

WNK bodies are large punctate membraneless cytosolic signaling foci that sequester WNK serine–threonine kinases and form in renal distal tubular epithelial cells during shifts in total body potassium balance. The assembly of these structures requires KS-WNK1, a truncated isoform of the WNK1 gene that is exclusively expressed in the distal tubule.
Abstract
With-no-lysine (WNK) kinases coordinate volume and potassium homeostasis by regulating renal tubular electrolyte transport. In the distal convoluted tubule (DCT), potassium imbalance causes WNK signaling complexes to concentrate into large discrete foci, which we call “WNK bodies.” Although these structures have been reported previously, the mechanisms that drive their assembly remain obscure. Here, we show that kidney-specific WNK1 (KS-WNK1), a truncated kinase-defective WNK1 isoform that is highly expressed in the DCT, is critical for WNK body formation. While morphologically distinct WNK bodies were evident in the distal tubules of mice subjected to dietary potassium loading and restriction, KS-WNK1 knockout mice were deficient in these structures under identical conditions. Combining in vivo observations in kidney with reconstitution studies in cell culture, we found that WNK bodies are dynamic membraneless foci that are distinct from conventional organelles, colocalize with the ribosomal protein L22, and cluster the WNK signaling pathway. The formation of WNK bodies requires an evolutionarily conserved cysteine-rich hydrophobic motif harbored within a unique N-terminal exon of KS-WNK1. We propose that WNK bodies are not pathological aggregates, but rather are KS-WNK1–dependent microdomains of the DCT cytosol that modulate WNK signaling during physiological shifts in potassium balance.
INTRODUCTION
With-no-lysine (WNK) kinases are a family of serine–threonine kinases that regulate blood pressure and potassium homeostasis. Gain-of-function mutations of WNK1 and WNK4 cause familial hyperkalemic hypertension (FHHt; pseudohypoaldosteronism type II, Gordon syndrome), a thiazide-sensitive disorder of hypertension and hyperkalemia (Wilson et al., 2001). WNK1 and WNK4 are key regulators of the thiazide-sensitive Na-Cl cotransporter (NCC; SLC12A3) in the distal convoluted tubule (DCT), a short segment of the nephron positioned between the loop of Henle and the collecting system (Subramanya and Ellison, 2014). The activated form of both WNKs stimulates NCC via a serine–threonine kinase cascade. The kinase-active forms of WNK1 and WNK4 phosphorylate and activate two downstream homologous kinases, the Ste20/SpS1-related proline alanine-rich kinase (SPAK, STK39) and oxidative stress responsive kinase 1 (OSR1, OXSR1; Vitari et al., 2005). SPAK and OSR1 then directly phosphorylate and activate NCC (Moriguchi et al., 2005), increasing sodium reabsorption in the DCT, which causes decreased potassium secretion by the downstream nephron, by limiting distal NaCl delivery and/or inducing structural changes (Sorensen et al., 2013; Hunter et al., 2014; Grimm et al., 2017). Current evidence indicates that WNK kinases are chloride sensors that are regulated by changes in extracellular potassium (Piala et al., 2014; Terker et al., 2015, 2016b).
In the DCT, regulation of the WNK-SPAK/OSR1 pathway is linked to changes in its subcellular distribution. Several papers have reported that WNK-SPAK/OSR1 pathway components strikingly concentrate into large discrete foci in the DCT in response to changes in potassium balance. During hypokalemia, for example, WNK4, SPAK, and OSR1 transition from a diffuse to punctate distribution in the DCT (Terker et al., 2015). Additionally, WNK1 can form discrete DCT-specific particles during either dietary K+ loading or restriction (Al-Qusairi et al., 2016, 2017). Studies using genetically modified mice have also visualized these structures. SPAK knockout (KO) mice, which develop a Gitelman syndrome-like potassium wasting phenotype, develop WNK1 and OSR1 puncta in the absence of dietary K+ maneuvers (McCormick et al., 2011; Grimm et al., 2012). Moreover, mice with an FHHt-associated mutation in Cullin 3, a key component of the cognate E3 ubiquitin ligase for WNK kinases, develop hyperkalemia and WNK4-positive DCT puncta (Schumacher et al., 2015). The common findings among these studies are that the puncta are almost entirely restricted to the DCT, and are only present during maneuvers that alter total body K+ balance, either through dietary manipulation or via genetic lesions that perturb the WNK-SPAK/OSR1 signaling pathway.
Despite numerous reports that have described large WNK-SPAK/OSR1 pathway puncta in the DCT, a unifying mechanism that explains how and why these peculiar structures form has been elusive. This led us to investigate the molecular composition and dynamics of these particles, and to identify mechanisms that lead to their formation during potassium loading and depletion. Our studies reveal that the formation of WNK-SPAK/OSR1 pathway puncta requires kidney-specific WNK1 (KS-WNK1), a truncated WNK1 isoform known to exhibit high and localized expression in the DCT (Delaloy et al., 2003; O’Reilly et al., 2003; Vidal-Petiot et al., 2012). Notably, a unique, evolutionarily conserved hydrophobic N-terminal domain of KS-WNK1 is essential for puncta formation, both in vivo and in vitro. Our results suggest that these KS-WNK1–dependent structures, which we call WNK bodies, retain WNK-SPAK/OSR1 pathway components, likely to control DCT-specific WNK-dependent signaling processes during periods of distal nephron stress.
RESULTS
WNK1 is located in discrete puncta in the DCT in response to dietary potassium maneuvers
To investigate WNK1 localization in the kidney in response to shifts in total body potassium, we used a C-terminal “pan-WNK1” antibody that detects all isoforms. Germ line WNK1 deletion leads to embryonic lethality (Zambrowicz et al., 2003); thus, we confirmed its specificity in a gene-edited WNK1 knockout cell line (Figure 1A; Roy et al., 2015b). To characterize the effect of dietary potassium on WNK1 puncta formation, C57BL/6 mice were fed low-K+ (LK), control, or high-K+ (HK) diets for 10 d. As shown in Figure 1B, mice fed a LK diet had a whole blood potassium concentration that was lower than that in mice on control or HK diets. The HK diet did not significantly increase serum potassium levels, likely due to compensatory urinary excretion (O’Reilly et al., 2006; Castaneda-Bueno et al., 2014; Terker et al., 2016b). In immunohistochemistry studies, WNK1 puncta were apparent in mice subjected to either potassium restriction or loading; in contrast, puncta were rarely visible in mice on the control diet (Figure 1C). Tubules positive for WNK1 puncta were confirmed to be DCTs by staining contiguous slices for NCC. Similar results were observed in the 129/Sv mouse strain (Supplemental Figure S1). In potassium-loaded mice, WNK1 also assumed a punctate distribution in a small population of NCC-negative cells in the early portion of the connecting tubule (CNT; Supplemental Figure S2). As shown in Figure 1D, the puncta present during K+ restriction were perinuclear and positioned toward the basal pole. In contrast, the HK WNK1 puncta were almost half the size of the LK puncta and were oriented subapically (Figure 1, D and E). In addition, LK diet-treated mice exhibited a greater number of puncta, averaging four per cell, whereas HK diet-treated mice averaged two puncta per cell (Figure 1F). Together, these observations confirm that diet-induced potassium stress triggers WNK1 puncta formation in the DCT, and that the puncta that form under high and low-K+ conditions exhibit measurable differences in size, location, and number.
FIGURE 1:

Dietary potassium maneuvers stimulate WNK1 puncta formation in mouse distal convoluted tubules. (A) pan-WNK1 antibody validation using gene-edited WNK1 KO and unedited (UE) cell lines, compared with a previously validated WNK1 antibody against exon 28 (Roy et al., 2015b). (B) Whole blood potassium ([K+]WB) in mice treated with LK, control, or HK diet for 10 d. (n = 8 mice per condition; **: p < 0.0001; ANOVA with Tukey posttest). (C) Representative immunohistochemical staining of kidney tissue from mice on LK, control, or HK diet. [K+]WB, measured by cardiac puncture at the time of kidney harvest, is indicated for each condition. DCTs were identified by NCC/nuclear costaining in contiguous sections. DCT in 2.5× zoom indicated by a dashed line. (n = 5 mice per condition; scale bar = 50 μm in 1× images, 10 μm in 2.5× images). (D–F) Quantification of puncta distance (D), diameter (E), and number per cell (F) under LK and HK conditions (n = 3 mice and more than 59 cells from five tubules per condition; **: p < 0.0001, *: p = 0.02, unpaired t test).
In contrast to L-WNK1, KS-WNK1 forms large puncta in vitro
The WNK1 gene yields two major products due to alternative promoter usage: a full-length kinase-active “long” isoform (L-WNK1), and a truncated kinase-dead “kidney-specific” isoform, termed KS-WNK1 (Delaloy et al., 2003; O’Reilly et al., 2003). While L-WNK1 is ubiquitously expressed, a distal nephron-specific promoter in intron 4 generates KS-WNK1 (Supplemental Figure S3A). The renal promoter that drives KS-WNK1 expression replaces the first four exons of L-WNK1 with exon 4a, which encodes 30 unique amino acids (Supplemental Figure S3B). As shown in Supplemental Figure S3C, L-WNK1 transcripts are modestly expressed along the entire length of the nephron. In contrast, although KS-WNK1 is the more highly represented isoform in whole kidney mRNA extracts, its transcript is tightly restricted to the DCT, where its expression is 80-fold higher than L-WNK1 (Vidal-Petiot et al., 2013).
The localized expression pattern of KS-WNK1 transcripts led us to hypothesize that KS-WNK1 is responsible for the punctate distribution of WNK1 in the distal tubule. Because the pan-WNK1 antibody is directed to the WNK1 C-terminus, it does not distinguish between L-WNK1 and KS-WNK1. Efforts to develop KS-WNK1–specific exon 4a antisera for kidney immunofluorescence studies were unsuccessful, and antibodies against the N-terminus of L-WNK1 were nonspecific in kidney tissue. Therefore, we moved to an in vitro model to determine the role of WNK1 isoforms in puncta formation. In immunofluorescence studies in HEK-293 cells transfected with WNK1 isoforms, overexpressed L-WNK1 adopted a diffuse granular appearance, whereas KS-WNK1 was located in large discrete puncta, reminiscent of the micron-sized puncta found in the DCTs of mice subjected to dietary K+ maneuvers (Figure 2A). In transmission electron microscopy (TEM) studies in KS-WNK1-HA transfected cells, immunogold particles directed against hemagglutinin (HA) epitopes collected in electron hypodense regions of the cytosol, and were not contained in membrane-bound organelles (Figure 2B). The difference in L- and KS-WNK1 distribution was also apparent in supernatant-pellet assays that distinguished between Triton X-100–sensitive and –resistant cellular fractions (Figure 2C). In these studies, L-WNK1 was enriched in a Triton X-100–soluble supernatant, whereas KS-WNK1 was predominantly found in the Triton-resistant, SDS–soluble pellet (Figure 2, D and E). When we summed these two fractions, we noted no significant differences in total L- and KS-WNK1 protein abundance, suggesting that the discrepancy in protein localization was not associated with changes in protein stability (Figure 2F). These findings support the observed differences in L- and KS-WNK1 subcellular localization by microscopy, indicating that the two isoforms are stably distributed in biochemically distinct fractions in cells.
FIGURE 2:

In contrast to L-WNK1, KS-WNK1 forms large puncta in vitro. (A) Immunofluoresence of HEK-293 cells transiently transfected with either L-WNK1-HA or KS-WNK1-HA. (n = 5 transfections; scale bar = 10 μm). (B) Immunogold electron micrographs of HEK-293 cells transiently transfected with KS-WNK1-HA, labeled with anti-HA antibody. Note the concentration of gold particles (arrows) in an electron hypodense region of the cytosol. M = mitochondria; Nuc = nucleus. Scale bar = 100 nm. (C) Supernatant/pellet (SP) assay. Cell lysates were separated into Triton-soluble and Triton-resistant, SDS–soluble fractions. (D) Immunoblots of HEK-293 cells transiently transfected with either L-WNK1-HA or KS-WNK1-HA, subjected to SP assay. Blots were probed with HA antibody revealing a band at ∼250 kDa, corresponding to the MW of L-WNK1 and slightly lower band for KS-WNK1. L-WNK1-HA Sup also contains several other bands, presumably degradation products. (E) Relative protein abunance of L-WNK1 vs. KS-WNK1 in the SP assay. Data were normalized to the L-WNK1 protein abundance in the Sup fraction. (n = 7 transfections; **: p = 0.0021, paired t test). (F) Comparison of the summed supernatant plus pellet protein abunance of L-WNK1 vs. KS-WNK1 in transiently transfected HEK-293 cells (n = 7 transfections; NS by unpaired t test).
Similar to WNK1 puncta in the kidney, KS-WNK1 clusters the WNK-SPAK/OSR1 pathway in cells
Several laboratories have reported that WNK1, WNK4, SPAK, and OSR1 form large micron-sized puncta in the DCT during dietary K+ maneuvers (van der Lubbe et al., 2013; Terker et al., 2015; Al-Qusairi et al., 2016, 2017), suggesting that they may be colocalized in DCT-specific WNK signaling complexes. To further test this, we developed a guinea pig anti-WNK4 antibody for colocalization studies, and confirmed its specificity in WNK4 knockout animals (Supplemental Figure S4; Castaneda-Bueno et al., 2012). In immunofluorescence studies of kidneys from mice subjected to LK diets, the WNK1-positive puncta also contained SPAK and WNK4 (Figure 3A), confirming that they are assemblages of several proteins involved in WNK-dependent signaling. We hypothesized that the KS-WNK1–specific puncta observed in HEK-293 cells (Figure 2A) and the punctate WNK1-positive structures detected in the DCT are similar in nature. Consistent with this prediction, immunofluorescence studies of transiently transfected HEK-293 cells revealed that L-WNK1 and endogenous WNK4 and SPAK partially redistributed into KS-WNK1 puncta (Figure 3, A–C). Live cell imaging studies of HEK-293 cells cotransfected with L-WNK1-eGFP and KS-WNK1-mRuby revealed that some of the puncta were mobile, and the L-WNK1 fraction confined within these structures tracked with KS-WNK1, suggesting that its movement was restricted (Supplemental Movie S1). Supernatant-pellet assays in HEK-293 cells transiently transfected with KS-WNK1 indicated that the fraction of L-WNK1 that was redistributed into the pellet was not phosphorylated (Supplemental Figure S5), a finding consistent with previous work suggesting that DCT WNK1 puncta are deficient in active phosphorylated WNK kinases (Al-Qusairi et al., 2016, 2017). These findings indicate that in our cellular model, KS-WNK1 controls the subcellular localization of the WNK-SPAK-OSR1 pathway.
FIGURE 3:

WNK1 puncta colocalize with the WNK-SPAK pathway in vivo and in vitro. (A) Immunofluorescence of kidney sections from mice on LK diet × 10 d ([K+]WB = 3.5 mEq/L) costained for WNK4 (both panel sets), pan-WNK1 (left), and SPAK (right). (n = 4 mice per condition; scale bar = 10 μm in 1× images, 5 μm in 4× images). (B) HEK-293 cells were transiently transfected with KS-WNK1-HA and were costained for HA epitopes (all panel sets), transiently transfected myc-L-WNK1 (with anti-myc antibody [left]), endogenous WNK4 (middle), or endogenous SPAK (right) (n = 4 transfections). (C) Percent colocalization in HEK-293 cells of transiently transfected KS-WNK1-HA with exogenous myc-L-WNK (n = 8 images obtained at 60× magnification with an average of four kidney tubules per field), endogenous WNK4 (n = 6 images), or endogenous SPAK (n = 7 images). Pearson correlation coefficients were calculated with Imaris (Bitplane).
Movie S1.
KS-WNK1 puncta are dynamic and cluster L-WNK1. Movie of a HEK-293 cell transfected with C-terminally tagged L-WNK1-eGFP (green) and KSWNK1- mRuby (red). Live cell images of L-WNK1 and KS-WNK1 trafficking were performed using a Nikon Ti-E-2000 confocal microscope, as described in the Methods. The arrow denotes a mobile KS-WNK1 punctate structure. Note how L-WNK1 is concentrated within this structure and tracks with KS-WNK1.
The DCT-specific puncta are KS-WNK1 dependent
The observation that KS-WNK1 is almost exclusively expressed in the distal tubule and can redistribute the WNK-SPAK/OSR1 pathway to discrete foci in cells suggests that it mediates the punctate localization of WNK kinases and SPAK in the DCT. To definitively test this hypothesis, we performed studies in KS-WNK1 KO mice (Supplemental Figure S6; Liu et al., 2011). In KS-WNK1 KO animals subjected to dietary K+ restriction, the characteristic subnuclear punctate signal recognized by the pan-WNK1, WNK4, and SPAK antibodies was absent (Figure 4A). In a rare subset of DCTs treated with HK and LK diets, WNK1 puncta were detected by immunohistochemistry (Figure 4B); however, compared with diet and whole blood K-matched controls, these structures were 13- to 40-fold less abundant, exhibited significant differences in diameter compared with controls, and were mislocalized to the basal pole (Figure 4C–E). Given the observations that these punctate structures are concentrated assemblages of WNK-SPAK/OSR1 components, and are dependent on KS-WNK1 for proper formation, we henceforth refer to them as “WNK bodies.”
FIGURE 4:

The DCT-specific puncta are KS-WNK1 dependent. (A) Immunofluorescence of mouse DCT from either WT or KS-WNK1−/− (KO) mice maintained on LK diet for 10 d. [K+]WB levels were similar for both WT and KS-WNK1 KO mice. DCTs were identified by NCC costaining. Pan-WNK1, WNK4, and SPAK antibodies detected puncta in WT mice, whereas puncta were nearly absent in KS-WNK1 KO mice. (n = 4 mice per genotype; scale bar = 10 μm in 1× images, 5 μm in 4× images). (B) Representative immunohistochemical staining of DCTs from KS-WNK1 KO mice maintained on either LK or HK diet for 10 d. Indicated with arrows are rare punctate structures that were detected in a small subset of DCTs with the pan-WNK1 antibody under both LK and HK conditions. (n = 3 mice per condition; DCT in 2.5× zoom indicated by a dashed line). (C–E) Comparison of WT and KS-WNK1 KO mice on LK and HK diets. KS-WNK1 KO mice exhibited dramatically reduced puncta abundance (C) compared with WT mice. These foci were positioned farther from the lumen (D) and demonstrated a normalization of diameter relative to WT (E; i.e., in KO mice, puncta diameter averaged 1.9 µM under both LK and HK conditions) (n = 3 mice per condition, and due to the scarcity of puncta more than 200 cells from 20 tubules were analyzed per condition. WT data from Figure 1 are presented alongside KO data for comparison. **: p < 0.0001; *: p = 0.0045, #: p = 0.03 by ANOVA, Tukey’s post hoc test).
The ribosomal subunit RPL22 specifically localizes to WNK bodies
Next, we sought to identify cellular components that colocalize with WNK bodies. In contrast to previous reports noting an association between L-WNK1 and membrane-bound organelles (Zagorska et al., 2007), KS-WNK1-HA did not colocalize with markers of early endosomes (EEA1), late endosomes and lysosomes (LAMP1, LAMP2, lysotracker), autophagosomes (P62, LC3B), the endoplasmic reticulum (PDI), and lipid droplets (BODIPY; Supplemental Figure S7A and Supplemental Movies S2 and S3). These findings are consistent with the electron micrographs in Figure 2B, and confirm that the transfected KS-WNK1 puncta are not endosomes, autophagosomes, or phospholipid-bound organelles. In addition, under unstressed conditions, KS-WNK1 did not colocalize with markers for membraneless organelles and proteotoxic aggregates, including stress granules (TIAR, G3BP1, eIF4E), P-bodies (eIF4E, DCP2), aggresomes (ubiquitin, vimentin), and mediators of chaperone-mediated autophagy (Hsc70; Supplemental Figure S7B).
Movie S2.
KS-WNK1 puncta do not colocalize with lysosomes. Movie of a HEK-293 cell transfected with C-terminally tagged KSWNK1-mRuby (red), and incubated with LysoTracker Green (green) to stain lysosomes. Live cell images of KS-WNK1 and LysoTracker trafficking were performed using a Zeiss 700 LSM confocal microscope, as described in the Methods. No apparent colocalization, or co-trafficking is observed with KSWNK1 and LysoTracker.
Movie S3.
KS-WNK1 puncta do not colocalize or traffic with lipid droplets. Movie of a HEK-293 cell transfected with C-terminally tagged KSWNK1-mRuby (red), and incubated with BODIPY FL (green) to stain lipid droplets. Live cell images of KS-WNK1 and BODIPY trafficking were performed using a Zeiss 700 LSM confocal microscope, as described in the Methods. No apparent colocalization or co-trafficking is observed with KS-WNK1 and BODIPY.
A recent study reported that the 60S ribosomal subunit L22 (RPL22) aggregates in the distal tubule cytosol of aldosterone-infused rats (Cheema et al., 2014). Although serum potassium levels were not measured in their study, these animals were presumably hypokalemic. Interestingly, the RPL22 aggregates were morphologically similar to the hypokalemic WNK bodies observed in Figure 1C. Moreover, immuno-TEM studies in kidney localized RPL22 to membraneless electron hypodense regions of the cytosol, which appeared strikingly similar to the electron micrographs in Figure 2B (Cheema et al., 2014). Thus, we tested the hypothesis that RPL22 is incorporated into WNK bodies in the setting of potassium stress. In kidney tissues from mice fed a LK diet, RPL22 concentrated into subnuclear puncta that were greater than 80% colocalized with WNK bodies, identified by pan-WNK1 immunostaining (Figure 5, A and B). Under high-K+ dietary conditions, RPL22 was also partially colocalized with the pan-WNK1 signal in DCT subapical puncta (Figure 5, A and B). As reported previously (Cheema et al., 2014), RPL22 did not assume a punctate localization pattern under control dietary conditions (unpublished data). In KS-WNK1–transfected HEK cells, endogenous RPL22 was also highly colocalized in KS-WNK1–positive puncta (Figure 5C and Supplemental Movie S4). Collectively, these findings identify RPL22 as a WNK body constituent. Importantly, they also further support the notion that distal tubule WNK bodies and transfected KS-WNK1 puncta in cells share the same molecular composition.
FIGURE 5:
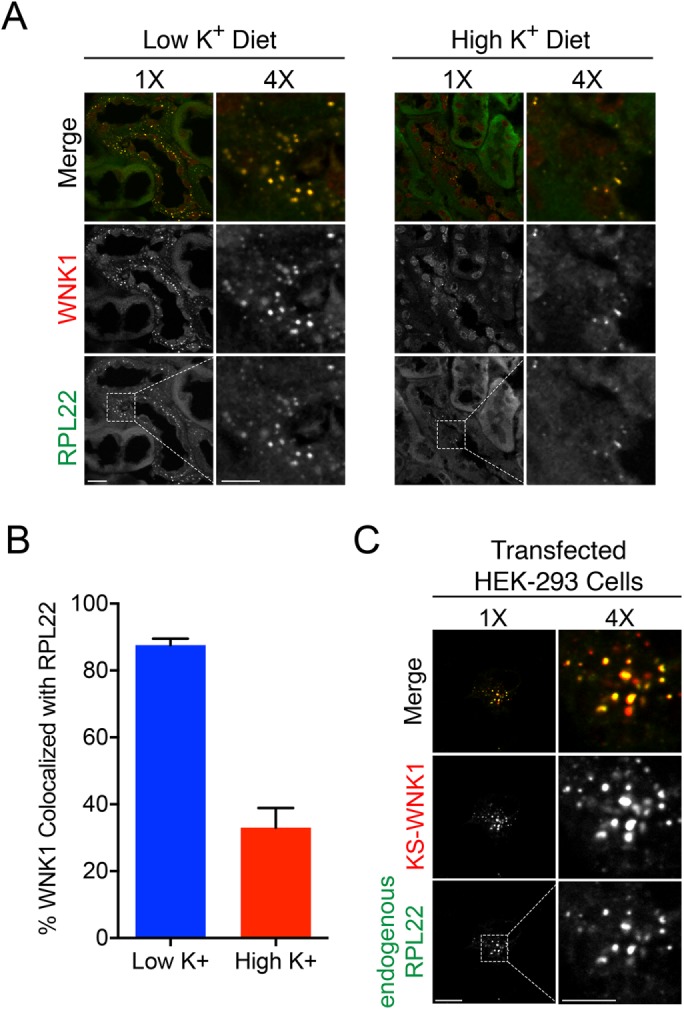
WNK bodies colocalize with RPL22. (A) Immunofluorescence of kidney sections from WT mice on LK or HK diets × 10 d, showing colocalization of pan-WNK1 puncta with RPL22. Box indicates area magnified (bar 1× 10 μm, bar 4× 5 μm). (B) Percent colocalization of between WNK1 and RPL22 under LK and HK dietary conditions. In kidney tissue, on LK diet, WNK1 was 85% colocalized with RPL22, whereas on a HK diet WNK1 was less colocalized with RPL22 (n = 3 mice per condition and 10 images obtained at 60× magnification with an average of four kidney tubules per field). (C) Immunofluorescence of HEK-293 cells transiently transfected with KS-WNK1-HA and costained for HA epitopes and endogenous RPL22 (n = 3 transfections; scale bar = 10 μm in 1× images, 5 μm in 4× images).
Movie S4.
3-dimensional colocalization of KS-WNK1 and RPL22. 3-dimensional Z-stack movie of a HEK-293 cell transfected with C-terminal HA tagged KSWNK1, fixed and stained using antibodies against HA epitopes (red) and endogenous RPL22 (green). Fifty four Z stack confocal slices were taken at Nyquist resolution, then rendered in 3D in Imaris. Movie created in Imaris.
A cysteine-rich hydrophobic motif in KS-WNK1 mediates WNK body formation
The N-terminus of KS-WNK1 consists of 30 unique amino acids, encoded by exon 4a (Supplemental Figure S3B). The primary amino acid sequence of exon 4a is highly conserved across evolution (Figure 6A). Based on an analysis of available genomic databases, exon 4a emerged in coelacanths, a sarcopterygian that evolved into land-dwelling tetrapods (Nikaido et al., 2013). This suggests that KS-WNK1 is involved in facilitating the transition of life from water to land, a function that has been ascribed to the kidney tubule (Rossier et al., 2015). An analysis of phylogenetically conserved residues in KS-WNK1 revealed an unusual cluster of cysteines interspaced by a block of five bulky hydrophobic residues (Figure 6A). To test the relevance of these sequences to WNK body formation, we conducted a mutagenic analysis (Figure 6B), comparing the subcellular localization of exon 4a mutants with wild-type (WT) KS-WNK1 in supernatant-pellet assays, and by immunofluorescence confocal microscopy. Deletion of the 30 amino acids encoded by exon 4a (Δ30) shifted KS-WNK1 from the pellet to the supernatant fraction (Figure 6C) and from puncta to the cytosol (Figure 6D). Mutation of the six cysteines within exon 4a to serine (C6→S) also forced KS-WNK1 from the pellet into the supernatant; the transition to cytosol was confirmed by immunofluorescence (Figure 6, C and D). All six cysteines were required for full redistribution to the cytosol, as mutation of the outer two cysteines (C2→S) did not influence KS-WNK1 localization, and mutation of the inner four residues (C4→S) only had a modest effect (Figure 6, C and D). Finally, we mutated the block of bulky hydrophobic residues to neutral glutamines (5Φ→5Q), and again saw similar results to the Δ30 and C6→S constructs: the hydrophobic mutant was predominantly expressed in the supernatant fraction and distributed diffusely on immunofluorescence. These data indicate that a cysteine-rich hydrophobic (CRH) motif in KS-WNK1 is required for the formation of WNK bodies.
FIGURE 6:

A CRH motif localizes KS-WNK1 to WNK bodies. (A) Amino acid sequence of exon 4a. The sequence emerged in coelacanths and is highly conserved to humans, including a putative CRH motif (highlighted in blue). (B) Exon 4a mutagenesis. Five constructs were created including 1) Δ30, lacking the entire exon 4a, 2) C6→S, mutating all of the cysteines to serines, 3) C2→S, mutating the two outer cysteines to serines, 4) C4→S, mutating the four inner cysteines to serines, and 5) 5Φ→5Q, mutating the hydrophobic cluster to glutamines. (C, D) SP assay (anti-HA immunoblot) of HEK-293 cells transiently transfected with KS-WNK1-HA mutants. Representative immunofluorescence of HEK-293 cells transiently transfected with either KS-WNK1-HA or N-terminal mutants, probed with HA antibody (n = 3 transfections). (E) Fluorescence recovery after photobleaching (FRAP) experiment. HEK-293 cells were transiently transfected with L-WNK1-GFP, KS-WNK1-GFP, or C6→S-KS-WNK1-GFP. Representative time series of WNK1-GFP recovery after photobleaching (area bleached circled in blue and recovery indicated by yellow arrows). (F) Percent recovery of the mobile fraction over 20 s. L-WNK1 and the C6→S mutant exhibited a similar rate of recovery (n = 10 replicates for both). In contrast, KS-WNK1 puncta (n = 3 replicates) exhibited limited mobility, with only 5% recovery by 20 s. Error bars = SEM.
Fluorescence recovery after photobleaching (FRAP) with C-terminal eGFP tagged constructs revealed a large difference in the recovery kinetics of L-WNK1 and KS-WNK1. While L-WNK1 exhibited rapid recovery post photobleaching, WNK body-localized KS-WNK1 recovered slowly over time (Figure 6, E and F). These findings suggest that WNK bodies restrict the diffusion of individual KS-WNK1 proteins. FRAP experiments with the KS-WNK1-C6→S-GFP construct revealed that the absence of cysteines freed the KS-WNK1 protein from WNK bodies, resulting in a greatly enhanced rate of diffusion relative to WT KS-WNK1 (Figure 6, E and F). Thus, although WNK bodies are distinct entities that are capable of dynamic trafficking in cells (Supplemental Movie S1), movement of KS-WNK1 within a given WNK body appears to be limited and dependent on the CRH motif.
DISCUSSION
WNK kinases regulate electrolyte transport in the distal tubule in response to physiological changes in extracellular potassium (Terker et al., 2015, 2016b; Boyd-Shiwarski and Subramanya, 2017). Consistent with unique regulation of WNK signaling in the DCT, several studies have reported that the WNKs, SPAK, and OSR1 coalesce into large puncta in the DCT during dietary K+ maneuvers, or in mouse models where regulation of the pathway is perturbed through genetic modifications (Grimm et al., 2012; Schumacher et al., 2015; Terker et al., 2015; Al-Qusairi et al., 2016, 2017). Although the formation of these peculiar structures is clearly linked to WNK signaling pathway activity in the DCT, the mechanisms that drive their assembly have been mysterious. Here, we determined that these “WNK bodies” are dependent on KS-WNK1 for their formation. A unique CRH motif within exon 4a of KS-WNK1 is critical for their assembly, and consistent with this, mice lacking KS-WNK1 were unable to form puncta. Further, we show that WNK bodies are compositionally unique, as they are membraneless and lack the classical signatures of stress granules, processing bodies, or proteotoxicity. Finally, our data indicate that WNK bodies are dynamic and restrict the movement of WNK pathway components, suggesting that they compartmentalize WNK-SPAK/OSR1 signaling in the DCT.
According to current models, potassium restriction causes basolateral hyperpolarization of the DCT, and phosphoactivation of NCC via WNK kinases (Terker et al., 2015; Cuevas et al., 2017). In addition, KS-WNK1 mRNA abundance is decreased during K+ restriction, which has been hypothesized to lessen the antagonistic effects of KS-WNK1 on active WNK kinases (Lazrak et al., 2006; O’Reilly et al., 2006; Subramanya et al., 2006). Conversely, excess dietary potassium is associated with NCC dephosphorylation, and increased KS-WNK1 mRNA expression (Wade et al., 2006; Cheng et al., 2012; Penton et al., 2016). On the basis of this congruent model, we initially hypothesized that KS-WNK1 would be necessary for puncta formation on a high-K+ diet, and were surprised to find that KS-WNK1 also drives WNK body formation during K+ depletion. It is possible that high- and low-K+ WNK bodies, although both KS-WNK1 dependent, form via distinct mechanisms that are tied to their morphological differences. Consistent with this idea, we recently found that the formation of WNK bodies during dietary K+ loading requires local expression of the canonical aldosterone-induced kinase SGK1, as these high-K+ WNK puncta were absent in inducible renal tubule-specific SGK1 knockout mice despite severe hyperkalemia (Al-Qusairi et al., 2016). On the other hand, under volume-replete low-K+ conditions, circulating aldosterone concentrations are low (Castaneda-Bueno et al., 2014) and its downstream signal should therefore be switched off in the renal distal tubule. Indeed, both SGK1 and mineralocorticoid receptors are dispensable for the strong up-regulation of NCC phosphorylation caused by dietary K+ restriction (Vallon et al., 2009; Terker et al., 2016a), an effect known to be dependent on WNK kinases (Castaneda-Bueno et al., 2014). These observations suggest that the formation of high-K+ WNK bodies requires aldosterone, whereas the formation of low-K+ WNK bodies does not. Although additional work is needed to understand how WNK body assembly and function differs during hyper- and hypokalemia, our data indicate that these unique structures serve the common goal of responding to potassium imbalance.
Several studies have reported that endogenous and overexpressed L-WNK1 can adopt a diffuse expression pattern in cell culture (Xu et al., 2000; Zagorska et al., 2007), or localize to innumerable submicron-sized intracellular puncta, either at baseline (Tu et al., 2011; Sengupta et al., 2012), or under conditions of hyperosmotic stress (Zagorska et al., 2007; Sengupta et al., 2012). Studies that localized L-WNK1 to these small puncta have reported that it partially overlaps with a variety of structures, including clathrin-coated vesicles, mitotic spindles, and regulators of autophagy (Zagorska et al., 2007; Tu et al., 2011; Gallolu Kankanamalage et al., 2016). These findings support the notion that L-WNK1 has pleiotropic kinase-dependent regulatory functions that affect membrane traffic, growth, and cell division, and is consistent with its ubiquitous expression in mammalian cells (Xu et al., 2000). Motivated by these previous studies, we sought to determine whether the large KS-WNK1–dependent WNK bodies observed here colocalize with similar intracellular markers, and found that they are clearly distinct from them. Nevertheless, when both WNK1 isoforms are present in the same cell, KS-WNK1 can sequester L-WNK1 and other members of the WNK-SPAK/OSR1 pathway into WNK bodies. Studies presented here and in previously published work indicate that these sequestered WNKs are dephosphorylated, both in heterologous expression systems (Supplemental Figure S5) and in the DCT (Al-Qusairi et al., 2016, 2017). Thus, KS-WNK1 may function as a DCT cell-specific regulator of WNK pleiotropy.
In addition to identifying WNK-SPAK/OSR1 pathway components as WNK body constituents, we localized RPL22 within these structures. Previous work by (Cheema et al., 2014) provided the critical clue that this ribosomal subunit might colocalize with WNK bodies. In their study, RPL22 foci were identified following aldosterone infusion, a condition that induces negative K+ balance. Matching our own observations presented here, these structures were subnuclear, and failed to colocalize with membrane markers, including lysosomes and autophagosomes. In addition, immuno-TEM studies in kidney clearly demonstrated RPL22 accumulation in membraneless electron hypodense regions of the cytosol (Cheema et al., 2014), similar to our own electron micrographs of KS-WNK1 expression in HEK-293 cells. RPL22 is a ubiquitously expressed ribosomal protein, and is an external component of the 60s subunit (Fahl et al., 2015). Although it is an RNA-binding protein, the 60s particle is not thought to be associated with membraneless organelles that process RNA, including stress granules and P-bodies (Anderson and Kedersha, 2006); indeed, we were unable to identify colocalization between RPL22-associated WNK bodies and these structures. More recently, RPL22 has been reported to participate in extraribosomal biological processes, including lymphocyte development and carcinogenesis (Fahl et al., 2015). Thus, it is conceivable that WNK bodies could influence DCT function through effects on ribosomal function, unconventional RNA processing, or currently undefined extraribosomal processes.
Several important observations strongly suggest that WNK bodies are not inert or dysfunctional protein aggregates. First, these structures require KS-WNK1, which when knocked out disrupts potassium-regulated WNK body formation, and causes NCC overactivity and hypertension (Hadchouel et al., 2010; Liu et al., 2011). This suggests that WNK bodies control DCT functional status and provide links between potassium balance and blood pressure regulation. Second, exon 4a, which appears to be critical for WNK body formation through its CRH motif, is highly conserved across kidney evolution, indicating that WNK bodies form due to selection pressures that require their presence in the distal tubule. Third, immunolocalization studies by ourselves and others (Cheema et al., 2014) indicate that WNK bodies are not conventional vimentin-caged aggresomes that are subject to disposal by macroautophagy. And perhaps most importantly, WNK bodies form (and by inference, disperse) during physiological changes in plasma potassium, suggesting they embody a natural, productive response to potassium stress. Indeed, it seems more likely to us that WNK bodies are demixed hydrophobic microdomains of the cytosol that arise to partition signaling processes linked to potassium-dependent ion transport in the DCT. Although we were unable to detect colocalization between WNK bodies and components of stress granules or P-bodies, they may be kidney-specific stress-induced assemblies that operate in a similar manner.
In conclusion, we have shown that WNK bodies are unique KS-WNK1–dependent structures that form in the DCT during changes in potassium balance. We propose that WNK body formation is linked to the normal physiology of the distal nephron, as an evolutionarily conserved manifestation of the renal response to potassium stress.
MATERIALS AND METHODS
Molecular methods
All L-WNK1 and KS-WNK1 clones used for this study were subcloned to the EcoRI and XbaI sites of pcDNA3.1 and were derived from the original untagged rat L-WNK1 cDNA (AAF74258.1), with an S2368G mutation, which corrects a variant serine present in the original cDNA back to an evolutionarily conserved glycine (Xu et al., 2000; Chavez-Canales et al., 2014; Roy et al., 2015a). To generate untagged KS-WNK1, an EcoRI-PacI fragment from a previously generated rat KS-WNK1 cDNA (Subramanya et al., 2006) was swapped with the 5′ end of L-WNK1. All mutations were designed as synthetic gBlocks (Integrated DNA Technologies) and 5′ EcoRI and PacI restriction fragments were swapped with the 5′ end of KS-WNK1 using standard methods. All clones contain an in-frame HA tag at the C-terminus. Additional in-frame C-terminal eGFP or mRuby tags were fused to WNK1 cDNAs and used for live cell imaging. Primer and gBlock sequences are available on request.
Antibodies
The rabbit pan-WNK1 antibody was purchased from Atlas antibodies, and is directed to residues 1779–1869 of human WNK1 (NP_061852.3). To generate the guinea pig WNK4 antibody, a GST fusion protein cDNA encoding the first 167 amino acids of mouse WNK4 was expressed in BL21 E.coli, purified using a HiTrap glutathione S-transferase (GST) column and eluted with 10 mM reduced glutathione. The free glutathione was removed by overnight dialysis and purity was confirmed by SDS–PAGE. Antibody production in guinea pigs was carried out by Pocono Rabbit Farm and Laboratory (Canadensis, PA). For a complete list of antibodies used for this study, see Supplemental Table S1.
Animal studies
All animal protocols were performed in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh IACUC. C57Bl/6 and 129/Sv mice (aged 8–10 wk, 25–30 g) were from Jackson Laboratories. KS-WNK1-KO mice (129/Sv background) were reported previously (Liu et al., 2011). Mice were fed low-K+ (<0.03%; Teklad TD.88239), normal K+ (1%), or high-K+ citrate diets (5%; Teklad TD.07278). After 10 d, mice were anesthetized with isoflurane, whole blood was collected with a heparinized syringe via cardiac puncture and analyzed by iSTAT (Abbot), and kidneys were then harvested and fixed in either 10% Formalin for immunohistochemistry or 4% paraformaldehyde for immunofluorescence. Animals were killed by cervical dislocation.
Kidney immunohistochemistry
Formalin-fixed kidney tissue was embedded in paraffin, and sections (4 μm) were deparaffinized. After inactivation of endogenous peroxidases and a blocking step, primary antibodies were incubated overnight, and subsequent incubation with biotinylated secondary antibodies (Jackson Immunoresearch) and ABC reagent were used to visualize staining with diaminobenzadine (Vector Laboratories). Images were acquired with a Leica DM6000B widefield microscope with a Retiga 4000R Fast 1394 camera. Images were obtained with Volocity 6.3, and analysis was performed using ImageJ (NIH).
Kidney immunofluorescence
Paraformaldehyde-fixed kidney tissue was cryoprotected with 0.9 M sucrose overnight at 4°, then embedded in blocks and frozen at −20°. Sections (4.5 μm) were cut on a cryostat (Leica Microsystems) and collected onto microscope slides (Fisher Scientific). Sections of fixed kidney were rehydrated and treated with 1% SDS for 10 min for retrieval of antigenic sites. Sections were washed with high salt buffer + bovine serum albumin (BSA) before addition of primary antibody. Primary antibodies were incubated overnight, and subsequent incubation with secondary antibodies was used to visualize staining.
Immunoelectron microscopy
Cells were fixed in 2% paraformaldehyde in phosphate-buffered saline (PBS) and stored at 4°C for 1 h. Cells were scraped from the dish and resuspended in a small amount of 3% gelatin in PBS, solidified at 4°C, then fixed an additional 15 min. Gelatin–cell block was cryoprotected in poly(vinylpyrrolidone) (PVP) overnight at 4°C (25% PVP, 2.3 M sucrose, 0.055 M Na2CO3, pH 7.4). Cell blocks were frozen on ultracryotome stubs under liquid nitrogen and stored in liquid nitrogen until use. Ultrathin sections (70–100 nm) were cut using a Reichert Ultracut UC7 ultramicrotome with cryoattachment, lifted on a small drop of 2.3 M sucrose, and mounted on Formvar-coated copper grids. Sections were washed three times with PBS and then three times with PBS containing 0.5% BSA and 0.15% glycine (PBG buffer) followed by a 30-min blocking incubation with 5% normal goat serum in PBG. Sections were labeled with mouse anti-HA (1:100) in PBG for 1 h. Sections were washed four times in PBG and labeled with goat anti-mouse (5 nm) gold conjugated secondary antibodies (Sigma), at a dilution of 1:25 for 1 h. Sections were washed three times in PBG, three times in PBS, then fixed in 2.5% glutaraldehyde in PBS for 5 min, washed two times in PBS and then washed six times in ddH2O. Sections were poststained in 2% neutral uranyl acetate, for 7 min, washed three times in ddH2O, stained 2 min in 4% uranyl acetate, and then embedded in 1.25% methyl cellulose. Labeling was observed on a JEOL JEM 1011 electron microscope (Peabody, MA) at 80 kV fitted with a side mount AMT 2k digital camera (Advanced Microscopy Techniques, Danvers, MA).
Cell culture and transfection
HEK-293 cells (American Type Culture Collection) were cultured and transient transfections with Lipofectamine 2000 were performed as described previously (Donnelly et al., 2013). Cells were studied within a 20-passage window, and tested quarterly for Mycoplasma using the MycoAlert Mycoplasma Detection Kit (Lonza). Cells were subjected to analysis with either Western blot or immunofluorescence 48–72 h posttransfection.
Preparation of cell lysates and immunoblot analysis
Cells were washed with ice-cold PBS, and pelleted by low speed centrifugation. For the supernatant-pellet assay, the supernatant lysate was extracted using cold Triton extraction with HEENG buffer in 1% Triton X-100 (Subramanya et al., 2009), incubating the samples on ice for 20 min, and pelleting the insoluble material by centrifugation in a microcentrifuge at max speed, 4°C for 7 min. Following removal of the supernatant fraction, Triton-insoluble pellets were then resuspended in SDS buffer (2% SDS, 60 mM Tris-HCl), boiled at 95° for 5 min, and centrifuged at max speed, 4°C for 7 min. The supernatant was removed and collected as the pellet fraction. To obtain the whole cell lysate, cells were washed with ice-cold PBS, then isolated by centrifugation at 4500 G, 4° C for 5 min. The lysates were obtained using SDS buffer (2% SDS, 60 mM Tris-HCl) and boiled at 95° for 5 min. The insoluble fraction was pelleted by centrifugation at max speed, 4°C for 7 min, and the supernatant was collected as the whole cell lysate. Protein quantifications were determined using the Pierce BCA Protein Assay Kit (Thermo Scientific). SDS–PAGE and immunoblotting were performed as described previously (Donnelly et al., 2013).
Preparation of fixed cells on coverslips for immunofluorescence
Cells grown on coverslips were fixed in 4% paraformaldehyde. The cells were washed in PBS, and then blocked for 1 h in PBS containing 5% fetal bovine serum (FBS), 5% 1 mM, 0.75% Triton X-100. Slides were subjected to immunofluorescence analysis with antibodies described in Supplemental Table S1. Primary antibodies were incubated 2 h, then washed with PBS containing 5% FBS and 5% 1 mM glycine. Primary antibodies raised in different species were used for dual immunostaining. Subsequent incubation with secondary antibodies was used to visualize staining.
Immunofluorescence confocal microscopy
Confocal imaging of the mounted cells or kidney tissue was performed using a confocal imaging system (Revolution XDi spinning disk; Andor Technology) at 40× (Nikon CFI Plan APO Lambda 20X, 0.75 NA) and 120× magnification (Nikon CFI APO TIRF 60X, 1.49 NA) with a 2× magnification lens on a Nikon Ti-E-2000 inverted microscope to achieve Nyquist resolution. Additionally, the imaging setup contained temperature and humidity controlled imaging capabilities, a mechanical Piezo XYZ-stage (Nikon), iXon 897 Ultra back-illuminated camera (Andor Technology), a laser combiner (Andor Technology) containing 405, 488, 515, 568, and 647 nm excitation capabilities, a Dell 5400 Workstation optimized for IQ2 imaging software (Andor Technology), and an active isolation air table (TMC). Images of representative fields were taken. A minimum of three biological replicates were performed to confirm the results. Confocal z-stacks were acquired at Nyquist resolution, which for our system is 0.19 µm when using our 60× objective. Image processing was performed using ImageJ software. 3D z-stack reconstruction and colocalization was performed using Imaris Coloc (Bitplane) image analysis software (Shiwarski et al., 2017).
Live cell imaging
To visualize L and KS-WNK1 trafficking simultaneously in live cells, eGFP and mRuby were C-terminally tagged to L-WNK1 and KS-WNK1, respectively. HEK-293 cells were transfected as described above in cell culture. Before imaging, cells were transferred to glass coverslips (Corning). Cells were imaged at 60× magnification (Nikon CFI APO TIRF) using a Nikon Ti-E-2000 inverted confocal microscope described above using the Nikon Perfect Focus System with a 37° heated enclosure in Leibovitz’s L-15 media + 1% FBS. eGFP and mRuby were imaged using 488 nm laser excitation with 525/50 nm emission filter, and 561 nm laser excitation with 620/60 nm emission filter, respectively. Images were acquired on an iXon 897 Ultra back-illuminated camera (Andor Technology) with 100 ms exposures every 200 ms for 12 s, and/or IQ2 software was used for image acquisition and postimaging processing.
FRAP
Confocal images were acquired on Nikon TI Eclipse with perfect focus system live at 37°C with 60× objective every 300 ms for 1 min. FRAP experiments were performed using a 488-nm laser at 100% power with an Andor FRAPPA unit with 200 µs dwell time and two repeats for each spot. The percent recovery after photobleaching for the mobile fraction was calculated by normalizing the bleached region to a control nonbleached region for each time point. The average of the baseline fluorescence was calculated and used for normalization. The minimum fluorescence signal for each bleached region was calculated to determine fluorescence remaining after bleaching. The minimum fluorescence intensity was subtracted from the normalized fluorescence intensity. Data were normalized a final time to the prebleached baseline values. These calculations constrain the prebleach fluorescence to 100% and the postbleach fluorescence to 0%, which isolates the percent mobile fraction recovery by taking into account the amount of the signal that was bleached and looking at its recovery over time. All images were analyzed and quantified using ImageJ and graphed in GraphPad Prism.
Data analysis
For Western blot quantification, densitometry was determined using NIH ImageJ software. Graph Pad Prism software was used for statistical analyses. Data are presented as mean ± SE. Comparisons between two groups were determined by a Student’s t test. Comparisons between multiple groups were determined using one-way analysis of variance (ANOVA), followed by the appropriate post hoc test as indicated. Significance was assumed to be p < 0.05.
Supplementary Material
Acknowledgments
This work was supported, in whole or in part, by National Institutes of Health grants R01DK098145 (to A.R.S.), R01DK038470 (to T.R.K.), P30DK79307 (to the Pittsburgh Center for Kidney Research), R01DK111542 (to C.-L.H.), and R01GM117425 (to M.A.P.); National Science Foundation grant NSF-1517776 (to M.A.P.); American Heart Association grant 13FTF16990086 (to R.J.T.); the UPMC Competitive Medical Research Fund (to R.J.T.); and VA VISN4 Competitive Pilot Project Funds (to A.R.S.). C.R.B.-S. is supported by NIH T32DK061296. H.N.N. was supported by the University of Pittsburgh School of Medicine Short-Term Medical Student Training Program in Renal, GI, Endocrine, and Epithelial Biology (NIH-T35DK065521). We thank Kelly Connolly and Brittney Rush for technical assistance, the Andrew Van Demark laboratory for advice and assistance with GST protein purification, David Ellison for antibodies and WNK4 KO lysates, and Udai Pandey, Jim Wade, Nathan Clark, Jeff Brodsky, Gerard Apodaca, Becky Hughey, and Ora Weisz for antibodies, reagents, and/or helpful discussions.
Abbreviations used:
- DCT
renal distal convoluted tubule
- KS-WNK1
kidney-specific (kinase deficient) WNK1 isoform
- L-WNK1
long (kinase-active) WNK1 isoform
- NCC
thiazide-sensitive Na-Cl cotransporter (gene symbol: SLC12A3)
- OSR1
oxidative stress responsive kinase 1 (gene symbol: OXSR1)
- RPL22
60S ribosomal protein L22 (gene symbol: RPL22)
- SPAK
Ste20/SpS1-related proline alanine-rich kinase (gene symbol: STK39)
- WNK
with-no-lysine (amino acid = K) kinase
- WNK1
with-no-lysine kinase 1 (gene symbol: WNK1)
- WNK4
with-no-lysine kinase 4 (gene symbol: WNK4).
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-08-0529) on December 13, 2017.
REFERENCES
- Al-Qusairi L, Basquin D, Roy A, Stifanelli M, Rajaram RD, Debonneville A, Nita I, Maillard M, Loffing J, Subramanya AR, Staub O. Renal tubular SGK1 deficiency causes impaired K+ excretion via loss of regulation of NEDD4-2/WNK1 and ENaC. Am J Physiol Renal Physiol. 2016:F330–F342. doi: 10.1152/ajprenal.00002.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Qusairi L, Basquin D, Roy A, Rajaram RD, Maillard MP, Subramanya AR, Staub O. Renal tubular ubiquitin-protein ligase NEDD4-2 is required for renal adaptation during long-term potassium depletion. J Am Soc Nephrol. 2017:2431–2442. doi: 10.1681/ASN.2016070732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. RNA granules. J Cell Biol. 2006:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd-Shiwarski CR, Subramanya AR. The renal response to potassium stress: integrating past with present. Curr Opin Nephrol Hypertens. 2017:411–418. doi: 10.1097/MNH.0000000000000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda-Bueno M, Cervantes-Perez LG, Rojas-Vega L, Arroyo-Garza I, Vazquez N, Moreno E, Gamba G. Modulation of NCC activity by low and high K(+) intake: insights into the signaling pathways involved. Am J Physiol Renal Physiol. 2014:F1507–F1519. doi: 10.1152/ajprenal.00255.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda-Bueno M, Cervantes-Perez LG, Vazquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+:Cl− cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA. 2012:7929–7934. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez-Canales M, Zhang C, Soukaseum C, Moreno E, Pacheco-Alvarez D, Vidal-Petiot E, Castaneda-Bueno M, Vazquez N, Rojas-Vega L, Meermeier NP, et al. WNK-SPAK-NCC cascade revisited: WNK1 stimulates the activity of the Na-Cl cotransporter via SPAK, an effect antagonized by WNK4 Hypertension 2014. 1047 1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheema MU, Damkier HH, Nielsen J, Poulsen ET, Enghild JJ, Fenton RA, Praetorius J. Distal renal tubules are deficient in aggresome formation and autophagy upon aldosterone administration. PLoS One. 2014:e101258. doi: 10.1371/journal.pone.0101258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CJ, Truong T, Baum M, Huang CL. Kidney-specific WNK1 inhibits sodium reabsorption in the cortical thick ascending limb. Am J Physiol Renal Physiol. 2012:F667–F673. doi: 10.1152/ajprenal.00290.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang CL, Ellison DH, Wang WH. Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol. 2017:1814–1825. doi: 10.1681/ASN.2016090935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaloy C, Lu J, Houot AM, Disse-Nicodeme S, Gasc JM, Corvol P, Jeunemaitre X. Multiple promoters in the WNK1 gene: one controls expression of a kidney-specific kinase-defective isoform. Mol Cell Biol. 2003:9208–9221. doi: 10.1128/MCB.23.24.9208-9221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly BF, Needham PG, Snyder AC, Roy A, Khadem S, Brodsky JL, Subramanya AR. Hsp70 and Hsp90 multichaperone complexes sequentially regulate thiazide-sensitive cotransporter endoplasmic reticulum-associated degradation and biogenesis. J Biol Chem. 2013:13124–13135. doi: 10.1074/jbc.M113.455394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahl SP, Wang M, Zhang Y, Duc AC, Wiest DL. Regulatory roles of Rpl22 in hematopoiesis: an old dog with new tricks. Crit Rev Immunol. 2015:379–400. doi: 10.1615/critrevimmunol.v35.i5.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallolu Kankanamalage S, Lee AY, Wichaidit C, Lorente-Rodriguez A, Shah AM, Stippec S, Whitehurst AW, Cobb MH. Multistep regulation of autophagy by WNK1. Proc Natl Acad Sci USA. 2016:14342–14347. doi: 10.1073/pnas.1617649113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm PR, Coleman R, Delpire E, Welling PA. Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J Am Soc Nephrol. 2017:2597–2606. doi: 10.1681/ASN.2016090948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm PR, Taneja TK, Liu J, Coleman R, Chen YY, Delpire E, Wade JB, Welling PA. SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. J Biol Chem. 2012:37673–37690. doi: 10.1074/jbc.M112.402800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadchouel J, Soukaseum C, Busst C, Zhou XO, Baudrie V, Zurrer T, Cambillau M, Elghozi JL, Lifton RP, Loffing J, Jeunemaitre X. Decreased ENaC expression compensates the increased NCC activity following inactivation of the kidney-specific isoform of WNK1 and prevents hypertension. Proc Natl Acad Sci USA. 2010:18109–18114. doi: 10.1073/pnas.1006128107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RW, Craigie E, Homer NZ, Mullins JJ, Bailey MA. Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. Am J Physiol Renal Physiol. 2014:F457–F467. doi: 10.1152/ajprenal.00339.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazrak A, Liu Z, Huang CL. Antagonistic regulation of ROMK by long and kidney-specific WNK1 isoforms. Proc Natl Acad Sci USA. 2006:1615–1620. doi: 10.1073/pnas.0510609103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Xie J, Wu T, Truong T, Auchus RJ, Huang CL. Downregulation of NCC and NKCC2 cotransporters by kidney-specific WNK1 revealed by gene disruption and transgenic mouse models. Hum Mol Genet. 2011:855–866. doi: 10.1093/hmg/ddq525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab. 2011:352–364. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriguchi T, Urushiyama S, Hisamoto N, Iemura S, Uchida S, Natsume T, Matsumoto K, Shibuya H. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J Biol Chem. 2005:42685–42693. doi: 10.1074/jbc.M510042200. [DOI] [PubMed] [Google Scholar]
- Nikaido M, Noguchi H, Nishihara H, Toyoda A, Suzuki Y, Kajitani R, Suzuki H, Okuno M, Aibara M, Ngatunga BP, et al. Coelacanth genomes reveal signatures for evolutionary transition from water to land Genome Res 2013. 1740 1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly M, Marshall E, Macgillivray T, Mittal M, Xue W, Kenyon CJ, Brown RW. Dietary electrolyte-driven responses in the renal WNK kinase pathway in vivo. J Am Soc Nephrol. 2006:2402–2413. doi: 10.1681/ASN.2005111197. [DOI] [PubMed] [Google Scholar]
- O’Reilly M, Marshall E, Speirs HJ, Brown RW. WNK1, a gene within a novel blood pressure control pathway, tissue-specifically generates radically different isoforms with and without a kinase domain. J Am Soc Nephrol. 2003:2447–2456. doi: 10.1097/01.asn.0000089830.97681.3b. [DOI] [PubMed] [Google Scholar]
- Penton D, Czogalla J, Wengi A, Himmerkus N, Loffing-Cueni D, Carrel M, Rajaram RD, Staub O, Bleich M, Schweda F, Loffing J. Extracellular K+ rapidly controls NaCl cotransporter phosphorylation in the native distal convoluted tubule by Cl−-dependent and independent mechanisms. J Physiol. 2016:6319–6331. doi: 10.1113/JP272504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal. 2014:ra41. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossier BC, Baker ME, Studer RA. Epithelial sodium transport and its control by aldosterone: the story of our internal environment revisited. Physiol Rev. 2015:297–340. doi: 10.1152/physrev.00011.2014. [DOI] [PubMed] [Google Scholar]
- Roy A, Al-Qusairi L, Donnelly BF, Ronzaud C, Marciszyn AL, Gong F, Chang YP, Butterworth MB, Pastor-Soler NM, Hallows KR, et al. Alternatively spliced proline-rich cassettes link WNK1 to aldosterone action J Clin Invest 2015a. 3433 3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Goodman JH, Begum G, Donnelly BF, Pittman G, Weinman EJ, Sun D, Subramanya AR. Generation of WNK1 knockout cell lines by CRISPR/Cas-mediated genome editing. Am J Physiol Renal Physiol. 2015b:F366–F376. doi: 10.1152/ajprenal.00612.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher FR, Siew K, Zhang J, Johnson C, Wood N, Cleary SE, Al Maskari RS, Ferryman JT, Hardege I, Yasmin, et al. Characterisation of the Cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia EMBO Mol Med 2015. 1285 1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Tu SW, Wedin K, Earnest S, Stippec S, Luby-Phelps K, Cobb MH. Interactions with WNK (with no lysine) family members regulate oxidative stress response 1 and ion co-transporter activity. J Biol Chem. 2012:37868–37879. doi: 10.1074/jbc.M112.398750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiwarski DJ, Tipton A, Giraldo MD, Schmidt BF, Gold MS, Pradhan AA, Puthenveedu MA. A PTEN-regulated checkpoint controls surface delivery of delta opioid receptors. J Neurosci. 2017:3741–3752. doi: 10.1523/JNEUROSCI.2923-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int. 2013:811–824. doi: 10.1038/ki.2013.14. [DOI] [PubMed] [Google Scholar]
- Subramanya AR, Ellison DH. Distal convoluted tubule. Clin J Am Soc Nephrol. 2014:2147–2163. doi: 10.2215/CJN.05920613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanya AR, Liu J, Ellison DH, Wade JB, Welling PA. WNK4 diverts the thiazide-sensitive NaCl cotransporter to the lysosome and stimulates AP-3 interaction. J Biol Chem. 2009:18471–18480. doi: 10.1074/jbc.M109.008185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanya AR, Yang CL, Zhu X, Ellison DH. Dominant-negative regulation of WNK1 by its kidney-specific kinase-defective isoform. Am J Physiol Renal Physiol. 2006:F619–F624. doi: 10.1152/ajprenal.00280.2005. [DOI] [PubMed] [Google Scholar]
- Terker AS, Yarbrough B, Ferdaus MZ, Lazelle RA, Erspamer KJ, Meermeier NP, Park HJ, McCormick JA, Yang CL, Ellison DH. Direct and indirect mineralocorticoid effects determine distal salt transport. J Am Soc Nephrol. 2016a:2436–2445. doi: 10.1681/ASN.2015070815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int. 2016b:127–134. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terker AS, Zhang C, McCormick JA, Lazelle RA, Zhang C, Meermeier NP, Siler DA, Park HJ, Fu Y, Cohen DM, et al. Potassium modulates electrolyte balance and blood pressure through effects on distal cell voltage and chloride Cell Metab 2015. 39 50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu SW, Bugde A, Luby-Phelps K, Cobb MH. WNK1 is required for mitosis and abscission. Proc Natl Acad Sci USA. 2011:1385–1390. doi: 10.1073/pnas.1018567108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol. 2009:F704–F712. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lubbe N, Moes AD, Rosenbaek LL, Schoep S, Meima ME, Danser AH, Fenton RA, Zietse R, Hoorn EJ. K+-induced natriuresis is preserved during Na+ depletion and accompanied by inhibition of the Na+-Cl− cotransporter. Am J Physiol Renal Physiol. 2013:F1177–F1188. doi: 10.1152/ajprenal.00201.2013. [DOI] [PubMed] [Google Scholar]
- Vidal-Petiot E, Cheval L, Faugeroux J, Malard T, Doucet A, Jeunemaitre X, Hadchouel J. A new methodology for quantification of alternatively spliced exons reveals a highly tissue-specific expression pattern of WNK1 isoforms. PLoS One. 2012:e37751. doi: 10.1371/journal.pone.0037751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Petiot E, Elvira-Matelot E, Mutig K, Soukaseum C, Baudrie V, Wu S, Cheval L, Huc E, Cambillau M, Bachmann S, et al. WNK1-related familial hyperkalemic hypertension results from an increased expression of L-WNK1 specifically in the distal nephron Proc Natl Acad Sci USA 2013. 14366 14371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J. 2005:17–24. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade JB, Fang L, Liu J, Li D, Yang CL, Subramanya AR, Maouyo D, Mason A, Ellison DH, Welling PA. WNK1 kinase isoform switch regulates renal potassium excretion. Proc Natl Acad Sci USA. 2006:8558–8563. doi: 10.1073/pnas.0603109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, et al. Human hypertension caused by mutations in WNK kinases Science 2001. 1107 1112 [DOI] [PubMed] [Google Scholar]
- Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem. 2000:16795–16801. doi: 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- Zagorska A, Pozo-Guisado E, Boudeau J, Vitari AC, Rafiqi FH, Thastrup J, Deak M, Campbell DG, Morrice NA, Prescott AR, Alessi DR. Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J Cell Biol. 2007:89–100. doi: 10.1083/jcb.200605093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrowicz BP, Abuin A, Ramirez-Solis R, Richter LJ, Piggott J, BeltrandelRio H, Buxton EC, Edwards J, Finch RA, Friddle CJ, et al. Wnk1 kinase deficiency lowers blood pressure in mice: a gene-trap screen to identify potential targets for therapeutic intervention Proc Natl Acad Sci USA 2003. 14109 14114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.