

Key Points
TRAIL is upregulated in LGL leukemia and TRAIL-DcR2 signaling drives constitutive activation of NF-κB.
Proteasome inhibitors (bortezomib and ixazomib) effectively interrupt TRAIL-induced activation of NF-κB and induce apoptosis.
Abstract
Large granular lymphocyte (LGL) leukemia results from clonal expansion of CD3+ cytotoxic T lymphocytes or CD3− natural killer (NK) cells. Chronic antigen stimulation is postulated to promote long-term survival of LGL leukemia cells through constitutive activation of multiple survival pathways, resulting in global dysregulation of apoptosis and resistance to activation-induced cell death. We reported previously that nuclear factor κB (NF-κB) is a central regulator of the survival network for leukemic LGL. However, the mechanisms that trigger constitutive activation of NF-κB in LGL leukemia remain undefined. Tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) is known to induce apoptosis in tumor cells but can also activate NF-κB through interaction with TRAIL receptors 1, 2, and 4 (also known as DR4, DR5, and DcR2, respectively). The role of TRAIL has not been studied in LGL leukemia. In this study, we hypothesized that TRAIL interaction with DcR2 contributes to NF-κB activation in LGL leukemia. We observed upregulated TRAIL messenger RNA and protein expression in LGL leukemia cells with elevated levels of soluble TRAIL protein in LGL leukemia patient sera. We also found that DcR2 is the predominant TRAIL receptor in LGL leukemia cells. We demonstrated that TRAIL-induced activation of DcR2 led to increased NF-κB activation in leukemic LGL. Conversely, interruption of TRAIL-DcR2 signaling led to decreased NF-κB activation. Finally, a potential therapeutic application of proteasome inhibitors (bortezomib and ixazomib), which are known to inhibit NF-κB, was identified through their ability to decrease proliferation and increase apoptosis in LGL leukemia cell lines and primary patient cells.
Visual Abstract
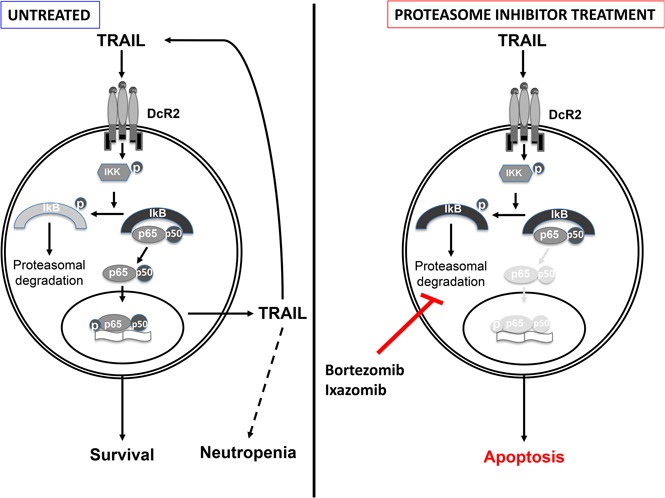
Introduction
Large granular lymphocyte (LGL) leukemia is a lymphoproliferative disorder characterized by clonal expansion of either cytotoxic T lymphocytes (CTLs) or natural killer (NK) cells.1 The majority of T-LGL (CD3+/CD8+/CD57+) and NK-LGL (CD3−/CD16+/CD56+) leukemia patients have a clinically indolent course.2 Chronic antigen stimulation is proposed to promote long-term survival of LGLs through constitutive activation of multiple survival pathways that contribute to global dysregulation of apoptosis and resistance to activation-induced cell death (AICD).3 Additionally, genetic alterations, such as signal transducer and activator of transcription 3 (STAT3) somatic activating mutations, have been identified in up to 40% of LGL leukemia patients and are thought to be central to the pathogenesis of this disease.4,5
We reported previously that leukemic T- and NK-LGLs are resistant to CD95/Fas-mediated apoptosis despite high expression levels of Fas/Fas ligand.3 We have also shown that nuclear factor κ-B (NF-κB) is constitutively active in T-LGL leukemia and mediates survival of leukemic T-LGLs.6 T-LGL leukemia cells, but not cells from normal donors, were sensitive to inhibitors of NF-κB signaling.6 The activation of NF-κB signaling in LGL leukemia has not been extensively studied, and mechanisms of activation are incompletely defined.
Tumor necrosis factor (TNF)–related apoptosis-inducing ligand (TRAIL; Apo-2 ligand, CD253, or TNF-SF10) is a TNF superfamily cytokine that induces apoptosis through binding a series of death receptors.7 TRAIL typically binds to death receptor 4 (DR4; also referred to as TRAILR1) or DR5 (TRAILR2) and induces caspase-8–dependent apoptosis through a functional cytoplasmic death domain. However, 2 other TRAIL receptors exist: decoy receptor 1 (DcR1; also referred to as TRAILR3) and DcR2 (TRAILR4). DcR1 functions as a TRAIL-neutralizing decoy receptor that is devoid of signaling capabilities. DcR2 contains a cytoplasmic domain that lacks a functional death domain; therefore, TRAIL binding to DcR2 fails to induce apoptosis. Conversely, TRAIL binding to DcR2 activates NF-κB and leads to transcription of genes that promote cell survival and resistance to apoptosis.8,9
Resting CD8+ CTLs and NK cells exhibit surface expression of DcR2, but not DR4, DR5 or DcR1; thus, they are resistant to TRAIL-induced killing.10 After activation, both CD8+ CTLs and NK cells upregulate DR5 and DcR1. These cells remain resistant to TRAIL-induced apoptosis through the high expression of both isoforms of cellular Fas-associated death domain protein-like interleukin-1 (IL-1)–converting enzyme inhibitor protein (c-FLIP). Knockdown of c-FLIP sensitized activated CTLs and NK cells to TRAIL-induced apoptosis.10
Here, we focus on the ability of TRAIL to activate NF-κB and promote leukemic LGL survival. We demonstrate for the first time that TRAIL messenger RNA (mRNA) and protein expression levels are elevated in LGL leukemia patient cells, and soluble TRAIL is elevated in patient sera. We also show that TRAIL contributes to survival of both T- and NK-LGLs. We identify DcR2 as the predominant TRAIL receptor on the LGL leukemia cell surface. Furthermore, TRAIL initiates and sustains NF-κB activation specifically within DcR2-positive leukemic cells. Inhibition of NF-κB with the proteasome inhibitors bortezomib and ixazomib leads to cell death in LGL leukemia cells.
Materials and methods
Reagents
All chemicals were purchased from Sigma-Aldrich unless otherwise specified. Recombinant human (rh) TRAIL, TNF-α, IL-15, and platelet-derived growth factor (PDGF) BB were purchased from ProSpec-TANY TechnoGene, and rhIL-2 was from the NIH AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health (rhIL-2 was from Maurice Gately, Hoffmann-La Roche). Antibodies and inhibitors from the following sources were used as recommended by the manufacturers: Canonical and noncanonical NF-κB detection antibody kits; antibodies for human TRAIL, TRAF2, c-FLIP, Bcl-2, Bcl-XL, caspase-3, caspase-8, poly (ADP-ribose) polymerase (PARP), β-actin, and GAPDH (Cell Signaling); c-FLIP antibody (clone NF6; Enzo Life Sciences); NF-κB family protein p50 and p65 antibodies used for immunocytochemical staining and goat anti-mouse immunoglobulin G (IgG) antibody (Santa Cruz Biotechnology); NF-κB (p65 and p50) Transcription Factor Assay Kit (Cayman Chemical); and the proteasome inhibitors bortezomib (PS-341) and ixazomib (MLN2238) (Selleckchem).
Patient characteristics and preparation of PBMCs
All patients met the clinical criteria of T- or NK-LGL leukemia with increased numbers of CD3+, CD8+/CD57+ T lymphocytes or CD3−, CD16+/CD56+ NK cells in the peripheral blood.2 Patients were not on treatment at the time of sample acquisition. Peripheral blood specimens were obtained and informed consents signed for sample collection according to the Declaration of Helsinki using a protocol approved by the institutional review board of the University of Virginia or Penn State Cancer Institute. Patient samples were screened for somatic activating mutations in STAT3 by Sanger sequencing as previously described.11 Buffy coats from age- and gender-matched normal donors were obtained from Virginia Blood Services. Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll-Hypaque gradient separation, as previously described.2 Cell viability was determined by trypan blue exclusion assay with more than 95% viability in all the samples. Normal PBMCs were stimulated with phytohemagglutinin at 1 μg/mL concentration in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) (Invitrogen) for 24 hours. Phytohemagglutinin was then replaced by rhIL-2 at 500 IU/mL concentration in RPMI-1640 culture medium with 10% FBS. For DcR2+ and DcR2− cell sorting, PBMC samples were prestained with anti-CD3-eFluor-450 and CD8-phycoerythrin (PE)-Cy7 antibodies (BD Pharmingen) as well as DcR2-PE antibody (LifeSpan BioScience). DcR2-positive and negative cells were separated from the CD3+/CD8+ cell population using fluorescence-activated cell sorting (FACSAria Fusion Cell Sorter). CD3−/CD16+/CD56+ NK cells were isolated by a negative selection process (Rosette Sep; StemCell Technologies), as previously described.12 Purified CD8+ T cells were isolated using Dynal CD8 antibody–positive isolation Kit (Invitrogen Life Technologies). Neutrophil counts were obtained from T-LGL leukemia patient complete blood count reports closest in date to the date of serum sample collection.
Cell culture
Culture of freshly isolated PBMCs, NK cells, and CD8+ T lymphocytes was carried out using RPMI-1640 medium supplemented with 10% FBS (Invitrogen). NKL cells, a leukemic LGL NK cell line,13 and TL-1 cells, a leukemic LGL T-cell line,14 were cultured in RPMI-1640 medium supplemented with 10% FBS and rhIL-2 (100 IU/mL for NKL; 200 IU/mL for TL-1). Jurkat cells were cultured in RPMI-1640 medium supplemented with 10% FBS. Cells were grown in 5% CO2 at 37°C. To test the efficacy of rhIL-15– and rh-PDGF-BB–mediated TRAIL production in TL1 and NKL cells, rhIL-2 was withdrawn from culture medium overnight, and cells were then treated with rhIL-15 (5 IU/mL) and rhPDGF-BB (50 ng/mL). Samples were harvested at designated time points. TRAIL expression in these samples was determined by western blot assay.
TRAIL determination by enzyme-linked immunosorbent assay (ELISA)
TRAIL protein levels in serum samples from LGL leukemia patients and normal controls were determined using a Human TRAIL Quantikine ELISA kit (R&D Systems). Serially diluted rhTRAIL protein (15.6 to 1000 pg/mL) was used to generate a standard curve to calculate the TRAIL levels in each sample. Samples were assayed in triplicate.
TRAIL receptor expression detection by flow cytometry
Cell surface expression of TRAIL receptor 1 (DR4; CD261), receptor 2 (DR5; CD262), receptor 3 (DcR1; CD263), and receptor 4 (DcR2; CD264) was determined using the Anti-TRAIL receptor 1 to 4 flow cytometry set (catalog no. ALX-850-273-KI01, Alexis).9 Purified mouse IgG1 served as an isotype control. Additional samples were used for DcR2 expression in specific cell populations as defined by CD3, CD8, and CD57 status. Briefly, PBMC samples were stained with anti-CD3-eFluor 450 (Invitrogen), CD8-PE-Cy7, CD57-fluorescein isothiocyanate, and DcR2-PE antibodies. 7-AAD was used as a dead cell marker for this refined profiling study to allow gating to remove all dead cells, which exhibited nonspecific DcR2 staining. Therefore, slightly different DcR2+ values were detected in the refined DcR2 profiling vs the initial profiling of all TRAIL receptors. Apoptosis was determined by staining cells with Annexin-V-APC. The percentage of DcR2+ and apoptosis in each cell fraction was determined by flow cytometry assay.
Electrophoretic mobility shift assay (EMSA)
Nuclear and cytoplasmic extracts from PBMC samples were prepared using NE-PER Nuclear and Cytoplasmic Extraction (Life Technologies). EMSA was performed using the LightShift Chemiluminescent EMSA Kit (Life Technologies). Alternatively, NF-κB transcription factors (human p50/p65) in nuclear protein samples were determined using the Transcription Factor Assay Kit (Cayman Chemicals). Probes for NF-κB (5′-GATCCGGCAGGGGAATCTCCCTCTC-3′) were biotinylated at the 5′ end. Corresponding nonbiotinylated oligonucleotides were used as competitive oligonucleotides.
NKL and TL-1 cell small interfering RNA (siRNA) transfection
NKL and TL-1 cells (1 million cells per well) were transfected using a NEON Transfection System (Invitrogen) with either DcR2 siRNA or scramble siRNA at 100 nM (GE Dharmacon) in a total volume of 10 μL according to the manufacturer’s protocol. Cells were cultured for 72 hours, and total protein was harvested.
Immunocytochemistry (ICC) staining
For ICC staining, the purity of freshly isolated CD8+ cells from patients with T-LGL leukemia and from normal donors was determined by flow cytometry using fluorescein isothiocyanate–conjugated anti-human CD8 antibody (clone SK1) to verify the isolation method. DcR+ and DcR2− cells were isolated from CD3+/CD8+ cells by fluorescence-activated cell sorting as described above. Purity was more than 95%. ICC staining was carried out using the Vectastain Elite ABC HRP kit (Vector Laboratories). Briefly, slides with a monolayer of cells were fixed with 3.7% paraformaldehyde phosphate-buffered saline at 37°C and permeabilized in ice-cold methanol-acetone (1:1) for 10 minutes at −20°C. Cells were incubated with polyclonal anti-p65, p50, or TRAIL antibodies (1:200 dilution) overnight at 4°C. This was followed by 1-hour incubation at room temperature with biotinylated goat anti-rabbit secondary antibody (1:250 dilution) and then by an additional 5 to 10 minutes in ImmPACT DAB chromagen solution. Cells were visualized using light microscopy (Leica Microsystems). Nonspecific staining was not detected upon incubation with secondary antibody alone.
3-(4,5 dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl-2-(4-sulfophenyl)-2H-tetrazolium (MTS) cell viability assays
Cells were treated with varying concentrations of bortezomib, ixazomib, or vehicle for 24 or 48 hours. Cell viability was determined using CellTiter 96 Aqueous One Solution assay kit (Promega). Refer to supplemental Materials and methods (available on the Blood Web site) for further details.
In vitro apoptosis assays
Apoptosis induced by bortezomib, ixazomib, or SuperKillerTRAIL (Enzo Life Science) was detected by flow cytometry with Annexin V-PE (BD Pharmingen) and 7-amino-actinomycin D (7-AAD) staining using 5 × 105 cells per sample in triplicate and normalized as previously described.15
Quantitative real-time polymerase chain reaction (PCR)
Quantitative real-time PCR to determine expression of TRAIL levels was performed using a CFX384 Real-Time PCR Detection System followed by data analysis with CFX Manager 3.1 software system (Bio-Rad) as previously described.16 Refer to supplemental Materials and methods for further details.
Western blot analysis
Western blotting was completed as previously described.17 Refer to supplemental Materials and methods for further details.
Statistical analysis
GraphPad Prism was used for statistical analyses (GraphPad Software). All data are expressed as mean ± standard error of the mean (SEM). Student t test, analysis of variance (ANOVA), and 2-way ANOVA tests were used to determine the statistical significance, and a value of P ≤ .05 was considered statistically significant.
Results
TRAIL mRNA and protein expression levels are elevated in LGL leukemia
Our recent prospective phase 2 study included gene expression microarray analysis of LGL leukemia patient samples vs normal donors.18 The data set (GSE42664) included PBMC samples isolated from T-LGL leukemia patients (n = 37) and normal control CD8+ T cells (n = 5) or terminal-effector memory CD45RA+ CD8+ T cells (Temra) (n = 3). Expression of TRAIL gene transcripts was significantly higher in T-LGL leukemia samples compared with both normal CD8+ (3.5-fold increase) and Temra (6.5-fold increase), which are the most representative normal cell type for LGL leukemia19,20 (Figure 1A). These results were verified by real-time PCR. TRAIL mRNA levels in purified CD8+ T cells from T-LGL leukemia patients (n = 11) were 15-fold higher than levels observed in CD8+ cells from normal donors (n = 3) (Figure 1B).
Figure 1.

TRAIL is overexpressed in LGL leukemia. (A) TRAIL gene expression levels in PBMC from T-LGL patients (triangles; n = 37), CD8+ cells from normal subjects (circles, n = 5), and Temra cells from normal subjects (squares, n = 3) in the GSE42664 Affymetrix data set.18 (B) Quantitative real-time PCR was performed to measure levels of TRAIL mRNA in CD8+ cells from T-LGL patients (n = 11) or purified CD8+ from normal donors (n = 3). Relative TRAIL mRNA expression was normalized to 18S. Data are presented as mean ± SEM. *P < .05 indicates significant difference between LGL leukemia patients and normal donors (Student t test). (C) Immunoblot analysis of TRAIL protein in purified CD8+ cells from patients with T-LGL leukemia (n = 6) vs CD8+ cells from normal (n) donors (n = 6) and purified NK cells from patients with NK-LGL leukemia (n = 4) vs purified NK cells isolated from normal donors (n = 4) and PBMCs from normal donors (n = 2). Loading of protein was confirmed by probing for GAPDH or β-actin. Vertical lines within the leukemic groups indicate regions where samples were removed from the image based on poor protein extract quality as indicated by loading controls. (D) CD8+ cells from a T-LGL patient or normal (NL) donor were stained with TRAIL antibodies and visualized using light microscopy (original magnification ×400). Rabbit IgG antibody was used as a negative control. Data are representative of 3 experiments conducted with cells from 3 independent patients. (E) Serum levels of TRAIL were determined using an ELISA assay. Sera were tested from T-LGL leukemia patients (squares, n = 24; ANOVA, *P < .0001 T-LGL vs normal donor), NK-LGL leukemia patients (triangles, n = 10; ANOVA, *P < .0001, NK-LGL vs normal donor), or normal donors (circles, n = 24). cDNA, complementary DNA.
Immunoblot analysis showed elevated levels of TRAIL protein in both purified CD8+ T cells from T-LGL leukemia patients vs normal donors and purified NK cells from NK-LGL leukemia patients vs normal donors. TRAIL protein expression levels were 2.0-fold and 1.6-fold increased in T- or NK-LGL patient cells, respectively, as compared with normal CD8+ or normal NK cells (P = .079 and P = .212, respectively) (Figure 1C). Additional measurements of TRAIL protein levels were pursued since these data were suggestive but fell short of statistical significance.
Next, ICC staining demonstrated that CD8+ T cells from normal donors had undetectable levels of TRAIL, whereas CD8+ T cells from patients with T-LGL leukemia had strong positive staining (brown) in cytoplasmic compartments (Figure 1D). We also detected significantly increased levels of soluble TRAIL in serum samples from T- and NK-LGL leukemia patients when compared with normal controls (Figure 1E). This finding extends and confirms our previous observations of elevated serum TRAIL levels in an independent cohort of exclusively T-LGL patients.18 TL-1 and NKL LGL leukemia cell lines treated with IL-15 or PDGF-BB, which are known to be key mediators of LGL leukemia survival,6,21-24 demonstrated that TRAIL expression is strongly induced by IL-15 (supplemental Figure 1). There was also a significant correlation between elevated levels of TRAIL and lower neutrophil counts (supplemental Figure 2). Since LGL leukemia cells are thought to arise from activated T and NK cells that have resisted AICD, we investigated TRAIL mRNA expression levels in activated cells from normal donors. We observed that activated PBMCs and CD8+ cells from normal donors have four- to sixfold higher levels of TRAIL mRNA than unactivated PBMCs and CD8+ cells, respectively (supplemental Figure 3). This increase is similar to the increase seen in LGL leukemia cells. Taken together, these findings are indicative of elevated expression and secretion of TRAIL in a large proportion of T- and NK-LGL leukemia patient samples.
LGL leukemia cells are resistant to TRAIL-induced apoptosis
NKL and TL-1 LGL leukemia and Jurkat T-cell lines were treated with SuperKiller-TRAIL, which is known to induce apoptosis in cancer cells, including Jurkat.25 TL-1 and NKL cells were resistant to TRAIL-induced apoptosis. (Figure 2A). PBMCs from patients with T-LGL and NK-LGL leukemia and normal donors treated with TRAIL were also resistant to apoptosis (Figure 2B). Next, we used flow cytometry to determine TRAIL receptor expression on the surface of LGL leukemia cells. There was no difference in expression of DR4, DR5, and DcR1 on leukemic T-LGL or NK-LGL relative to normal PBMCs (Figure 2C). DcR2 levels were elevated in LGL leukemia samples and generally correlated with the percentage of T-LGLs in the PBMC samples. Therefore, we more fully characterized DcR2 expression within the CD3+/CD8+ population, which is known to include the leukemic LGLs. This analysis revealed that CD3/CD8 double-positive cells from T-LGL leukemia samples exhibit a significantly higher percentage of DcR2 positivity relative to normal and activated normal controls (Figure 2D; P = .001). In addition, leukemic samples demonstrated relatively equal DcR2 levels in both CD3+/CD8+/CD57− and CD3+/CD8+/CD57+ subpopulations that are proposed to represent progenitor and mature populations of leukemic cells, respectively.26,27 Therefore, leukemic LGLs lack an apoptotic response to TRAIL treatment and exhibit DcR2 expression that is substantially higher than normal controls.
Figure 2.

LGL leukemia cells are resistant to TRAIL-induced apoptosis and primarily express TRAIL receptor DcR2. (A) NKL, TL-1, and Jurkat cells were treated with either vehicle (normal saline) or rhTRAIL (10 ng/mL) for 48 hours. Cells were stained with Annexin-V and 7-AAD and analyzed by flow cytometry to identify apoptotic cells. Data are presented as mean ± SEM and representative of 3 separate experiments (ANOVA, *P < .0001 Jurkat cells vs TL-1 and NKL). (B) PBMCs isolated from normal donors (n = 4), NK-LGL patients (n = 4), and T-LGL patients (n = 9) were treated with either vehicle (normal saline) or rhTRAIL (10 ng/mL) for 48 hours. Cells were stained with Annexin-V and 7-AAD and analyzed by flow cytometry to identify apoptotic cells. Data represent mean ± SEM. (C) PBMCs from normal donors or LGL leukemia patients were assessed for their cell surface expression of TRAIL receptors using flow cytometry. Percentage of LGL cells from pathology flow cytometry report is shown below the horizontal axis. (D) PBMCs from normal donors, without or with activation (Act), or LGL leukemia patients were assessed for their cell surface expression of DcR2 receptor using flow cytometry to stratify samples based on CD3, CD8, and CD57 expression.
TRAIL activates NF-κB in leukemic LGL
We previously showed that NF-κB is constitutively active in LGL leukemia and contributes to survival and resistance to apoptosis.6 TRAIL binding to TRAIL receptors, especially DcR2, has been shown to activate NF-κB.8 We observed that rhTRAIL treatment increased nuclear NF-κB DNA binding activity in PBMCs from T-LGL and NK-LGL leukemia (Figure 3A). Using ICC staining, we next examined the cellular localization of both subunits of NF-κB, p50 and p65. Vehicle-treated CD8+ cells from both T-LGL patients and normal donors exhibited cytoplasmic NF-κB p50 and p65. rhTRAIL treatment of CD8+ cells from T-LGL patients led to nuclear translocation of NF-κB p50 and p65 in a manner similar to rhTNF-α, a known NF-κB–activating cytokine (Figure 3B). In contrast, rhTRAIL treatment of CD8+ cells from normal donors did not cause translocation of NF-κB p50 and p65. TRAIL-mediated p65 translocation was specifically demonstrated within leukemic CD3+/CD8+/DcR2+ cells, but not CD3+/CD8+/DcR2− counterparts isolated from the same patient (Figure 3C). This DcR2-dependent response to TRAIL was observed in both CD8+/CD57+ and CD8+/CD57− cells from LGL leukemia samples (data not shown). We hypothesized that LGL patient sera, which contain high levels of TRAIL (Figure 1E), would also activate NF-κB. PBMCs from T-LGL patients were cultured with pooled sera from 3 normal donors to establish basal levels of nuclear NF-κB p50. Treatment with pooled sera from 3 LGL leukemia patients increased nuclear NF-κB p50 levels. This effect was significantly blocked by a neutralizing antibody to TRAIL, which suggests that soluble TRAIL in patient sera activates NF-κB in leukemic LGLs (Figure 3D).
Figure 3.

TRAIL induces NF-κB activation and nuclear translocation in LGL leukemia cells that are blocked by proteasome inhibitors. (A) EMSA demonstrating NF-κB activity in nuclear extracts from T-LGL patient PBMCs (n = 4) and NK-LGL patient PBMCs (n = 3) treated with either saline or rhTRAIL (10 ng/mL) for 2 hours. (B) NF-κB p50 or p65 protein ICC staining as visualized by light microscopy in CD8+ cells from an LGL leukemia patient compared with CD8+ cells from a normal donor (original magnification ×1000). Cells were treated with control (NTC), TNF-α (positive control, 10 ng/mL), or rhTRAIL (10 ng/mL). Brown staining represents NF-κB p50 or p65 protein. Data are representative of 3 experiments conducted with cells from 3 independent patients. (C) NF-κB p65 protein ICC staining as visualized by light microscopy in CD3+/CD8+/DcR2− and CD3+/CD8+/DcR2+ cells from an LGL leukemia patient (original magnification ×400). Cells were treated with either vehicle control (NTC) or rhTRAIL (10 ng/mL). Brown staining represents NF-κB p65 protein. Data are representative of experiments conducted with cells from 2 independent patients. (D) NF-κB p50 ELISA of PBMC nuclear protein extracts from T-LGL leukemia patients (n = 4) that were treated with sera from normal donor or sera from LGL patients. Patient cells treated with LGL sera were cotreated with vehicle, TRAIL neutralizing antibody (Neut Ab), or IgG control antibody. (E) EMSA demonstrating NF-κB activity in nuclear extracts from T-LGL patient PBMCs (n = 3) treated with vehicle (DMSO), rhTRAIL (10 ng/mL), or TRAIL (10 ng/mL) plus bortezomib (5 nM). (F) EMSA demonstrating NF-κB activity in nuclear extracts from patient cells from a T-LGL or NK-LGL patient treated with rhTRAIL (10 ng/mL) or rhTRAIL (10 ng/mL) plus increasing doses of ixazomib (0-100 nM). (G) NF-κB p50 or p65 protein ICC staining as visualized by light microscopy in CD8+ cells from normal control or LGL leukemia (original magnification ×400). Cells were pretreated with either bortezomib (5 nM) or DMSO for 2 hours followed with the treatment of rhTRAIL (10 ng/mL) or rhTNF-α (10 ng/mL positive control). Brown staining represents NF-κB p50 or p65 protein. Data are representative of 3 experiments conducted with cells from 3 independent patients.
Proteasome inhibitors block TRAIL-induced activation of NF-κB in leukemic LGLs
Bortezomib and ixazomib are proteasome inhibitors that block degradation of IκB and the proteolytic processing of p105, thereby inhibiting NF-κB nuclear translocation.28 Pre-treatment with bortezomib blocked the TRAIL-induced increase in NF-κB activity (Figure 3E). This effect was confirmed by ICC staining of CD8+ cells from patients with T-LGL leukemia (Figure 3G). In both T-LGL and NK-LGL patient cells, the orally available proteasome inhibitor ixazomib exerted similar effects and dose-dependently blocked TRAIL-induced NF-κB activity (Figure 3F).
DcR2 knockdown and proteasome inhibitor treatment decrease NF-κB activation in leukemic LGL
It has been shown that DR4, DR5, and DcR2-induced NF-κB activation is mediated by a TRAF2/NF-κB–inducing kinase/IκB kinase (IKK) α/β signaling cascade.29 DcR2 contains a truncated death domain but retains a functional cytoplasmic domain that can activate NF-κB upon TRAIL binding. To establish a role for DcR2 in the activation of NF-κB signaling in LGL leukemia, we transfected TL-1 and NKL cells with siRNA targeting DcR2 or scrambled control (Figure 4A). DcR2 knockdown led to decreased phosphorylation of NF-κB p65, without affecting total protein levels, while TRAF2 and NF-kB p50 were also downregulated. To further characterize the pathway of TRAIL-induced NF-κB activation, we treated TL-1 and NKL cells with bortezomib (5 nM) or ixazomib (100 nM). Both proteasome inhibitors downregulated TRAF2, IKK α/β phosphorylation, p65 phosphorylation, and the expression of IKKβ and NF-κB p50 (Figure 4B-C). Similarly, treatment with proteasome inhibitors resulted in decreased IKK α/β and p65 phosphorylation and decreased levels of TRAF2 and NF-κB p50 in PBMCs from patients with T-LGL leukemia (Figure 4D). Overall, these findings are consistent with TRAIL/DcR2-mediated NF-κB activation and target gene expression in leukemic LGL that is abrogated by proteasome inhibitors.
Figure 4.

TRAIL DcR2 knockdown or proteasome inhibitor treatment inhibits NF-κB activation in LGL leukemia cells. (A) TL-1 and NKL cells were transfected with DcR2-specific siRNA (100 nM) or scramble siRNA by electroporation. Cells were kept in culture for 72 hours. The expression of DcR2, TRAF2, NF-κB p65, phosphorylated p65, and NF-κB p50 was determined by western blot immunoassay. (B-C) TL-1 cells (B) and NKL cells (C) were treated with bortezomib (5 nM), ixazomib (100 nM), or vehicle (dimethyl sulfoxide [DMSO]), and protein samples were harvested at different time points as indicated. The expression of TRAF2, phosphorylated IKK α/β, IKK β, NF-κB p65, phosphorylated p65, and NF-κB p50 was determined by western blot immunoassay. β-Actin was used as a control for equal loading. (D) PBMCs from T-LGL leukemia patients were treated with bortezomib (5 nM) or ixazomib (100 nM) for 24 hours. The expression of TRAF2, phosphorylated IKK α/β, NF-κB p65, phosphorylated p65, and NF-κB p50 was determined by western blot immunoassay. β-Actin antibody was used as a control for equal loading.
Proteasome inhibitors decrease cell proliferation and induce apoptosis in LGL leukemia cells through caspase-3 and PARP cleavage
To further elucidate the role of TRAIL and NF-κB in LGL leukemia cell survival and resistance to apoptosis, we treated LGL cell lines with proteasome inhibitors and assessed their effects on proliferation and cell death. TL-1 and NKL cells treated with varying concentrations of bortezomib and ixazomib exhibited a dose-dependent decrease in cell viability at low nanomolar concentrations (Figure 5A-B). Treated cells also exhibited dose- and time-dependent increased apoptosis (Figure 5C-D). c-FLIP is upregulated in LGL leukemia and is a prosurvival protein regulated by NF-κB.3 It has been shown to play a role in resistance to TRAIL-induced apoptosis.30-34 In both TL-1 (Figure 5E) and NKL (Figure 5F) cells, we observed downregulation of c-FLIP coupled with increased caspase-3 and PARP cleavage after proteasome inhibitor treatment. Next, PBMCs from T- and NK-LGL leukemia patients and normal donors were treated with proteasome inhibitors for 48 hours (Figure 6A). Bortezomib and ixazomib induced apoptosis in primary patient cells with similar efficacy as in TL-1 and NKL cells. Both proteasome inhibitors exhibited increased efficacy in leukemic PBMCs vs PBMCs from normal donors. Notably, we did not observe differences in proteasome inhibitor responsiveness between wild-type and STAT3-mutant leukemic samples (supplemental Figure 4). Mechanistically, bortezomib and ixazomib treatment led to decreased expression of c-FLIP coupled with increased caspase-3 and PARP cleavage (Figure 6B). Proteasome inhibitor–mediated apoptosis was strongly induced within the CD3+/CD8+ subpopulation of leukemic samples, but not normal controls (Figure 6C). The apoptotic response was observed in CD57+ leukemic LGLs as well as CD57− cells. Therefore, LGL cell lines and primary samples are susceptible to proteasome inhibitor–mediated apoptosis.
Figure 5.

Proteasome inhibitors decrease viability and induce apoptosis in LGL leukemia cell lines through increased caspase-3 and PARP cleavage and downregulation of c-FLIP. (A) Bortezomib decreases viability in LGL leukemia cell lines. TL-1 and NKL cells were treated with bortezomib at varying concentrations for 48 hours, and cell viability was assessed using an MTS assay. (B) Ixazomib decreases viability in LGL leukemia cell lines. TL-1 and NKL cells were treated with ixazomib at varying concentrations for 48 hours, and cell viability was assessed using an MTS assay. (C) Proteasome inhibitors induce apoptosis in TL-1 cells. TL-1 cells were treated with bortezomib or ixazomib at varying concentrations for 24 or 48 hours, and cells were stained for apoptosis with Annexin-V and 7-AAD and analyzed by flow cytometry. (D) Proteasome inhibitors induce apoptosis in NKL cells. NKL cells were treated with bortezomib or ixazomib at varying concentrations for 24 or 48 hours, and cells were stained for apoptosis with Annexin-V and 7-AAD and analyzed by flow cytometry. (E) Proteasome inhibitors decrease expression of the NF-κB target c-FLIP and induce caspase-3 and PARP cleavage in TL-1 cells. TL-1 cells were treated with bortezomib (5 nM) or ixazomib (100 nM), and protein was harvested at various time points. Western blot analysis was performed for c-FLIP, caspase-3, and PARP. (F) NKL cells were treated with bortezomib (5 nM) or ixazomib (100 nM) and protein was harvested at various time points. Western blot analysis was performed for c-FLIP, caspase-3, and PARP expression. 50% effective concentration (EC50) values were determined by nonlinear regression in GraphPad Prism.
Figure 6.

Proteasome inhibitors induce apoptosis in LGL leukemia samples. (A) PBMCs from normal donors (n = 9), T-LGL patients (n = 16), and NK-LGL patients (n = 6) were treated with DMSO or bortezomib (2.5 or 5 nM) for 48 hours, and cells were stained for apoptosis with Annexin-V and 7-AAD and analyzed by flow cytometry (left). PBMCs from normal donors (n = 9), T-LGL patients (n = 12), and NK-LGL patients (n = 6) were treated with DMSO or ixazomib (100 or 200 nM) for 48 hours, and cells were stained for apoptosis with Annexin-V and 7-AAD and analyzed by flow cytometry (right). (B) PBMCs from patients with T-LGL leukemia were treated with proteasome inhibitor bortezomib (5 nM) or ixazomib (100 nM) for 24 hours, and c-FLIP, caspase-3, and PARP cleavage was determined by western blot assay. Equal loading for all western blot assays was confirmed by probing with a β-actin antibody. (C) PBMCs from normal donors (n = 3) and T-LGL patients (n = 4) were treated with DMSO, bortezomib, or ixazomib for 48 hours. Cells were stained for CD3, CD8, CD57 and apoptosis markers. *P < .05, **P < .01, ***P < .001 (1-way ANOVA) significant differences between T-LGL patients and normal donors.
Proteasome inhibitors downregulate NF-κB–mediated TRAIL gene expression and protein levels in leukemic LGLs
The levels of TRAIL and TRAIL receptors are closely regulated by NF-κB,35,36 and the NF-κB pathway is constitutively activated in LGL leukemia.6 Since LGL leukemia cells are the major source of TRAIL production (Figure 1), we hypothesized that inhibition of NF-κB would affect TRAIL expression in leukemic LGLs. Bortezomib and ixazomib treatment significantly downregulated TRAIL mRNA levels in LGL cell lines (Figure 7A-B). Similarly, both proteasome inhibitors decreased TRAIL mRNA levels in PBMCs from patients with T-LGL or NK-LGL leukemia (Figure 7C). We verified that TRAIL protein levels in LGL leukemia cell lines and patient samples were also downregulated by bortezomib or ixazomib in a time-dependent manner (Figure 7D-F). Taken together, these findings indicate that proteasome inhibitors suppress NF-κB activation and signaling via multiple mechanisms in leukemic LGLs.
Figure 7.

Proteasome inhibitors downregulate TRAIL expression in leukemic LGLs. (A-C) TL-1 (A), NKL (B), and LGL PBMCs (C; T-LGL n = 7, NK-LGL n = 3) were treated with bortezomib, ixazomib, or DMSO vehicle for 6 hours. Relative TRAIL mRNA expression was determined by quantitative real-time PCR. Values are presented as mean ± SEM. *P < .05 indicates significant difference between DMSO and proteasome inhibitor treatments (Student t test). In panel C, **P = 1.84E−5 and ***P = .0015. (D-F) LGL leukemia cell line TL-1 (D), NKL (E), or LGL patient PBMCs (F; T-LGL n = 4; NK-LGL n = 2) were treated with bortezomib (5 nM), ixazomib (100 nM), or DMSO, and total protein samples were collected to assess TRAIL protein levels by immunoblotting assay. Equal loading for western blot assay was confirmed by probing with β-actin antibody. Vertical lines within some patient blots indicate regions where lanes were removed from the image in order to show identical time points for all samples.
Discussion
Here, we demonstrate TRAIL-mediated NF-κB activation in T- and NK-leukemic LGLs and further define downstream components involved in this process. NF-κB consists of a group of inducible transcription factors that regulate immune and inflammatory responses and protect cells from undergoing apoptosis in response to cellular stress and chemotherapeutic agents as well as AICD in lymphocytes.37 Constitutive activation of the NF-κB pathway contributes to development of various autoimmune, inflammatory, and malignant disorders, including rheumatoid arthritis, atherosclerosis, and malignant tumors.38-40 In this study, we demonstrated that LGL leukemia cells have increased TRAIL mRNA and protein expression and increased levels of soluble TRAIL in patient sera. High levels of TRAIL correlated with lower neutrophil counts. Together with previous reports of TRAIL-induced neutrophil apoptosis, these finding suggest that elevated TRAIL may contribute to the neutropenia that is a frequent indication for treatment in LGL leukemia patients.41-43 We further demonstrate that LGL leukemia cells are resistant to TRAIL-induced apoptosis and instead exhibit increased NF-κB activation in response to TRAIL treatment.
In this study, we identified DcR2 as the predominant TRAIL receptor in T- and NK-LGL leukemia cells and showed that levels are substantially higher in leukemic samples relative to normal controls. Similar to TNF-α, TRAIL is a cytokine that executes its biological functions by binding to its receptors, which initiates a trimerization process for activation.44 The binding of TRAIL to DR4 or DR5 can induce death-induced signaling complex (DISC) formation and subsequent caspase-8–dependent apoptosis. Alternatively, TRAIL can activate an NF-κB pathway through the TRAF2/NF-κB–inducing kinase/IKK α/β signaling cascade, which leads to IKKα/β phosphorylation, and subsequently p65 phosphorylation, in cells that are resistant to TRAIL-mediated apoptosis.29,45 DcR1 acts as a TRAIL-neutralizing decoy receptor, as it does not contain a functional cytoplasmic domain. In this sense, it acts as a competitor for TRAIL binding to its death receptors and prevents DR4- and/or DR5-associated DISC assembly. DcR2 acts similarly to DR4 and DR5 but has a truncated cytoplasmic domain, negating its ability to induce DISC formation while retaining its ability to activate NF-κB.8,44,46 Therefore, the resistance of LGL leukemia cells to TRAIL-induced apoptosis prompted us to further elucidate the significance of high DcR2 expression in LGL leukemia.
We demonstrated that strong DcR2 expression, combined with its unique signaling characteristics, has important biological implications in LGL leukemia. We observed that rhTRAIL induces dramatic increases of NF-κB nuclear translocation in PBMCs from patients with T- and NK-LGL leukemia and that TRAIL activates NF-κB specifically in DcR2-positive leukemic cells. TRAIL neutralizing antibody abolished LGL leukemia patient sera-mediated elevation of p50 nuclear levels in PBMCs from patients with T-LGL leukemia, highlighting that the increased TRAIL in patient sera is capable of inducing NF-κB activation. We also showed that knockdown of DcR2 in TL-1 and NKL cells decreased phosphorylation of IKKα/β and NF-κB p65 and expression of TRAF2 and NF-κB p50. These data indicate that DcR2 is essential in TRAIL-mediated IKK complex formation and subsequent NF-κB activation in leukemic LGLs.
In the current study, bortezomib and ixazomib inhibited phosphorylation of IKK α/β and p65 and downregulated the expression of NF-κB p50 in both LGL leukemia cell lines and PBMCs from patients with T-LGL leukemia. In addition, treatment with these proteasome inhibitors dramatically decreased NF-κB nuclear DNA-binding activity in leukemic T- and NK-LGLs. Notably, bortezomib and ixazomib treatment also mediated dramatic decreases in cell viability and increased apoptosis in TL-1 and NKL cells. More importantly, these proteasome inhibitors mediated leukemia-selective apoptosis in PBMCs from patients with T- and NK-LGL leukemia through caspase-3 and PARP cleavage. We previously discovered somatic mutations in the STAT3 gene Src homology 2 (SH2) dimerization and activation domain in ∼40% of patients with T- and NK-LGL leukemia.4,5 Bortezomib and ixazomib showed no differential efficacy in induction of apoptosis between patients with either wild-type or mutated STAT3 genes. Thus STAT3 activation does not confer resistance to these compounds. Finally, both proteasome inhibitors induced apoptosis in CD57− and CD57+ cells from patients, which have been proposed to represent progenitor and mature populations of leukemic LGLs, respectively.26,27 Taken together, these data highlight the potential of inhibiting NF-κB with proteasome inhibitors for the treatment of LGL leukemia.
High expression levels of c-FLIP have been detected in LGL leukemia cells. Interestingly, knockdown of c-FLIP abrogated the resistance to TRAIL-mediated apoptosis in activated NK and CD8+ cells.10 In the current study, we observed that bortezomib or ixazomib treatment reduced c-FLIP protein levels in LGL leukemia cell lines and in PBMC from LGL leukemia patients. These results indicate that the persistent NF-κB activation contributes to the high expression of c-FLIP and apoptotic resistance in leukemic LGLs.
IL-15 is a known driver of LGL leukemia.6,21-23 STAT3 also lies downstream of IL-15 receptor activation, and STAT3 activation, through somatic activating mutations or other mechanisms, is central to the pathogenesis of LGL leukemia.4,5,47 We observed that IL-15 treatment of LGL leukemia cell lines resulted in upregulation of TRAIL. In IL-15 transgenic mice that develop LGL leukemia, a liposomal formulation of bortezomib effectively induced remission.21 Furthermore, it has been shown that IL-15 can decrease proapoptotic Bid protein levels in LGL leukemia via proteasome-mediated degradation.48 Hence, it is possible that IL-15 may directly or indirectly contribute to some of the effects shown to result from TRAIL stimulation of DcR2 and subsequent activation of NF-κB signaling. Therefore, extending and integrating these studies into IL-15, TRAIL, and NF-κB interaction warrants further investigation.
Collectively, our data suggest that expression of DcR2 and constitutive activation of NF-κB are responsible for TRAIL resistance in leukemic LGLs. We demonstrated that increased TRAIL levels trigger constitutive NF-κB activation through interaction with DcR2. Activated NF-κB in turn promotes further TRAIL production in leukemic LGLs through NF-κB, creating a TRAIL autocrine regulatory loop.35,36 We observed that inhibition of NF-κB activity with bortezomib or ixazomib interrupts this loop, resulting in decreased expression of TRAIL. These preclinical findings provide a solid framework for the future clinical evaluation of proteasome inhibitors in the treatment of LGL leukemia.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
LGL leukemia patient samples and clinical information were obtained from the LGL Leukemia Registry at the University of Virginia with the assistance of Kendall Baab, Holly Davis, Bryna Shemo, and Andrea Hines. Alexander Wendling and Matthew Schmachtenberg provided excellent technical support while processing patient samples. The authors thank the University of Virginia School of Medicine Flow Cytometry Core Facility for their assistance. Qing Zhong (Penn State University, Department of Comparative Medicine) and Patcharin Pramoonjago (Biorepository and Tissue Research Facility, University of Virginia School of Medicine) helped with ICC staining. rhIL-2 was kindly provided by the National Institutes of Health (NIH) AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases. rhIL-2 was from Maurice Gately (Hoffmann-La Roche). NKL cells, a leukemic LGL NK-cell line, were kindly provided by Howard Young (NIH National Cancer Institute).
This research was funded by the Leukemia & Lymphoma Society (TRP-6159-14) (T.P.L.) and the National Cancer Institute, National Institutes of Health (R01CA098472, R01CA178393, and P30CA044579) (T.P.L.). Additional funding was provided by the Bess Family Charitable Fund, the LGL Leukemia Foundation, and a generous anonymous donor.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.Y. and F.R.L. designed and performed research, analyzed data, and wrote the paper; S.A.D. performed research; C.E.H. and T.L.O. contributed to STAT3 mutational sequencing; D.J.F. designed research, analyzed data, and wrote the paper; and T.P.L. designed research, analyzed data, and reviewed the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Thomas P. Loughran Jr, University of Virginia Cancer Center, P.O. Box 800334, Charlottesville, VA 22908-0334; e-mail: tl7cs@hscmail.mcc.virginia.edu.
References
- 1.Loughran TP., Jr Clonal diseases of large granular lymphocytes. Blood. 1993;82(1):1-14. [PubMed] [Google Scholar]
- 2.Lamy T, Loughran TP Jr. How I treat LGL leukemia. Blood. 2011;117(10):2764-2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang J, Epling-Burnette PK, Painter JS, et al. Antigen activation and impaired Fas-induced death-inducing signaling complex formation in T-large-granular lymphocyte leukemia. Blood. 2008;111(3):1610-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jerez A, Clemente MJ, Makishima H, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood. 2012;120(15):3048-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koskela HL, Eldfors S, Ellonen P, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366(20):1905-1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang R, Shah MV, Yang J, et al. Network model of survival signaling in large granular lymphocyte leukemia. Proc Natl Acad Sci USA. 2008;105(42):16308-16313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiley SR, Schooley K, Smolak PJ, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3(6):673-682. [DOI] [PubMed] [Google Scholar]
- 8.Hall MA, Cleveland JL. Clearing the TRAIL for Cancer Therapy. Cancer Cell. 2007;12(1):4-6. [DOI] [PubMed] [Google Scholar]
- 9.Sanlioglu AD, Dirice E, Aydin C, Erin N, Koksoy S, Sanlioglu S. Surface TRAIL decoy receptor-4 expression is correlated with TRAIL resistance in MCF7 breast cancer cells. BMC Cancer. 2005;5(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mirandola P, Ponti C, Gobbi G, et al. Activated human NK and CD8+ T cells express both TNF-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors but are resistant to TRAIL-mediated cytotoxicity. Blood. 2004;104(8):2418-2424. [DOI] [PubMed] [Google Scholar]
- 11.Olson KC, Kulling PM, Olson TL, et al. Vitamin D decreases STAT phosphorylation and inflammatory cytokine output in T-LGL leukemia. Cancer Biol Ther. 2017;18(5):290-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Warren HS, Rana PM. An economical adaptation of the RosetteSep procedure for NK cell enrichment from whole blood, and its use with liquid nitrogen stored peripheral blood mononuclear cells. J Immunol Methods. 2003;280(1-2):135-138. [DOI] [PubMed] [Google Scholar]
- 13.Robertson MJ, Cochran KJ, Cameron C, Le JM, Tantravahi R, Ritz J. Characterization of a cell line, NKL, derived from an aggressive human natural killer cell leukemia. Exp Hematol. 1996;24(3):406-415. [PubMed] [Google Scholar]
- 14.Ren T, Yang J, Broeg K, Liu X, Loughran TP Jr, Cheng H. Developing an in vitro model of T cell type of large granular lymphocyte leukemia. Leuk Res. 2013;37(12):1737-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.LeBlanc FR, Liu X, Hengst J, et al. Sphingosine kinase inhibitors decrease viability and induce cell death in natural killer-large granular lymphocyte leukemia. Cancer Biol Ther. 2015;16(12):1830-1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schacter JL, Henson ES, Gibson SB. Estrogen regulation of anti-apoptotic Bcl-2 family member Mcl-1 expression in breast cancer cells. PLoS One. 2014;9(6):e100364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watters RJ, Fox TE, Tan SF, et al. Targeting glucosylceramide synthase synergizes with C6-ceramide nanoliposomes to induce apoptosis in natural killer cell leukemia. Leuk Lymphoma. 2013;54(6):1288-1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loughran TP Jr, Zickl L, Olson TL, et al. Immunosuppressive therapy of LGL leukemia: prospective multicenter phase II study by the Eastern Cooperative Oncology Group (E5998). Leukemia. 2015;29(4):886-894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kallemeijn MJ, de Ridder D, Schilperoord-Vermeulen J, et al. Dysregulated signaling, proliferation and apoptosis impact on the pathogenesis of TCRγδ+ T cell large granular lymphocyte leukemia. PLoS One. 2017;12(4):e0175670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wlodarski MW, Nearman Z, Jankowska A, et al. Phenotypic differences between healthy effector CTL and leukemic LGL cells support the notion of antigen-triggered clonal transformation in T-LGL leukemia. J Leukoc Biol. 2008;83(3):589-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mishra A, Liu S, Sams GH, et al. Aberrant overexpression of IL-15 initiates large granular lymphocyte leukemia through chromosomal instability and DNA hypermethylation. Cancer Cell. 2012;22(5):645-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Petrus M, Bamford R, et al. Increased serum soluble IL-15Rα levels in T-cell large granular lymphocyte leukemia. Blood. 2012;119(1):137-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zambello R, Facco M, Trentin L, et al. Interleukin-15 triggers the proliferation and cytotoxicity of granular lymphocytes in patients with lymphoproliferative disease of granular lymphocytes. Blood. 1997;89(1):201-211. [PubMed] [Google Scholar]
- 24.Yang J, Liu X, Nyland SB, et al. Platelet-derived growth factor mediates survival of leukemic large granular lymphocytes via an autocrine regulatory pathway. Blood. 2010;115(1):51-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Monian P, Jiang X. The cellular apoptosis susceptibility protein (CAS) promotes tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis and cell proliferation. J Biol Chem. 2016;291(5):2379-2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melenhorst JJ, Eniafe R, Follmann D, et al. T-cell large granular lymphocyte leukemia is characterized by massive TCRBV-restricted clonal CD8 expansion and a generalized overexpression of the effector cell marker CD57. Hematol J. 2003;4(1):18-25. [DOI] [PubMed] [Google Scholar]
- 27.Melenhorst JJ, Sorbara L, Kirby M, Hensel NF, Barrett AJ. Large granular lymphocyte leukaemia is characterized by a clonal T-cell receptor rearrangement in both memory and effector CD8(+) lymphocyte populations. Br J Haematol. 2001;112(1):189-194. [DOI] [PubMed] [Google Scholar]
- 28.Wang H, Guan F, Chen D, Dou QP, Yang H. An analysis of the safety profile of proteasome inhibitors for treating various cancers. Expert Opin Drug Saf. 2014;13(8):1043-1054. [DOI] [PubMed] [Google Scholar]
- 29.Hu WH, Johnson H, Shu HB. Tumor necrosis factor-related apoptosis-inducing ligand receptors signal NF-kappaB and JNK activation and apoptosis through distinct pathways. J Biol Chem. 1999;274(43):30603-30610. [DOI] [PubMed] [Google Scholar]
- 30.Siegmund D, Hadwiger P, Pfizenmaier K, Vornlocher HP, Wajant H. Selective inhibition of FLICE-like inhibitory protein expression with small interfering RNA oligonucleotides is sufficient to sensitize tumor cells for TRAIL-induced apoptosis. Mol Med. 2002;8(11):725-732. [PMC free article] [PubMed] [Google Scholar]
- 31.MacFarlane M, Harper N, Snowden RT, et al. Mechanisms of resistance to TRAIL-induced apoptosis in primary B cell chronic lymphocytic leukaemia. Oncogene. 2002;21(44):6809-6818. [DOI] [PubMed] [Google Scholar]
- 32.Kim SY, Park SE, Shim SM, et al. Bay 61-3606 sensitizes TRAIL-induced apoptosis by downregulating Mcl-1 in breast cancer cells. PLoS One. 2015;10(12):e0146073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy AC, Weyhenmeyer B, Noonan J, et al. Modulation of Mcl-1 sensitizes glioblastoma to TRAIL-induced apoptosis. Apoptosis. 2014;19(4):629-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park SH, Lee DH, Kim JL, et al. Metformin enhances TRAIL-induced apoptosis by Mcl-1 degradation via Mule in colorectal cancer cells. Oncotarget. 2016;7(37):59503-59518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ravi R, Bedi GC, Engstrom LW, et al. Regulation of death receptor expression and TRAIL/Apo2L-induced apoptosis by NF-kappaB. Nat Cell Biol. 2001;3(4):409-416. [DOI] [PubMed] [Google Scholar]
- 36.Baetu TM, Kwon H, Sharma S, Grandvaux N, Hiscott J. Disruption of NF-kappaB signaling reveals a novel role for NF-kappaB in the regulation of TNF-related apoptosis-inducing ligand expression. J Immunol. 2001;167(6):3164-3173. [DOI] [PubMed] [Google Scholar]
- 37.Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013;12(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karin M. NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol. 2009;1(5):a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun SC, Chang JH, Jin J. Regulation of nuclear factor-κB in autoimmunity. Trends Immunol. 2013;34(6):282-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makarov SS. NF-kappa B in rheumatoid arthritis: a pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res. 2001;3(4):200-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGrath EE, Marriott HM, Lawrie A, et al. TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J Leukoc Biol. 2011;90(5):855-865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Renshaw SA, Parmar JS, Singleton V, et al. Acceleration of human neutrophil apoptosis by TRAIL. J Immunol. 2003;170(2):1027-1033. [DOI] [PubMed] [Google Scholar]
- 43.Liu X, Loughran TP Jr. The spectrum of large granular lymphocyte leukemia and Felty’s syndrome. Curr Opin Hematol. 2011;18(4):254-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kimberley FC, Screaton GR. Following a TRAIL: update on a ligand and its five receptors. Cell Res. 2004;14(5):359-372. [DOI] [PubMed] [Google Scholar]
- 45.Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274(43):30353-30356. [DOI] [PubMed] [Google Scholar]
- 46.Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7(6):813-820. [DOI] [PubMed] [Google Scholar]
- 47.Rajala HL, Porkka K, Maciejewski JP, Loughran TP Jr, Mustjoki S. Uncovering the pathogenesis of large granular lymphocytic leukemia-novel STAT3 and STAT5b mutations. Ann Med. 2014;46(3):114-122. [DOI] [PubMed] [Google Scholar]
- 48.Hodge DL, Yang J, Buschman MD, et al. Interleukin-15 enhances proteasomal degradation of bid in normal lymphocytes: implications for large granular lymphocyte leukemias. Cancer Res. 2009;69(9):3986-3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.