

Abstract
The discovery of the CRISPR-Cas9 endonuclease has enabled facile genome editing in living cells and organisms. Catalytically inactive Cas9 (dCas9) retains the ability to bind DNA in an RNA-guided fashion, and has additionally been explored as a tool for transcriptional modulation, epigenetic editing, and genomic imaging. This review highlights recent progress and challenges in the development of dCas9 for imaging genomic loci. The emergence and maturation of this technology offers the potential to answer new mechanistic questions about chromosome dynamics and three-dimensional genome organization in vivo.
Graphical abstract
Bringing Genomes to Light
The expression of genes that govern an organism’s response to environmental stimuli must be carefully regulated[1]. Of particular importance is the spatiotemporal organization of the genome, which plays a direct role in controlling transcription, cellular differentiation, and development[2,3]. Examples include long-range (50+ kilobases) enhancer-promoter interactions that activate gene expression, compartmentalization of transcriptionally silent DNA into heterochromatin, and interchromosomal tethering of distant genes mediated by proteins and non-coding RNAs[4–8].
Given the profound effect that genome organization has on phenotype, there has been great interest in developing tools for probing the dynamic spatial organization of chromatin in vivo (Figure 1). Early efforts towards this end focused on fluorescence in situ hybridization (FISH, Figure 1a), whereby a fluorescent oligonucleotide probe is selectively hybridized to a locus of interest and visualized under a microscope[9]. By combining multiple probes with spectrally resolved emission profiles, DNA FISH could be used to visualize multiple genomic loci within cells[10]. As such, it provided some of the earliest insights into genomic structure[11,12].
Figure 1. Methods for probing and visualizing genome architecture.
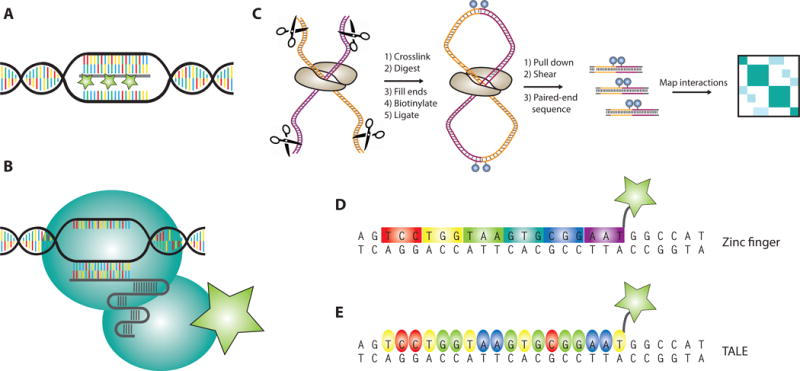
a) Schematic of single-locus imaging in fixed cells using DNA fluorescence in situ hybridization (FISH)
b) General pipeline for a Hi-C genomics experiment
c-e) Live-cell technologies for visualizing single genomic loci including zinc fingers (ZFs, Panel C), transcription activator-like effectors (TALEs, Panel D), and CRISPR-Cas9 (Panel E)
Despite its utility, FISH suffers from a number of drawbacks[12,13]. First, it requires fixation and cannot be used to visualize dynamic processes in living cells. Second, FISH typically requires several fluorescent probes per genomic locus to achieve sufficient signal, rendering it prohibitively expensive for certain applications. Most importantly, the harsh conditions required for hybridization– namely heat and formamide–pose a risk of disrupting the structural integrity of the genome, calling into question some of the conclusions about 3D organization drawn using this technique[13].
The disadvantages of FISH led several researchers to explore alternative, live-cell techniques to image genomic loci (Figure 1). Some of the earliest successes came from imaging localized, highly repetitive sequences such as telomeres, LacO repeats, and H-NS motifs with fluorescently-tagged proteins that natively bind to these sequences[14–22]. Collectively, these experiments offered the first insights into chromosome dynamics and provided critical, time-resolved information about cell division and translocation events associated with cancer[17].
A major breakthrough in our understanding of genome organization came from the maturation of chromatin conformation capture technologies (especially Hi-C) (Figure 1b) [23]. Hi-C allowed for the first time a relatively unbiased view of the entire 3D genome based on ligation and sequencing of proximal interchromosomal fragments. Over the last decade, conformation capture techniques have provided rich information about conserved structural features within the genome (e.g. loops and topologically associating domains) as well as the core elements that regulate maintenance of these structures.
Complementary to Hi-C, which provided genome-wide structural snapshots, the near-simultaneous emergence of zinc fingers (ZFs) and transcription activator-like effectors (TALEs) stimulated interest in dynamically imaging non-repetitive single loci[24–27] (Figures 1c,d). Early crystal structures and biochemical studies of ZFs and TALEs revealed a modular, programmable mode of DNA recognition by individual subunits within these proteins[26–28]. In the context of genome editing, nuclease fusions of TALEs and ZFs could be engineered to cleave arbitrary genomic sequences[24,29–32]. Later experiments showed that GFP-fusions of ZFs and TALEs could be programmed to image repetitive DNA elements such as telomeres and centromeres[18,22,33–35]. Unfortunately, imaging non-repetitive genomic regions with ZFs and TALEs proved less tractable. Specificity issues and cloning challenges associated with tiling a non-repetitive sequence rendered these technologies largely impractical for single-locus imaging despite their initial promise[36–38].
A CRISPR View of Chromatin
An unlikely game changer for genomic imaging emerged from biochemical investigations of Cas9 proteins derived from the bacterial CRISPR immune system[39–42]. Early experiments identified Cas9 as a site-specific endonuclease that cleaves DNA based on complementarity to a single guide RNA (sgRNA)[43,44]. The protein additionally requires a 3-nt PAM (NGG) on the non-complementary DNA strand proximal to the cleavage site, which allows it to discriminate between self versus non-self DNA. Rather than cloning a new protein for every new target, Cas9 achieves site-specific DNA cleavage by adjusting the sgRNA sequence. Because of the ease of short RNA synthesis, CRISPR-Cas9 has been successfully adapted for high-throughput knockout experiments across multiple eukaryotic cell lines and living organisms[40–42,45]. To date, it is unrivaled by any other genome editing technology.
Cas9 maintains its specific DNA binding properties in the absence of its nuclease activity[44,46]. As such, the catalytically dead version of the protein has been extensively explored for non-editing applications[47–55]. In a seminal paper, Chen et al. reported the first use of dCas9 for genomic imaging in 2013[49]. Fusion of dCas9 to a nuclear localization sequence (NLS) and eGFP tag allowed for dynamic visualization of telomeres in living cells upon expression of the appropriate sgRNA (Figure 1e). This system was further extended to visualize a non-repetitive genomic region within the MUC4 gene by tiling 36-73 sgRNAs across the locus. Labeling of this locus was intense and robust enough such that individual MUC4 alleles could be tracked during cell cycling to highlight the dynamic behavior of chromosomes during mitosis.
The early success of CRISPR-based imaging inspired a number of follow-up studies to investigate and improve upon this technology[56–63]. Important considerations include: (1) the relative kinetics of on- versus off-target binding; (2) chromatin effects on Cas9 targeting; (3) the generalizability of dCas9 imaging to other cell lines; (4) the optimal number of sgRNAs; (5) the optimal identity and placement of the fluorescent tag; and (6) two-color controls to ensure correct locus labeling.
A Bacterial Dance with Eukaryotic DNA
Several studies have attempted to address the kinetics and thermodynamics of Cas9 binding at target and off-target sequences[44,59,62,64–80]. Early biochemical and chromatin immunoprecipitation (ChIP) experiments established a tight (0.25 nM) target affinity and provided evidence for off-target binding being driven by the PAM and the 8-12 nucleotide “seed” region immediately upstream of it[44,46,64,65,74,81–83]. Measurements of in vitro dissociation rates have been reported using single-molecule imaging, bio-layer interferometry, and competitive ChIP[46,73,78,79,84]. Estimates vary widely depending on the experimental method, but it is generally accepted that dCas9 dissociates extremely slowly (perhaps on the order of hours) from target and near-target sequences bearing >13 nucelotides of complementarity. In contrast, single mutations within the seed region correlate with dramatic increases in koff. Cleavage at stably bound near-target sequences is further mitigated by a second kinetic proofreading step that involves complementarity-gated conformational activation of the HNH catalytic domain[77,85].
How in vitro kinetic measurements translate to in vivo binding times remains somewhat unclear. Within a living eukaryotic cell, the bacterial Cas9 protein faces unique challenges outside of its native evolutionary context, including nucleosomes, epigenetic modifications, compartmentalization of DNA, vastly larger genomes, and numerous competing DNA-binding proteins[5,86,87]. Several groups have measured residence times of dCas9 in eukaryotes using single-molecule imaging and fluorescence recovery after photobleaching (FRAP)[59,62,72,80]. Single-molecule data provide evidence for a diffusion-dominated search mechanism with generally short-lived residence times at PAMs and short seed sequences[72]. With respect to target loci, FRAP experiments performed at both tandem (e.g. centromeres and telomeres) and interspersed (e.g. SINEs) repeats have consistently resulted in extremely slow recovery rates, on the order of tens of minutes to several hours[59,62,72,80]. Conversely, FRAP experiments performed at random genomic sequences, off-target puncta, or with mismatched guides have resulted in recovery rates that are 10-1000-fold faster (seconds to a few minutes).
Does Cas9 target DNA in a chromatin-blind fashion, or is its activity dependent on the epigenetic and nucleosomal signature of the target locus? Genome occupancy and single-molecule data suggest that Cas9 undersamples heterochromatin, and in vitro binding assays demonstrated a strong binding bias against nucleosomal DNA[74,81–83,88,89]. Within a living cell, this effect appears more nuanced, as robust cleavage activity of Cas9 has been observed at heterochromatic loci[90]. Moreover, dCas9 occupancy at transcriptionally silenced loci has been shown to locally increase chromatin accessibility[91,92].
What do these data mean in the context of genomic imaging? Collectively, they suggest that careful consideration should be given to off-target and chromatin effects when designing sgRNAs for imaging a target locus. If binding at near-cognate sequences is indeed long-lived, guides should be algorithmically chosen to minimize off-target homology[93–99]. With respect to chromatin, the epigenetic state of the target locus may dictate the minimum number of guides that is required to achieve sufficient signal. Whether Cas9 can be rationally engineered to circumvent some of these challenges is the topic of ongoing research[100–102].
Focusing the Tools
Single-locus visualization requires a delicate balance of maximizing target signal while minimizing noise, and a number of studies have explored different tagging strategies and expression systems to optimize this ratio[49,56–62] (Figure 2). Regardless of labeling method, low expression of Cas9 and high expression of sgRNA appear critical to visualize single loci above background levels. This is typically achieved via gating dCas9 mRNA expression through a doxycycline-inducible promoter. At the sgRNA level, expression can be enhanced via stabilizing stem-loop motifs as well as multiple-guide expression constructs[49,103,104].
Figure 2. CRISPR-Cas9 imaging platforms.
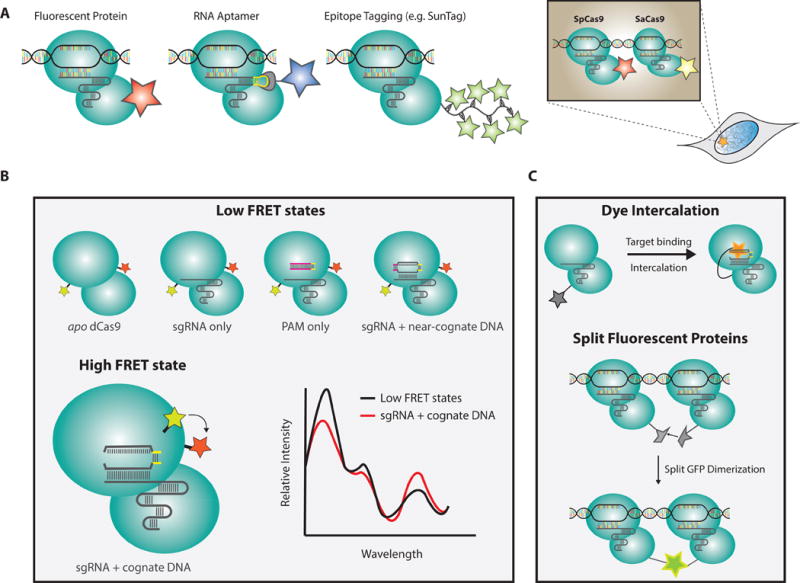
a) Schematic depicting existing methods for fluorescently labeling dCas9-sgRNA ribonucleoprotein complexes (RNP) including fluorescent fusion proteins, SunTag, aptamer-conjugated sgRNA scaffolds, and multicolored orthogonal Cas9 proteins
b) FRET diagram illustrating different conformational states of Cas9 and how they might be exploited for imaging
c) Proposed labeling strategies to improve imaging signal-to-noise ratios
Nucleofection/electroporation has been studied as a means of delivering Cas9 to cells[84,105]. Using this method, Cas9 can be pre-loaded with sgRNA in an in vitro environment to prevent apo protein aggregation/off-target labeling in vivo. While remarkably effective for genome editing, the utility of nucleofection for cellular imaging is likely much more limited for the following reasons: (1) the initial shock to the cells is harsh and may considerably alter morphology in the short-term; (2) RNP levels will likely vary wildly at the single-cell level. A more reliable strategy to achieve uniformly low expression is to generate a stably integrated, inducible dCas9 from a clonal line[49,63].
Single-locus visualization requires tiling several Cas9 particles in tandem at the target site, and the minimal number of sgRNAs required to achieve sufficient signal has been the subject of ongoing research[49,57,59,62]. Imaging the non-repetitive region of MUC4 with dCas9-eGFP requires minimally 26-36 sgRNAs to achieve sufficient signal[49]. Importantly, signal strength does not necessarily scale with the number of single guide RNAs, as recently illustrated by imaging experiments that achieved higher signal at a ribosomal locus with 17 repeats than at another locus with 70 repeats[57]. Collectively, these data hint that other factors– including nucleosomes, epigenetic landscape, sequence preferences within the guide, delivery/expression efficiency, and cell cycling– may strongly influence efficacy of targeting.
An important consideration for genomic imaging is how to ensure that a fluorescent punctum represents a desired locus and not an off-target sequence. One of the most effective approaches to control for this is to label multiple imaging components and then check for multicolor co-localization[56,58–60,62,106,107] (Figure 2a). The tetraloop and 3′-terminal regions of the Cas9 sgRNA scaffold have proven remarkably amenable to modification, and they have been extensively explored as handles to append fluorescent proteins for dual-color labeling[59,60,62,108]. In a recent study, up to 14 MS2-aptamers were fused to the sgRNA allowing for ultrabright labeling of Cas9 RNPs in two colors using fluorescently tagged, MS2-coat proteins[62]. An alternative approach has been the use of Cas9 species variants with orthogonal PAM specificities to label the same locus with multiple colors[56,58,109,110]. Whether other RNA-guided single-subunit effectors from other CRISPR systems (e.g. Cpf1) can also be utilized for this same purpose remains to be determined[111].
Beyond two-color controls, can we engineer a CRISPR imaging system that is selectively fluorescent at a target locus and otherwise quenched (Figures 3b)? Such a tool would considerably lower background fluorescence and increase labeling confidence. Förster resonance energy transfer (FRET) experiments suggest a distinct conformational state of target-bound dCas9 that can be optically resolved from near-cognate binding events in vitro[77,85]. An in vivo extension of this assay could prove promising for imaging single loci. Other possibilities include constitutively quenched HaloTag dyes that fluoresce upon DNA intercalation or split-fluorescent protein systems that become especially bright when bound in high concentration at a target locus[112,113]. Concomitant with improvements to microscopy, exploration of these approaches may greatly improve the robustness of this technology.
Figure 3. Future applications of single-locus imaging.
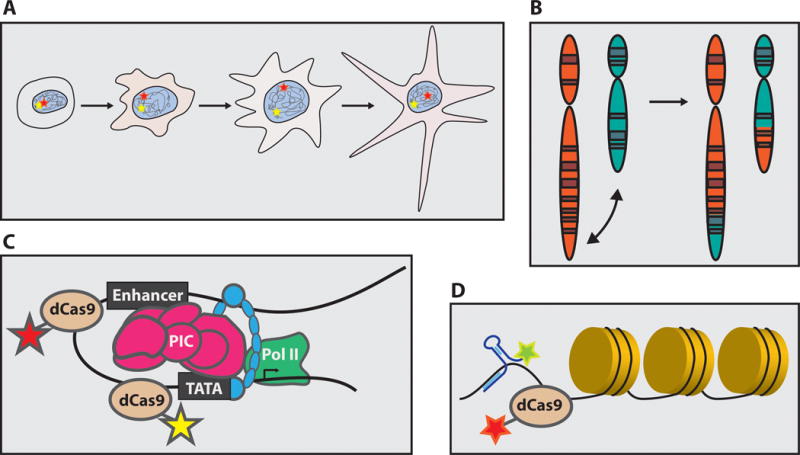
A mature CRISPR-based imaging system could be used to visualize allelic segregation (Panel A), chromosomal translocations (Panel B), enhancer-promoter looping (Panel C), and lncRNA-chromatin interactions (Panel D) in vivo.
A Bright Future
CRISPR-based imaging is still in its early days, and there are a number of hurdles that must be overcome in order for this technology to become broadly accessible and applicable to the larger biophysical community. Sequence- and chromatin-based constraints that govern the efficacy of dCas9 targeting must be more quantitatively resolved; background fluorescence must be reduced via continued technological developments; and sgRNA delivery/expression systems must be optimized to maximize targeting efficiency[41,102].
Once refined, CRISPR-based imaging systems offer extraordinary potential to visualize genome architecture within living cells. Examples include: (1) monitoring interchromosomal contact shuffling during cellular differentiation[114]; (2) visualizing aberrant enhancer-promoter interactions and chromatin loops associated with a particular disease state[5,87,115]; (3) tracking allelic segregation during cell cycling and development; (4) and real-time kinetic measurements of mis-segregation, recombination, and DNA repair events in vivo[17,116] (Figure 3). Forging beyond the test tube, refinement of single-locus imaging technologies will allow us to quantitatively probe dynamic biochemical processes at the single-cell, single-molecule level.
Yet another exciting avenue of research is the use of CRISPR-based imaging to explore the interface between long, non-coding RNAs (lncRNA) and chromatin during disease and development (Figure 3). Explosive growth in lncRNA research has shed light on their importance in mediating enhancer-promoter interactions, interchromosomal contacts, and chromosome condensation[117]. The emergence of mature genomic and RNA imaging tools offers the potential to provide a comprehensive, time-resolved picture of these processes[6]. Cas9 can be repurposed to target RNA and has already been adapted for live-cell RNA imaging[118,119]. Alternatively, CRISPR-based genomic imaging might be combined with other RNA-imaging platforms to visualize the dynamic interplay between genomes and lncRNAs[120].
The future of genomic imaging is bright. While there are still technical hurdles to be overcome, the discovery of RNA-programmable CRISPR proteins has expanded the toolkit for imaging non-repetitive loci in vivo. In combination with high-throughput genomics, live-cell imaging will quantitatively enhance our spatiotemporal understanding of macromolecular processes associated with disease and development. Insights gained from these experiments will be profoundly consequential to both biomedical and clinical research.
Acknowledgments
We thank members of the Doudna and Tjian labs for helpful discussions and comments on the manuscript. This work was supported in part by the Howard Hughes Medical Institute.
Biographies
Spencer Knight obtained his PhD from UC Berkeley and currently works as a data scientist at Foresite Capital Management, a healthcare growth equity group in San Francisco, CA. As a graduate student, he worked under Robert Tjian and Jennifer Doudna on single-molecule imaging experiments to study the search mechanism of CRISPR-Cas9 proteins within mammalian cells.
Dr. Jennifer Doudna is a professor of molecular and cell biology and of chemistry at the University of California, Berkeley, and is also an HHMI Investigator. Her laboratory aims to discover how RNA molecules and ribonucleoprotein complexes control synthesis and protein expression. Their research on CRISPR biology led to the discovery of the mechanism by which small RNAs direct Cas9 to bind and cleave specific DNA sequences. This work is a major breakthrough in biology, providing a simple and effective means of making targeted changes in the genomes of virtually any cell type. Dr. Doudna has been recognized with many awards for her efforts. She is the recipient of the Breakthrough Prize in the Life Sciences in 2014, listed among the Time 100 in 2015, and winner of the Japan Prize in 2016.
Dr. Robert Tjian, Professor of Molecular and Cell Biology since 1979, recently also served as President of the Howard Hughes Medical Institute from 2009-2016. Trained as a biochemist at Cal and Harvard, he was a pioneer in studying how genetic information in our DNA is decoded to produce mRNA and proteins. During nearly 4 decades on the faculty at Cal, he taught 1000’s of undergrads while doing biomedical discovery research. He has received many scientific awards including election to the National Academy of Sciences, American Philosophical Society, was California Scientist of the Year and he has been an HHMI investigator since 1987. He also founded Tularik, Inc. and launched several biotech companies as part of a venture capital firm called TCG.
References
- 1.Rockman MV, Kruglyak L. Nature Reviews Genetics. 2006;7:862–872. doi: 10.1038/nrg1964. [DOI] [PubMed] [Google Scholar]
- 2.Misteli T. Cell. 2007;128:787–800. doi: 10.1016/j.cell.2007.01.028. [DOI] [PubMed] [Google Scholar]
- 3.Misteli T. Cell. 2013;152:1209–1212. doi: 10.1016/j.cell.2013.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levine M, Tjian R. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- 5.Levine M, Cattoglio C, Tjian R. Cell. 2014;157:13–25. doi: 10.1016/j.cell.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hacisuleyman E, Goff LA, Trapnell C, Williams A, Henao-Mejia J, Sun L, McClanahan P, Hendrickson DG, Sauvageau M, Kelley DR, et al. Nature Structural & Molecular Biology. 2014;21:198–206. doi: 10.1038/nsmb.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hathaway NA, Bell O, Hodges C, Miller EL, Neel DS, Crabtree GR. Cell. 2012;149:1447–1460. doi: 10.1016/j.cell.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eissenberg JC, Elgin SCR. Current Opinion in Genetics Development. 2015;10:204–210. doi: 10.1016/s0959-437x(00)00058-7. [DOI] [PubMed] [Google Scholar]
- 9.Langer-Safer PR, Levine M, Ward DC. Proc Natl Acad Sci USA. 1982;79:4381–4385. doi: 10.1073/pnas.79.14.4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Speicher MR, Ballard SG, Ward DC. Nat Genet. 1996;12:368–375. doi: 10.1038/ng0496-368. [DOI] [PubMed] [Google Scholar]
- 11.Trask BJ. Trends in Genetics. 1991;7:149–154. doi: 10.1016/0168-9525(91)90378-4. [DOI] [PubMed] [Google Scholar]
- 12.Swiger RR, Tucker JD. Environ Mol Mutagen. 1996;27:245–254. doi: 10.1002/(SICI)1098-2280(1996)27:4<245::AID-EM1>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 13.Levsky JM, Singer RH. Journal of Cell Science. 2003;116:2833–2838. doi: 10.1242/jcs.00633. [DOI] [PubMed] [Google Scholar]
- 14.Gasser S. Science. 2002;296:1412–1416. doi: 10.1126/science.1067703. [DOI] [PubMed] [Google Scholar]
- 15.Matzke AJM. PLANT PHYSIOLOGY. 2005;139:1586–1596. doi: 10.1104/pp.105.071068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinett CC, Straight A, Li G, Willhelm C, Sudlow G, Murray A, Belmont AS. The Journal of Cell Biology. 1996;135:1685–1700. doi: 10.1083/jcb.135.6.1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roukos V, Voss TC, Schmidt CK, Lee S, Wangsa D, Misteli T. Science. 2013;341:660–664. doi: 10.1126/science.1237150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindhout BI, Fransz P, Tessadori F, Meckel T, Hooykaas PJJ, van der Zaal BJ. Nucleic Acids Research. 2007;35:e107–e107. doi: 10.1093/nar/gkm618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bandaria JN, Qin P, Berk V, Chu S, Yildiz A. Cell. 2016;164:735–746. doi: 10.1016/j.cell.2016.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt JC, Zaug AJ, Cech TR. Cell. 2016;166:1188–1197.e9. doi: 10.1016/j.cell.2016.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang W, Li GW, Chen C, Xie S, Zhuang X. Science. 2011;333:1445–1449. doi: 10.1126/science.1204697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma H, Reyes-Gutierrez P, Pederson T. Proc Natl Acad Sci USA. 2014;110:21048–21053. doi: 10.1073/pnas.1319097110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dekker J, Marti-Renom MA, Mirny LA. Nat Rev Microbiol. 2013:1–14. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bibikova M, Carroll D, Segal DJ, Trautman JK, Smith J, Kim YG, Chandrasegaran S. Molecular and Cellular Biology. 2001;21:289–297. doi: 10.1128/MCB.21.1.289-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Nat Rev Microbiol. 2010;11:636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 26.Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 27.Moscou MJ, Bogdanove AJ. Science. 2009;326:1501–1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 28.Pavletich NP, Pabo CO. Science. 1991;252:809–817. doi: 10.1126/science.2028256. [DOI] [PubMed] [Google Scholar]
- 29.Bibikova M, Golic M, Golic KG, Carroll D. Genetics. 2002;161:1169–1175. doi: 10.1093/genetics/161.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith J, Bibikova M, Whitby FG, Reddy AR, Chandrasegaran S, Carroll D. Nucleic Acids Research. 2000;28:3361–3369. doi: 10.1093/nar/28.17.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Genetics. 2010;186:757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al. Nature Biotechnology. 2010;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 33.Miyanari Y, Ziegler-Birling C, Torres-Padilla ME. Nature Structural & Molecular Biology. 2013;20:1321–1324. doi: 10.1038/nsmb.2680. [DOI] [PubMed] [Google Scholar]
- 34.Thanisch K, Schneider K, Morbitzer R, Solovei I, Lahaye T, Bultmann S, Leonhardt H. Nucleic Acids Research. 2014;42:e38–e38. doi: 10.1093/nar/gkt1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren R, Deng L, Xue Y, Suzuki K, Zhang W, Yu Y, Wu J, Sun L, Gong X, Luan H, et al. Nat Rev Microbiol. 2017;27:483–504. doi: 10.1038/cr.2017.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, Nowrouzi A, Bartholomae CC, Wang J, Friedman G, et al. Nature Biotechnology. 2011;29:816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- 37.Pattanayak V, Ramirez CL, Joung JK, Liu DR. Nat Meth. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun N, Zhao H. Biotechnol Bioeng. 2013;110:1811–1821. doi: 10.1002/bit.24890. [DOI] [PubMed] [Google Scholar]
- 39.Hsu PD, Lander ES, Zhang F. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Terns RM, Terns MP. Trends in Genetics. 2014;30:111–118. doi: 10.1016/j.tig.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Doudna JA, Charpentier E. Science. 2014;346:1258096–1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 42.Wright AV, Nuñez JK, Doudna JA. Cell. 2016;164:29–44. doi: 10.1016/j.cell.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 43.Gasiunas G, Barrangou R, Horvath P, Siksnys V. Proc Natl Acad Sci USA. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsu PD, Lander ES, Zhang F. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Larson MH, Gilbert LA, Wang X, Lim WA, Weissman JS, Qi LS. Nat Protoc. 2013;8:2180–2196. doi: 10.1038/nprot.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, et al. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zalatan JG, Lee ME, Almeida R, Gilbert LA, Whitehead EH, La Russa M, Tsai JC, Weissman JS, Dueber JE, Qi LS, et al. Cell. 2015;160:339–350. doi: 10.1016/j.cell.2014.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, Gersbach CA. Nature Biotechnology. 2015;33:510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, Jaenisch R. Cell. 2016;167:233–235.e17. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dominguez AA, Lim WA, Qi LS. Nat Rev Microbiol. 2015;17:5–15. doi: 10.1038/nrm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu SJ, Horlbeck MA, Cho SW, Birk HS, Malatesta M, He D, Attenello FJ, Villalta JE, Cho MY, Chen Y, et al. Science. 2017;355:eaah7111–16. doi: 10.1126/science.aah7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ma H, Naseri A, Reyes-Gutierrez P, Wolfe SA, Zhang S, Pederson T. Proc Natl Acad Sci USA. 2015;112:3002–3007. doi: 10.1073/pnas.1420024112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen B, Hu J, Almeida R, Liu H, Balakrishnan S, Covill-Cooke C, Lim WA, Huang B. Nucleic Acids Research. 2016;44:e75–e75. doi: 10.1093/nar/gkv1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D, Pederson T. Nature Biotechnology. 2016;34:528–530. doi: 10.1038/nbt.3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shao S, Zhang W, Hu H, Xue B, Qin J, Sun C, Sun Y, Wei W, Sun Y. Nucleic Acids Research. 2016;44:e86–e86. doi: 10.1093/nar/gkw066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang S, Su JH, Zhang F, Zhuang X. Sci Rep. 2016;6:1–7. doi: 10.1038/srep26857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fu Y, Luo VM, Raviram R, Deng Y, Mazzoni EO, Rocha PP, Skok JA. Nature Communications. 2016;7:1–8. doi: 10.1038/ncomms11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qin P, Parlak M, Kuscu C, Bandaria J, Mir M, Szlachta K, Singh R, Darzacq X, Yildiz A, Adli M. Nature Communications. 2017;8:1–10. doi: 10.1038/ncomms14725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen B, Guan J, Huang B. Annu Rev Biophys. 2016;45:1–23. doi: 10.1146/annurev-biophys-062215-010830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. Nature Biotechnology. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. Nature Biotechnology. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. Genome Research. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, Alt FW. Nature Biotechnology. 2014;33:179–186. doi: 10.1038/nbt.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim D, Bae S, Park J, Kim E, Kim S, Yu HR, Hwang J, Kim JI, Kim JS. Nat Meth. 2015;12:237–243. doi: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
- 69.Singh R, Kuscu C, Quinlan A, Qi Y, Adli M. Nucleic Acids Research. 2015;43:e118–e118. doi: 10.1093/nar/gkv575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, et al. Nature Biotechnology. 2014;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang X, Wang Y, Wu X, Wang J, Wang Y, Qiu Z, Chang T, Huang H, Lin RJ, Yee JK. Nature Biotechnology. 2015;33:175–178. doi: 10.1038/nbt.3127. [DOI] [PubMed] [Google Scholar]
- 72.Knight SC, Xie L, Deng W, Guglielmi B, Witkowsky LB, Bosanac L, Zhang ET, El Beheiry M, Masson J-B, Dahan M, et al. Science. 2015;350:823–827. doi: 10.1126/science.aac6572. [DOI] [PubMed] [Google Scholar]
- 73.Szczelkun MD, Tikhomirova MS, Sinkunas T, Gasiunas G, Karvelis T, Pschera P, Siksnys V, Seidel R. Proc Natl Acad Sci USA. 2014;111:9798–9803. doi: 10.1073/pnas.1402597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Polstein LR, Perez-Pinera P, Kocak DD, Vockley CM, Bledsoe P, Song L, Safi A, Crawford GE, Reddy TE, Gersbach CA. Genome Research. 2015;25:1158–1169. doi: 10.1101/gr.179044.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Doench JG, et al. Nature Biotechnology. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Farasat I, Salis HM. PLoS Comput Biol. 2016;12:e1004724–33. doi: 10.1371/journal.pcbi.1004724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sternberg SH, LaFrance B, Kaplan M, Doudna JA. Nature. 2015;527:110–113. doi: 10.1038/nature15544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boyle EA, Andreasson JOL, Chircus LM, Sternberg SH, Wu MJ, Guegler CK, Doudna JA, Greenleaf WJ. bioRxiv. 2016:1–18. doi: 10.1073/pnas.1700557114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Singh D, Sternberg SH, Fei J, Doudna JA, Ha T. Nature Communications. 2016;7:1–8. doi: 10.1038/ncomms12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ma H, Tu LC, Naseri A, Huisman M, Zhang S, Grunwald D, Pederson T. The Journal of Cell Biology. 2016;214:529–537. doi: 10.1083/jcb.201604115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kuscu C, Arslan S, Singh R, Thorpe J, Adli M. Nature Biotechnology. 2014;32:677–682. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- 82.Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, et al. Nature Biotechnology. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.O’Geen H, Henry IM, Bhakta MS, Meckler JF, Segal DJ. Nucleic Acids Research. 2015;43:3389–3404. doi: 10.1093/nar/gkv137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Nature Biotechnology. 2016;34:339–344. doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- 85.Dagdas YS, Chen JS, Sternberg SH, Doudna JA, Yildiz A. bioRxiv. 2017:1–17. doi: 10.1126/sciadv.aao0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kadonaga JT. Cell. 1998;92:307–313. doi: 10.1016/s0092-8674(00)80924-1. [DOI] [PubMed] [Google Scholar]
- 87.Voss TC, Hager GL. Nat Rev Microbiol. 2013;15:69–81. doi: 10.1038/nrg3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horlbeck MA, Witkowsky LB, Guglielmi B, Replogle JM, Gilbert LA, Villalta JE, Torigoe SE, Tjian R, Weissman JS. eLife. 2016;5:e12677. doi: 10.7554/eLife.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Isaac RS, Jiang F, Doudna JA, Lim WA, Narlikar GJ, Almeida R. eLife. 2016;5:e13450. doi: 10.7554/eLife.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moreno-Mateos MA, Vejnar CE, Beaudoin JD, Fernandez JP, Mis EK, Khokha MK, Giraldez AJ. Nat Meth. 2015;12:982–988. doi: 10.1038/nmeth.3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barkal AA, Srinivasan S, Hashimoto T, Gifford DK, Sherwood RI. PLoS ONE. 2016;11:e0152683–8. doi: 10.1371/journal.pone.0152683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ding X, Feng Y, Seebeck T, Jiang Y, Davis GD, Chen F. Nature Communications. 2017;8:1–12. doi: 10.1038/ncomms14958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Naito Y, Hino K, Bono H, Ui-Tei K. Bioinformatics. 2015;31:1120–1123. doi: 10.1093/bioinformatics/btu743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. Nature Biotechnology. 2014:1–8. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lei Y, Lu L, Liu H-Y, Li Sen, Xing F, Chen L-L. Molecular Plant. 2014;7:1494–1496. doi: 10.1093/mp/ssu044. [DOI] [PubMed] [Google Scholar]
- 96.Zhu LJ. Front Biol. 2015;10:289–296. [Google Scholar]
- 97.Tycko J, Myer VE, Hsu PD. Molecular Cell. 2016;63:355–370. doi: 10.1016/j.molcel.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Perez AR, Pritykin Y, Vidigal JA, Chhangawala S, Zamparo L, Leslie CS, Ventura A. Nat Rev Microbiol. 2017;35:347–349. doi: 10.1038/nbt.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, et al. eLife. 2016;5:e19760. doi: 10.7554/eLife.19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Science. 2015;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, Joung JK. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tycko J, Myer VE, Hsu PD. Molecular Cell. 2016;63:355–370. doi: 10.1016/j.molcel.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nissim L, Perli SD, Fridkin A, Perez-Pinera P, Lu TK. Molecular Cell. 2014;54:698–710. doi: 10.1016/j.molcel.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sakuma T, Nishikawa A, Kume S, Chayama K, Yamamoto T. Sci Rep. 2014;4:2–6. doi: 10.1038/srep05400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lin S, Staahl BT, Alla RK, Doudna JA. eLife. 2014;3:1–13. doi: 10.7554/eLife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guan J, Liu H, Shi X, Feng S, Huang B. Biophys J. 2017;112:1077–1084. doi: 10.1016/j.bpj.2017.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Deng W, Shi X, Tjian R, Lionnet T, Singer RH. Proc Natl Acad Sci USA. 2015;112:11870–11875. doi: 10.1073/pnas.1515692112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen B, Hu J, Almeida R, Liu H, Balakrishnan S, Covill-Cooke C, Lim WA, Huang B. Nucleic Acids Research. 2016;44:e75–e75. doi: 10.1093/nar/gkv1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Nat Meth. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, et al. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Benvin AL, Creeger Y, Fisher GW, Ballou B, Waggoner AS, Armitage BA. J Am Chem Soc. 2007;129:2025–2034. doi: 10.1021/ja066354t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 114.Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. Nature. 2016;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bártová E, Krejčí J, Harničarová A, Galiová G, Kozubek S. Journal of Histochemistry & Cytochemistry. 2008;56:711–721. doi: 10.1369/jhc.2008.951251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Iyama T, Wilson DM., III DNA Repair. 2013;12:620–636. doi: 10.1016/j.dnarep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cech TR, Steitz JA. Cell. 2014;157:77–94. doi: 10.1016/j.cell.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 118.O’Connell MR, Oakes BL, Sternberg SH, East-Seletsky A, Kaplan M, Doudna JA. Nature. 2014;516:263–266. doi: 10.1038/nature13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nelles DA, Fang MY, O’Connell MR, Xu JL, Markmiller SJ, Doudna JA, Yeo GW. Cell. 2016;165:488–496. doi: 10.1016/j.cell.2016.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Buxbaum AR, Haimovich G, Singer RH. Nat Rev Microbiol. 2014:1–15. [Google Scholar]