Google Scholar]
- Reilly PL, Woska JR, Jr, Jenfavre DD, McNally E, Rothlein R, Bormann BJ. The native structure of intercellular adhesion molecule-1 (ICAM-1) is a dimmer. Correlation with binding to LFA-1. J Immunol. 1995:529–532. [PubMed] [Google Scholar]
- Robert P, Brechtefeld D, Walzog B. Intraluminal crawling versus interstitial neutrophil migration during inflammation. Mol Immunol. 2013:70–75. doi: 10.1016/j.molimm.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Shaw SK, Ma S, Kim MB, Rao RM, Hartman CU, Froio RM, Yang L, Jones T, Liu Y, Nusrat A, et al. Coordinated redistribution of leukocyte LFA-1 and endothelial cell ICAM-1 accompany neutrophil transmigration J Exp Med 2004. 1571 1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon SI, Green CE. Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu Rev Biomed Eng. 2005:151–185. doi: 10.1146/annurev.bioeng.7.060804.100423. [DOI] [PubMed] [Google Scholar]
- Sircar M, Bradfield PF, Aurrand-Lions M, Fish RJ, Alcaide P, Yang L, Newton G, Lamont D, Sehrawat S, Mayadas T, et al. Neutrophil transmigration under shear flow conditions in vitro is junctional adhesion molecule-C independent J Immunol 2007. 5879 5887 [DOI] [PubMed] [Google Scholar]
- Tees DFJ, Waugh RE, Hammer DA. A microcantilever device to assess the effect of force on the lifetime of selectin-carbohydrate bonds. Biophys J. 2001:668–682. doi: 10.1016/S0006-3495(01)76047-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voisin MB, Nourshargh S. Neutrophil transmigration: emergence of an adhesive cascade within venular walls. J Innate Immun. 2013:336–347. doi: 10.1159/000346659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcikiewicz EP, Abdulreda MH, Zhang XH, Moy VT. Force spectroscopy of LFA-1 and its ligands, ICAM-1 and ICAM-2. Biomacromolecules. 2006:3188–3195. doi: 10.1021/bm060559c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfin A, Voisin MB, Nourshargh S. Recent developments and complexities in neutrophil transmigration. Curr Opin Hematol. 2010:9–17. doi: 10.1097/MOH.0b013e3283333930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Zhu J, Chen X, Mi L, Nishida N, Springer TA. Structure of an integrin with an alpha I domain, complement receptor type 4. EMBO J. 2010:666–679. doi: 10.1038/emboj.2009.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HY, Yu JP, Fu G, Shi XL, Xiao L, Chen YZ, Fang XH, He C. Interaction between single molecules of Mac-1 and ICAM-1 in living cells: an atomic force microscopy study. Exp Cell Res. 2007:3497–3504. doi: 10.1016/j.yexcr.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Yang Y, Jun CD, Liu JH, Zhang R, Joachimiak A, Springer TA, Wang JH. Structural basis for dimerization of ICAM-1 on the cell surface. Mol Cell. 2004:269–276. doi: 10.1016/s1097-2765(04)00204-7. [DOI] [PubMed] [Google Scholar]
- Zarbock A, Ley K. Neutrophil adhesion and activation under flow. Microcirculation. 2009:31–42. doi: 10.1080/10739680802350104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan DY, Zhang Y, Long M. Spreading of human neutrophils onto ICAM-1-immobilized substrate under shear flow. Chin Sci Bull. 2012:769–775. [Google Scholar]
- Zhang F, Marcus WD, Goyal NH, Selvaraj P, Springer TA, Zhu C. Two-dimensional kinetics regulation of alpha(L)beta(2)-ICAM-1 interaction by conformational changes of the alpha(L)-inserted domain. J Biol Chem. 2005:42207–42218. doi: 10.1074/jbc.M510407200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wojcikiewicz E, Moy VT. Force spectroscopy of the leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction. Biophys J. 2002:2270–2279. doi: 10.1016/S0006-3495(02)73987-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sun GY, Lü SQ, Li N, Long M. Low spring constant regulates P-selectin-PSGL-1 bond rupture. Biophys J. 2008:5439–5488. doi: 10.1529/biophysj.108.137141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.