

Abstract
Background
Dentinogenesis imperfecta (DI) is a rare debilitating hereditary disorder affecting dentin formation and causing loss of the overlying enamel. Clinically, DI sufferers have a discolored and weakened dentition with an increased risk of fracture.
The aims of this study were to assess genotype‐phenotype findings in three families with DI–II with special reference to mutations in the DSPP gene and clinical, histological, and imaging manifestations.
Methods
Nine patients participated in the study (two from family A, four from family B, and three from family C). Buccal swab samples were collected from all participants and extracted for genomic DNA. Clinical and radiographic examinations had been performed longitudinally, and the dental status was documented using photographic images. Four extracted and decalcified tooth samples were prepared for histological analysis to assess dysplastic manifestations in the dentin. Optical coherence tomography (OCT) was applied to study the health of enamel tissue from in vivo images and the effect of the mutation on the function and structure of the DSPP gene was analyzed using bioinformatics software programs.
Results
The direct DNA sequence analysis revealed three distinct mutations, one of which was a novel finding. The mutations caused dominant phenotypes presumably by interference with signal peptide processing and protein secretion. The clinical and radiographic disturbances in the permanent dentition indicated interfamilial variability in DI–II manifestations, however, no significant intrafamilial variability was observed.
Conclusion
The different mutations in the DSPP gene were accompanied by distinct phenotypes. Enamel defects suggested deficit in preameloblast function during the early stages of amelogenesis.
Keywords: bioinformatics, dentinogenesis imperfecta, genetics
1. INTRODUCTION
Dentinogenesis imperfecta (DI) is a rare hereditary disorder characterized primarily by defective dentin formation and resulting in early loss of the overlying enamel with high risk of tooth loss. DI has an estimated incidence of 1:8000 (Witkop, 1975) and can be classified into three subtypes: DI–I, DI–II, and DI–III (Shields, Bixler, & el‐Kafrawy, 1973). These subtypes can either appear in association with osteogenesis imperfecta (DI–I) or as an isolated finding associated with mutations in the dentin sialophosphoprotein gene DSPP (DI–II and DI–III) (Hart & Hart, 2009).
The DSPP gene contains five exons and four introns and encodes a precursor protein that is cleaved to form two mature proteins: dentin sialoprotein (DSP) and dentin phosphoprotein (DPP) (Bai et al., 2010). Exons 1–4 encode the DSP, while exon 5 encodes the carboxy terminus of the DSP and the entire DPP (Xiao et al., 2001; Zhang et al., 2007). The DSP and DPP are proteins that constitute major noncollagenous components in the dentin and have important functions in its mineralization. Studies with mutant mice have suggested that DSP regulates the initiation of dentin mineralization, while the DPP is associated with maturation of dentin mineralization (Suzuki et al., 2009).
DI–II (MIM 125490) is an autosomal anomaly of dental development with complete penetrance (Bixler, Conneally, & Christen, 1969; Malmgren, Lindskog, Elgadi, & Norgren, 2004). In the period 2001–2017 more than 40 different mutations in the DSPP gene have been identified (Bloch‐Zupan et al., 2016; Li et al., 2017; Liu et al., 2016). Clinically, DI–II is characterized by discolored deciduous and permanent teeth of which the primary dentition is often the most severely affected (Bixler et al., 1969). Teeth in patients with DI–II exhibit an opalescent hue, ranging from amber brown to grayish‐blue. Radiographically, the teeth have bulbous crowns with narrow roots, and a constriction at the cervix (Hart & Hart, 2007; Leal, Martins, Verli, de Souza, & Ramos‐Jorge, 2010). Pulpal obliteration occurs soon after eruption and sometimes even before eruption (Shields et al., 1973). Histologically, the dentin is characterized by a dysplastic appearance with reduced mineralization, irregular dentin tubules, and occasionally interglobular dentin (Barron, McDonnell, MacKie, & Dixon, 2008; Hart & Hart, 2007). Davis, Fearne, Sabel, and Norén (2015) found that although the pulp in teeth affected with D‐II appears to be completely obliterated, a network of interconnected vessels may remain, and they suggested that the presence of large dentinal tubules and the remnants of blood vessels can provide a pathway for bacteria from the oral cavity. The enamel has been reported to be normal in structure and mineral content (Malmgren, Lundberg, & Lindskog, 1998). However, Lee et al. (2013), Wang et al. (2011) and Bloch‐Zupan et al. (2016) reported the enamel to be hypoplastic. Furthermore, in teeth with DI–II, the supportive dentin layer is dysplastic and has been associated with a brittle enamel that is more prone to fracture (Finn, 1962). This exposes the underlying dentin and leads to severe attrition with risk of pulp exposure (Hart & Hart, 2007).
To the best of our knowledge, no previous studies have investigated the structure of enamel tissue in DI–II patients. In this study, we aimed to examine the dental phenotype, including the microstructure of the enamel and dentin, and discuss genotype‐phenotype findings in three affected families.
2. MATERIALS AND METHODS
2.1. Subjects
During a 50‐year‐period, three Danish families from Zealand with DI–II were known to the Department of Odontology, Pediatric Dentistry and Clinical Genetics, Faculty of Health and Medical Sciences, University of Copenhagen, and the three probands had received extensive dental treatment. The family pedigrees are presented in Figure 1. A total of 9 individuals from these families were included in this study (two from Family A, four from Family B, and three from family C). However, Case B‐I.2 was not available for clinical examination, but participated in the genetic investigation. All patients were of Caucasian descent.
Figure 1.
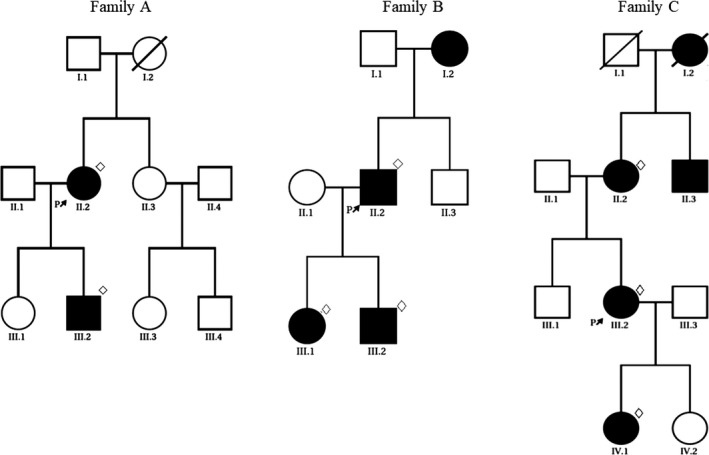
Pedigrees of the families A, B, and C. Unaffected individuals are shown as empty symbols and affected members are shown as solid symbols. Investigated patients are marked with a rhombus and the probands are marked with an arrow
The distribution of the individuals according to gender and age is given in Table 1. Five of the 8 examined subjects had been seen for the first time prior to the age of three years, and another two had been examined prior to the age of 5 years. All subjects had been examined longitudinally.
Table 1.
Age and gender distribution for the 8 examined patients and listing of available material in the individual patient
| Patients photographs | Gender | Age at 1st examination | Age at last examination | Dental x‐rays | Intraoral |
|---|---|---|---|---|---|
| A‐II.2 | F | 4.75 | 53 | + | + |
| A‐III.2 | M | 2.50 | 17 | + | + |
| B‐II.2 | M | 2.75 | 36 | + | + |
| B‐III.1 | F | 2.75 | 16 | + | + |
| B‐III.2 | M | 1.5 | 11 | + | + |
| C‐II.2 | F | 11.5 | 18 | − | + |
| C‐III.2 | F | 3.5 | 16 | + | + |
| C‐IV.1 | F | 1.5 | 3 | + | + |
Informed written consent had been obtained from all participating patients or their legal guardians prior to examinations.
2.2. Methods
2.2.1. Primer design, polymerase chain reaction, and DNA sequencing
Buccal swab samples were collected from all participants of the families. Two swabs were obtained from each patient from the left and right side of the buccal mucosa. The samples were subjected to mutational analysis at Department of Oral and Maxillofacial Diseases, Biomedicum, University of Helsinki, Finland.
As described in a previously published paper (Nieminen et al., 2011) the genomic DNA was isolated from buccal swab cells using Qiagen's kit (QiaAmp DNA mini kit) according to the manufacturer′s instructions (Qiagen, Chatsaworth, Calif., USA). PCR was used to amplify the immediate promoter and exons 1 through 5 with immediately flanking sequences of the DSPP gene (NM_014208.3). The PCR products were purified from primers and nucleotides with ExoSAP method and sequenced with ABI BigDye 3.1 reagent followed by capillary electrophoresis at the Institute for Molecular Medicine Finland Technology Center, Helsinki (for primer sequences and reaction conditions, see Table S1).
2.2.2. Bioinformatics analysis of the mutations
Various software programs were used to investigate how or whether the identified mutations affected the function and structure of the DSPP gene.
PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/) was used to predict the impact of the amino acid substitution on the protein structure and function in the missense mutations. Spliceport (http://spliceport.cbcb.umd.edu/), Netgene2 Server (http://www.cbs.dtu.dk/services/NetGene2/), and BGDP (http://www.fruitfly.org/seq_tools/splice.html) was used to predict effects on pre‐mRNA splicing. SignalP 4.1 (http://www.cbs.dtu.dk/services/SignalP/) was used to predict effects on the presence and location of signal peptide cleavage sites.
2.2.3. Clinical and radiographic investigation
All investigated family members were examined both clinically and radiographically, except for Case B‐I.2 (see above) and Case C‐II.1 for whom only longitudinal intraoral photographs were available for study (Table 2). The clinical and radiographic examinations included assessment of tooth formation, maturation, eruption, discoloration, and attrition. In addition, assessment of periapical radiolucencies and the number of extracted teeth due to periapical conditions was carried out. Deviations in tooth formation were assessed as deviations in tooth number and malformations (e.g., cervical constriction and abnormally large or obliterated pulp chambers and root canals). In addition to the radiographic examination of teeth, a few extracted primary teeth were CBCT scanned and three dimensional (3D) images were produced to visualize the size of the pulp chamber. Dental maturity was scored according to the assessment system of Haavikko, and compared with available normative data (Haavikko, 1970). Discoloration of teeth was graded into: amber‐brown, light‐brown, yellow‐brown, and grayish‐blue. The degree of attrition was assessed from intraoral photographs according to Broca′s tooth wear index (Bardow, Pallesen, & Bakke, 2013).
Table 2.
Clinical and radiographic dental findings in the affected and investigated members of the three families with DI
| Family Patient | A III.2 | A IV.2 | B II.2 | B III.1 | B III.2 | C II.1 | C III.2 | C IV.1a |
|---|---|---|---|---|---|---|---|---|
| Primary teeth | ||||||||
| Enamel defects | + | + | + | + + | + | + | + | |
| Discoloration | AB | AB | AB | AB | AB | AB | AB | AB |
| Attrition (score) | 4 | 4 | 4 | 3 | 4 | 3 | 4 | 4 |
| Cervical constriction | + | + | + | + | + | NA | + | + |
| Obliterated pulp chambers | + | + | + | + | + | NA | + | − |
| Obliterated root canals | + | + | + | + | + | NA | + | − |
| Periapical radiolucencies | + | + | + | + | + | NA | NA | − |
| Extracted teeth due to periapical conditions | 2 | 3 | 2 | 3 | 2 | 2 | NA | 0 |
| Permanent teeth | ||||||||
| Enamel defects | + | + | + | + | + | + | + | + |
| Discoloration | YB | YB | LB | LB | LB | GB | GB | NA |
| Attrition (score)b | 3 | 3 | 3 | 2 | 3 | NA | 2 | NA |
| Cervical constriction | + | + | + | + | + | NA | + | NA |
| Obliterated pulp chambers | + | + | + | + | + | NA | + | NA |
| Obliterated root canals | + | + | + | + | + | NA | + | NA |
| Periapical radiolucencies | − | + | NA | + | − | NA | − | NA |
| Extracted teeth due to periapical conditions | 1 | 1 | 2 | 1 | 0 | NA | NA | NA |
AB, amber brown; GB, grayish‐blue; LB, light‐brown; NA, not available; YB, yellow‐brown.
The patient was only examined at the age of 3 years.
Attrition patterns are based on Broca′s tooth wear index: a clinical measurement for tooth wear (Bardow et al., 2013).
All adult patients had received comprehensive dental treatment, including composite fillings, crowns, dental bridges or dental implants, prior to this study. A few affected subjects were still undergoing dental treatment.
2.2.4. Histological investigation
Four tooth samples from Family B were available for histological investigation. The teeth were sent to Oral Pathology and Medicine, Department of Odontology, Faculty of Health and Medical Sciences, University of Copenhagen, over a 12‐year‐period. The teeth were fixed in 10% formalin and decalcified in ethylenediaminetetraacetic acid (EDTA). The teeth were embedded in paraffin, cut longitudinally, and stained with hematoxylin and eosin (HE). The stained sections were observed under the microscope. The dentin in each tooth was examined for: the appearance and branching of the dentin tubules, dentin mineralization, and the presence of areas void of dentin tubules.
2.2.5. Tomographic imaging investigation
In a single subject, Case B‐III.2, the enamel in the permanent dentition was examined in vivo with optical coherence tomography (OCT) to make a qualitative and quantitative assessment of the tissue. An optical hand‐piece was placed on the tooth and its corresponding gingiva, and the tooth parts without dental restorations were scanned. Application and system setup have been described in a previously published paper (Hsieh et al., 2013).
3. RESULTS
3.1. Mutational analysis
Direct DNA sequence analysis revealed that all affected and investigated members of the three families had a heterozygous mutation in the DSPP gene. Mutation analysis of Family A revealed a missense mutation involving a C→T transition at nucleotide 49 in exon 2 (c.49C>T; Figure 2a). The transition causes a single amino acid substitution of a proline residue with a serine residue (p.Pro17Ser). A novel missense mutation was identified in Family B involving a G→T transition at nucleotide 135 in exon 3 (c.135G>T; Figure 2b), which results in an amino acid substitution of a glutamine residue by a histidine residue (p.Gln45His). Finally, mutation analysis of Family C revealed a splice site mutation involving an A→G transition at c.52‐2 in an acceptor splice site of intron 2 (c.52‐2 A>G; Figure 2c).
Figure 2.
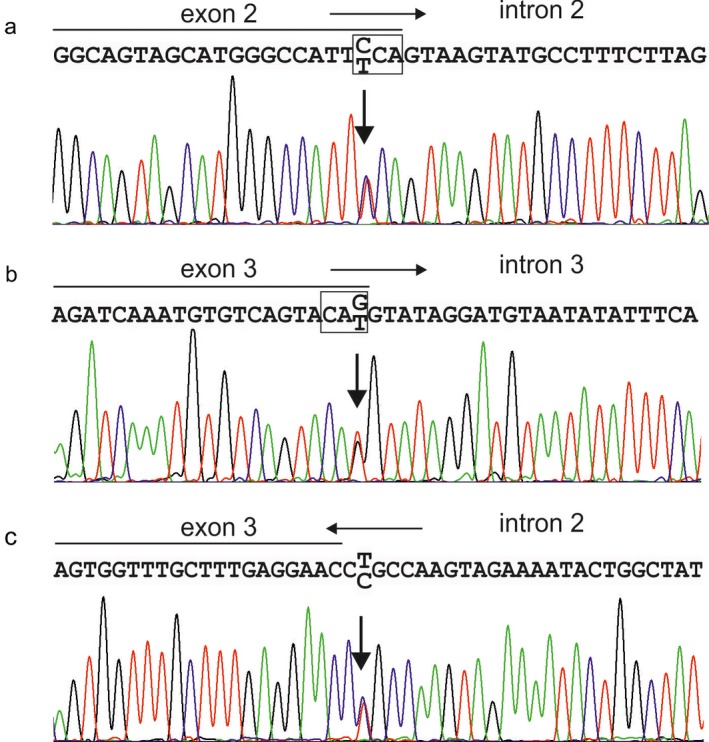
Mutational analysis. (a) A missense mutation involving a C→T transition at nucleotide 49 (p.Pro17Ser) was identified in Family A. (b) A novel missense mutation revealed a G→T transition at nucleotide 135 (p.Gln45His) in Family B. (c) A splice site mutation involving a A→G transition at c.52‐2 was identified in an acceptor splice site of intron 2 (c.52‐2 A>G) in Family C. Horizontal arrows indicate the direction of transcription. Boxes indicate the affected codons
3.2. Bioinformatics findings
p.Pro17Ser and p.Gln45His impact on the protein structure and function were predicted as Probably Damaging (score: 0.998) by PolyPhen‐2 (see Table S2). For the normal sequence, the results of SignalP 4.1 suggested a most likely cleavage site was between position 15–16 and a max cleavage site probability of 0.724 (Table S2). However, the max cleavage site probability decreased to .600 at the same position for the substitution of p.Pro17Ser. Assuming excluding the whole exon 3, the same probability decreased to 0.622. Splicing prediction tools did not indicate a significant effect on the exon 2 donor site (junction of exon 2 and intron 2) by c.49C>T. However, they suggested loss of the exon 3 acceptor (junction of intron 2 and exon 3) and exon 3 donor (junction of exon 3 and intron 3) sites by the mutations c.52‐2A>G and c.135G>T, respectively (see Table S3), both of which have been associated with omission of exon 3 from the mature mRNA (Lee et al. 2011).
3.3. Clinical and radiographic findings
The clinical and radiographic dental findings in the 8 examined subjects are summarized in Table 2, and intraoral photographs of the representative proband from each family are shown in Figure 3.
Figure 3.
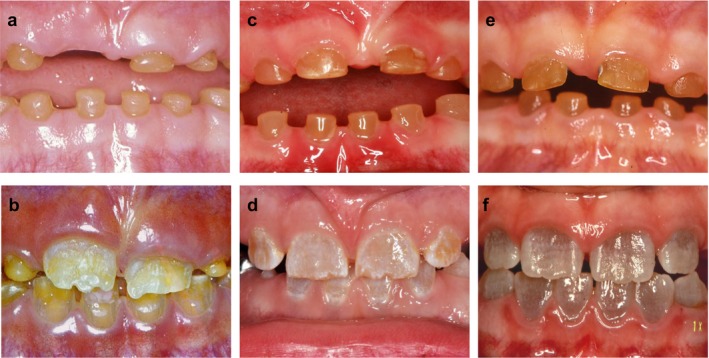
Clinical photographs of the primary and permanent teeth of the probands. Left row: Intraoral photographs of Case A‐II.2 (A: 6 years; B: 7½ years). Middle row: Intra oral photographs of Case B‐III.2 (C: 3½ years; D: 7½ years). Right row: Intraoral photographs of Case C‐III.2 (E: 3½ years; F: 9 years). Note the similar amber brown color of the primary teeth in the all cases (a–c), whereas there is a marked difference in color of the permanent teeth; in Case A the discolored teeth are yellow brown (b) Case B the discoloration is light‐brown (d), and in Case C it is grayish‐blue (f)
3.3.1. Primary dentition
The clinical and radiographic findings in the primary dentition showed limited variability both within and between the three families. In general, tooth morphology was characterized by cervical constriction. The pulp chambers and root canals were initially abnormally large, during tooth eruption, (see Figure 4a), but obliterated before the age of 5 years. The timing of dental maturity and eruption was unremarkable. The tooth crowns were, in general, amber brown and showed excessive attrition. In several cases attrition score 4 was seen already at 2.5–3 years of age (see Figure 4b). Periapical radiolucencies were observed in nearly all patients, and most patients had 2–3 teeth extracted due to periapical conditions.
Figure 4.
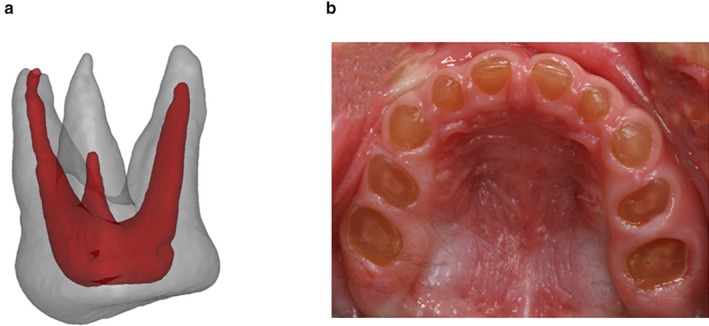
(a) 3D picture of an extracted maxillary first molar in the primary dentition of Case B‐III.2 at the age of 2 years. The size of the enlarged pulp chamber is illustrated. (b) Intraoral photograph of Case C‐IV.1 at the age of 2.5 years exemplifying the excessive attrition with pulp exposure in DI–II patients
3.3.2. Permanent dentition
Tooth number was normal in all cases but one Case B‐III.2 had agenesis of the left mandibular second premolar. In general, the teeth were characterized by cervical constriction and obliteration of pulp chambers and root canals. Similar to the findings in the primary dentition, the pulp chambers and root canals were initially abnormally large, but soon after emergence the size of the pulp chambers and root canals decreased rapidly and soon obliterated completely. The timing of dental maturity and eruption was unremarkable. A qualitative clinical and radiographic assessment of the enamel gave the impression that was thin and hypoplastic in most cases and in all families. In addition, the teeth were discolored in all patients. The intrafamilial variability in tooth color was small. However, there was a marked interfamilial variability in tooth color: in Family A the teeth were yellow‐brown, in Family B the teeth were light‐brown, and in Family C they were grayish‐blue (see Figure 3). All permanent dentitions showed excessive attrition, but not nearly to the degree observed in the primary dentition. Periapical radiolucencies were observed in a few cases and a few teeth had been extracted due to periapical conditions, but not to the extent that was observed in the primary dentition.
3.4. Histological findings
The histological analysis of the teeth in Case B‐III.I revealed dysplastic changes in the dentin in all the examined teeth (Figure 4). The dentino‐enamel junction was characterized by lack of scalloping, although the underlying mantle dentin appeared normal in structure. Progressing inward to the circumpulpal dentin the pattern was, however, very atypical. The dysplastic appearance was characterized by abnormal granular dentin matrix due to interglobular mineralization and the irregular dentin tubules were reduced in number, arranged haphazardly, and of larger size.
3.5. Tomographic imaging findings
In vivo OCT images of the teeth in Case B‐III.2 revealed delineated areas of hypomineralization adjacent to the dentino‐enamel junction (Figure 6b).
Figure 6.
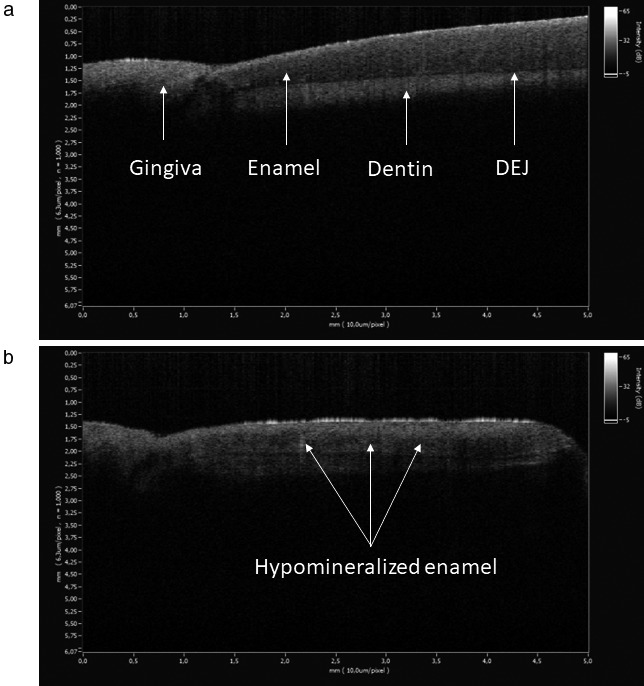
Imaging results of 2‐ from the author K.T and from Case B‐III.2. (a) The healthy enamel had a normal homogeneous appearance. (b) The dysplastic enamel was characterized by dark horizontally arranged areas of hypomineralization adjacent to the dentino‐enamel junction
4. DISCUSSION
This study identified three different mutations in the DSPP gene in the three DI–II affected families, one of which was novel. All mutations were localized to regions in which all previous DI–II mutations in the DSP coding part of the gene have been found. The different mutations in the DSPP gene possibly provide a molecular explanation to the distinct phenotypic features in the investigated families.
It is assumed that all missense, nonsense, and splice‐site DSPP mutations associated with exon 2 and 3 result in dominant phenotypes due to biochemical events that can interfere with signal peptide cleavage and effects on protein secretion (McKnight, Suzanne Hart, et al., 2008). The identified missense mutation (p.Pro17Ser) in Family A, was earlier reported in three affected DI–II families (Hart & Hart, 2007; McKnight, Simmer, Hart, Hart, & Fisher, 2008; Zhang et al., 2007) with similar phenotypic features as regards to enamel loss, attrition, discoloration and periapical radiolucency. The Pro17 residue was also mutated to a threonine (Pro17Thr) (Xiao et al., 2001) and leucine (Pro17Leu) (Li et al., 2012) residue in two different DI–II families. Pro17 is evolutionarily conserved in mammalian species, suggesting that it is critical for the function of the DSPP gene and thus a mutational hotspot (McKnight, Suzanne Hart, et al., 2008; Zhang et al., 2007). The exact molecular mechanism which caused DI–II is unclear. However, previous studies propose possible explanations (Rajpar et al., 2002; Xiao et al., 2001; Zhang et al., 2007). Pro17 is the second amino acid residue in the mature DSP protein. It falls within the conserved isoleucine‐proline‐valine (IPV) domain assumed necessary for efficient processing of the DSPP′s signal peptide (McKnight, Suzanne Hart, et al., 2008). The conservation of the chemical properties is therefore crucial for maintaining the biological functions of the protein (Li et al., 2012; Zhang et al., 2007). The hydrophobic proline is replaced with a hydrophilic serine altering the hydrophobic region and the identified mutation might therefore, interfere with signal peptide cleavage and lead to reduced translocation of DSP and DPP into the endoplasmic reticulum (ER) during translation (McKnight, Suzanne Hart, et al., 2008; Rajpar et al., 2002). This is in accordance with the results obtained from PolyPhen‐2, suggesting a Probably Damaging and SignalP suggesting defective signal peptide cleavage site recognition. The findings are supported by functional studies (Hart & Hart, 2007; Lee et al., 2013) indicating a retention of the Pro17 mutated protein in the ER and proposing a dominant negative effect due to cellular stress and reduction in protein export.
The identified novel missense mutation (p.Gln45His) in Family B affects the amino acid with a previously described nonsense mutation (Gln45stop) in a DI–II Chinese family (Zhang et al., 2001). The affected family had similar clinical manifestations of the disease, with special reference to attrition and discoloration, as observed in Family B in this study. The identified missense mutation is reported as Probably Damaging and according to Spliceport, Netgene2 Server, and BDGP the mutation may affect pre‐mRNA splicing in the donor splice site of exon 3. It has been shown that loss of this site causes a partial or complete skipping of exon 3 (Lee, Lee, Jung, Lee, & Kim, 2011) which would change the IPV motif to IPD. According to SignalP such change has a slight effect on signal peptide cleavage site recognition. Experimental evidence suggests that loss of exon 3 donor site also causes ER retention (Hart & Hart, 2007).
The identified splice site mutation in Family C (c.52‐2A>G) was previously reported in a Chinese DI–III patient (Li et al., 2017) with similar clinical features as regards discoloration, attrition, and enlarged pulps in the primary dentition, although data from the permanent dentition is not available due to the young age of the patient. The overlapping clinical findings are consistent with the DI–II and DI–III representing different phenotypic expression of the same disease, and not separate entities (Lee et al., 2009). Spliceport predicted that the mutation may affect recognition of exon 2 acceptor splice site shown to cause skipping of exon 3 and retention of the protein in the ER (Hart & Hart, 2007; Lee et al., 2011).
The consequence of mutations in the DSPP gene is presumably defective formation of enamel and dentin, since DSPP is expressed in odontoblasts and transiently in preameloblasts (Bègue‐Kirn, Bartlett, & Butler, 1998). Enamel and dentin are formed as a result of various interactions between the ectoderm and ectomesenchyme, partly consisting of enamel formation on the dentin matrix (Thesleff, 2003). The enamel is in general of normal thickness in DI–II patients, but it fractures easily, and splits from the dentino‐enamel junction (Finn, 1962) suggesting a poor attachment of the enamel to the dentin either due to lack of scalloping at the dentino‐enamel junction, a poorly mineralized dentin, or a defect in the enamel (Finn, 1962; Lee et al., 2011). This study identified a dysplastic circumpulpal dentin, lack of scalloping of the dentino‐enamel junction (Figure 5), and a hypomineralized enamel of reduced strength (Figure 6b) proposing a deficiency of the epithelial structures as well in DI–II patients. Although wild‐type Dspp‐knockout mice had apparently normal enamel (Sreenath et al., 2003), it is possible that a severe form of mutant DSPP expressed transiently may disturb ameloblast differentiation and influence enamel formation (Lee, Lee, Hwang, et al., 2011).
Figure 5.
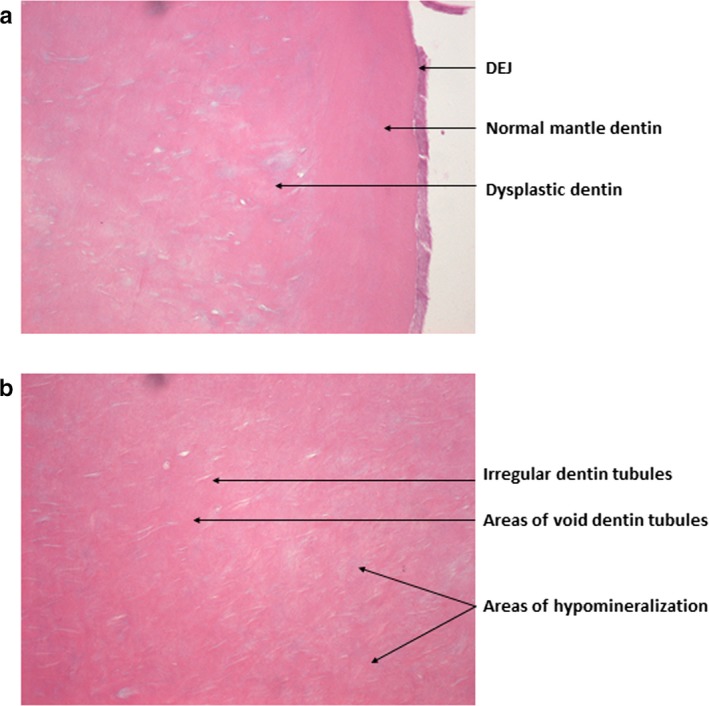
The histological analysis of the teeth in Case B‐III.1 revealed the presence of (a) A dentino‐enamel junction with an even appearance suggesting lack of scalloping, a normal mantle dentin, and an underlying layer of dysplastic dentin with incomplete mineralization, recognized as pale or blue staining areas. (b) The dentin tubules were irregular and enlarged with frequent branching in the circumpulpal dentin in the crown
Several similar phenotypic features were shared in the affected individuals, although interfamilial variability was observed in the permanent dentition (see Figure 3) as regards to discoloration. Clinically, the primary teeth were, in general, more severely affected than the permanent teeth. A possible explanation is that in primary teeth, the enamel is thinner and can expose the discolored dentin very early, leading to extreme tooth wear (Malmgren et al., 2004). Obliteration of pulps and root canals could be a consequence of the attrition. However, pulpal obliteration can also occur before tooth eruption which indicates that the phenomenon is a rather a manifestation of the disease (Finn, 1962). Although the teeth seemed completely obliterated radiographically, a network of interconnected vessels may remain, providing a pathway for bacteria from the oral cavity (Davis et al., 2015). This may account for the presence of pathologic periapical conditions despite obliterated pulps in both dentitions.
In general, early diagnosis is of considerable importance for the treatment of DI–II in order to obtain a favorable prognosis. Although the treatment of the investigated patients varied, all treatments aimed to ensure removal of infectious sites and/or prevention of pain, establishing a good occlusion by preventing loss of vertical facial dimension, and restoring aesthetics (Barron et al., 2008; Leal et al., 2010; Sapir & Shapira, 2001).
5. CONCLUSION
In summary, the enamel and dentin were examined, and the described DI–II families were assessed clinically, radiographically, and histologically. Three distinct mutations were identified in the DSP region of the DSPP gene, one of which was novel. The molecular pathogenesis by which the DSPP mutations have caused DI–II was characterized in order to elucidate the genotype‐phenotype correlation. The present data indicate that DI–II might affect both dentin and enamel. However, further studies are needed to elucidate the underlying mechanisms of enamel defects in DI–II patients.
CONFLICT OF INTEREST
None declared.
Supporting information
ACKNOWLEDGMENTS
We thank all the family members for their cooperation in this research. The authors are grateful to Professor Jesper Reibel, Department of Odontology, Oral Pathology and Medicine, Faculty of Health and Medical Sciences, University of Copenhagen, Denmark, for his work on the histological analysis. We also express our gratitude to Hiba Al‐Imam DDS., Department of Odontology, Oral Rehabilitation, Dental Materials and Oral Diagnostics, Faculty of Health and Medical Sciences, University of Copenhagen, for her assistance on the tomographic imaging investigation.
Taleb K, Lauridsen E, Daugaard‐Jensen J, Nieminen P, Kreiborg S. Dentinogenesis imperfecta type II‐ genotype and phenotype analyses in three Danish families. Mol Genet Genomic Med. 2018;6:339–349. https://doi.org/10.1002/mgg3.375
REFERENCES
- Bai, H. , Agula, H. , Wu, Q. , Zhou, W. , Sun, Y. , Qi, Y. , … Qiu, C. (2010). A novel DSPP mutation causes dentinogenesis imperfecta type II in a large Mongolian family. BMC Medical Genetics, 11, 23 https://doi.org/10.1186/1471-2350-11-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardow, A. , Pallesen, U. , & Bakke, M. (2013). Erosion og andet tandslid – ajourført viden og forebyggelse. Aktuel Nordisk Odontologi, 3, 11–32. [Google Scholar]
- Barron, M. J. , McDonnell, S. T. , MacKie, I. , & Dixon, M. J. (2008). Hereditary dentine disorders: Dentinogenesis imperfecta and dentine dysplasia. Orphanet Journal of Rare Diseases, 3, 31 https://doi.org/10.1186/1750-1172-3-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bègue‐Kirn, C. K. P. , Bartlett, J. D. , & Butler, W. T. (1998). Dentin sialoprotein, dentin phosphoprotein, enamelysin and ameloblastin, tooth‐ specific molecules that are distinctively expressed during murine dental differentiation. European Journal of Oral Sciences, 5, 963–970. https://doi.org/10.1046/j.0909-8836.1998.eos106510.x [DOI] [PubMed] [Google Scholar]
- Bixler, D. , Conneally, P. M. , & Christen, A. G. (1969). Dentinogenesis imperfecta: Genetic variations in a six‐generation family. Journal of Dental Research, 48, 1196–1199. https://doi.org/10.1177/00220345690480061701 [DOI] [PubMed] [Google Scholar]
- Bloch‐Zupan, A. , Huckert, M. , Stoetzel, C. , Meyer, J. , Geoffroy, V. , Razafindrakoto, R. W. , … Ramanampamaharana, R. H. (2016). Detection of a Novel DSPP Mutation by NGS in a Population Isolate in Madagascar. Frontiers in Physiology, 7, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis, G. R. , Fearne, J. M. , Sabel, N. , & Norén, J. G. (2015). Microscopic study of dental hard tissues in primary teeth with dentinogenesis imperfecta type ii: Correlation of 3D imaging using X‐ray microtomography and polarising microscopy. Archives of Oral Biology, 60, 1013–1020. https://doi.org/10.1016/j.archoralbio.2015.03.010 [DOI] [PubMed] [Google Scholar]
- Finn, S. D. (1962). Dentin and enamel anomalies (pp. 219–246). New York, NY: McGraw‐Hill Book Company. [Google Scholar]
- Haavikko, K. (1970). The formation and the alveolar and clinical eruption of the permanent teeth. An orthopantomographic study. Suomen Hammaslaakariseuran Toimituksia, 66, 103–170. [PubMed] [Google Scholar]
- Hart, P. S. , & Hart, T. C. (2007). Disorders of human dentin. Cells Tissues Organs, 186, 70–77. https://doi.org/10.1159/000102682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart, T. C. , & Hart, P. S. (2009). Genetic studies of craniofacial anomalies: Clinical implications and applications. Orthodontics and Craniofacial Research, 12, 212–220. https://doi.org/10.1111/j.1601-6343.2009.01455.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh, Y. S. , Ho, Y. C. , Lee, S. Y. , Chuang, C. C. , Tsai, J. C. , Lin, K. F. , & Sun, C. W. (2013). Dental optical coherence tomography. Sensors (Basel), 13, 8928–8949. https://doi.org/10.3390/s130708928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal, C. T. , Martins, L. D. , Verli, F. D. , de Souza, M. A. L. , & Ramos‐Jorge, M. L. (2010). Case report: Clinical, histological and ultrastructural characterization of type II dentinogenesis imperfecta. European Archives Of Paediatric Dentistry, 11, 306–309. https://doi.org/10.1007/BF03262769 [DOI] [PubMed] [Google Scholar]
- Lee, S. K. , Lee, K. E. , Hwang, Y. H. , Kida, M. , Tsutsumi, T. , Ariga, T. , … Kim, J. W. (2011). Identification of the DSPP mutation in a new kindred and phenotype‐genotype correlation. Oral Diseases, 17, 314–319. https://doi.org/10.1111/j.1601-0825.2010.01760.x [DOI] [PubMed] [Google Scholar]
- Lee, S. K. , Lee, K. E. , Jeon, D. , Lee, G. , Lee, H. , Shin, C. U. , … Kim, J. W. (2009). A novel mutation in the DSPP gene associated with dentinogenesis imperfecta type II. Journal of Dental Research, 88, 51–55. https://doi.org/10.1177/0022034508328168 [DOI] [PubMed] [Google Scholar]
- Lee, K. E. , Lee, S. K. , Jung, S. E. , Lee, Z. H. , & Kim, J. W. (2011). Functional splicing assay of DSPP mutations in hereditary dentin defects. Oral Diseases, 17, 690–695. https://doi.org/10.1111/j.1601-0825.2011.01825.x [DOI] [PubMed] [Google Scholar]
- Lee, S. K. , Lee, K. E. , Song, S. J. , Hyun, H. K. , Lee, S. H. , & Kim, J. W. (2013). A DSPP mutation causing dentinogenesis imperfecta and characterization of the mutational effect. BioMed Research International, 2013, 948181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Du, X. , Zhang, R. , Shen, B. , Huang, Y. , Valenzuela, R. K. , … Xu, Z. (2012). Mutation identification of the DSPP in a Chinese family with DGI‐II and an up‐to‐date bioinformatic analysis. Genomics, 99, 220–226. https://doi.org/10.1016/j.ygeno.2012.01.006 [DOI] [PubMed] [Google Scholar]
- Li, F. , Liu, Y. , Liu, H. , Yang, J. , Zhang, F. , & Feng, H. (2017). Phenotype and genotype analyses in seven families with dentinogenesis imperfecta or dentin dysplasia. Oral Diseases, 23, 360–366. https://doi.org/10.1111/odi.12621 [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Huang, Y. , Gao, J. , Li, S. , Zhao, X. , & Zhang, X. (2016). [Identification of a novel mutation of DSPP gene in a Chinese family affected with dentinogenesis imperfecta shields type II]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi, 33, 34–37. [DOI] [PubMed] [Google Scholar]
- Malmgren, B. , Lindskog, S. , Elgadi, A. , & Norgren, S. (2004). Clinical, histopathologic, and genetic investigation in two large families with dentinogenesis imperfecta type II. Human Genetics, 114, 491–498. https://doi.org/10.1007/s00439-004-1084-z [DOI] [PubMed] [Google Scholar]
- Malmgren, B. , Lundberg, M. , & Lindskog, S. (1998). Dentinogenesis imperfecta in a six‐generation family. Swedish Dental Journal, 12, 73–84. [PubMed] [Google Scholar]
- McKnight, D. A. , Simmer, J. P. , Hart, P. S. , Hart, T. C. , & Fisher, L. W. (2008). Overlapping DSPP mutations cause dentin dysplasia and dentinogenesis imperfecta. Journal of Dental Research, 87, 1108–1111. https://doi.org/10.1177/154405910808701217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKnight, D. A. , Suzanne Hart, P. , Hart, T. C. , Hartsfield, J. K. , Wilson, A. , Wright, J. T. , & Fisher, L. W. (2008). A comprehensive analysis of normal variation and disease‐causing mutations in the human DSPP gene. Human Mutation, 29, 1392–1404. https://doi.org/10.1002/humu.20783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminen, P. , Papagiannoulis‐Lascarides, L. , Waltimo‐Siren, J. , Ollila, P. , Karjalainen, S. , Arte, S. , … Holappa, H. (2011). Frameshift mutations in dentin phosphoprotein and dependence of dentin disease phenotype on mutation location. Journal of Bone and Mineral Research, 26, 873–880. https://doi.org/10.1002/jbmr.276 [DOI] [PubMed] [Google Scholar]
- Rajpar, M. H. , Koch, M. J. , Davies, R. M. , Mellody, K. T. , Kielty, C. M. , & Dixon, M. J. (2002). Mutation of the signal peptide region of the bicistronic gene DSPP affects translocation to the endoplasmic reticulum and results in defective dentine biomineralization. Human Molecular Genetics, 11, 2559–2565. https://doi.org/10.1093/hmg/11.21.2559 [DOI] [PubMed] [Google Scholar]
- Sapir, S. , & Shapira, J. (2001). Dentinogenesis imperfecta: An early treatment strategy. Pediatric Dentistry, 23, 232–237. [PubMed] [Google Scholar]
- Shields, E. D. , Bixler, D. , & el‐Kafrawy, A. M. (1973). A proposed classification for heritable human dentine defects with a description of a new entity. Archives of Oral Biology, 18, 543–553. https://doi.org/10.1016/0003-9969(73)90075-7 [DOI] [PubMed] [Google Scholar]
- Sreenath, T. , Thyagarajan, T. , Hall, B. , Longenecker, G. , D'Souza, R. , Hong, S. , … Kulkarni, A. B. (2003). Dentin sialophosphoprotein knockout mouse teeth display widened predentin zone and develop defective dentin mineralization similar to human dentinogenesis imperfecta type III. Journal of Biological Chemistry, 278, 24874–24880. https://doi.org/10.1074/jbc.M303908200 [DOI] [PubMed] [Google Scholar]
- Suzuki, S. , Sreenath, T. , Haruyama, N. , Honeycutt, C. , Terse, A. , Cho, A. , … Kulkarni, A. B. (2009). Dentin sialoprotein and dentin phosphoprotein have distinct roles in dentin mineralization. Matrix Biology, 28, 221–229. https://doi.org/10.1016/j.matbio.2009.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thesleff, I. (2003). Epithelial‐mesenchymal signalling regulating tooth morphogenesis. Journal of Cell Science, 116(9), 1647–1648. https://doi.org/10.1242/jcs.00410 [DOI] [PubMed] [Google Scholar]
- Wang, S. K. , Chan, H. C. , Rajderkar, S. , Milkovich, R. N. , Uston, K. A. , Kim, J. W. , … Hu, J. C. C. (2011). Enamel malformations associated with a defined dentin sialophosphoprotein mutation in two families. European Journal of Oral Sciences, 119(Suppl 1), 158–167. https://doi.org/10.1111/j.1600-0722.2011.00874.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkop, C. J. Jr (1975). Hereditary defects of dentin. Dental Clinics of North America, 19, 25–45. [PubMed] [Google Scholar]
- Xiao, S. , Yu, C. , Chou, X. , Yuan, W. , Wang, Y. , Bu, L. , … Hu, L. (2001). Dentinogenesis imperfecta 1 with or without progressive hearing loss is associated with distinct mutations in DSPP. Nature Genetics, 27, 201–204. https://doi.org/10.1038/84848 [DOI] [PubMed] [Google Scholar]
- Zhang, X. , Chen, L. , Liu, J. , Zhao, Z. , Qu, E. , Wang, X. , … Liu, M. (2007). A novel DSPP mutation is associated with type II dentinogenesis imperfecta in a Chinese family. BMC Medical Genetics, 8, 52 https://doi.org/10.1186/1471-2350-8-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Zhao, J. , Li, C. , Gao, S. , Qiu, C. , Liu, P. , … Shen, Y. (2001). DSPP mutation in dentinogenesis imperfecta Shields type II. Nature Genetics, 27, 151–152. https://doi.org/10.1038/84765 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials