

TORC1 modulates proteosynthesis, nitrogen metabolism, stress responses, and autophagy. Here it is shown that the Sch9 branch of TORC1 signaling depends specifically on vacuolar membranes and that this specificity allows the cells to regulate selectively the outputs of divergent downstream pathways in response to oxidative stress.
Abstract
Target of rapamycin complex 1 (TORC1) is a central cellular signaling coordinator that allows eukaryotic cells to adapt to the environment. In the budding yeast, Saccharomyces cerevisiae, TORC1 senses nitrogen and various stressors and modulates proteosynthesis, nitrogen uptake and metabolism, stress responses, and autophagy. There is some indication that TORC1 may regulate these downstream pathways individually. However, the potential mechanisms for such differential regulation are unknown. Here we show that the serine/threonine protein kinase Sch9 branch of TORC1 signaling depends specifically on the integrity of the vacuolar membrane, and this dependency originates in changes in Sch9 localization reflected by phosphatidylinositol 3,5-bisphosphate. Moreover, oxidative stress induces the delocalization of Sch9 from vacuoles, contributing to the persistent inhibition of the Sch9 branch after stress. Thus, our results establish that regulation of the vacuolar localization of Sch9 serves as a selective switch for the Sch9 branch in divergent TORC1 signaling. We propose that the Sch9 branch integrates the intrinsic activity of TORC1 kinase and vacuolar status, which is monitored by the phospholipids of the vacuolar membrane, into the regulation of macromolecular synthesis.
INTRODUCTION
Target of rapamycin (TOR) is a phosphatidylinositol 3-kinase–related kinase conserved in eukaryotes (Cafferkey et al., 1993) that transmits signals downstream by phosphorylating a set of downstream targets, including proteins involved in ribosomal synthesis and translation (Burnett et al., 1998; Jorgensen et al., 2004). Previous studies have revealed that TOR is composed of two structurally and functionally distinct protein complexes, namely TOR complex 1 (TORC1) and TOR complex 2 (TORC2) (Loewith et al., 2002; Jacinto et al., 2004). TORC1 serves important functions in nutrient sensing and stress response (Hara et al., 1998; Urban et al., 2007). In addition to the catalytic subunit TOR, TORC1 is composed of several associated proteins that contribute to the regulation of its kinase activity. In the budding yeast Saccharomyces cerevisiae, TORC1 consists of Tor1/2, Kog1, Lst8, and Tco89 protein subunits (Loewith et al., 2002; Reinke et al., 2004).
Yeast TORC1 is involved in the regulation of multiple physiological functions, including proteosynthesis, nitrogen metabolism, nitrogen permeability, and autophagy (Calvo et al., 1992; Noda and Ohsumi, 1998; Schmidt et al., 1998; Beck and Hall, 1999; Loewith et al., 2002). These physiologically diverse branches of the TORC1 signaling pathway are regulated separately downstream of TORC1. Sch9, the functional homologue of mammalian protein kinase S6K, activates proteosynthesis on direct phosphorylation of TORC1 (Urban et al., 2007), and the phosphorylation of Atg13 by TORC1 inhibits autophagy (Kamada et al., 2010). In addition, regulation of amino acid uptake and nitrogen metabolism by TORC1 is achieved via the inhibition of protein phosphatase 2A (Di Como and Arndt, 1996; Schmidt et al., 1998; Beck et al., 1999).
Amino acids are the primordial activators of TORC1 activity (Hara et al., 1998). Heterodimeric Rag GTPases Gtr1 and Gtr2 mediate amino acid signaling and regulate TORC1 activity (Dubouloz et al., 2005; Binda et al., 2009). A combination of the GTP-bound form of Gtr1 and the GDP-bound form of Gtr2 activates TORC1, whereas a complex of the GDP-bound form of Gtr1 and the GTP-bound form of Gtr2 represses TORC1 (Dubouloz et al., 2005; Binda et al., 2009). In addition to the Gtr proteins, Ego1, Ego2, and Ego3 constitute the EGO (exit from G0) complex, which is thought to be important in TORC1 regulation, because a loss of these factors or a mutation affecting the GTPase activity of the Gtr proteins leads to delocalization of TORC1 and a defective response of TORC1 activity to nutrient availability (Stracka et al., 2014; Powis et al., 2015; Kira et al., 2016).
Rapamycin is a drug that selectively inhibits TORC1 activity (Heitman et al., 1991; Loewith et al., 2002). Screening for hypersensitive mutants to rapamycin enriched those defective in TORC1 activity (Dubouloz et al., 2005). Similar screens also searched for a subset of genes involved in the vacuolar protein-sorting pathway. When this pathway is deficient, phosphorylation of a TORC1 substrate is decreased; moreover, such a mutation shows synthetic lethality with TOR1 (Xie et al., 2005; Zurita-Martinez et al., 2007). Previous studies have indicated that TORC1 is localized on the cytoplasmic surface of lysosomes and vacuoles in mammals and yeast, respectively, and that this localization is important for its activation (Wedaman et al., 2003; Yan et al., 2006; Urban et al., 2007; Sancak et al., 2008; Kira et al., 2016). Among the proteins in the vacuolar protein-sorting pathway, components of the homotypic fusion and vacuole protein-sorting complex (HOPS) impact the TORC1 pathway (Kingsbury et al., 2014). HOPS mediates molecular transport from the Golgi apparatus and late endosomes to vacuoles. Although a HOPS component, Vam6, reportedly acts as a guanine nucleotide exchange factor (GEF) for Gtr1 of the EGO complex (Binda et al., 2009), the mechanisms of action of other HOPS components on TORC1 activity remain elusive.
Under conditions suitable for growth (i.e., rich in nutrients and no stressors), TORC1 activity contributes to cell cycle progression and proteosynthesis (Calvo et al., 1992; Barbet et al., 1996). By contrast, when cells encounter a condition that is inappropriate for growth, TORC1 activity is repressed, which in turn suppresses protein synthesis and proliferation (Urban et al., 2007; Loewith and Hall, 2011). Decreased TORC1 activity also induces Sch9-mediated activation of stress response genes (Wanke et al., 2008; Wei et al., 2008). Thus, TORC1 senses numerous environmental conditions and transmits signals to distinct branches of the TORC1 pathway. Therefore, TORC1 is considered a cellular signaling hub (Goberdhan et al., 2016). However, compared with its physiological importance in amino acid sensing, the role of TORC1 in other stress response pathways is still unclear.
Although TORC1 plays a role as a signaling hub, its downstream branches may not always respond uniformly to changes in the environment. For example, under amino acid or nitrogen depletion, the materials for constructing proteins are insufficient. In this situation, proteosynthesis, and thus proliferation, are suppressed, and general amino acid uptake and autophagy are induced, all of which are mediated by the inhibition of TORC1 activity (Barbet et al., 1996; Noda and Ohsumi, 1998; Schmidt et al., 1998; MacGurn et al., 2011). On the other hand, cells can respond to stresses such as glucose starvation, phosphate starvation, osmotic stress, oxidative stress, high temperature, or DNA damage by decreasing protein synthesis and activating stress response genes, both of which are mediated through TORC1 (Beck and Hall, 1999; Mahfouz et al., 2006; Urban et al., 2007; Wanke et al., 2008; Budanov et al., 2010; Loewith and Hall, 2011; Takahara and Maeda, 2012; Thedieck et al., 2013). However, such stressors do not necessarily affect the activities of other branches of the TORC1 pathway other than proteosynthesis (Hughes Hallett et al., 2014). These observations suggest that whereas TORC1 integrates environmental information, the outputs of the TORC1 pathway may be optimized such that different branches of the TORC1 pathway are regulated separately, and only certain branches are modulated by certain environmental signals. To date, the mechanism underlying such regulation remains an open question.
How does TORC1 separately regulate the activity of different downstream pathway branches based on specific environmental cues? To reveal this mechanism, we investigated the functions of TORC1 regulators in heterogeneous downstream signaling. In this study, we show that a deficiency in HOPS components specifically affects signaling to the Sch9 branch of the TORC1 pathway. Moreover, we show that changes in the localization of phosphatidylinositol 3,5-bisphosphate [PI(3,5)P2] are important for the specific inhibition of the Sch9 branch under oxidative stress. Our results suggest that branches of the TORC1 downstream pathway are differentially regulated by their dependence on vacuolar membrane localization. The Sch9 branch is highly dependent on vacuoles, which enables the selective inhibition of this signaling branch in response to a specific subset of stressors.
RESULTS
Signal outputs of the TORC1 pathway are differentially regulated in an input signal–dependent manner
To investigate the mechanisms underlying TORC1 downstream signaling diversity, we first examined the status of each branch of the TORC1 pathway under several stress conditions by evaluating the phosphorylation of Sch9 (Sch9 branch), Npr1, whose phosphorylation is maintained by activated TORC1 via suppression of protein phosphatase 2A (PP2A) activity, Par32 (phosphorylated after rapamycin 32), which is phosphorylated by Npr1 when TORC1 is suppressed (PP2A branch), and Atg13 (Atg13 branch) (Schmidt et al., 1998; Urban et al., 2007; Huber et al., 2009; Kamada et al., 2010; Hughes Hallett et al., 2014).
Consistent with a previous report (Hughes Hallett et al., 2014), phosphorylation of Sch9, Npr1, and Atg13 was robustly decreased following nitrogen deprivation as well as the addition of rapamycin, a specific inhibitor of TORC1 (Figure 1). By contrast, hyperosmotic stress induced by treatment with 1 M NaCl caused a significant reduction in Sch9 phosphorylation and slightly impacted Atg13 phosphorylation status but did not significantly change Npr1 or Par32 modifications (Figure 1). These observations confirmed that different stressors elicit differential downstream responses of TORC1 signaling (Hughes Hallett et al., 2014), where nitrogen deprivation or rapamycin treatment effectively turned off TORC1 signaling to most branches of the TORC1 pathway.
FIGURE 1:
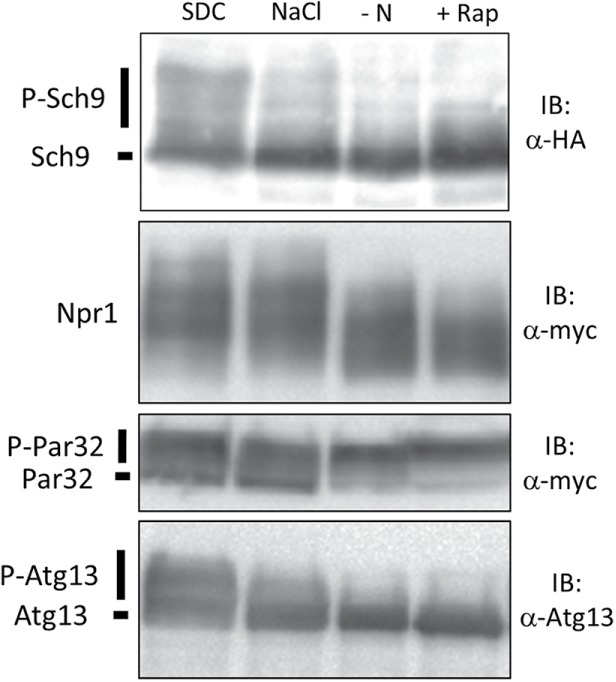
Signal outputs of the TORC1 pathway are differentially regulated in an input signal–dependent manner. Western blot analysis of the C-terminal fragment of Sch9-5HA (yet515), Npr1-13myc (yet610), Atg13 (yet562), and Par32-13myc (yet515) was performed. Cells in log phase were treated with SDC (control), SDC + 1 M NaCl (hyperosmotic stress), SD−N (nitrogen depletion), or SDC + 200 ng/ml rapamycin (TORC1 inhibitor) for 30 min, and the lysates were subjected to Western blotting.
The HOPS complex is specifically required for transmitting the TORC1 signal to the Sch9 branch
We hypothesized that previously identified TORC1 signaling–related factors contribute to the differential regulation of downstream branches in the TORC1 pathway. To confirm this hypothesis, we focused on three factors known to be involved in TORC1 activity. Vps41 is a component of HOPS that mediates endosome-vacuole fusion and vacuole-vacuole fusion (Seals et al., 2000; Balderhaar and Ungermann, 2013), and its deficiency confers rapamycin hypersensitivity (Xie et al., 2005; Zurita-Martinez et al., 2007). Ego1 is a component of the EGO complex (Dubouloz et al., 2005), the counterpart of mammalian Ragulator, which contributes to the targeting of mTORC1 to lysosomal surfaces and its amino acid–dependent activation (Sancak et al., 2010; Kira et al., 2016). Tco89 is a nonessential component of TORC1, but its deletion decreases TORC1 activity (Reinke et al., 2004).
First, we estimated the activity of TORC1 in each mutant under nutrient-rich conditions with a rapamycin recovery assay (Dubouloz et al., 2005). As shown in Figure 2A, the three mutants exhibited increased sensitivity to rapamycin, indicating that TORC1-related signals were noticeably attenuated by these mutations (Figure 2A). Next, we investigated the downstream transduction of TORC1 signaling in these mutants by assessing the phosphorylation status of target proteins in each branch. The phosphorylation status of Sch9 in ∆vps41 mutant cells was lower than that in ∆ego1 mutant cells (Figure 2, B and C). This was consistent with a previous report in which the effects of HOPS disruption on the Sch9 branch were more severe than those of the EGO complex (Kingsbury et al., 2014).
FIGURE 2:

The HOPS complex is specifically required for transmitting the TORC1 signal to the Sch9 branch. (A) Spot assay investigating growth recovery after rapamycin treatment in wild-type (WT) (BY4741), ∆vps41 (yet567), ∆ego1 (yet576), and ∆tco89 (yet701) cells. Cells in log phase were cultured in the presence or absence of 200 ng/ml rapamycin for 3 h at 30°C. The cell cultures were serially diluted 10-fold and then spotted onto YPD medium. Colonies were photographed after 1 d (YPD) or 2 d (Rap recovery). (B) Western blot analysis of C-terminal fragment of Sch9-5HA (yet515, 569, 577, and 729), Npr1-13myc (yet610, 618, 619, and 780), Atg13 (yet562, 574, 580, and 726), and Par32-13myc (yet515, 569, 577, and 729). Lysates of WT (yet515, 610, and 562), ∆vps41 (yet569, 618, and 574), ∆ego1 (yet577, 619, and 577), and ∆tco89 (yet729, 780, and 726) cells grown in SDC medium, or WT cells treated with 200 ng/ml rapamycin in SDC medium, were subjected to Western blotting. (C) Quantification of the band shift data of ∆vps41 and ∆ego1 cells for Sch9 from B (n = 5). To calculate the p value, we applied the Brunner-Munzel test. Error bars represent 95% confidence intervals. *p < 0.005. (D) Western blot analysis of C-terminal fragment of Sch9-5HA and Par32-13myc. WT (yet515), ∆vps41 (yet569), ∆ego1 (yet577), and ∆tco89 (yet729) cells under nitrogen starvation for 30 min were resuspended in SDC medium. Cells grown in SDC medium were collected at each time point, and cell lysates were subjected to Western blotting. (E) Western blot analysis of C-terminal fragment of Sch9-5HA. Lysates of VPS41 ∆gtr1 ∆gtr2 and ∆vps41 ∆gtr1 ∆gtr2 cells harboring empty vector (yet639, 755), GTR1WT GTR2WT (yet640, 756), GTR1GDP GTR2GTP (yet645, 761), or GTR1GTP GTR2GDP (yet647, 763) grown in SDC medium were subjected to Western blotting.
In ∆ego1 or ∆tco89 cells, dephosphorylated forms of Npr1 accumulated even under nutrient-rich conditions (Figure 2B). Surprisingly, the phosphorylation status of Npr1 was not affected in the HOPS deletion mutant ∆vps41 (Figure 2B). The effects of HOPS deletion on the PP2A branch of the TORC1 pathway were further confirmed by the phosphorylation status of Par32. Par32 was constantly phosphorylated in ∆ego1 and ∆tco89 cells but not in ∆vps41 cells. We also performed the same experiments using other HOPS mutants, ∆vam6 and ∆pep3, and the results were similar to those in ∆vps41 cells (Supplemental Figure S1).
Interestingly, the phosphorylation status of Atg13 was essentially unchanged in any mutant (Figure 2B). Thus, the downstream transduction of TORC1 signaling to Atg13 is relatively resistant to genetic perturbation. This observation is consistent with our previous finding that autophagy was not activated by the deletion of an Ego subunit in nutrient-rich conditions (Kira et al., 2014), as TORC1-dependent phosphorylation of Atg13 inhibited the induction of autophagy (Kamada et al., 2010).
We further addressed the effects of HOPS deletion on TORC1 downstream signaling in response to nutrient input. As shown in Figure 2D, when wild-type (WT) cells were transferred from nutrient-depleted to nutrient-rich media, activated TORC1 transduced the signal to its branches, thereby inducing Sch9 phosphorylation and Par32 dephosphorylation within 2−10 min. In ∆vps41 cells, the PP2A branch still responded to nitrogen sources, as revealed by the reduced phosphorylation of Par32 observed in a similar time course to WT cells. However, Sch9 phosphorylation was barely induced in these mutants. On the other hand, ∆ego1 and ∆tco89 cells did not show any changes in the phosphorylation status of Sch9 or Par32. Taken together, these results indicate that HOPS is specifically required for TORC1-dependent activation of the Sch9 branch. This specified role of HOPS in the TORC1 pathway is in striking contrast with the roles of the EGO complex and TORC1 component, which are critical for activation of the two downstream branches.
A previous report showed that activation of Gtr partially rescued the rapamycin sensitivity of HOPS mutants (Kingsbury et al., 2014). Thus, we investigated whether activation of the EGO complex rescues the defect in Sch9 signaling caused by disrupting HOPS. To achieve this, we first constructed a ∆gtr1 ∆gtr2 ∆vps41 triple mutant, after which we introduced WT Gtr1 and Gtr2, the activated forms Gtr1GTP and Gtr2GDP, or the inactivated forms Gtr1GDP and Gtr2GTP (Kira et al., 2014). As shown in Figure 2E, the ∆gtr1 ∆gtr2 deletion reduced Sch9 phosphorylation, which was rescued by Gtr1-Gtr2 or Gtr1GTP-Gtr2GDP but not by Gtr1GDP-Gtr2GTP or empty vector. In the ∆vps41 background, the effects of these alleles on recovery were minimal (Figure 2E). Although Vam6, a subunit of HOPS, acts as a GEF for Gtr1, the insufficient suppression of ∆vps41 by activated Gtr implies that HOPS has an additional function other than Gtr1 activation in TORC1 signaling via the Sch9 branch.
The HOPS complex is necessary for normal Sch9 localization
The HOPS mutant was slightly resistant to rapamycin compared with the ego mutant and still stimulated TORC1 activity on nutrient input (Figure 2, A and D). To determine how HOPS specifically affects the Sch9 branch, we focused on the localization of Sch9 and TORC1, because we previously reported that localization of Sch9 and TORC1 was regulated independently (Kira et al., 2016) and that the integrity of the Sch9 branch depends on the formation of mature vacuoles, which contributes to the membrane localization of Sch9 (Jin and Weisman, 2015).
We expressed a C-terminal Sch9-2GFP fusion protein from the SCH9 promoter encoded on a centromeric plasmid and observed intracellular localization using fluorescence microscopy. Consistent with previous studies (Jorgensen et al., 2004; Jin et al., 2014), Sch9 was localized to both the cytoplasm and vacuolar membranes (Figure 3A). Similarly to WT cells, ∆ego1 and ∆tco89 cells showed Sch9 localization on vacuolar membranes and throughout the cytoplasm (Figure 3A). By contrast, we found that membrane localization of Sch9 disappeared in ∆vps41 cells (Figure 3B), which had fragmented, immature vacuoles stained with the vacuolar marker 7-amino-4-chloromethylcoumarin (CMAC), as previously reported (Nakamura et al., 1997; Seals et al., 2000). These results indicate that vacuolar membrane localization of Sch9 requires the HOPS complex. We also observed the localization of Tor1, the catalytic subunit of TORC1 (Loewith et al., 2002). As shown in Figure 3C, GFP-tagged Tor1 was localized to vacuolar membranes in WT cells. In ∆vps41 cells, TORC1 appeared to be associated with immature vacuoles stained by CMAC (Figure 3C). This indicates that the differential localization of TORC1 and Sch9 may be the cause of the reduced phosphorylation of Sch9 due to disrupted HOPS. Thus, these results suggest that HOPS disruption causes abnormal localization of Sch9 by changing the structure or quality of vacuolar membranes that facilitate Sch9 localization to vacuoles.
FIGURE 3:
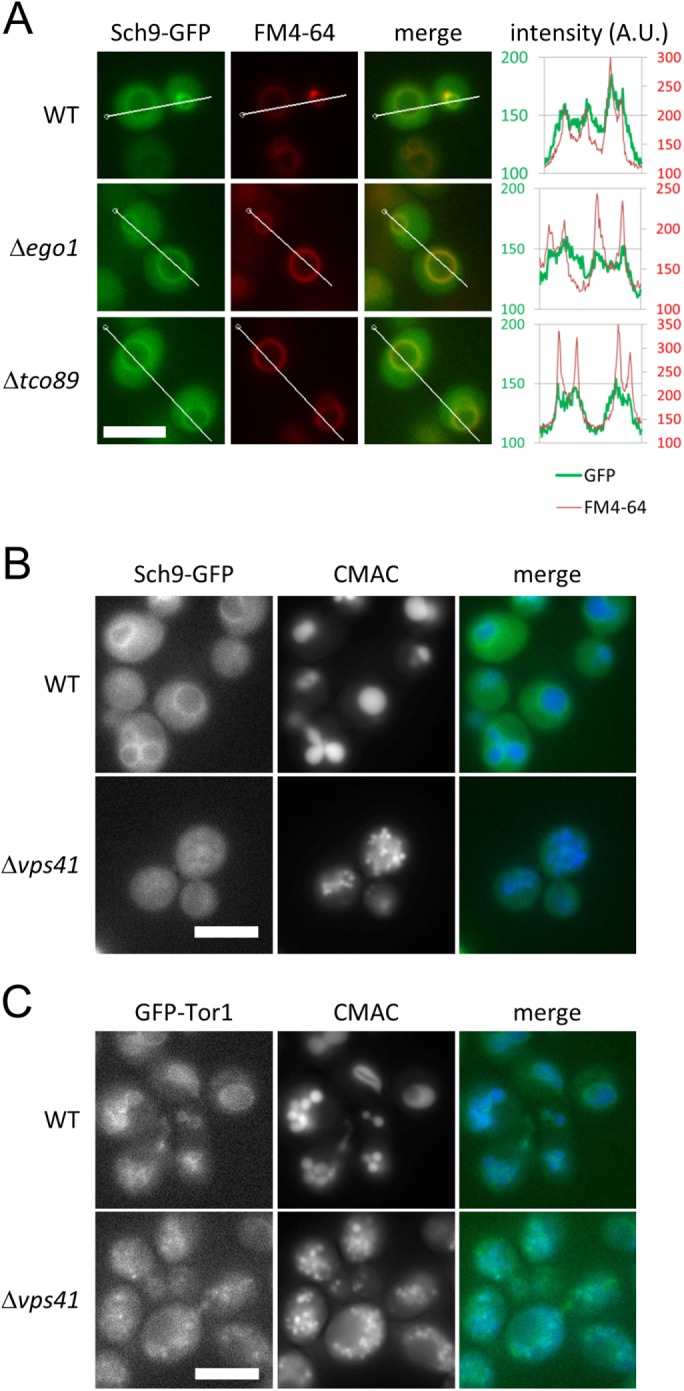
The HOPS complex is necessary for normal localization of Sch9. (A) Representative images of WT (yet120), ∆ego1 (yet691), and ∆tco89 (yet727) cells expressing Sch9-2GFP from the SCH9 promoter encoded on a centromeric plasmid. Cells at mid–log phase grown in SDC medium were stained with FM4-64 as a vacuolar marker. The signal intensities of Sch9-2GFP and FM4-64 along the indicated lines were measured by softWoRx software. Scale bar = 5 µm. (B) Representative images of WT (yet120) and ∆vps41 (yet732) cells expressing Sch9-2GFP. Cells at mid–log phase grown in SDC medium were stained with CMAC as a vacuolar marker. Scale bar = 5 µm. (C) Representative images of WT (SKY374-A) and ∆vps41 (yet665) cells expressing Tor1-GFP. Cells at mid–log phase grown in SDC medium were stained with CMAC as a vacuolar marker. Scale bar = 5 µm.
Disruption of the HOPS complex decreases TORC1 output to the Sch9 branch by inducing the delocalization of Sch9 from vacuolar membranes
We hypothesized that the specific reduction of TORC1-Sch9 signaling in HOPS mutants may be attributable to a deficiency in scaffolds for Sch9 localization to a specific membrane site. To test this possibility, we artificially tethered Sch9 to intracellular membranes by fusing a FYVE (Fab1, YOTB, Vac1, and EEA1) domain from mammalian EEA1 (Burd and Emr, 1998) and investigated whether TORC1-Sch9 signaling was rescued. The FYVE domain has been implicated in binding with phospholipid phosphatidylinositol 3-phosphate (PI3P), which is 20 times as abundant as PI(3,5)P2 in yeast cells and is concentrated on vacuolar membranes (Duex et al., 2006). As shown in Figure 4A, GFP-FYVE-Sch9 was mainly localized on vacuolar membranes in WT cells, whereas in ∆vps41 cells, it was localized to the surfaces of immature vacuoles (Figure 4A).
FIGURE 4:

Disruption of HOPS complex decreases TORC1 output to the Sch9 branch by inducing Sch9 delocalization from vacuolar membranes. (A) Representative images of WT (yet234) and ∆vps41 (yet606) cells expressing GFP-FYVE-Sch9. Cells were analyzed during mid–log phase in SDC (+ uracil) medium. Scale bar = 5 µm. (B) Western blot analysis of the C-terminal fragment of Sch9-5HA. Lysates of WT (yet628, 629), and ∆vps41 (yet661, 662) cells in SDC (+ uracil) medium and/or WT cells treated with 200 ng/ml rapamycin + SDC (+ uracil) medium were subjected to Western blotting. GFP-Sch9 (yet628, 661) or GFP-FYVE-Sch9 (yet629, 662) were expressed from the endogenous SCH9 promoter. (C) Spot assay investigating growth recovery after rapamycin treatment in VPS41 GFPSCH9 (yet36), VPS41 GFP-FYVESCH9 (yet234), ∆vps41 GFPSCH9 (yet605), and ∆vps41 GFP-FYVESCH9 (yet606) cells. Cells in mid–log phase were cultured in the presence of absence of 200 ng/ml rapamycin for 3 h. The cell cultures were serially diluted 10-fold and then spotted onto YPD medium. Colonies were photographed after 1 d (YPD) or 3 d (Rap recovery).
Next, we investigated the effects of Sch9 membrane tethering on TORC1 signaling. Interestingly, the tethering caused robust phosphorylation of Sch9 in WT and ∆vps41 cells, and this phosphorylation was canceled by the addition of rapamycin, indicating both vacuolar membrane localization dependence and TORC1 dependence (Figure 4B). This result clearly shows that membrane localization stimulates TORC1-dependent phosphorylation of Sch9 and that defective membrane localization of Sch9 restricts phosphorylation by TORC1 in ∆vps41 cells. Moreover, we found that membrane-tethered Sch9 is functional for growth regulation, as the expression of GFP-FYVE-Sch9 in WT cells did not cause any growth defects and substantially rescued rapamycin sensitivity caused by HOPS disruption (Figure 4C). These data suggest that the specific involvement of HOPS in TORC1-Sch9 signaling is achieved by regulating Sch9 intracellular membrane localization.
Oxidative stress alters the localization of Sch9 and inhibits TORC1-Sch9 signaling
What is the physiological role of membrane-assisted signaling to the Sch9 branch? On the basis of the analysis of HOPS mutants, we expected that the regulation of Sch9 localization may be important in controlling TORC1-Sch9 signaling under specific physiological conditions.
Previous studies have indicated that TORC1-Sch9 signaling was suppressed under oxidative stress, heat stress, or hyperosmotic stress conditions (Urban et al., 2007), whereas these stresses only moderately affected the PP2A branch (Hughes Hallett et al., 2014). We found that phosphorylation of Atg13 was decreased under heat stress or hyperosmotic stress conditions but not under oxidative stress (Figures 1 and 5A). Consistent with previous reports (Hughes Hallett et al., 2014), we observed that Npr1 phosphorylation was not significantly changed, but that of Sch9 was robustly decreased under oxidative conditions (Figure 5B). These data demonstrate that oxidative stress suppresses the signal to the Sch9 branch from TORC1 but not to the Atg13 or PP2A branch. Moreover, we observed slight and transient dephosphorylation of Npr1 10 min after oxidative stress induction, in contrast with the gradual decline of Sch9 phosphorylation (Figure 5C). Thus, in addition to the specific suppression of TORC1-Sch9 signaling, TORC1 activity itself may be suppressed by oxidative stress, but this suppression occurred only transiently.
FIGURE 5:

Oxidative stress alters Sch9 localization and inhibits TORC1-Sch9 signaling. (A) Western blot analysis of the C-terminal fragment of Sch9-5HA (yet515) and Atg13 (yet562). Cells in mid–log phase were treated with SDC (control), SDC + 2 mM H2O2 (oxidative stress), SDC at 42°C (heat stress), SD−N (nitrogen depletion), or SDC + 200 ng/ml rapamycin (TORC1 inhibitor) for 30 min, and the lysates were subjected to Western blotting. (B) Western blot analysis of the C-terminal fragment of Sch9-5HA (yet515), Npr1-13myc (yet610), and Atg13 (yet562). Cells in mid–log phase were treated with 2 mM H2O2 in SDC, and the lysates were subjected to Western blotting at each time point. (C) Quantification of the band shift data of Npr1 and Sch9 from B (Npr1: n = 5, Sch9: n = 3). Error bars represent 95% confidence intervals. (D) Representative images of control or oxidative stress-induced (treated with 2 mM H2O2 for 30 min) cells expressing Sch9-2GFP and Ego3-3mCherry (yet765) during mid–log phase in SDC medium. Ego3-3mCherry was used as a vacuolar marker. Scale bar = 5 µm. (E) Quantification of the ratio of Sch9 fluorescence intensity of vacuolar membranes to that of the cytoplasm. Related to D. A ratio of 1.00 means no enrichment on vacuolar membranes. To calculate the p value, we applied the Brunner-Munzel test. *p < 0.005.
How is the Sch9 branch specifically suppressed in this situation? Fluorescence microscopic observation showed that Sch9 localization on vacuolar membranes was decreased on oxidative stress as time proceeded (Figure 5, D and E). The ratio of fluorescence intensity of Sch9-GFP on vacuolar membranes to that of the cytoplasm suggested that the amount of Sch9 on vacuolar membranes at 30 or 60 min was decreased to half of that at 0 min (Figure 5E). The results led us to hypothesize that TORC1-Sch9 signaling is specifically suppressed under oxidative stress through changes in Sch9 subcellular localization. To address this possibility, we examined the phosphorylation state of the FYVE-Sch9 fusion protein, which was more associated with vacuolar membranes than Sch9, even under oxidative stress conditions (Figure 6A; compare Figure 5E with Supplemental Figure S2). As shown in Figure 6, B and C, the phosphorylated form of FYVE-Sch9 was still apparent after treatment, although the proportion of the phosphorylated protein was reduced. Moreover, this phosphorylation was suppressed by simultaneous addition of rapamycin, revealing its TORC1 dependency (Figure 6B). Protein levels of normal and FYVE-fused alleles of SCH9 were comparable before treatment, and the amounts were not significantly changed following oxidative stress (Supplemental Figure S3). Thus, the localization, not the abundance, of Sch9 was responsible for TORC1-dependent Sch9 phosphorylation on oxidative stress.
FIGURE 6:

The localization of Sch9 is responsible for TORC1-dependent Sch9 phosphorylation on oxidative stress. (A) Representative images of control and oxidative stress-induced (treated with 2 mM H2O2 for 30 min) cells (yet234) expressing GFP-FYVE-Sch9 during mid–log phase in SDC (+ uracil) medium. Scale bar = 5 µm. (B) Western blot analysis of the C-terminal fragment of Sch9-5HA. Cells expressing Sch9-5HA (yet628) or FYVE-Sch9-5HA (yet629) were exposed to oxidative stress (2 mM H2O2) or exposed to oxidative stress and treated with rapamycin (200 nM) during mid–log phase in SDC medium, and the lysates were subjected to Western blotting at each time point. (C) Quantification of the band shift data of Sch9 and FYVE-Sch9 from B (n = 3). Error bars represent 95% confidence intervals. (D) Representative images of control and oxidative stress-induced (treated with 2 mM H2O2 for 30 min) cells (yet857) expressing Vac8-GFP-cSch9-5HA (cSch9 = C-terminal 116 amino acids of Sch9) during mid–log phase in SDC (+ uracil) medium. Scale bar = 5 µm. (E) Western blot analysis of Vac8-cSch9-5HA. Cells expressing Vac8-cSch9-5HA (yet859) were exposed to oxidative stress (2 mM H2O2) or exposed to oxidative stress and treated with rapamycin (200 nM) during mid–log phase in SDC medium, and the lysates were subjected to Western blotting at each time point. (F) Quantification of the band shift data of Vac8-cSch9 from E (n = 3). Error bars represent 95% confidence intervals.
As the abundance of FYVE-Sch9 on vacuolar membranes was gradually declined following oxidative stress (Supplemental Figure S2), we further tethered Sch9 to the vacuoles in a PI3P/PI(3,5)P2-independent manner by fusing C-terminal 116 amino acids of Sch9 (cSch9) to Vac8 C-terminus (Urban et al., 2007; Jin et al., 2014). Vac8-GFP-cSch9-5HA protein was substantially localized to vacuolar membranes under normal conditions, and its localization was essentially unchanged on oxidative stress (Figure 6D and Supplemental Figure S4). As shown in Figure 6, E and F, Vac8-cSch9 remained phosphorylated until 30 min after H2O2 treatment, but the phosphorylation was decreased at 60 min. These results indicate that cells suppress TORC1-Sch9 signaling in a Sch9 localization–dependent manner during early response to oxidative stress. In addition, the incomplete rescue of Sch9 phosphorylation at later time points suggests the presence of additional mechanism to suppress TORC1-Sch9 signaling without affecting that to the other branches.
PI(3,5)P2 localization changes in HOPS mutants and under oxidative stress
How is Sch9 localization regulated in HOPS mutants or in cells under oxidative stress? Our previous results indicated that PI(3,5)P2 is a regulator of Sch9 localization (Jin et al., 2014). We hypothesized that localization changes in PI(3,5)P2 delocalize Sch9 from vacuolar membranes. To address this possibility, we investigated the intracellular localization of PI(3,5)P2 in HOPS mutant cells or under oxidative stress using GFP-tagged Atg18, which harbors a PI(3,5)P2-binding domain (Dove et al., 2004). As shown in Figure 7A, GFP-Atg18 was localized to vacuolar membranes in WT cells. By contrast, Atg18 localization exhibited a dotlike pattern in ∆vps41 cells. This pattern of localization was similar to that observed in ∆vac7 cells (Figure 7A), in which the abundance of PI(3,5)P2 was reduced (Bonangelino et al., 2002). This result suggests that PI(3,5)P2 localization is abnormal in HOPS mutant cells.
FIGURE 7:

Subcellular localization of PI(3,5)P2 localization is altered in HOPS mutants and under oxidative stress conditions. (A) Representative images of WT (yet681), ∆vps41 (yet709), and ∆vac7 (yet782) cells expressing GFP-Atg18 at mid–log phase in SDC (+ uracil) medium. Scale bar = 5 µm. (B) Representative images of control and oxidative stress-induced (treated with 2 mM H2O2 for 30 min) cells (yet777) expressing GFP-Atg18 and Ego3-3mCherry during mid–log phase in SDC (+ uracil) medium. Scale bar = 5 µm. (C) Quantification of the ratio of Atg18 fluorescence intensity of vacuolar membranes to that of the cytoplasm. Related to B. (D) Representative images of control and oxidative stress-induced (treated with 2 mM H2O2 for 30 min) cells (yet793) expressing Fab1-GFP and Ego3-3mCherry during mid–log phase in SDC (+ uracil) medium. Scale bar = 5 µm. (E) Quantification of the ratio of Fab1 fluorescence intensity of vacuolar membranes to that of the cytoplasm. Related to D. In C and E, we applied the Brunner-Munzel test to calculate p values. *p < 0.005. (F) Representative images of control and oxidative stress-induced (treated with 2 mM H2O2 for 30 min) cells (yet838) expressing GFP-FYVE and mCherry-Atg18 during mid–log phase in SDC (+ uracil) medium. Scale bar = 5 µm.
We further investigated the effects of stress on the localization of PI(3,5)P2 using GFP-Atg18. After oxidative stress induction, vacuolar localization of Atg18 was decreased, similarly to that of Sch9 (Figures 5E and 7, B and C). These observations are consistent with the model that oxidative stress alters PI(3,5)P2, which causes delocalization of Sch9 from vacuolar membranes. In addition, the total level of PI(3,5)P2 was not significantly altered under oxidative stress (Supplemental Table 1), suggesting that the local distribution is changed by oxidative stress.
How are local levels of PI(3,5)P2 regulated on stress? Our previous report indicated that Fab1, the sole PI3P-5 kinase in yeast, and Sch9 did not localize to premature vacuolar membranes that were newly synthesized (Jin and Weisman, 2015). We assessed whether the localization of Fab1-GFP changed under oxidative stress. As shown in Figure 6, D and E, Fab1-GFP was substantially localized to vacuolar membranes under normal conditions, and localization decreased on oxidative stress. This result suggests that the loss of Fab1 localization after oxidative stress may be related to the reduction in the local amounts of PI(3,5)P2 on vacuolar membranes. Moreover, the fraction of vacuolar membrane-localized GFP-FYVE-Sch9 or GFP-FYVE was decreased under the oxidative stress condition, although the total amount of PI3P was increased (Figure 7F, Supplemental Figure 2, and Supplemental Table 1). These results suggest that subcellular localization of PI3P is also changed on oxidative stress.
DISCUSSION
The fundamental survival strategy of living beings is metabolic regulation. The cell, the basic unit of life, activates its own anabolic activity to stimulate growth and division under nutrient-rich conditions suitable for cellular proliferation. By contrast, the cell inactivates these activities and represses its growth and division under nutrient-deficient or stressful conditions. It is thought that only living beings capable of properly performing these regulatory processes are able to effectively utilize limited resources and survive to the present day. TOR has been implicated as one of the master players in these regulatory processes.
As the biochemical activity of TORC1 does not necessarily reflect intracellular activity after its isolation from cells (Kamada, 2017; Tanigawa and Maeda, 2017), in vivo TORC1 activity, including its changes due to physiological conditions or genetic manipulation, has been assessed by estimating the phosphorylation state of its downstream factors. This study shows that the signaling strength of each branch of the TORC1 signaling pathway can be differentially regulated, indicating the necessity of investigating more than two individual branches in parallel to effectively estimate the in vivo activity of TORC1.
In this study, we reevaluated the regulation of signal transduction in the TORC1 pathway under several different types of stress and discovered that this regulation is composed of two different mechanisms, namely the regulation of TORC1 activity itself and that of the specific interaction between TORC1 and downstream substrates. Evidence has accumulated for the former regulation of TORC1, which is enacted by evolutionarily conserved positive and negative factors of TORC1 (Garami et al., 2003; Inoki et al., 2003; Gwinn et al., 2008; Sancak et al., 2008; Binda et al., 2009; Bonfils et al., 2012; Takahara and Maeda, 2012; Bar-Peled et al., 2013; Panchaud et al., 2013; Thedieck et al., 2013; Tsun et al., 2013; Hughes Hallett et al., 2014, 2015; Kim and Cunningham, 2015; Péli-Gulli et al., 2015; González and Hall, 2017; Michel et al., 2017). By contrast, the latter form of regulation can cause a branch-specific switch in signaling, which may contribute to physiological responses to stress by optimizing a pathway by modulating a particular downstream branch. The presence of two modes of regulation is reasonable, because the central control by the main regulator, TORC1, may lead to futile changes in a specific branch under a particular condition.
Vacuoles as scaffolds for TORC1 activation and signal transmission
Numerous studies have investigated the mechanisms underlying the regulation of TORC1 by nutrient signals, showing that TORC1 and its regulators in yeast are localized to vacuolar membranes (Dubouloz et al., 2005; Kira et al., 2016). Moreover, disruption of the genes related to vacuolar morphogenesis or sorting resulted in rapamycin hypersensitivity (Xie et al., 2005; Zurita-Martinez et al., 2007). These results have established the importance of vacuoles and lysosomes in the TORC1 pathway.
Our previous report pointed out the differences in the localization of TORC1 and Sch9 or Fab1: TORC1 was localized to both mature and premature vacuoles, but Sch9 and Fab1 were associated exclusively with mature ones (Jin and Weisman, 2015). Partial colocalization of TORC1 and Sch9 suggests that the signal output of the Sch9 branch reflects TORC1 activity itself and the integrity of vacuoles.
In this study, we showed that the importance of vacuoles in transducing downstream signals differs in each branch of the TORC1 pathway, such that vacuolar deficiency via HOPS mutation severely affects the Sch9 branch but only minimally impacts the PP2A and Atg13 branches. One possible explanation for this mechanism is that the difference in vacuolar dependency is due to the level of TORC1 activity required for signal transduction. A lack of normal vacuoles may reduce the catalytic activity of TORC1, and the Sch9 branch may require higher TORC1 activity for its activation than the other branches. Indeed, the signaling diversity downstream of TORC1 is achieved by the inherent capacity of phosphorylation sites to serve as TORC1 substrates in mammals (Kang et al., 2013). However, our data suggest that differential preferences for phosphorylation by TORC1 do not always explain branch-specific signal transduction. The PP2A branch is greatly influenced by a deficiency of the EGO complex (Figure 2B). Previous studies support our observation that the activity of TORC1 is reduced in these mutants, but these observations suggest that the influence of an EGO complex deficiency on the Sch9 branch is less than that of a deficiency of the HOPS complex (Figure 2, B and C) (Kingsbury et al., 2014). This phenomenon is not simply explained by the “substrate quality” model (Kang et al., 2013), leading us to consider the possibility that vacuolar dysfunction affects not only TORC1 itself but also its interaction with a specific downstream target. This possibility is supported by our observation that mutations causing vacuolar dysfunction resulted in a loss in vacuolar localization of Sch9 and reduction in its TORC1-mediated phosphorylation, both of which were rescued by artificially tethering Sch9 to vacuoles (Figure 4B).
On the basis of these results, we propose that the Sch9 branch is more dependent on vacuolar function than other branches, and this characteristic may be utilized for specific inhibition of the Sch9 branch for particular physiological responses. As the main role of TORC1-Sch9 signaling is the regulation of proteosynthesis, the effects of endomembrane status on Sch9 activity may contribute to the coordination of the synthesis of two important macromolecules, proteins and phospholipids.
Branch-specific cross-talk may generate different outputs of the TORC1 pathway under stress conditions
On carbon starvation or hyperosmotic stress, changes in the localization of Sch9 were evident: vacuolar membrane localization of Sch9 was increased on hyperosmotic stress (our unpublished observations), whereas it was decreased on carbon starvation (Jorgensen et al., 2004). The significance of these changes was unclear because TORC1 itself seems to be inhibited under these stress conditions, as Atg13 and FYVE-Sch9 were both dephosphorylated in these situations (Supplemental Figures S5 and S6). Under these conditions, although TORC1 signaling to other branches was inhibited, signaling to the PP2A branch was only moderately or minimally inhibited (Hughes Hallett et al., 2014). One possible explanation is that the PP2A branch may also have an alternative, TORC1-independent regulatory mechanism in which PKA and other signaling molecules are involved (Castermans et al., 2012).
We showed previously that localization of TORC1 was affected by nitrogen starvation; however, localization of Sch9 was not significantly affected, suggesting that regulation of Sch9 localization is not involved in the response of TORC1 signaling to nitrogen sources (Kira et al., 2016). Under conditions of heat stress, sequestration of TORC1 into stress granules contributes to inhibition of TORC1 (Takahara and Maeda, 2012). We noticed that, on heat stress, Sch9 delocalized from vacuoles and colocalized to stress granules, similarly to TORC1 (our unpublished observations). In this situation, the activity of TORC1 itself was inhibited because Atg13 was dephosphorylated (Figure 5A). A previous study indicated that different stressors drive the TORC1 pathway into distinct sets of states (Hughes Hallett et al., 2014). Although TORC1 activity likely has several different inhibitory mechanisms (Hughes Hallett et al., 2014, 2015), all of our results suggest that diverse cross-talk of TORC1 with other downstream signaling pathways contributes to differential outputs of the TORC1 pathway.
Selective inhibition of the Sch9 branch may contribute to adaptation to oxidative stress
Oxidative stress stimulates mTORC1 activity by inhibiting the TSC complex in mammalian cells (Bae et al., 1999; Sarbassov and Sabatini, 2005; Yoshida et al., 2011). By contrast, the TORC1 pathway is relatively repressed under oxidative stress in yeast; TORC1 signaling to the Sch9 branch is halted, but signaling to PP2A or Atg13 is only slightly affected (Urban et al., 2007; Hughes Hallett et al., 2014) (Figure 5, A and B). These results suggest that the regulation of TORC1 activity itself is less prominent under oxidative stress and that modulation of signaling output to a specific substrate is required for adjusting yeast physiology to stress conditions. Indeed, the inhibition of Sch9 leads to the activation of Rim15 and Sod2, which contribute to oxidative stress resistance (Fabrizio et al., 2001; Wei et al., 2008).
The role of PI(3,5)P2 in the TORC1 signaling pathway
PI(3,5)P2 is less abundant than other phosphoinositides in yeast and mammalian cells (McCartney et al., 2014; Hasegawa et al., 2017) and is synthesized by Fab1, the sole PI(3,5)P2 synthase in yeast (Yamamoto et al., 1995; Gary et al., 1998). Previous studies have revealed the roles of PI(3,5)P2 in vesicle fragmentation and trafficking (Yamamoto et al., 1995; Gary et al., 1998; Odorizzi et al., 1998; Dove et al., 2009; Hasegawa et al., 2017). We previously showed that a PI(3,5)P2 deficiency caused a reduction in the TORC1 signaling pathway, with greater influence on the Sch9 branch (Jin et al., 2014). In this study, we demonstrated that local changes in PI(3,5)P2 levels under oxidative stress contributed to specific suppression of the Sch9 branch. This type of regulation may be important for linking vacuolar integrity with signals driving cellular proliferation, as the vacuole is a dynamic, highly regulated organelle in its morphology, responsive to extracellular and intracellular signals in budding yeast (Li and Kane, 2009).
Recent studies have reported that PI(3,5)P2 fulfills a regulatory function in the TORC1 pathway in species other than budding yeast. In fission yeast, disruption of Ste12 (PI3P-5 kinase) caused hypersensitivity to a TORC1 inhibitor, without inducing apparent dephosphorylation of downstream substrates (Cobley et al., 2017). This result suggests that PI(3,5)P2 abundance does not have a major effect on TORC1 catalytic activity but does affect the activity of the TORC1 network as a whole in this organism. In mammals, Raptor, a component of mTORC1, binds to PI(3,5)P2, and this interaction is required for mTORC1 to respond to insulin and amino acids in adipocytes (Bridges et al., 2012). On the other hand, PI(3,5)P2 also affects nutrient availability-driven mTORC1 activation indirectly, rather than directly, in mammary epithelial cells (Krishna et al., 2016). Thus, the role of PI(3,5)P2 in the regulation of TORC1 signaling may be plastic, with its effects varying from tissue to tissue in mammalian cells. Different sensitivities of mTORC1 to PI(3,5)P2 among tissues may generate tissue-specific regulation of the mTORC1 network, whereas different sensitivities to PI(3,5)P2 among branches of the TORC1 pathway may contribute to the differential responsiveness of each branch in budding yeast.
In budding yeast, PI(3,5)P2 is increased under hyperosmotic or heat stress conditions (Dove et al., 1997 , 2009; Gary et al., 1998). Detailed regulation under these conditions remains unclear, although the regulation of Fab1 and its regulatory proteins (Vac7, Vac14, Fig4, and Atg18) has been implicated (McCartney et al., 2014; Hasegawa et al., 2017). As PI(3,5)P2-binding proteins exhibit specific subcellular localization, the detailed spatial regulation of PI(3,5)P2 abundance remains to be clarified. The change in Fab1 localization on oxidative stress (Figure 7D) implies that regulation of the local distribution of Fab1 might be one mechanism. Furthermore, we showed that the local distribution of PI3P was also affected under oxidative conditions (Figure 7F and Supplemental Figure S2). As Fab1 contains the FYVE domain (Burd and Emr, 1998), the change in its localization may be ascribed to that of PI3P. Further studies are required to elucidate the spatial regulation of phosphatidylinositol metabolism and its contribution to the TORC1 pathway.
Where is activated TORC1 localized, and where are TORC1 targets phosphorylated?
Although lysosomal localization of mTORC1 is important for its activation by Rheb in mammalian cells (Garami et al., 2003; Sancak et al., 2008), the Rheb homologue in yeast does not seem to be involved in the regulation of TORC1 (De Virgilio and Loewith, 2006; Tanigawa and Maeda, 2017). However, the importance of vacuolar context in TORC1 signaling has been established; TORC1 phosphorylates Sch9 on vacuolar membranes (Urban et al., 2007; Jin et al., 2014; Jin and Weisman, 2015). Consistently, we showed that FYVE domain–dependent tethering of Sch9 increased its phosphorylation in WT cells (Figures 4B and 6B).
In HOPS mutants, fragmented vacuoles stained well with CMAC (Figure 3, B and C), but they did not contain Fab1 or Atg18 (Figure 7A and Supplemental Figure S7). This result suggests that vacuoles present in these mutants are essentially immature, with minimal levels of PI(3,5)P2. We found that nutrient-dependent activation of TORC1 was robust in a HOPS mutant (Figure 2D), indicating that vacuole maturation is not necessary for the activation of TORC1 itself. Furthermore, Tor1 is localized to immature vacuoles in HOPS mutants (Figure 3C), and FYVE-Sch9 on immature vacuoles is partially phosphorylated in HOPS mutants in a TORC1-dependent manner (Figure 4, A and B). Therefore, active TORC1 appears to localize on both immature and mature vacuoles. Indeed, our previous observations indicated that newly synthesized immature vacuoles contained Tor1 but not Fab1 or Sch9 (Jin and Weisman, 2015). Such nonselective vacuolar localization of TORC1 may contribute to selective transmission of signals, depending on diverse localization of substrates (i.e., substrates in one branch may prefer mature vacuoles while substrates in another branch prefer immature ones).
The site of TORC1-catalyzed phosphorylation of substrates other than Sch9 remains unclear, although it was reported that substrates of the PP2A branch interact with TORC1 in the membrane fraction (Yan et al., 2006). As shown in Supplemental Figure S8, GFP-Atg13 did not specifically localize to vacuolar membranes, showing that phosphorylation of Atg13 by TORC1 may not occur on vacuolar membranes. Thus, localization and substrate quality may be the factors determining phosphorylation. A recent study indicated that mTORC2 is regulated in a different manner depending on the location of the cell, and that differentially localized mTORC2 may phosphorylate different substrates (Ebner et al., 2017). Similarly, spatial differences in both TORC1 and its substrates may contribute to the specific regulation of TORC1 outputs.
MATERIALS AND METHODS
Growth conditions
Yeast cells were grown in YPD medium, consisting of 1% yeast extract (BD Biosciences, San Jose, CA), 2% hipolypepton (Nihon Pharmaceutical Corporation, Tokyo, Japan), and 2% glucose (Nacalai Tesque, Kyoto, Japan), or SDC consisting of 0.17% yeast nitrogen base without amino acids or ammonium sulfate (BD Biosciences), 0.5% (NH4)2SO4 (Wako Pure Chemical Industries, Ltd., Osaka, Japan), 0.5% casamino acid (BD Biosciences), and 2% glucose. When required, media were supplemented with 20 µg/ml uracil to compensate for auxotrophy.
Yeast strains and plasmids
The S. cerevisiae strains and plasmids used in this study are listed in Supplemental Table 2. To construct the GFP-FYVE-SCH9 allele (yet234), the FYVE domain was cloned from human EEA1 by PCR with oligonucleotide primers FYVE-up, 5′-TGGCAATCTAGTCAACGGAG-3′, and FYVE-L-Sch9-down, 5′-GATGTAAAAAAATTCATCATTCCACCTCCGCCTCCTCCTTGCAAGTCATTGAAAC-3′ (Burd and Emr, 1998). The resulting fragment was combined with the GFP fragment derived from pMaM173 (Khmelinskii et al., 2011) by performing a second PCR with oligonucleotide primers Sch9Ntag-S1, 5′-GAAGAATAAGTCTGAGAATTATACTCGTATAAGCAAGAAATAAAGATACGAATATACAATCGTACGCTGCAGGTCGAC-3′, and S4-FYVE-down, 5′-GTTTCTCAAGCTCACTAACTCTCCGTTGACTAGATTGCCACATCGATGAATTCTCTGTCG-3′, and then the PCR product was used for transformation of BY4741. The transformant expressed the GFP-FYVE-SCH9 gene, using the NOP1 promoter. To construct cells in which SCH9 promoter-driven GFP-FYVE-SCH9 was expressed, cells were cultured in YPD medium for several days and then were spread on SDC medium containing 1 mg/ml 5-fluoroorotic acid and uracil. Cells in which the URA3-NOP1 promoter sequence was popped out by homologous recombination were selected. Correct insertion of the fragment was confirmed by PCR.
Western blot analysis
Protein extraction and 2-nitro-5-thiocyanobenzoic acid (NTCB) cleavage of Sch9 were performed in accordance with a previous report (Urban et al., 2007), with some modifications. Cell cultures (5 ml) in mid–log phase or under stress conditions were treated with 6% trichloroacetic acid (TCA) and kept on ice for at least 30 min. Pelleted cells were washed with 4°C water and twice with –30°C acetone and then dried. The pellets were dissolved in 75 µl urea buffer (6 M urea, 5 mM EDTA, 1% SDS, 5 mM NaF, 5 mM NaN3, 5 mM 4-nitrophenylphosphate, 5 mM Na2P2O4, 5 mM β-glycerophosphate, 0.5 × Roche PI, 1.5 mM PMSF) and vortexed with the same volume of 0.5 mm low-alkaline glass beads (Yasui Kikai Corporation, Osaka, Japan) for 10 min at 4°C. Cell lysates were collected by piercing the bottom of a tube with a hot needle and centrifuging with a new tube as the receiver. Lysates were heated at 65°C for 10 min, and soluble proteins were separated by centrifugation at 15,000 rpm for 5 min at 4°C. For NTCB cleavage of Sch9, cell lysates were treated with 100 mM N-cyclohexyl-2-aminoethanesulfonic acid (CHES) buffer (pH 10.5) and 1 mM NTCB (in distilled water) for 12−16 h at room temperature in the dark. For SDS–PAGE, samples were heated to 95°C with 2× sample buffer (0.125 mM Tris pH 6.8, 4% SDS, 20% glycerol, 0.1 mg/ml bromophenol blue, 5 mM NaF, 5 mM NaN3, 5 mM 4-nitrophenylphosphate, 5 mM Na2P2O4, 5 mM β-glycerophosphate, 0.5 × Roche PI, 0.5 mM PMSF, 5% 2-mercaptoethanol) and electrophoresed on an 8% acrylamide gel (for Sch9, Atg13, and Par32) or a 6% acrylamide gel (for Npr1 and Vac8-cSch9). Sch9-5HA (C-terminal fragment), Npr1-13myc, Par32-13myc, and Atg13 were detected using mouse anti-HA (16B12; BioLegend, San Diego, CA; Figures 2E and 6E), rabbit anti-HA [as described previously (Itakura et al., 2016)], anti-myc (#2272; Cell Signaling, Beverly, MA), and anti-Atg13 (as described previously [ Kamada et al., 2010]) antibodies, respectively. To quantify phosphorylation levels of Sch9, we assessed bands that disappeared in rapamycin-treated cells as phosphorylated bands and then measured the integrated density of the rates of phosphorylated versus whole bands. To quantify phosphorylation levels of Npr1, we divided the control band at the center, and then the phosphorylation level was defined as the ratio of the upper band to total intensity. Relative phosphorylation levels were assessed as the ratio of the phosphorylation level at each time point normalized to that of cells at 0 min. Quantification of the band shift data was conducted using ImageJ software (National Institutes of Health, Bethesda, MD).
Fluorescence microscopy
Yeast cells grown to log phase were collected by centrifugation (3000 rpm for 30 s), and then the cells were observed under an inverted microscope (DeltaVision). Images were captured using softWoRx image acquisition and analysis software. Fab1-GFP was observed using a confocal laser microscope (FV1000 IX81; Olympus, Tokyo, Japan). For FM4-64 staining, pelleted cells in log phase were resuspended in 50 µl SDC medium containing 20 µM FM4-64 (T13320; Invitrogen, Carlsbad, CA) and incubated for 30 min. The cells then were washed with 500 µl fresh medium and incubated for 1 h before microscopic observation. For CMAC staining, CMAC (Y-7531 Yeast Vacuole Marker Sampler Kit; Molecular Probes, Eugene, OR) was added to cells in log phase (final concentration, 10 µM). The cells were incubated at 30°C for 30 min. Resuspended cells in fresh media were subjected to microscopic observation. To quantify membrane localization data, we calculated the ratio of fluorescence intensity of vacuolar membranes to cytoplasm with line plots and then defined it as the relative amount of protein on the vacuolar membranes.
Measurement of cellular levels of phosphoinositides
Cells grown in SDC + amino acids and uracil medium were labeled with [3H]inositol, and total cellular phosphoinositide was extracted. Extraction and measurement of phosphoinositides were performed as described previously (Duex et al., 2006).
Supplementary Material
Acknowledgments
We thank R. Loewith (University of Geneva) and M. Knop (University of Heidelberg) for providing materials; R. Hatakeyama (University of Fribourg), T. Maeda (University of Tokyo), T. Takahara (University of Nagoya), and Y. Sakai (Kyoto University) for valuable discussions and Textcheck for editing the manuscript. This work was supported in part by a grant to A. Matsuura from the Grants-in-Aid for Scientific Research program from the Japan Society for the Promotion of Science.
Abbreviations used:
- EGO
exit from G0
- HOPS
homotypic fusion and vacuole protein-sorting
- PP2A
protein phosphatase 2A
- TORC1
target of rapamycin complex 1
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-09-0553) on December 13, 2017.
REFERENCES
- Bae GU, Seo DW, Kwon HK, Lee HY, Hong S, Lee ZW, Ha KS, Lee HW, Han JW. Hydrogen peroxide activates p70(S6k) signaling pathway. J Biol Chem. 1999:32596–32602. doi: 10.1074/jbc.274.46.32596. [DOI] [PubMed] [Google Scholar]
- Balderhaar HJ, Ungermann C. CORVET and HOPS tethering complexes—coordinators of endosome and lysosome fusion. J Cell Sci. 2013:1307–1316. doi: 10.1242/jcs.107805. [DOI] [PubMed] [Google Scholar]
- Barbet NC, Schneider U, Helliwell SB, Stansfield I, Tuite MF, Hall MN. TOR controls translation initiation and early G1 progression in yeast. Mol Biol Cell. 1996:25–42. doi: 10.1091/mbc.7.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science. 2013:1100–1106. doi: 10.1126/science.1232044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck T, Hall MN. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature. 1999:689–692. doi: 10.1038/45287. [DOI] [PubMed] [Google Scholar]
- Beck T, Schmidt A, Hall MN. Starvation induces vacuolar targeting and degradation of the tryptophan permease in yeast. J Cell Biol. 1999:1227–1238. doi: 10.1083/jcb.146.6.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda M, Péli-Gulli MP, Bonfils G, Panchaud N, Urban J, Sturgill TW, Loewith R, De Virgilio C. The Vam6 GEF controls TORC1 by activating the EGO complex. Mol Cell. 2009:563–573. doi: 10.1016/j.molcel.2009.06.033. [DOI] [PubMed] [Google Scholar]
- Bonangelino CJ, Nau JJ, Duex JE, Brinkman M, Wurmser AE, Gary JD, Emr SD, Weisman LS. Osmotic stress-induced increase of phosphatidylinositol 3,5-bisphosphate requires Vac14p, an activator of the lipid kinase Fab1p. J Cell Biol. 2002:1015–1028. doi: 10.1083/jcb.200201002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfils G, Jaquenoud M, Bontron S, Ostrowicz C, Ungermann C, De Virgilio C. Leucyl-tRNA synthetase controls TORC1 via the EGO complex. Mol Cell. 2012:105–110. doi: 10.1016/j.molcel.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Bridges D, Ma JT, Park S, Inoki K, Weisman LS, Saltiel AR. Phosphatidylinositol 3,5-bisphosphate plays a role in the activation and subcellular localization of mechanistic target of rapamycin 1. Mol Biol Cell. 2012:2955–2962. doi: 10.1091/mbc.E11-12-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, Lee JH, Karin M. Stressin’ Sestrins take an aging fight. EMBO Mol Med. 2010:388–400. doi: 10.1002/emmm.201000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd CG, Emr SD. Phosphatidylinositol(3)-phosphate signaling mediated by specific binding to RING FYVE domains. Mol Cell. 1998:157–162. doi: 10.1016/s1097-2765(00)80125-2. [DOI] [PubMed] [Google Scholar]
- Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc Natl Acad Sci USA. 1998:1432–1437. doi: 10.1073/pnas.95.4.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferkey R, Young PR, McLaughlin MM, Bergsma DJ, Koltin Y, Sathe GM, Faucette L, Eng WK, Johnson RK, Livi GP. Dominant missense mutations in a novel yeast protein related to mammalian phosphatidylinositol 3-kinase and VPS34 abrogate rapamycin cytotoxicity. Mol Cell Biol. 1993:6012–6023. doi: 10.1128/mcb.13.10.6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo V, Crews CM, Vik TA, Bierer BE. Interleukin 2 stimulation of p70 S6 kinase activity is inhibited by the immunosuppressant rapamycin. Proc Natl Acad Sci USA. 1992:7571–7575. doi: 10.1073/pnas.89.16.7571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castermans D, Somers I, Kriel J, Louwet W, Wera S, Versele M, Janssens V, Thevelein JM. Glucose-induced posttranslational activation of protein phosphatases PP2A and PP1 in yeast. Cell Res. 2012:1058–1077. doi: 10.1038/cr.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobley D, Hálová L, Schauries M, Kaczmarek A, Franz-Wachtel M, Du W, Krug K, Maček B, Petersen J. Ste12/Fab1 phosphatidylinositol-3-phosphate 5-kinase is required for nitrogen-regulated mitotic commitment and cell size control. PLoS One. 2017:e0172740. doi: 10.1371/journal.pone.0172740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Virgilio C, Loewith R. Cell growth control: little eukaryotes make big contributions. Oncogene. 2006:6392–6415. doi: 10.1038/sj.onc.1209884. [DOI] [PubMed] [Google Scholar]
- Di Como CJ, Arndt KT. Nutrients, via the Tor proteins, stimulate the association of Tap42 with type 2A phosphatases. Genes Dev. 1996:1904–1916. doi: 10.1101/gad.10.15.1904. [DOI] [PubMed] [Google Scholar]
- Dove SK, Cooke FT, Douglas MR, Sayers LG, Parker PJ, Michell RH. Osmotic stress activates phosphatidylinositol-3,5-bisphosphate synthesis. Nature. 1997:187–192. doi: 10.1038/36613. [DOI] [PubMed] [Google Scholar]
- Dove SK, Dong K, Kobayashi T, Williams FK, Michell RH. Phosphatidylinositol 3,5-bisphosphate and Fab1p/PIKfyve underPPIn endo-lysosome function. Biochem J. 2009:1–13. doi: 10.1042/BJ20081950. [DOI] [PubMed] [Google Scholar]
- Dove SK, Piper RC, McEwen RK, Yu JW, King MC, Hughes DC, Thuring J, Holmes AB, Cooke FT, Michell RH, et al. Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors EMBO J 2004. 1922 1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell. 2005:15–26. doi: 10.1016/j.molcel.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Duex JE, Nau JJ, Kauffman EJ, Weisman LS. Phosphoinositide 5-phosphatase Fig 4p is required for both acute rise and subsequent fall in stress-induced phosphatidylinositol 3,5-bisphosphate levels. Eukaryot Cell. 2006:723–731. doi: 10.1128/EC.5.4.723-731.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner M, Sinkovics B, Szczygieł M, Ribeiro DW, Yudushkin I. Localization of mTORC2 activity inside cells. J Cell Biol. 2017:343–353. doi: 10.1083/jcb.201610060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Liou LL, Moy VN, Diaspro A, Valentine JS, Gralla EB, Longo VD. SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics. 2003:35–46. doi: 10.1093/genetics/163.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001:288–290. doi: 10.1126/science.1059497. [DOI] [PubMed] [Google Scholar]
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003:1457–1466. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- Gary JD, Wurmser AE, Bonangelino CJ, Weisman LS, Emr SD. Fab1p is essential for PtdIns(3)P 5-kinase activity and the maintenance of vacuolar size and membrane homeostasis. J Cell Biol. 1998:65–79. doi: 10.1083/jcb.143.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goberdhan DC, Wilson C, Harris AL. Amino acid sensing by mTORC1: intracellular transporters mark the spot. Cell Metab. 2016:580–589. doi: 10.1016/j.cmet.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González A, Hall MN. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017:397–408. doi: 10.15252/embj.201696010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, Yonezawa K, Weng QP, Kozlowski MT, Belham C, Avruch J. Amino acid sufficiency and mTOR regulate p70 S6 kinase and eIF-4E BP1 through a common effector mechanism. J Biol Chem. 1998:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- Hasegawa J, Strunk BS, Weisman LS. PI5P and PI(3,5)P2: minor, but essential phosphoinositides. Cell Struct Funct. 2017:49–60. doi: 10.1247/csf.17003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- Huber A, Bodenmiller B, Uotila A, Stahl M, Wanka S, Gerrits B, Aebersold R, Loewith R. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 2009:1929–1943. doi: 10.1101/gad.532109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes Hallett JE, Luo X, Capaldi AP. State transitions in the TORC1 signaling pathway and information processing in Saccharomyces cerevisiae. Genetics. 2014:773–786. doi: 10.1534/genetics.114.168369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes Hallett JE, Luo X, Capaldi AP. Snf1/AMPK promotes the formation of Kog1/Raptor-bodies to increase the activation threshold of TORC1 in budding yeast. Elife. 2015:e09181. doi: 10.7554/eLife.09181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Itakura E, Zavodszky E, Shao S, Wohlever ML, Keenan RJ, Hegde RS. Ubiquilins chaperone and triage mitochondrial membrane proteins for degradation. Mol Cell. 2016:21–33. doi: 10.1016/j.molcel.2016.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Rüegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- Jin N, Mao K, Jin Y, Tevzadze G, Kauffman EJ, Park S, Bridges D, Loewith R, Saltiel AR, Klionsky DJ, Weisman LS. Roles for PI(3,5)P2 in nutrient sensing through TORC1. Mol Biol Cell. 2014:1171–1185. doi: 10.1091/mbc.E14-01-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Weisman LS. The vacuole/lysosome is required for cell-cycle progression. Elife. 2015:e08160. doi: 10.7554/eLife.08160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P, Rupes I, Sharom JR, Schneper L, Broach JR, Tyers M. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 2004:2491–2505. doi: 10.1101/gad.1228804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada Y. Novel tRNA function in amino acid sensing of yeast Tor complex1. Genes Cells. 2017:135–147. doi: 10.1111/gtc.12462. [DOI] [PubMed] [Google Scholar]
- Kamada Y, Yoshino K, Kondo C, Kawamata T, Oshiro N, Yonezawa K, Ohsumi Y. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol. 2010:1049–1058. doi: 10.1128/MCB.01344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SA, Pacold ME, Cervantes CL, Lim D, Lou HJ, Ottina K, Gray NS, Turk BE, Yaffe MB, Sabatini DM. mTORC1 phosphorylation sites encode their sensitivity to starvation and rapamycin. Science. 2013:1236566. doi: 10.1126/science.1236566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khmelinskii A, Meurer M, Duishoev N, Delhomme N, Knop M. Seamless gene tagging by endonuclease-driven homologous recombination. PLoS One. 2011:e23794. doi: 10.1371/journal.pone.0023794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A, Cunningham KW. A LAPF/phafin1-like protein regulates TORC1 and lysosomal membrane permeabilization in response to endoplasmic reticulum membrane stress. Mol Biol Cell. 2015:4631–4645. doi: 10.1091/mbc.E15-08-0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury JM, Sen ND, Maeda T, Heitman J, Cardenas ME. Endolysosomal membrane trafficking complexes drive nutrient-dependent TORC1 signaling to control cell growth in Saccharomyces cerevisiae. Genetics. 2014:1077–1089. doi: 10.1534/genetics.114.161646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kira S, Kumano Y, Ukai H, Takeda E, Matsuura A, Noda T. Dynamic relocation of the TORC1-Gtr1/2-Ego1/2/3 complex is regulated by Gtr1 and Gtr2. Mol Biol Cell. 2016:382–396. doi: 10.1091/mbc.E15-07-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kira S, Tabata K, Shirahama-Noda K, Nozoe A, Yoshimori T, Noda T. Reciprocal conversion of Gtr1 and Gtr2 nucleotide-binding states by Npr2-Npr3 inactivates TORC1 and induces autophagy. Autophagy. 2014:1565–1578. doi: 10.4161/auto.29397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna S, Palm W, Lee Y, Yang W, Bandyopadhyay U, Xu H, Florey O, Thompson CB, Overholtzer M. PIKfyve regulates vacuole maturation and nutrient recovery following engulfment. Dev Cell. 2016:536–547. doi: 10.1016/j.devcel.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SC, Kane PM. The yeast lysosome-like vacuole: endpoint and crossroads. Biochim Biophys Acta. 2009:650–663. doi: 10.1016/j.bbamcr.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Hall MN. Target of rapamycin (TOR) in nutrient signaling and growth control. Genetics. 2011:1177–1201. doi: 10.1534/genetics.111.133363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- MacGurn JA, Hsu PC, Smolka MB, Emr SD. TORC1 regulates endocytosis via Npr1-mediated phosphoinhibition of a ubiquitin ligase adaptor. Cell. 2011:1104–1117. doi: 10.1016/j.cell.2011.09.054. [DOI] [PubMed] [Google Scholar]
- Mahfouz MM, Kim S, Delauney AJ, Verma DP. Arabidopsis TARGET OF RAPAMYCIN interacts with RAPTOR, which regulates the activity of S6 kinase in response to osmotic stress signals. Plant Cell. 2006:477–490. doi: 10.1105/tpc.105.035931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCartney AJ, Zhang Y, Weisman LS. Phosphatidylinositol 3,5-bisphosphate: low abundance, high significance. Bioessays. 2014:52–64. doi: 10.1002/bies.201300012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AH, Hatakeyama R, Kimmig P, Arter M, Peter M, Matos J, De Virgilio C, Kornmann B. Functional mapping of yeast genomes by saturated transposition. Elife. 2017:e23570. doi: 10.7554/eLife.23570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Hirata A, Ohsumi Y, Wada Y. Vam2/Vps41p and Vam6/Vps39p are components of a protein complex on the vacuolar membranes and involved in the vacuolar assembly in the yeast Saccharomyces cerevisiae. J Biol Chem. 1997:11344–11349. doi: 10.1074/jbc.272.17.11344. [DOI] [PubMed] [Google Scholar]
- Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- Odorizzi G, Babst M, Emr SD. Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell. 1998:847–858. doi: 10.1016/s0092-8674(00)81707-9. [DOI] [PubMed] [Google Scholar]
- Panchaud N, Péli-Gulli MP, De Virgilio C. Amino acid deprivation inhibits TORC1 through a GTPase-activating protein complex for the Rag family GTPase Gtr1. Sci Signal. 2013:ra42. doi: 10.1126/scisignal.2004112. [DOI] [PubMed] [Google Scholar]
- Péli-Gulli MP, Sardu A, Panchaud N, Raucci S, De Virgilio C. Amino acids stimulate TORC1 through Lst4-Lst7, a GTPase-activating protein complex for the Rag family GTPase Gtr2. Cell Rep. 2015:1–7. doi: 10.1016/j.celrep.2015.08.059. [DOI] [PubMed] [Google Scholar]
- Powis K, Zhang T, Panchaud N, Wang R, De Virgilio C, Ding J. Crystal structure of the Ego1-Ego2-Ego3 complex and its role in promoting Rag GTPase-dependent TORC1 signaling. Cell Res. 2015:1043–1059. doi: 10.1038/cr.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke A, Anderson S, McCaffery JM, Yates J, Aronova S, Chu S, Fairclough S, Iverson C, Wedaman KP, Powers T. TOR complex 1 includes a novel component, Tco89p (YPL180w), and cooperates with Ssd1p to maintain cellular integrity in Saccharomyces cerevisiae. J Biol Chem. 2004:14752–14762. doi: 10.1074/jbc.M313062200. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Sabatini DM. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J Biol Chem. 2005:39505–39509. doi: 10.1074/jbc.M506096200. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Beck T, Koller A, Kunz J, Hall MN. The TOR nutrient signalling pathway phosphorylates NPR1 and inhibits turnover of the tryptophan permease. EMBO J. 1998:6924–6931. doi: 10.1093/emboj/17.23.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DF, Eitzen G, Margolis N, Wickner WT, Price A. A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc Natl Acad Sci USA. 2000:9402–9407. doi: 10.1073/pnas.97.17.9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracka D, Jozefczuk S, Rudroff F, Sauer U, Hall MN. Nitrogen source activates TOR (target of rapamycin) complex 1 via glutamine and independently of Gtr/Rag proteins. J Biol Chem. 2014:25010–25020. doi: 10.1074/jbc.M114.574335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahara T, Maeda T. Transient sequestration of TORC1 into stress granules during heat stress. Mol Cell. 2012:242–252. doi: 10.1016/j.molcel.2012.05.019. [DOI] [PubMed] [Google Scholar]
- Tanigawa M, Maeda T. An in vitro TORC1 kinase assay that recapitulates the Gtr-independent glutamine-responsive TORC1 activation mechanism on yeast vacuoles. Mol Cell Biol. 2017:e00075-17. doi: 10.1128/MCB.00075-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thedieck K, Holzwarth B, Prentzell MT, Boehlke C, Kläsener K, Ruf S, Sonntag AG, Maerz L, Grellscheid SN, Kremmer E, et al. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells Cell 2013. 859 874 [DOI] [PubMed] [Google Scholar]
- Tsun ZY, Bar-Peled L, Chantranupong L, Zoncu R, Wang T, Kim C, Spooner E, Sabatini DM. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol Cell. 2013:495–505. doi: 10.1016/j.molcel.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, Wanke V, Anrather D, Ammerer G, Riezman H, et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae Mol Cell 2007. 663 674 [DOI] [PubMed] [Google Scholar]
- Wanke V, Cameroni E, Uotila A, Piccolis M, Urban J, Loewith R, De Virgilio C. Caffeine extends yeast lifespan by targeting TORC1. Mol Microbiol. 2008:277–285. doi: 10.1111/j.1365-2958.2008.06292.x. [DOI] [PubMed] [Google Scholar]
- Wedaman KP, Reinke A, Anderson S, Yates J, McCaffery JM, Powers T. Tor kinases are in distinct membrane-associated protein complexes in Saccharomyces cerevisiae. Mol Biol Cell. 2003:1204–1220. doi: 10.1091/mbc.E02-09-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Fabrizio P, Hu J, Ge H, Cheng C, Li L, Longo VD. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 2008:e13. doi: 10.1371/journal.pgen.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie MW, Jin F, Hwang H, Hwang S, Anand V, Duncan MC, Huang J. Insights into TOR function and rapamycin response: chemical genomic profiling by using a high-density cell array method. Proc Natl Acad Sci USA. 2005:7215–7220. doi: 10.1073/pnas.0500297102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, DeWald DB, Boronenkov IV, Anderson RA, Emr SD, Koshland D. Novel PI(4)P 5-kinase homologue, Fab1p, essential for normal vacuole function and morphology in yeast. Mol Biol Cell. 1995:525–539. doi: 10.1091/mbc.6.5.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan G, Shen X, Jiang Y. Rapamycin activates Tap42-associated phosphatases by abrogating their association with Tor complex 1. EMBO J. 2006:3546–3555. doi: 10.1038/sj.emboj.7601239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S, Hong S, Suzuki T, Nada S, Mannan AM, Wang J, Okada M, Guan KL, Inoki K. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J Biol Chem. 2011:32651–32660. doi: 10.1074/jbc.M111.238014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurita-Martinez SA, Puria R, Pan X, Boeke JD, Cardenas ME. Efficient Tor signaling requires a functional class C Vps protein complex in Saccharomyces cerevisiae. Genetics. 2007:2139–2150. doi: 10.1534/genetics.107.072835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.