

Abstract
Background
Multiple osteochondromas is a dysplasia characterized by growth of two or more osteochondromas. It is genetically heterogeneous, caused by pathogenic variants in EXT1 or EXT2 genes in 70%–90% of patients. The EXT1 is more often mutated than EXT2 gene, with a variable prevalence between populations. There are no data about EXT1 and EXT2 pathogenic variants in patients with multiple osteochondromas in Brazilian population. The aim of this survey is to characterize these to determine the genotype profile of this population.
Methods
DNA sequencing (Sanger Method) and MLPA analysis were performed to identify point mutations and deletions/duplications in the sample of 153 patients in 114 families.
Results
Germline variants were identified in 83% of families in which EXT2 variants were detected in 46% and EXT1 in 37% of cases. No variants were detected in 17% of them. We identified 50 different variants, 33 (13 frameshift, 11 nonsense, 5 missense, 2 splice site mutation, and 2 large deletions) in EXT1 and 17 (6 frameshift, 6 splice site mutation, 3 nonsense, 1 missense, and 1 large deletion) in EXT2. Of all 50 variants, 31 (62%) were novel, including 20 out of 33 (60,6%) EXT1 and 11 out of 17 (64.7%) EXT2 alleles. The vast majority of variants (88%) were “loss‐of‐function” and two novel hotspots in EXT2 gene were observed in our study.
Conclusion
The prevalence of variants detected in the EXT2 gene differs from other researches from Latin America, European, and Asian population. This uncommon prevalence could be related with the newly characterized variant hotspot sites detected in EXT2 gene (p.Ala409Profs*26 and p.Ser290*). A high number of novel variants were also identified indicating that Brazilian population has a unique genetic profile. Characterizing this population and establishing its genotype is essential to understand the molecular pathogenesis of this disease in Brazil.
Keywords: Brazilian population, chondrosarcoma, EXT1, EXT2, genotype, multiple osteochondromas
1. INTRODUCTION
Multiple osteochondromas (MO, OMIM 133700 and 133701) are a skeletal dysplasia characterized by development of two or more osteochondromas (cartilage‐capped bone tumors, always benign, which protrude from the metaphysis of long bone or flat bone surface; Hennekam, 1991). The median age of diagnosis is 3 years and it is based on clinical and radiological signs (Schmale, Conrad, & Raskind, 1994). Osteochondromas increase in number and size during growth and frequently cause short stature, bone deformity, pain, joint limitation, early osteoarthritis, peripheral nerve or tendon or blood vessel compression (Pedrini et al., 2011; Porter et al., 2004). The most serious complication is malignant transformation in chondrosarcoma, occurring in 1%–5% of cases (Pedrini et al., 2011; Porter et al., 2004; Schmale et al., 1994).
MO are genetically heterogeneous autosomal dominant disorder, with 100% of age‐related penetrance. The prevalence is estimated to be one in 50,000 North Americans, which is likely to be a conservative estimation due to a significant proportion of undiagnosed MO patients with no or mild unidentified symptoms (Schmale et al., 1994). MO are caused by pathogenic variants in two ubiquitously expressed tumor suppressor genes, EXT1 (OMIM 608177, chr 8q24.11) and EXT2 (OMIM 608210, chr 11p11.2) (Ahn et al., 1995; Stickens et al., 1996). The EXT1 gene is composed of 11 exons and encodes for a protein of 746 amino acids (Ahn et al., 1995). The EXT2 gene comprises 16 exons, but only exons 2 to 14 code for a product of 718 amino acids (Stickens et al., 1996). EXT1 and EXT2 are highly similar, specially in the carboxy terminal region that contains significantly less pathogenic variants (Ciavarella et al., 2013; Wuyts & Van Hul, 2000)). In the EXT1 gene, missense mutations are common in or nearby regions that encode the highly conserved arginine residues at codons 280 and 340, which are located in the d‐glucuronic acid glycosyltransferase (d‐GlcA‐TII) catalytic domain of EXT1. Similarly, in EXT2, a recurrent pathogenic variant in exon 4 affecting an asparagine residue (p.Asp227Asn) is known and most pathogenic variants are found in the first eight exons (Heinritz et al., 2009; Philippe et al., 1997). EXT1 and EXT2 glycoproteins form a heterooligomeric complex that catalyzes the polymerization of the polysaccharide, heparan sulfate. This complex is an essential factor for multiple cellular functions, as growth signaling pathway, anchorage of extracellular matrix, signal transduction cascade for regulation of chondrocyte differentiation, ossification, and apoptosis (Hameetman et al., 2007; McCormick, Duncan, Goutsos, & Tufaro, 2000; Samuel, Costa, & Lindskog, 2014). The precise function of EXT1 and EXT2 in the development of MO has not been entirely understood (Jones, Pacifici, & Hilton, 2014; Samuel et al., 2014). Previous molecular analysis of the coding regions of both EXT1 and EXT2 genes described pathogenic variants in 46%–95% of affected individuals (Guo, Deng, & Liu, 2014; Jennes et al., 2009). The relative frequencies differed in EXT1 and EXT2 pathogenic variants among various ethnic groups. The frequency of pathogenic variants in EXT1 was characterized to be 40%–75% versus 20%–40% in EXT2 in Caucasian (French, British, German, Spanish, Belgian, Dutchman, Italian, Chilean, Argentine Caucasian) and Japanese populations (Delgado et al., 2014; Francannet et al., 2001; Heinritz et al., 2009; Ishimaru et al., 2016; Jennes et al., 2008; Lonie et al., 2006; Pedrini et al., 2011; Sarrión et al., 2013). In contrast, in Canadian and Chinese populations, the frequency of pathogenic variants in EXT1 was 14%–30% versus 28%–50% in EXT2 (Alvarez, Tredwell, De Vera, & Hayden, 2006; Guo et al., 2014; Xu et al., 1999). The 10 Canadian families that were studied by Alvarez had their ancestral origins in the UK (5), Germany (1), France (1), Japan (1), Austria (1), and India (1). Less than 20% of patients have a “de novo” mutation (Jennes et al., 2009; Schmale et al., 1994) and most of the identified pathogenic variants are inactivating mutations, including nonsense, frameshift, and splice site mutations (Jennes et al., 2009; Wuyts & Van Hul, 2000).
To date, the profile of mutations in EXT1 and EXT2 genes in Brazilian patients with MO is unknown. The objective of this survey is to identify, describe, and analyze variants in EXT1 and EXT2 genes of 153 Brazilian patients with MO recruited at our rehabilitation center.
2. MATERIALS AND METHODS
2.1. Ethical compliance
This study was approved by the National Committee of Ethics in Research, SARAH Network of Rehabilitation Hospitals. All the patients or their parents (if the patient was underage) signed informed consent and approved the anonymous use of clinical and molecular data for the present diagnostic study.
2.2. Patients and clinical data
We identified 153 patients in 114 families with a clinical diagnosis of MO (89 unrelated and 25 families) attended at the SARAH Network of Rehabilitation Hospitals in Brasília from 2012 to 2014.
The inclusion criteria for this study were to be affected by MO, to be SARAH's patient, and to consent to the research. A retrospective study of clinical, surgical, and radiological data of these patients was made. The diagnosis of MO was established when radiologically at least two osteochondromas of the juxta‐epiphyseal region of long bones were observed. The diagnostic of malignant transformation was obtained by radiological investigation and histopathological examination.
Brazilians form one of the most heterogeneous populations in the world, the result of 5 centuries of interethnic crosses between peoples from three continents: the European immigration, represented mainly by the Portuguese; African slaves; and the autochthonous Amerindians (Alves‐Silva et al., 2000). The total sample showed nearly equal amounts of Native American, African, and European matrilineal genetic contribution but with regional differences (Alves‐Silva et al., 2000). This study included patients from all regions of the country, reducing ethnic differences. The age of patients ranged from 3 to 75 years old and the median age was 20. The prevalence was higher in males (ratio = 1.22/1) and familial cases (63%). Seven unrelated patients aged between 21 and 54 presented malignant degeneration to chondrosarcoma. Histological analyses were made in six of them: four had grade I and two had grade II according to World Health Organization criteria.
2.3. Genotyping and mutation analysis
Genomic DNA was extracted from peripheral blood samples according to a standard protocol (Miller, Dykes, & Polesky, 1988). The codifying and flanking regions of all exons of EXT1 and EXT2 genes were amplified by PCR. Primers were designed by Primer Blast Designing Tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast) and are listed in Table S1. Exon 1 of EXT1 and exon 2 and 10 of EXT2 were split into several overlapping fragments in order to obtain amplification products that did not exceed 300 bp. PCR was performed in a 50 μl reaction volume, containing 50 ng of DNA genomic, 1× PCR buffer, 1.5–2 mmol/L MgCl2, 0.2 mmol/L of each dNTP, 0.25 μmol/L of each forward and reverse primer, and 0.5 U of platinum Taq polymerase (Invitrogen, Carlsbad, CA). All PCR programs included an initial denaturation of 5 min at 95°C, followed by 35 cycles of 30 s at 95°C, 30 s at annealing temperature, and 1 min at 72°C. The annealing temperature was 62°C for all primer combinations. We first analyzed 153 patients in 114 families for EXT1 variants by denaturing high‐performance liquid chromatography (dHPLC) using WAVE DNA Fragment Analysis System (Transgenomic, Crewe, UK). Samples with abnormal elution profile were directly sequenced (Big Dye Terminator Cycle Sequence kit v.3.0 and ABI3130xl automated Sequencer, Applied Biosystems) in both directions with forward and reverse primers on the original amplicon and on different PCR product. Negative cases were then screened for EXT2 variants using the same approach. Negative cases for both genes were directly sequenced. If no mutation was detected, negative cases were analyzed by MLPA (Multiplex ligation‐dependent probe amplification—SALSA MLPA kit P215‐B1 EXT, MCR‐Holland, Amsterdam, The Netherlands). Variants were searched in the database Osteochondromas Mutation Database (MOdb) (http://medgen.ua.ac.be/LOVD) (last accessed on October 2016).
The reference EXT1 and EXT2 sequences were obtained from GenBank with accession numbers NM_000127.2, NP_000118.2, NM_001178083.1, NP_001171554.1, respectively. Mutation numbering is based on cDNA CCDS6324.1 and CCDS53618.1, respectively.
The pathogenic role of novel missense mutations that were neither found in MOdb nor ExAC or 1,000 Genomes (https://www.ncbi.nlm.nih.gov/variation/tools/reporter) was evaluated by testing other family members, when available, and by three in silico online tools: PolyPhen‐2 (Polymorphism Phenotyping v2; http://genetics.bwh.harvard.edu/pph2/, last accessed on October 2016), MutPred (v.1.2; http://mutpred.mutdb.org/, last accessed on October 2016), and Mutation Taster2 (http://www.mutationtaster.org, last accessed on October 2016). The Human Splice Finder (http://www.umd.be/HSF/, last accessed October 2016) online tool was used to assess the possible effect of novel intronic variants on splicing sites.
The nomenclature of novel variants was given according to the official HGVS guideline (http://www.hgvs.org) and based on the GenBank accession number.
3. RESULTS
Exons and flanking regions of the EXT1 and EXT2 genes were analyzed from the genomic DNA of 153 patients in 114 families with MO and MLPA analyses were performed in DNA samples with negative results for sequencing analysis. A mutant allele was found in 83% (95/114) of unrelated MO patients in which EXT2 variants were detected in 46% (53/114) and EXT1 variants were detected in 37% (42/114) of cases. We identified 50 different variants: 33 (13 frameshift, 11 nonsense, 5 missense, 2 splice site mutation, and 2 large deletions) in EXT1 and 17 (6 frameshift, 6 splice site mutation, 3 nonsense, 1 missense, and 1 large deletion) in EXT2 gene. Of all 50 variants, 31 (62%) were novel with no description in the Multiple Osteochondroma Mutation Database (MOdb) (http://medgen.ua.ac.be/LOVDDv.2.0/home.php). Twenty out of 33 (60.6%) EXT1 variants were novel as were 11 of 17 (64.7%) of EXT2 variants (Table 1).
Table 1.
(a) List of mutations in EXT1 in MO patients (HPO id 0002762). (b) List of mutations in EXT2 in MO patients (HPO id 0002762)
| Family number⊤ | Exon–intron | DNA | Deduced protein change | Mutation type | Publication |
|---|---|---|---|---|---|
| (a) | |||||
| P01 | Exon 1 | c.9insA | p.Lys6Thrfs*29 | Frameshift | This study |
| P02 | Exon 1 | c.125delG | p.Gly42Valfs*93 | Frameshift | This study |
| P03 | Exon 1 | c.173_174insG | p.Phe58Leufs*72 | Frameshift | This study |
| c.186_192delCGCCC | |||||
| P05,P06, P07 | Exon 1 | c.651_672del22 | p.Lys218Alafs*27 | Frameshift | This study |
| P08 | Exon 1 | c.859delC | p.His287Ilefs* | Frameshift | This study |
| P10 | Exon 1 | c.774_775delCT | p.Pro258Profs*29 | Frameshift | This study |
| P20, P21, F8, F24 | Exon 6 | c.1469delT | p.Leu490Argfs*9 | Frameshift | Signori et al. (2007) |
| P22, P23 | Exon 6 | c.1468_1469insC | p.Leu490Profs*31 | Frameshift | Signori et al. (2007) |
| P25 | Exon 8 | c.1642delA | p.Ser548Alafs*73 | Frameshift | Lonie et al. (2006) |
| F21 | Exon 8 | c.1680_1682insGC | p.Val561Profs*16 | Frameshift | This study |
| P26 | Exon 8 | c.1718_1719delCA | p.Thr573Argfs*53 | Frameshift | This study |
| F14 | Exon 10 | c.2040_2041delGA | p.Thr681Aspfs*? | Frameshift | This study |
| P27 | Exon 10 | c.2001delG | p.Leu667Phefs*5 | Frameshift | This study |
| F22 | Exon 10 | c.1896_1898delCCT | p.Tyr632* | Nonsense | This study |
| F12 | Exon 1 | c.289A>T | p.Lys97* | Nonsense | This study |
| P04 | Exon 1 | c.385G>T | p.Glu129* | Nonsense | This study |
| F15, P16 | Exon 4 | c.1236G>A | p.Trp412* | Nonsense | This study |
| P18 | Exon 5 | c.1320insT | p.Asp441* | Nonsense | Gigante et al. (2001) |
| P19 | Exon 5 | c.1335G>A | p.Trp445* | Nonsense | This study |
| F17 | Exon 6 | c.1522C>T | p.Gln508* | Nonsense | Pedrini, not published |
| F7 | Exon 7 | c.1550G>A | p.Trp517* | Nonsense | Jennes, not published |
| P24 | Exon 8 | c.1696G>T | p.Glu566* | Nonsense | Vink et al. (2005) |
| F23 | Exon 10 | c.1911C>G | p.Tyr637* | Nonsense | This study |
| P29 | Exon 10 | c.1902C>G | p.Tyr634* | Nonsense | This study |
| P14 | Intron 2 | c.1056+1G>A | Splice Site | Jennes et al. (2008) | |
| P28 | Intron 9 | c.1884‐2A>G | Splice Site | This study | |
| F04 | Exon 1 | c.803G>A | p.Gly268Glu | Missense | This study |
| P09 | Exon 1 | c.791T>G | p.Leu264Arg | Missense | This study |
| P11, P12, P13 | Exon 2 | c.1019G>T | p.Arg340Leu | Missense | Jennes et al. (2008) |
| F9 | Exon 2 | c.1019G>A | p.Arg340His | Missense | Pedrini et al. (2005) |
| P15 | Exon 2 | c.1037G>C | p.Arg346Thr | Missense | Delgado et al. (2014) |
| P70 | Del EXT1 | Deletion | Hall et al. (2002) | ||
| P71 | Exon 1 | ex1del | Deletion | Jennes, not published | |
| (b) | |||||
| P34 | Exon 2 | c.202delG | p.Val68Leufs*43 | Frameshift | This study |
| P35 | Exon 2 | c.453insT | p.Gly152Trpfs*13 | Frameshift | This study |
| P37 | Exon 3 | c.539_540insTC | p.Asp181Glyfs*89 | Frameshift | This study |
| P38 | Exon 3 | c.550_551dupGT | p.Thr184Valfs*86 | Frameshift | This study |
| P51 | Exon 5 | c.760delC | p.Leu255Cysfs*15 | Frameshift | This study |
| F1, F2, F5, F18, F19, F20, F25, P56, P57, P58, P59, P17, P60, P61, P62, P63, P64, P65, P66 | Exon 8 | c.1225delG | p.Ala409Profs*26 | Frameshift | This study |
| F10, F13, P30, P31, P32, P33, | Exon 2 | c.151G>T | p.Glu51* | Nonsense | Lonie et al. (2006) |
| F11, F16, P41, P42, P43, P44, P45, P46, P47, P48, P49 | Exon 5 | c.869C>A | p.Ser290* | Nonsense | This study |
| F6, P67, P69 | Exon 8 | c.1182G>A | p.Trp394* | Nonsense | Delgado et al. (2014) |
| P36 | Intron 3 | c.626+1G>A | Splice Site | Vink et al. (2005) | |
| P39 | Intron 3 | c.626+2delT | Splice Site | This study | |
| P40 | Intron 4 | c.743+1G>A | Splice Site | Vink et al. (2005) | |
| P50 | Intron 4 | c.744‐13del14insATTC | Splice Site | This study | |
| P52, P53 | Intron 6 | c.1079+1G>A | Splice Site | Jennes, not published | |
| P54 | Intron 6 | c.1173+1dupG | Splice Site | Francannet et al. (2001) | |
| P55 | Exon 7 | c.1110G>T | p.Met370Ile | Missense | This study |
| F26 | Exon 6‐8 | ex6_ex8_del | Deletion | This study | |
⊤: P means individual case and F, familial nucleus.
All identified variants were submitted to ClinVar with ACMG 2015 classification criteria (Richards et al., 2015).
Of the 50 different variants identified, 39 (78%) were found in only one individual. Recurrent pathogenic variants observed in patients of Brazil included two novel hotspots in EXT2 gene. The most frequent pathogenic variant detected in EXT1 gene was p.Leu490Argfs*9 occurring in four families, followed by p.Arg340Leu and p.Lys218Alafs*27 both occurring in three families. Only, p.Lys218Alafs*27 was novel. Two novel hotspots in EXT2 gene were observed in our study: p.Ala409Profs*26 detected in 19 families, and p.Ser290Ter detected in 11 families (Table 1).
The vast majority of variants (88%) were “loss‐of‐function” mutation (frameshift, nonsense, splicesite, and large deletions) responsible for the production of truncated protein products. Two out of five missense mutations detected in EXT1 gene were novel. Bioinformatic predictions suggested a pathogenic role for these alterations. In particular, p.Leu264Arg change from a nonpolar amino acid (Leu) to a basic amino acid (Arg) was considered “probably damaging” by Polyphen2 (score of 0.997; sensitivity: 0.27; specificity: 0.98), “deleterious mutation” by MutPred (score: 0.851, with a confident hypothesis of a gain of methylation at Leu264Arg; p = .0089), and “disease causing” by the Mutation Taster (score: 0.999, amino acid sequence changed, protein features (might be) affected, splicesite changes). Also, this variant was neither found in ExAC nor 1,000 Genomes (https://www.ncbi.nlm.nih.gov/variation/tools/reporter); a variant at same codon (p.Leu264Pro) was previously described (Delgado et al., 2014). Similarly, the missense mutation, p.Gly268Glu, changed from a nonpolar amino acid (Gly) to a polar amino acid (Glu) was predicted as “possibly damaging” by Polyphen2 (score: 0.856; sensitivity: 0.72; specificity: 0.88). In addition, it was predicted as “deleterious mutation” by MutPred (score: 0.927, with a confident hypothesis of a loss of a MoRF binding; p = .0188) and as “disease causing” by the Mutation Taster (score: 0.999, amino acid sequence changed, protein features (might be) affected, splice site changes). Also, it was submitted as likely pathogenic at ClinVar (https://preview.ncbi.nlm.nih.gov/clinvar/variation/265126/). Only one novel missense mutation was detected in EXT2 gene. There is no amino acid classification change in the p.Met370Ile mutation detected where both Methionine (Met) and Isoleucine (Ile) are nonpolar amino acids. As expected, bioinformatics predictions suggested a benign role for this variant in all three tools tested (Polyphen2, MutPred, and Mutation Taster) as well was described as likely benign according to ClinVar criteria (https://www.ncbi.nlm.nih.gov/clinvar/variation/134217/).
Only one novel intronic variant in EXT1 gene was detected. In silico analyses of c.1884‐2A>G predicted the use of cryptic acceptor splice sites by Human Splice Finder (score: −83.68%) located between 11 nucleotides upstream and three nucleotides downstream from the wild‐type sequence with an alteration of the WT acceptor site, most likely affecting splicing. On the other hand, two novel intronic variants were found in EXT2 gene. The Human Splice Finder analysis of c.626+2delT variant predicted the use of cryptic donor splice site (score: −157.2%) located 88 nucleotides downstream from the wild‐type sequence with an alteration of the WT donor site, possibly affecting splicing. The other intronic variant in EXT2 gene, c.744‐13del14insATTC, was predicted by Human Splice Finder as an alteration of the WT acceptor site, most probably affecting splicing located (score −50.05%) 12 nucleotides upstream from the wild‐type sequence.
The position of the pathogenic variants in EXT1 gene showed that the same number of EXT1 pathogenic variants were located in regions that encode the exostosin domain (45.2%—from amino acid 111 to 396) and the glycosyltransferase domain (45.2%—from amino acid 480 to 729) of the EXT1 protein (Figure 1). In contrast, 87.5% (14/16) of EXT2 variants were located in regions that encode the exostosin domain (from exon 2 to 7) and only 12.5% (2/16) were located in regions that encode the middle segment of the EXT2 gene, both specifically in the exon 8, considering that no variants were located in glycosyltransferase domain (from exon 10 to 14, Figure 1). Deletions in EXT1 or EXT2 genes were not considered in these frequencies.
Figure 1.
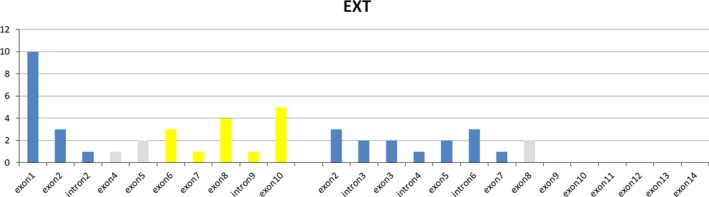
The position and frequency of the pathogenic variants in EXT1 and EXT2 genes
Large deletions were detected by the MLPA technique in 3 of 114 (2.6%) families. In the EXT1 gene, we found one patient with a deletion of exon 1 and the other with a complete deletion of the gene. The deletion of 3 exons in the EXT2 gene (from exon 6 to exon 8) was detected in one family. No mutation was found in EXT1 or neither EXT2 gene in 19 (17%) families.
Seven individuals unrelated of 114 families (6%) presented malignant transformation to chondrosarcoma (Table 2). The degree of anaplasia was not determined in F24 family, who presented p.Leu490Argfs*9 variant in EXT1 gene. Chondrosarcoma grade I was diagnosed in four families. The P03 and F14 families bore novel and no recurrent frameshift mutations (p.Phe58Leufs*7 and p.Thr681Aspfs*, respectively) in EXT1 gene; P74 and P75 families had no EXT1/EXT2 variants identified. Malignant transformation to chondrosarcoma grade II was detected in F19 and F6 families, who presented pathogenic variants in EXT2 gene (p.Ala409Profs*26 and p.Trp394*, respectively).
Table 2.
Characteristics of patients with chondrosarcoma
| Gender | Family number | Gene | Age at chondrosarcoma diagnosis | Location | Mutation type | Exon | DNA | Deduced protein change | Publication | Histopathologic grading of anaplasia |
|---|---|---|---|---|---|---|---|---|---|---|
| Female | F14 | EXT1 | 29 | Humerus | Frameshift | 10 | c.2040_2041delGA | p.T681DfsX | This study | Grade I chondrosarcoma |
| Male | F19 | EXT2 | 28 | Iliac | Frameshift | 8 | c.1225delG | p.A409PfsX26 | This study | Grade II chondrosarcoma |
| Female | — | Non‐ EXT1/Non‐ EXT2 | 22 | Femur | Grade I chondrosarcoma | |||||
| Female | — | EXT1 | 54 | Humerus | Frameshift | 1 | c.173_174insG, c.186_192delCGCCC | p.F58LfsX72 | This study | Grade I chondrosarcoma |
| Female | F6 | EXT2 | 30 | Iliac | Nonsense | 8 | c.1182G>A | p.W384X | Delgado et al. (2014) | Grade II chondrosarcoma |
| Male | F24 | EXT1 | 40 | Tibia | Frameshift | 6 | c.1469delT | p.L490RfsX9 | Signori et al. (2007) | |
| Female | — | Non‐ EXT1/Non‐ EXT2 | 21 | Iliac | Grade I chondrosarcoma |
4. DISCUSSION
This study is the first Brazilian research in MO, with a broad spectrum of variants detected in EXT1 and EXT2 genes affected. Variants were found in 83% of MO families. EXT2 variants were detected in 46% (53/114) and EXT1 variants were detected in 37% (42/114) (Figure 2). These results differ from those observed in other populations, in which EXT1 gene was responsible for the vast majority of MO cases (Delgado et al., 2014; Jennes et al., 2008; Pedrini et al., 2011; Sarrión et al., 2013). So far, the evaluation of EXT1 and EXT2 genes in Canadian and Chinese populations with MO were the only reports that identified a higher frequency of mutations in EXT2 than EXT1 gene (Alvarez et al., 2006; Guo et al., 2014; Jennes et al., 2009; Xu et al., 1999). Canadian group considered that one of the main limitations of their study was the small sample size (Alvarez et al., 2006). Although Xu et al. (1999) and Guo et al. (2014) attributed the differences in involvement of EXT1 and EXT2 genes in Chinese population to the genomic variance, the scope of gene segments tested, the inconsistency in variant screening methods, and the presence of hotspots in EXT1 was observed only in Caucasian and Japanese patients.
Figure 2.
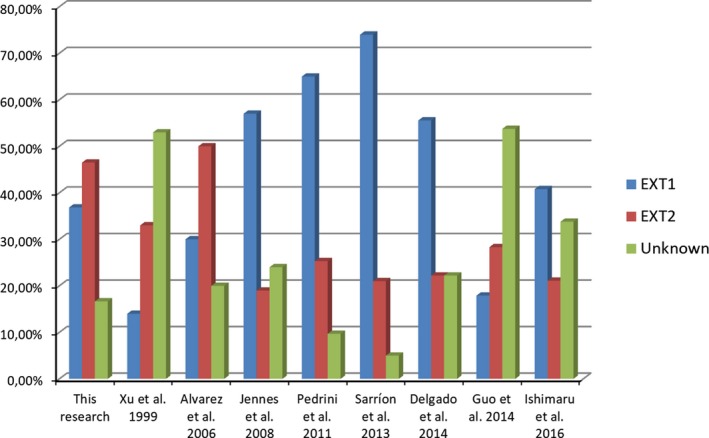
The frequency of pathogenic variants in EXT1 and EXT2 genes, and unknown cause observed in this research and other populations
In contrast to a Latin America study recently published, which evaluated Chileans and Argentines whose origins are basically European and indigenous (Delgado et al., 2014), we observed recurrent pathogenic variants in patients of Brazil. The two most frequent pathogenic variants in EXT2 gene have not been previously reported. Both of them may be considered new hotspots in EXT2, which may contribute for the fact that EXT2 mutations are more frequent in Brazilian patients. The frameshift mutation p.Ala409Profs*26 was detected in 19 families and the nonsense mutation p.Ser290Ter was detected in 11 families with EXT2 mutated allele.
On the other hand, the two most frequent pathogenic variants detected in EXT1 gene (p.Leu490Argfs*9 and p.Arg340Leu) have previously been reported in European populations and both of them were already mentioned to be located in mutation hotspots (Ciavarella et al., 2013; Francannet et al., 2001; Jennes et al., 2008; Philippe et al., 1997). The Brazilian population is a mixed population that consists of a high influence of Europeans in its colonization, which could be responsible for the presence of recurrent alterations in hotspots found in our study. Unfortunately, there are no similar researches about Amerindians or African population to compare with our results.
As previously reported in distinct populations (Ciavarella et al., 2013; Delgado et al.,2014; Heinritz et al., 2009; Ishimaru et al., 2016; Pedrini et al., 2005; Sarrión et al., 2013) a high number of novel pathogenic variants was also identified in Brazilian patients. Of all 50 different variants found in this study, 31 (62%) were novel with no description in the Multiple Osteochondroma Mutation Database (MOdb) (http://medgen.ua.ac.be/LOVDDv.2.0/home.php). Twenty out of 33 (60,6%) EXT1 variants were novel as were 11 of 17 (64.7%) of EXT2 variants (Table 1). This fact confirms the strong allelic heterogeneity of EXT1/2 genes in MO patients. Moreover, inactivating mutations (frameshift, splice, and nonsense mutations and large deletions), which results in the formation of nonfunctional EXT1 and EXT2 proteins and represent the most common MO‐causing variants, corresponded to 88% of variants found in this study concurring with other reports previously published (Ciavarella et al., 2013; Delgado et al., 2014; Guo et al., 2014; Jennes et al., 2008, 2009; Pedrini et al., 2005, 2011; Sarrión et al., 2013; Wuyts & Van Hul, 2000) (Table 1). Two out of five exonic missense mutations detected in EXT1 gene (p.Leu264Arg and p.Gly268Glu) and the only one exonic missense mutation detected in EXT2 gene (p.Met370Ile) were novel. Both variants in EXT1 are predicted to be deleterious, while the variant found in EXT2 was considered benign according to three different in silico analysis tools. Splice site variants were detected in two patients in EXT1 gene. One of them, c.1884‐2A>G, was a novel variant observed in one unrelated patient. Use of cryptic acceptor splice sites was predicted by in silico analysis. On the other hand, seven splice site variants were identified in EXT2 gene. Two of them were novels: c.626+2delT and c.744‐13del14insATTC, both in one unrelated patients (Table 1). In silico analysis predicted the use of alternative cryptic donor splice site to both variants, c.626+2delT and c.744‐13del14insATTC.
All missense mutations were present in exon 1 and 2 of EXT1 gene. Both of them are located in exons that encode the exostosin domain of EXT1 protein (from exon 1 to exon 3) suggesting that the amino acids involved are crucial for proper functioning of it.
Analysis of the distribution of variants over the EXT1 gene reveals that alterations in Brazilian patients are equally distributed over the exons 1 to 3 which encode aminoacids of exostosin domain (45.2%) and over the exons 6 to 10 which encode aminoacids of glycosyltransferase domain (45.2%) of EXT1 protein. The remaining variants were found in exons 4 and 5 (9.6%) which encode aminoacids of the middle segment of EXT1 protein. This pattern of distribution was not observed in studies previously reported where most of variants are located in exons 1 to 3 (Ciavarella et al., 2013; Delgado et al., 2014; Francannet et al., 2001; Signori et al., 2007; Wuyts & Van Hul, 2000; Xu et al., 1999). These data suggest that variants that modify the glycosyltransferase domain may be as relevant as variants that modify exostosin one despite the COOH‐terminal catalytic domains. In contrast, the vast majority of EXT2 variants (87.5%) found in this report are located in regions that are involved in encoding aminoacids of exostosin domain (exons 2 to part of 7), and no variation was found in regions that are involved in encoding aminoacids of glycosyltransferase domain (part of exon 9–14). Similar observations have been reported in different European and Asian populations analyzed by several authors (Ciavarella et al., 2013; Francannet et al., 2001; Pedrini et al., 2005; Sarrión et al., 2013; Signori et al., 2007; Wuyts & Van Hul, 2000; Xu et al.,1999) but not in a Latin American study in which EXT2 pathogenic variants were more frequent in the last exons (Delgado et al., 2014). A genotype–phenotype evaluation could provide a better understanding of the relevance and the role of exostosin and glycosyltransferase domains in MO patients.
MLPA analysis was able to detect three (2.6%) families with large deletions, two in EXT1 gene and one in EXT2 gene. Exon 1 has been previously reported as a hotspot in MO patients where a exon1 deletion has been found (Delgado et al., 2014; Signori et al., 2007; Vink et al., 2005) and a deletion of the entire EXT1 gene was previously reported as well (Hall, Cole, Haynes, & Hecht, 2002; Jennes‐not published). However, further analysis needs to be carried out to determine whether the breakpoints in all cases are the same. A novel deletion of three exons in EXT2 gene (from exon 6 to exon 8) was found in one family. These findings show a slightly lower frequency of large deletions found in other studies where 4% to 9% of deletions in one or more exons are observed (Delgado et al., 2014; Jennes et al., 2008; Pedrini et al., 2011; Signori et al., 2007; White et al., 2004).
No variant was found in EXT1 or EXT2 genes in 17% of families after a multistep variant screening was performed. The results are consistent with other studies reporting that 10%–30% of cases present no alterations in both EXT1 and EXT2 (Boveé, 2008; Ciavarella et al., 2013; Gigante et al., 2001; Jennes et al., 2008; Lonie et al., 2006; Pedrini et al., 2005, 2011). There are some plausible reasons for the lack of EXT gene mutations. First, genetic heterogeneity must be considered, and the involvement of other still unidentified genes cannot be ruled out (Ciavarella et al., 2013; Ishimaru et al., 2016; Jennes et al., 2012; Sarrión et al., 2013; Wuyts & Van Hul, 2000). Second, we have not evaluated the presence of variants in the promoter region, the 5′untranslated region (5′UTR) nor the 3′UTR of both genes. Previous studies have shown that variant analysis of the open reading frame of EXT1 did not detect pathogenic variants in several families known to be linked to the EXT1 locus (Hecht et al., 1997; Wells et al., 1997) which suggests the existence of some pathogenic variants outside the EXT1 coding region. Moreover, the description of a regulatory role of SNP (rs34016643) within transcription factor binding site of EXT1 gene that results in a relevant increase in EXT1 promoter activity was reported (Jennes et al., 2012). Furthermore, the presence of EXT mosaic pathogenic variants in a fraction of the patients must be considered as a putative cause of undetected EXT mutations in MO cases (Sarrión et al., 2013; Szuhai et al., 2011). Finally, intronic deletion and duplication in EXT1 detected only by array‐CGH was revealed as a new causative mechanism for MO (Waaijer et al., 2013). According to this, screening with array‐CGH and next‐generation sequencing to study additional causative mechanism should be performed.
The most serious complication in MO is malignant transformation to a chondrosarcoma, occurring in 1%–5% of cases. It is interesting to note that, in our cohort, malignant progression occurred in 6% of families affecting both sporadic cases and family nuclei which is not in agreement with other authors (Pedrini et al., 2011; Porter et al., 2004; Schmale et al., 1994). Irrespective of EXT1 pathogenic variant or no EXT1/EXT2 pathogenic variant identified, the histopathology classification of anaplasia of their tumors was grade I. The two patients who presented loss‐of‐function mutations in exon 8 of EXT2 gene underwent malignant degeneration to chondrosarcoma grade II, which may suggest that mutations in EXT2 gene may be responsible for a propensity of MO patients to develop tumors that are more aggressive. Our data did not agree with other studies in which an increased rate of malignant transformation in patients with EXT1 pathogenic variants was observed (Alvarez et al., 2006; Porter et al., 2004).
In conclusion, the genotype characterization of a wide number of MO patients suggests that Brazilian population has a unique genetic profile that differs from the European and Latin American populations. The identification of a high number of novel pathogenic variants, presence of a higher frequency of EXT2 than EXT1 pathogenic variants, detection of two novel hotspots in EXT2 gene, and the different pattern of distribution of pathogenic variants over the EXT1 and EXT2 protein indicate the uniqueness of our population.
Further research to detect mosaic alterations in mutation‐negative patients, to study the genotype–phenotype correlation of these patients and to perform the genotyping of chondrosarcoma will contribute to elucidate this complex disease.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest with any financial organization or corporation or individual that can inappropriately influence this work.
Supporting information
Santos SCL, Rizzo IMPO, Takata RI, Speck‐Martins CE, Brum JM, Sollaci C. Analysis of mutations in EXT1 and EXT2 in Brazilian patients with multiple osteochondromas. Mol Genet Genomic Med. 2018;6:382–392. https://doi.org/10.1002/mgg3.382
REFERENCES
- Ahn, J. , Lüdecke, H. J. , Lindow, S. , Horton, W. A. , Lee, B. , Wagner, M. J. , … Wells, D. E. (1995). Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1). Nature Genetics, 11, 137–143. https://doi.org/10.1038/ng1095-137 [DOI] [PubMed] [Google Scholar]
- Alvarez, C. , Tredwell, S. , De Vera, M. , & Hayden, M. (2006). The genotype‐phenotype correlation of hereditary multiple exostoses. Clinical Genetics, 70, 122–130. https://doi.org/10.1111/j.1399-0004.2006.00653.x [DOI] [PubMed] [Google Scholar]
- Alves‐Silva, J. , da Silva Santos, M. , Guimarães, P. E. , Ferreira, A. C. , Bandelt, H. J. , Pena, S. D. , & Prado, V. F. (2000). The ancestry of Brazilian mtDNA lineages. The American Journal of Human Genetics, 67(2), 444–461. https://doi.org/10.1086/303004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveé, J. V. M. G. (2008). Multiple osteochondromas. Orphanet Journal of Rare Diseases, 3, 3 https://doi.org/10.1186/1750-1172-3-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciavarella, M. , Coco, M. , Baorda, F. , Stanziale, P. , Chetta, M. , Bisceglia, L. , … Caldarini, C. (2013). 20 novel point mutations and one large deletion in EXT1 and EXT2 genes: Report of diagnostic screening in a large Italian cohort of patients affected by hereditary multiple exostosis. Gene, 515, 339–348. https://doi.org/10.1016/j.gene.2012.11.055 [DOI] [PubMed] [Google Scholar]
- Delgado, M. A. , Martinez‐Domenech, G. , Sarrión, P. , Urreizti, R. , Zecchini, L. , Robledo, H. H. , … Asteggiano, C. G. (2014). A broad spectrum of genomic changes in latinamerican patients with EXT1/EXT2‐CDG. Scientific Reports, 4, 6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francannet, C. , Cohen‐Tanugi, A. , Le Merrer, M. , Munnich, A. , Bonaventure, J. , & Legeai‐Mallet, L. (2001). Genotype‐phenotype correlation in hereditary multiple exostoses. Journal of Medical Genetics, 38, 430–434. https://doi.org/10.1136/jmg.38.7.430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigante, M. , Matera, M. G. , Seripa, D. , Izzo, A. M. , Venanzi, R. , Giannotti, A. , … Monteleone, M. (2001). Ext‐mutation analysis in Italian sporadic and hereditary osteochondromas. International Journal of Cancer, 95, 378–383. https://doi.org/10.1002/(ISSN)1097-0215 [DOI] [PubMed] [Google Scholar]
- Guo, X. L. , Deng, Y. , & Liu, H. G. (2014). Clinical characteristics of hereditary multiple exostoses: A retrospective study of mainland Chinese cases in recent 23 years. Journal of Huazhong University of Science and Technology. Medical Sciences, 34, 42–50. https://doi.org/10.1007/s11596-014-1230-3 [DOI] [PubMed] [Google Scholar]
- Hall, C. R. , Cole, W. G. , Haynes, R. , & Hecht, J. T. (2002). Reevaluation of a genetic model for the development of exostosis in hereditary multiple exostosis. The American Journal of Medical Genetics, 112, 1–5. https://doi.org/10.1002/(ISSN)1096-8628 [DOI] [PubMed] [Google Scholar]
- Hameetman, L. , David, G. , Yavas, A. , White, S. J. , Taminiau, A. H. , Cleton‐Jansen, A. M. , … Bovée, J. V. (2007). Decreased EXT expression and intracellular accumulation of heparan sulphate proteoglycan in osteochondromas and peripheral chondrosarcomas. Journal of Pathology, 211, 399–409. https://doi.org/10.1002/path.2127 [DOI] [PubMed] [Google Scholar]
- Hecht, J. T. , Hogue, D. , Wang, Y. , Blanton, S. H. , Wagner, M. , Strong, L. C. , … Wells, D. (1997). Hereditary multiple exostoses (EXT): Mutational studies of familial EXT1 cases and EXT‐associated malignancies. The American Journal of Human Genetics, 60, 80–86. [PMC free article] [PubMed] [Google Scholar]
- Heinritz, W. , Hüffmeier, U. , Strenge, S. , Miterski, B. , Zweier, C. , Leinung, S. , … Froster, U. G. (2009). New mutations of EXT1 and EXT2 genes in German patients with Multiple Osteochondromas. Annals of Human Genetics, 73, 283–291. https://doi.org/10.1111/j.1469-1809.2009.00508.x [DOI] [PubMed] [Google Scholar]
- Hennekam, R. C. (1991). Hereditary multiple exostoses. Journal of Medical Genetics, 28, 262–266. https://doi.org/10.1136/jmg.28.4.262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimaru, D. , Gotoh, M. , Takayama, S. , Kosaki, R. , Matsumoto, Y. , Narimatsu, H. , … Matsumoto, K. (2016). Large‐scale mutational analysis in the EXT1 and EXT2 genes for Japanese patients with multiple osteochondromas. BMC Genetics, 17, 52 https://doi.org/10.1186/s12863-016-0359-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennes, I. Retrieved from http://medgen.ua.ac.be/LOVDv.2.0/home.php
- Jennes, I. , Entius, M. M. , Van Hul, E. , Parra, A. , Sangiorgi, L. , & Wuyts, W. (2008). Mutation screening of EXT1 and EXT2 by denaturing high‐performance liquid chromatography, direct sequencing analysis, fluorescence in situ hybridization, and a new multiplex ligation‐dependent probe amplification probe set in patients with multiple osteochondromas. Journal of Molecular Diagnostics, 10, 85–92. https://doi.org/10.2353/jmoldx.2008.070086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennes, I. , Pedrini, E. , Zuntini, M. , Mordenti, M. , Balkassmi, S. , Asteggiano, C. G. , … Wuyts, W. (2009). Multiple Osteochondromas: Mutation update and description of the multiple osteochondromas mut Database (MOdb). Human Mutation, 12, 1620–1627. https://doi.org/10.1002/humu.21123 [DOI] [PubMed] [Google Scholar]
- Jennes, I. , Zuntini, M. , Mees, K. , Palagani, A. , Pedrini, E. , De Cock, G. , … Wuyts, W. (2012). Identification and functional characterization of the human EXT1 promoter region. Gene, 492, 148–159. https://doi.org/10.1016/j.gene.2011.10.034 [DOI] [PubMed] [Google Scholar]
- Jones, K. B. , Pacifici, M. , & Hilton, M. J. (2014). Multiple hereditary exostoses (MHE): Elucidating the pathogenesis of a rare skeletal disorder through interdisciplinary research. Connective Tissue Research, 55, 80–88. https://doi.org/10.3109/03008207.2013.867957 [DOI] [PubMed] [Google Scholar]
- Lonie, L. , Porter, D. E. , Fraser, M. , Cole, T. , Wise, C. , Yates, L. , … Ragoussis, J. (2006). Determination of the mutation spectrum of the EXT1/EXT2 genes in British Caucasian patients with multiple osteochondromas, and exclusion of six candidate genes in EXT negative cases. Human Mutation, 27, 1160 https://doi.org/10.1002/humu.9467 [DOI] [PubMed] [Google Scholar]
- McCormick, C. , Duncan, G. , Goutsos, K. T. , & Tufaro, F. (2000). The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the Golgi apparatus and catalyzes the synthesis of heparan sulfate. Proceedings of the National Academy of Sciences of the United States of America, 97, 668–673. https://doi.org/10.1073/pnas.97.2.668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, S. A. , Dykes, D. D. , & Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research, 16(3), 1215 https://doi.org/10.1093/nar/16.3.1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrini, E. , De Luca, A. , Valente, E. M. , Maini, V. , Capponcelli, S. , Mordenti, M. , … Dallapiccola, B. (2005). Novel EXT1 and EXT2 mutations identified by DHPLC in Italian patients with multiple osteochondromas. Human Mutation, 26, 280 https://doi.org/10.1002/humu.9359 [DOI] [PubMed] [Google Scholar]
- Pedrini, E. , Jennes, I. , Tremosini, M. , Milanesi, A. , Mordenti, M. , Parra, A. , … Pignotti, E. (2011). Genotype‐phenotype correlation study in 529 patients with multiple hereditary exostoses: Identification of “protective” and “risk” factors. Journal of Bone and Joint Surgery. American Volume, 93, 2294–2302. https://doi.org/10.2106/JBJS.J.00949 [DOI] [PubMed] [Google Scholar]
- Philippe, C. , Porter, D. E. , Emerton, M. E. , Wells, D. E. , Simpson, A. H. , & Monaco, A. P. (1997). Mutation screening of the EXT1 and EXT2 genes in patients with hereditary multiple exostoses. The American Journal of Human Genetics, 61, 520–528. https://doi.org/10.1086/515505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter, D. E. , Lonie, L. , Fraser, M. , Dobson‐Stone, C. , Porter, J. R. , Monaco, A. P. , & Simpson, A. H. (2004). Severity of disease and risk of malignant change in hereditary multiple exostoses. A genotype‐phenotype study. Journal of Bone and Joint Surgery. British Volume, 86, 1041–1046. https://doi.org/10.1302/0301-620X.86B7.14815 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … ACMG Laboratory Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. https://doi.org/10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel, A. M. , Costa, J. , & Lindskog, D. M. (2014). Genetic alterations in chondrosarcomas – keys to targeted therapies? Cellular Oncology (Dordrecht), 37, 95–105. https://doi.org/10.1007/s13402-014-0166-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarrión, P. , Sangorrin, A. , Urreizti, R. , Delgado, A. , Artuch, R. , Martorell, L. , … Nevado, J. (2013). Mutations in the EXT1 and EXT2 genes in Spanish patients with multiple osteochondromas. Scientific Reports, 3, 1346 https://doi.org/10.1038/srep01346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmale, G. A. , Conrad, E. U. 3rd , & Raskind, W. H. (1994). The natural history of hereditary multiple exostoses. Journal of Bone and Joint Surgery. American Volume, 76, 986–992. https://doi.org/10.2106/00004623-199407000-00005 [DOI] [PubMed] [Google Scholar]
- Signori, E. , Massi, E. , Matera, M. G. , Poscente, M. , Gravina, C. , Falcone, G. , … Fazio, V. M. (2007). A combined analytical approach reveals novel EXT1/2 gene mutations in a large cohort of Italian multiple osteochondromas patients. Genes Chromosomes Cancer, 46, 470–477. https://doi.org/10.1002/(ISSN)1098-2264 [DOI] [PubMed] [Google Scholar]
- Stickens, D. , Clines, G. , Burbee, D. , Ramos, P. , Thomas, S. , Hogue, D. , … Evans, G. A. (1996). The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nature Genetics, 14, 25–32. https://doi.org/10.1038/ng0996-25 [DOI] [PubMed] [Google Scholar]
- Szuhai, K. , Jennes, I. , de Jong, D. , Bovée, J. V. , Wiweger, M. , Wuyts, W. , & Hogendoorn, P. C. (2011). Tiling resolution array‐CGH shows that somatic mosaic deletion of the EXT gene is causative in EXT gene mutation negative multiple osteochondromas patients. Human Mutation, 32, E2036–E2049. https://doi.org/10.1002/humu.21423 [DOI] [PubMed] [Google Scholar]
- Vink, G. R. , White, S. J. , Gabelic, S. , Hogendoorn, P. C. , Breuning, M. H. , & Bakker, E. (2005). Mutation screening of EXT1 and EXT2 by direct sequence analysis and MLPA in patients with multiple osteochondromas: Splice site mutations and exonic deletions account for more than half of the mutations. European Journal of Human Genetics, 13, 470–474. https://doi.org/10.1038/sj.ejhg.5201343 [DOI] [PubMed] [Google Scholar]
- Waaijer, C. J. , Winter, M. G. , Reijnders, C. M. , de Jong, D. , John Ham, S. , Bovée, J. V. , & Szuhai, K. (2013). Intronic deletion and duplication proximal of the EXT1 gene: A novel causative mechanism for multiple osteochondromas. Genes Chromosomes Cancer, 52(4), 431–436. https://doi.org/10.1002/gcc.22041 [DOI] [PubMed] [Google Scholar]
- Wells, D. E. , Hill, A. , Lin, X. , Ahn, J. , Brown, N. , & Wagner, M. J. (1997). Identification of novel mutations in the human EXT1 tumor suppressor gene. Human Genetics, 99, 612–615. https://doi.org/10.1007/s004390050415 [DOI] [PubMed] [Google Scholar]
- White, S. J. , Vink, G. R. , Kriek, M. , Wuyts, W. , Schouten, J. , Bakker, B. , … den Dunnen, J. T. (2004). Two‐color multiplex ligation‐dependent probe amplification: Detecting genomic rearrangements in hereditary multiple exostoses. Human Mutation, 24, 86–92. https://doi.org/10.1002/(ISSN)1098-1004 [DOI] [PubMed] [Google Scholar]
- Wuyts, W. , & Van Hul, W. (2000). Molecular basis of multiple exostoses: Mutations in the EXT1 and EXT2 genes. Human Mutation, 15, 220–227. https://doi.org/10.1002/(ISSN)1098-1004 [DOI] [PubMed] [Google Scholar]
- Xu, L. , Xia, J. , Jiang, H. , Zhou, J. , Li, H. , Wang, D. , … Deng, H. X. (1999). Mutation analysis of hereditary multiple exostoses in the Chinese. Human Genetics, 105, 45–50. https://doi.org/10.1007/s004399900058 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials