

Abstract
Objectives
Alpha-sarcoglycan deficiency is a severe form of muscular dystrophy (LGMD2D) without treatment. Gene replacement represents a strategy for correcting the underlying defect. Questions related to this approach were addressed in this clinical trial, particularly the need for immunotherapy, and persistence of gene expression.
Methods
A double-blind, randomized controlled trial using rAAV1.tMCK.hSGCA injected into the extensor digitorum brevis (EDB) muscle was conducted. Control sides received saline. A three-day course of methylprednisolone accompanied gene transfer without further immune suppression.
Results
No adverse events were encountered. SGCA gene expression increased 4-5 fold over control sides when examined at 6 weeks (two subjects) and 3 months (one subject). The full sarcoglycan complex was restored in all subjects and muscle fiber size was increased in the 3-month subject. AAV1 neutralizing antibodies were seen as early as 2 weeks. Neither CD4+ nor CD8+ cells were increased over contralateral sides. Scattered foci of inflammation could be found but showed features of programmed cell death. ELISpot showed no IFN-γ response to α-SG or AAV1 capsid peptide pools with the exception of a minimal capsid response in one subject. Restimulation to detect low frequency capsid specific T cells by ELISpot assays was negative. Results of the first three subjects successfully achieved study aims precluding the need for additional enrollment.
Interpretation
The finding of this gene replacement study in LGMD2D has important implications for muscular dystrophy. Sustained gene expression was seen, but studies over longer time periods without immunotherapy will be required for design of vascular delivery gene therapy trials
Introduction
The sarcoglycans, alpha, beta, gamma, and delta, form a subcomplex of the transmembrane dystrophin-glycoprotein complex (DGC).1,2 Sarcoglycan mutations are inherited as autosomal recessive disorders, responsible for forms of limb-girdle muscular dystrophy (LGMD2C, delta-SG; LGMD2D, alpha-SG; LGMD2E, beta-SG; LGMD2F, gamma-SG).3-5 Overall, the clinical spectrum associated with sarcoglycan gene mutations overlaps the scope of severity associated with dystrophin mutations responsible for Duchenne and Becker muscular dystrophies.6 There is no treatment for sarcoglycan-related LGMDs with the exception of rare individuals reported to benefit from immunomodulatory agents.7,8
In the study reported herein, LGMD2D, alpha-sarcoglycan deficiency, was the target disease based on pre-clinical studies showing promise for gene therapy9,10, although one study raised issues of toxicity related to overexpression.11 It is the most common of the sarcoglycanopathies in North America and in other parts of the world.3,12 Novel findings in this phase I/II clinical gene therapy trial provide promising results for a therapeutic approach using adeno-associated virus serotype 1 (AAV1) to transfer the alpha-sarcoglycan gene (SGCA). The study employed a double-blind randomized controlled design, in order to remove bias in outcomes analyses. Muscle post gene transfer demonstrated sustained alpha-SG (α-SG) gene expression accompanied by full restoration of the sarcoglycan complex…Although, the study plan included low and high dose cohorts (n = 3 each), the findings in the low dose group, precluded the need to expose patients to higher viral doses. The immune response did not impose a barrier to prolonged gene expression in this study. It is also the first clinical report employing the muscle-specific creatine kinase (MCK) promoter13 to improve the safety profile of gene transfer targeting muscle. In addition, the study reinforces pre-clinical predictions that the AAV1 serotype provides relative persistence of genomes in dystrophic muscle.14
Methods
Study Design
Subject eligibility included established SGCA mutations of both alleles, ability to cooperate for testing, willingness to practice contraception during the study (if appropriate), negative pregnancy test (for females), and no evidence of cardiomyopathy, diabetes, or organ system abnormalities of bone marrow, liver, or kidney. HIV infection, hepatitis A, B, or C, or known autoimmune diseases were exclusion criteria. Subjects could not take immunosuppressive drugs or corticosteroids during the trial and were required to be off treatment for 3 months prior to enrollment.
IRB approved consent and assent (ages 9 to 17) forms were obtained by the principal investigator (JRM) and signed by parents and subjects. Pre-gene transfer immune studies included serum neutralizing antibodies to AAV1, interferon-gamma (IFN-γ) enzyme linked immunospot (ELISpot) assay for both AAV capsid proteins and the α-SG protein.
This was a double-blind, randomized controlled trial of rAAV1.tMCK.hSGCA vector injected into the extensor digitorum brevis (EDB) muscle. This is a small muscle on the foot that provides a unique site for gene expression because it is relatively spared from the dystrophic process even in the face of advanced limb muscle degeneration. The study was approved by the Recombinant DNA Advisory Committee (# 0610-815; October 31, 2006) and the FDA (IND # is BB-IND 13434). Approximately 4 hours prior to gene transfer, subjects received intravenous methylprednisolone, 2.0 mg/kg, used for its anti-inflammatory properties to reduce potential inflammation aroused by the needle manipulation at the time of gene transfer. The procedures were carried out in the pediatric intensive care unit at Nationwide Children’s Hospital under conscious sedation. The investigators received labeled syringes (left and right) from the pharmacy, monitored for temperature stability, containing either viral vector or phosphate buffered saline. Injection sides were based on computer-generated random numbers. Unblinding envelopes were held in the pharmacy. The gene delivery was guided by ultrasound and electromyographic recordings (37 mm Teca Myoject ® injection recording needle) to ensure that muscle was the destination of the delivered product. Following injection, the empty syringes were resealed and stored at -20°C. Repeat doses of methylprednisolone were given at 24 and 48 hours post injection. Patients received corticosteroids only during this peri-gene transfer period and received no other immunosuppressive drugs during the remainder of the trial.
EDB muscles were removed bilaterally from patients one and three at post injection days 42 and 51, respectively, and from patient two at 12 weeks (92 days). The muscle was immediately cut into blocks of approximately 1.5 × 1 cm, and frozen in isopentane cooled in liquid nitrogen.
Vector Production
rAAV1.tMCK.haSG was produced at the Harvard Gene Therapy Initiative according to current good manufacturing practices (cGMP). Vector production followed previously published methods using plasmid DNA tri-transfection of HEK293 cells, followed by iodixanol and anion exchange column chromatography purification.14 The vector was formulated in sterile PBS and passed all quality control acceptance criteria established by FDA for strength, identity, and purity.
Safety and Efficacy
Post gene transfer subjects were evaluated on the day of the EDB biopsies and days 1, 2, 7, 14, 30, 60, 120, 180. Photographs of the injection sites were taken immediately and at 8 to 12 hours post gene transfer and at each follow up visit. Efficacy was evaluated by blinded analyses of muscle tissue assessing α-SG gene expression by immune stains of muscle sections and western blot analysis with quantitation assessed by densitometry comparisons between sides. The number of α-SG positive fibers was expressed as the percent of the total number of fibers. “Total fibers” included only those from blocks of muscle showing α-SG gene expression. This included four of six blocks in each case with limitations imposed by spread of vector related to connective tissue barriers in the muscle and by the directional planes of injection illustrated in Figure 1. The results of the gene expression findings were presented to the pharmacy and to our oversight Data Safety Monitoring Board at NIH in a written report before the blind was broken. MHCI and MHCII antigens were assessed on muscle sections. CD4+ and CD8+ mononuclear cells were reported as number /mm2 area. Muscle morphometrics included fiber size histograms.
Figure 1.

A) Extensor digitorum brevis (EDB) muscle shown with dotted lines indicating plane of injection from apex to base (long axis) and across muscle from medial to lateral. B) The ultrasound picture shows the injection needle inserted through the long axis of the muscle (arrow). The arrowheads (two above and two below) define the margins of the muscle.
Statistical analyses were based on differences between the sides in the total number of cells per mm2 area expressing CD4+ and CD8+ mononuclear cells, MHCI and MHCII antigens, and muscle fiber size using a paired t test (p < 0.05).
Results
Patients and Dosing Regimen
Six potential LGMD2D subjects met inclusion criteria. The risks, potential lack of benefits and procedures were explained prior to requesting informed consent. The six subjects were equally divided into low and high dose cohorts. Gene delivery to the EDB with subsequent analysis has been completed in the three low dose subjects with the following characteristics: patient 1, age 13, non-ambulatory, homozygous for SGCA R77C substitution; patient 2, age 12 non-ambulatory, homozygous for SGCA I124T substitution; patient 3, age 14, non-ambulatory, compound heterozygote with substitutions I124T/E137K. These three subjects received 3.25 × 1011 vector genomes delivered in 1.5 ml in a 3 ml syringe. The needle of the syringe was inserted 0.5 cm below the fascia (guided by ultrasound) and pushed in the full length of the muscle (proximal to distal), parallel to the longitudinal orientation of the muscle fibers of the EDB. Additional confirmation of muscle placement was obtained by electromyographic recording in the muscle. The EDB lies in a groove on the proximal outer part of the foot. The lateral head is prominently displayed with the base proximal and apex pointing toward the small toe (Fig 1). The gene was delivered in equal amounts of fluid volume (0.75 ml) as the needle was withdrawn. The needle was then reinserted one-third the distance from base to apex with delivery of the remaining fluid to muscle upon needle withdrawal.
Safety of Vector
Patient monitoring included vital signs hourly for four hours and every four hours for the first 24 hours. The patients were discharged and stayed locally until re-examination at 48 hours after which time they were sent home. They returned for checkups according to the schedule provided (above). There were no adverse events of any kind. The site of injection never appeared swollen or erythematosus and no rash was encountered. There was no difference observed between the side receiving vector versus saline. There was no pain in the post procedure period. No organomegaly (liver, spleen) or lymphadenopathy (groin or axilla) were encountered.
Gene Expression
All analyses were done prior to breaking the blind. Relatively consistent gene expression findings were seen between subjects. Figure 2 shows findings by immune stains (A) and western blots (B). Robust staining was seen on only one side in each case that was easily distinguishable from low level or background staining observed on the contralateral side. Photographs were taken with adjustment of exposure times so that only the brightest fibers could be visualized, and counted. Quantitation revealed concordance between immune stained sections and western blots. On the side of robust staining, 57% of fibers were positive in subject 1, 69% in subject 2, and 62% in subject 3. Sections from these same blocks taken for western blots showed a 4 to 5 fold increase in all three cases. A further sign of muscle repair was illustrated by restoration of the full sarcoglycan complex including β-, γ-, and δ-sarcoglycan on the side of intense staining, easily differentiated from the contralateral side (Fig 2C). Subject 2 demonstrated gene expression for 3 months (biopsy was done 92 days post gene transfer). There was no diminution of staining comparing subject 2 (at 3 months) with subjects 1 and 3 (six-week studies). On the side of increased α-SG expression, quantitative PCR demonstrated viral DNA gene transfer. A vector specific primer probe set amplified a portion of the unique 5’ untranslated leader sequence of the α-SG cassette. SGCA transgene copies increased by an average of 48-fold over baseline (representing 3.6 copies per nucleus) comparing the side of increased gene expression to the contralateral side. The RNase P gene was used as an internal control to normalize for genomic input and confirm the absence of PCR inhibitors in the sample DNA.
Figure 2.

A) Post gene transfer tissue sections from EDB muscles for subjects 1, 2, and 3. The left panel shows α-SG gene expression on the side of vector injection (T) compared to muscle from opposite side (C)(scale bar = 100 μm). B) Western blots from all three cases show increased α-SG gene expression on the side of gene transfer compared to the contralateral side (treated on left, control on right) showing residual gene expression from mutant protein. α-SG is normalized to actin (lower band). C) β-Sarcoglycan staining demonstrates restoration on the side of gene transfer. Other sarcoglycans (γ and δ) were also restored (not shown). Muscle from subject (#2) is shown (scale bar = 100μm). D) Densitometry measurements show α-SG gene expression increased 4 to 5 fold on the side of vector injection.
Muscle fiber size was determined for α-SG positive fibers (n = 300 per side). After persistent gene expression for 3 months, Subject 2 showed an increase in mean fiber diameter from 32.4 ±12.9 μm to 45.4±10.2 μm, p < 0.001, comparing the side from which vector was recovered with the opposite side. The gene transfer experiments studied at six weeks failed to show an increase in fiber size (subject 1, control 57.6 ± 17.1 μm vs treated 54.5 ± 21.1 μm p = 0.18; subject 3, control 41.1 +/- 12.9 μm vs treated 38.2 ± 11.9 μm p = 0.07).
Immunologic Response to Vector mediated Gene Expression
Direct Muscle Assessment
MHC class I molecules were expressed on sarcolemma of most muscle fibers on the side of upregulated gene expression (Fig 3C). In contrast, MHC class II expression was not observed. Mononuclear cell infiltrates were evaluated on both sides in each of the three cases. The total numbers of CD8+ and CD4+ mononuclear cells per mm2 area of muscle was not different between sides in any of the cases, although numbers were slightly higher on the side demonstrating increased gene expression (Fig 3A,B). In subject 2, the biopsy done at 12 weeks showed a few scattered clusters of CD4+ and CD8+ mononuclear cells, in the endomysial connective tissue and perivascular. This raised the possibility of a focal reactive inflammatory infiltrate. Invasion of non-necrotic muscle fibers, a common finding in MHC class I directed T cell attack on muscle fibers in the inflammatory myopathies (inclusion body myositis and polymyositis)15, was not observed. In spite of the focal infiltrate, α-SG gene expression persisted without apparent loss as far out as the three month time point. This prompted us to further characterize the inflammatory cells using a TUNEL stain based on experimental gene transfer studies demonstrating programmed death of the antigen-specific effector cells in AAV-transduced muscle.16 The TUNEL stain showed that the majority of cells associated with these foci of inflammatory cells appeared to be undergoing apoptosis (Fig 3D).
Figure 3.
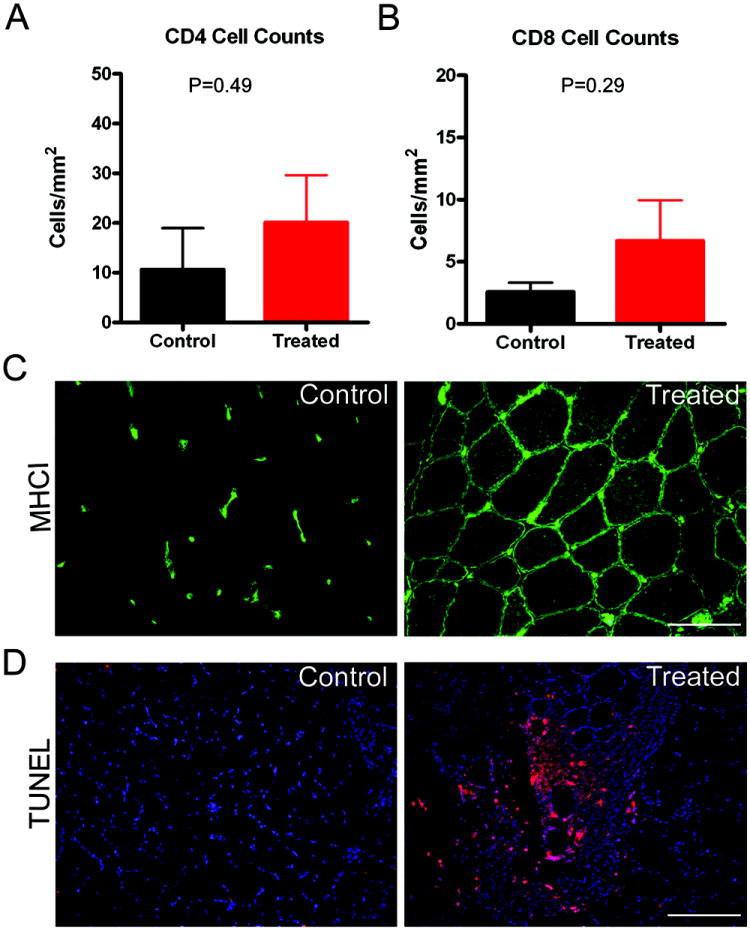
A) Compares the number of CD4+ mononuclear cells/ mm2 area between control and the side of gene transfer; B) Compares the number of CD8+ mononuclear cells/ mm2 area between control and the side of gene transfer; C) MHC I staining of muscle sections shows lack of staining of muscle fibers on control (left) side, while the side of gene transfer shows distinct staining of the sarcolemmal membrane in this subject (#3). Microvascular circulation in MHC I stained on both sides (scale bar = 100 μm). All three cases showed this same staining pattern on control and gene transfer sides; D) TUNEL positive mononuclear cells (red) seen in a perivascular location in a muscle section from subject 2. Other nuclei appear blue in DAPI stain (scale bar = 100 μm).
Neutralizing Antibody to AAV1
In all three subjects serum neutralizing antibody titers to AAV1 were very low (1:100 or below) prior to gene transfer. Follow up studies showed a slow rise in serum AAV1 titers that peaked at 6 to 12 weeks in Subjects 2 and 3. This contrasted with the rapid rise and peak elevation by week 1 in Subject 1, who also reached serum titers 8 fold higher than the others (Table 1). Neutralizing antibody titers did not influence gene expression.
Table 1.
Neutralizing antibody titers against AAV1
| Time Post Injection | Subject 1 | Subject 2 | Subject 3 |
|---|---|---|---|
| Pre -treatment | 1:100 | <1:50 | 1:100 |
| 1 weeks | 1:25600 | 1:400 | 1:200 |
| 2 weeks | 1:25600 | 1:3200 | 1:1600 |
| 6 weeks | 1:12800 | 1:3200 | 1:3200 |
| 12 weeks | ND | 1:3200 | 1:3200 |
| 26 weeks | 1:12800 | 1:3200 | - |
Endpoint titers determined using a cell based reporter vector infection assay as previously described.25
ND = not done because sample not available;
- = to be collected
ELISpot Assay for Detection of Interferon-γ (IFN-γ)
Peripheral blood mononuclear cells (PBMCs) were serially assayed by ELISpot for antigen specific production of IFN-γ secretion beginning pre-gene transfer for each subject (Fig 4). Patients 2 and 3 showed no IFN-γ response to α-SG peptides or AAV1 capsid peptide pools. Subject 1 showed a very minimal IFN-γ response specific to Pool 2 of the AAV1 capsid exceeding our confidence limits for a negative response (>50 spot forming cells /million PBMCs) at days 14 and 43. This suggests a very low, transient T cell mediated immune response to Capsid Pool 2 of AAV1 that was unsustained. PBMCs were further expanded by capsid peptide pool restimulation and no additional AAV capsid responses were detected. At no time was there evidence of a T cell response to the transgene product, α-SG. No anti-α-SG antibodies were detected using western strip blots at baseline or at follow up time points for any subject (data not shown).
Figure 4.
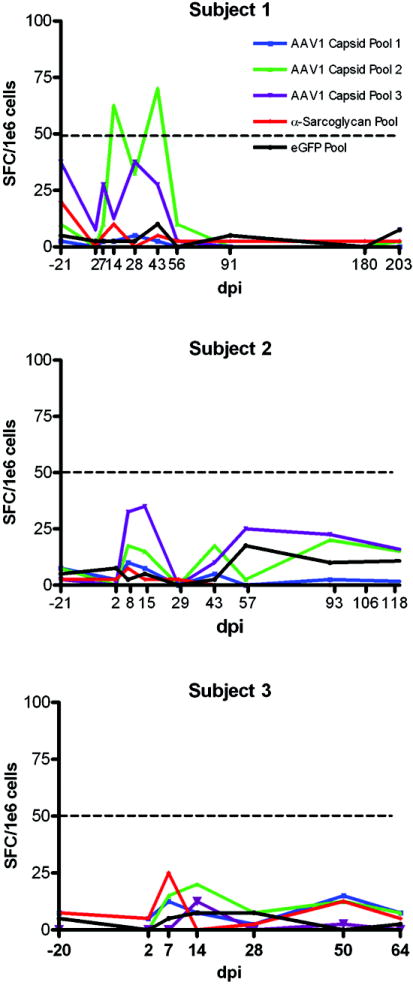
IFN-γ ELISpot assays for all three subjects. α-SG and control enhanced Green fluorescent protein (eGFP) peptide stimulation showed no increase in spot forming colonies (SFC) per million peripheral blood mononuclear cells (PBMCs) in any subject. In Subject 1 there was a very minimal IFN-γ response specific to Pool 2 of the AAV1 capsid exceeding our confidence limits for a negative response (>50 spot forming cells /million PBMCs) at days 14 and 43. ELISpot assays were negative for Subjects 2 and 3. (dpi = days post injection; red = α–SG; black = eGFP; blue = AAV1 capsid pool 1; green = AAV1 capsid pool 2; purple = AAV1 capsid pool 3).
Discussion
The double-blind, randomized controlled trial design employed in this study is uncommonly applied to phase I/II gene therapy trials. This provides added measure of confidence in our findings. Upon demonstration of successful gene expression without adverse events in cohort 1, the blind was broken after discussions with the data safety monitoring committee. By consensus, the findings in the first three subjects made it unnecessary to expose three additional subjects to a higher dose.
In the patients under study, direct muscle injections of AAV1 delivering the SGCA gene under control of the truncated muscle specific MCK promoter resulted in a 4-5-fold increase in α-SG expression for up to 3 months. While safety and gene expression were the primary outcome variables, α-SG replacement resulted in restoration of the full sarcoglycan complex in all subjects and an increase in muscle fiber size exclusive to the subject showing gene expression for three months. The findings must be interpreted cautiously but imply functional improvement based upon pre-clinical studies where increasing muscle fiber size, especially coupled with gene replacement, shows protection against contraction induced injury10,17 and increased grip strength18. Conventional functional measures of improvement cannot be assessed in single muscle human gene transfer trials and await gene delivery through the vasculature allowing gene expression in multiple muscle groups hopefully producing clinically meaningful results.
This is only the second completed trial using AAV119, a serotype with potential advantages for therapeutic strategies employing direct muscle injection20. A further novel aspect of this study is the use of the truncated muscle specific MCK promoter. In preparation for this clinical trial, we systematically compared SGCA gene expression using different promoters. The constitutive CMV promoter showed a trend toward reduced gene expression over time.10 In another study using CMV to express SGCA in the mouse, gene expression was lost over 4 to 6 weeks.11 For muscle gene therapy, the CMV promoter may have disadvantages because potential off target gene expression in antigen presenting cells could result in inadvertent augmentation of an immune response.
In light of the robust α-SG gene expression, the findings observed in the gamut of immune studies are particularly pertinent. Not surprisingly, AAV1 neutralizing antibodies in Subjects 2 and 3 appeared to peak at two weeks post gene transfer followed by a plateau. Subject 1, found to have significant pre-gene transfer neutralizing antibodies to AAV2 (not observed in subjects 2 or 3), generated a more rapid rise in AAV1 titers reaching levels at least 8-fold higher compared to other subjects. This we presume to be an anamnestic response related to cross reactivity with the AAV2 capsid.
The IFN-γ ELISpot assay showed low response levels considering the prediction based on other human gene therapy trials where spot forming units /106. peripheral blood mononuclear cells were far greater than 100, indicative of a cytotoxic T cell response. 21 Direct evidence of a transient AAV1 capsid induced response was seen in subject 1 that just exceeded our threshold for significance. Although, attempts to enhance this by restimulation with AAV1 peptide pools in subject 1, as well as parallel efforts in other subjects, did not show detectable capsid specific T cell immunity. The upregulation of MHCI in the muscle fibers on the side of gene transfer suggests antigen presentation for a recently acquired epitope. Based on previous studies of AAV2.Factor IX delivery through the portal vein21 where a T cell response was directed against the virus, it seems likely that the MHC response was a reflection of the same process. In fact, this seems to correlate with our findings of scattered foci of inflammatory cells seen on the side of gene transfer, especially in subject 2. Of particular interest is that this inflammation in no way precluded gene expression. Second, the infiltrating cells showed findings consistent with programmed cell death, suggesting that T cells were recruited to the scene but failed to invade the antigen presenting target. A similar scenario for silencing antigen specific T cells has been described in vector-transduced mouse muscle with the majority of antigen-specific CD8(+) T cells showing loss of function and programmed cell death resulting in stable gene expression.16 This could explain why T cell infiltration failed to clear transduced muscle fibers in this clinical study. In general, these findings contrast with loss of gene expression following AAV2.Factor IX delivery through the portal vein.21 Further studies will be required to sort out differences between observations, including the target tissue, route of delivery and dose and serotype of AAV. The need for more aggressive immunosuppression for alpha-SG gene transfer can only be assessed with clinical intramuscular or vascular delivery gene therapy trials of longer duration.
In summary, this is the first gene therapy trial in muscular dystrophy demonstrating promising findings, setting the stage for moving forward with treatment of this catastrophic group of diseases. Prior studies support this viewpoint showing both safety and persistence of AAV transcripts in non-dystrophic skeletal muscle.22 Nevertheless, clinically meaningful outcomes will require vascular delivery to multiple muscle groups. Relevance beyond muscular dystrophy is also illustrated because skeletal muscle offers a substrate for vector delivery of transgene products such as myostatin inhibitors that could benefit a variety of neuromuscular disorders.18 The value of the EDB muscle as a safe testing site for initial gene therapy trials and a potential delivery site for secretory gene products was also demonstrated. A detailed description and method of injection have been included in this discussion because the landmarks of the EDB are little known beyond the subspecialty of electromyography and could be of value to gene therapists. This target for gene therapy was previously advocated but never tested24.
Acknowledgments
William M. Fountain IV was responsible for stability assays on the vector; Hugh A. Allen, M.D., provided editorial assistance; Richard C. Mulligan, Ph.D. and Jeng-Shin Lee, M.D., Ph.D. produced the clinical grade AAV vector at the Harvard Gene Therapy Initiative, Department of Genetics, Harvard Medical School; Xiao Xiao, Ph.D., Department of Pharmacology, University of North Carolina kindly provided the truncated MCK promoter; Kevin P. Campbell, Ph.D., University of Iowa, originally cloned the 50 kDa SGCA used for the toxicology study to obtain the IND and used in the gene construct delivered to subjects in the clinical trial.
This work was supported by NIAMS, NIH 1U54NS055958 and the Muscular Dystrophy Association and performed under the FDA IND # is BB-IND 13434. This study has been registered at Clinicaltrials.gov NCT00494195.
Footnotes
Conflicts of Interest
There are no contributors to this manuscript who have any conflict of interest
References
- 1.Anastasi G, Cutroneo G, Rizzo G, Favaloro A. Sarcoglycan subcomplex in normal and pathological human muscle fibers. Eur J Histochem. 2007;51(Suppl 1):29–33. [PubMed] [Google Scholar]
- 2.Ozawa E, Mizuno Y, Hagiwara Y, et al. Molecular and cell biology of the sarcoglycan complex. Muscle Nerve. 2005;32:563–76. doi: 10.1002/mus.20349. [DOI] [PubMed] [Google Scholar]
- 3.Laval SH, Bushby KM. Limb-girdle muscular dystrophies--from genetics to molecular pathology. Neuropathol Appl Neurobiol. 2004;30:91–105. doi: 10.1111/j.1365-2990.2004.00555.x. [DOI] [PubMed] [Google Scholar]
- 4.Klinge L, Dekomien G, Aboumousa A, et al. Sarcoglycanopathies: Can muscle immunoanalysis predict the genotype? Neuromuscul Disord. 2008;18:934–41. doi: 10.1016/j.nmd.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Bueno MR, Moreira ES, Vainzof M, et al. A common missense mutation in the adhalin gene in three unrelated Brazilian families with a relatively mild form of autosomal recessive limb-girdle muscular dystrophy. Hum Mol Genet. 1995;4:1163–7. doi: 10.1093/hmg/4.7.1163. [DOI] [PubMed] [Google Scholar]
- 6.Carrié A, Piccolo F, Leturcq F. Mutational diversity and hot spots in the alpha-sarcoglycan gene in autosomal recessive muscular dystrophy (LGMD2D) J Med Genet. 1997;34:470–75. doi: 10.1136/jmg.34.6.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Angelini C, Fanin M, Menegazzo E, et al. Homozygous alpha-sarcoglycan mutation in two siblings: one asymptomatic and one steroid-responsive mild limb-girdle muscular dystrophy patient. Muscle Nerve. 1998;21:769–75. doi: 10.1002/(sici)1097-4598(199806)21:6<769::aid-mus9>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Connolly AM, Pestronk A, Mehta S, Al-Lozi M. Primary alpha-sarcoglycan deficiency responsive to immunosuppression over three years. Muscle Nerve. 1998;21:1549–53. doi: 10.1002/(sici)1097-4598(199811)21:11<1549::aid-mus30>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 9.Fougerousse F, Bartoli M, Poupiot J, et al. Phenotypic correction of alpha-sarcoglycan deficiency by intra-arterial injection of a muscle-specific serotype 1 rAAV vector. Mol Ther. 2007;15:53–61. doi: 10.1038/sj.mt.6300022. [DOI] [PubMed] [Google Scholar]
- 10.Rodino-Klapac LR, Lee J-S, Mulligan RC, et al. Lack of toxicity of alpha-sarcoglycan overexpression supports clinical gene transfer trial in LGMD2D. Neurology. 2008;71:240–47. doi: 10.1212/01.wnl.0000306309.85301.e2. [DOI] [PubMed] [Google Scholar]
- 11.Dressman D, Araishi K, Imamura M, et al. Delivery of alpha- and beta-sarcoglycan by recombinant adeno-associated virus: efficient rescue of muscle, but differential toxicity. Hum Gene Ther. 2002;13:1631–46. doi: 10.1089/10430340260201725. [DOI] [PubMed] [Google Scholar]
- 12.Moore SA, Shilling CJ, Westra S, et al. Limb-girdle muscular dystrophy in the United States. J Neuropathol Exp Neurol. 2006;65:995–1003. doi: 10.1097/01.jnen.0000235854.77716.6c. [DOI] [PubMed] [Google Scholar]
- 13.Shield MA, Haugen HS, Clegg CH, Hauschka SD. E-box sites and a proximal regulatory region of the muscle creatine kinase gene differentially regulate expression in diverse skeletal muscles and cardiac muscle of transgenic mice. Mol Cell Biol. 1996;16:5058–68. doi: 10.1128/mcb.16.9.5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rabinowitz JE, et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002;76:791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chahin N, Engel AG. Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology. 2008;70:418–24. doi: 10.1212/01.wnl.0000277527.69388.fe. [DOI] [PubMed] [Google Scholar]
- 16.Velazquez VM, Bowen DG, Walker CM. Silencing of T lymphocytes by antigendriven programmed death in recombinant adeno-associated virus vector (rAAV)-mediated gene therapy. Blood. 2008 Jun 19; doi: 10.1182/blood-2008-01-131375. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abmay S, Gregorevic P, Allen JM, et al. Phenotypic improvement of dystrophic muscles by rAAV/microdystrophin vectors Is augmented by Igf1 codelivery. Mol Ther. 2005;12:441–54. doi: 10.1016/j.ymthe.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 18.Haidet AM, Rizo L, Handy C, et al. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc Natl Acad Sci USA. 2008 Mar 18;105:4318–22. doi: 10.1073/pnas.0709144105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stroes Erik S, Nierman Melchior C, Meulenberg Janneke J, et al. Intramuscular administration of AAV1-Lipoprotein LipaseS447X lowers triglycerides in lipoprotein Lipase–Deficient Patients. Arterioscler Thromb Vasc Biol. 2008;28:2303–04. doi: 10.1161/ATVBAHA.108.175620. [DOI] [PubMed] [Google Scholar]
- 20.Pacak CA, Conlon T, Mah CS, Byrne BJ. Relative persistence of AAV serotype 1 vector genomes in dystrophic muscle. Genet Vaccines Ther. 2008;6:14. doi: 10.1186/1479-0556-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manno CS, Pierce GF, Arruda VR, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–47. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 22.Manno CS, Chew AJ, Hutchison S, et al. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101:2963–72. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- 23.Rodino-Klapac L, Haidet AM, Kota J, Handy C, Kaspar BK, Mendell JR. Inhibiton of myostatin with emphasis on follistatin as a therapy for muscle disease. Muscle Nerve. 2008;39:283–96. doi: 10.1002/mus.21244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stedman H, Wilson JM, Finke R, Klenke A-L, Mendell JR. Clinical Protocol. Phase I clinical trial utilizing gene therapy for limb girdle muscular dystrophy: α–, β–,γ–, or δ–sarcoglycan gene delivered with intramuscular instillations of adeno-associated vectors. Human Gene Therapy. 2000;11:777–90. doi: 10.1089/10430340050015671. [DOI] [PubMed] [Google Scholar]
- 25.Johnson PR, Schnepp BC, Connell MJ, et al. Novel Adeno-Associated Virus Vector Vaccine Restricts Replication of Simian Immuno-deficiency Virus in Macaques. J Virol. 2005;79:955–65. doi: 10.1128/JVI.79.2.955-965.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]